

Abstract
Purpose
We determined whether a novel combination of field defect DNA methylation markers could predict the presence of prostate cancer using histologically normal transrectal ultrasound guided biopsy cores.
Materials and Methods
Methylation was assessed using quantitative Pyrosequencing® in a training set consisting of 65 nontumor and tumor associated prostate tissues from University of Wisconsin. A multiplex model was generated using multivariate logistic regression and externally validated in blinded fashion in a set of 47 nontumor and tumor associated biopsy specimens from University of Washington.
Results
We observed robust methylation differences in all genes at all CpGs assayed (p <0.0001). Regression models incorporating individual genes (EVX1, CAV1 and FGF1) and a gene combination (EVX1 and FGF1) discriminated nontumor from tumor associated tissues in the original training set (AUC 0.796-0.898, p <0.001). On external validation uniplex models incorporating EVX1, CAV1 or FGF1 discriminated tumor from nontumor associated biopsy negative specimens (AUC 0.702, 0.696 and 0.658, respectively, p <0.05). A multiplex model (EVX1 and FGF1) identified patients with prostate cancer (AUC 0.774, p = 0.001) and had a negative predictive value of 0.909. Comparison between 2 separate cores in patients in this validation set revealed similar methylation defects, indicating detection of a widespread field defect.
Conclusions
A widespread epigenetic field defect can be used to detect prostate cancer in patients with histologically negative biopsies. To our knowledge this assay is unique, in that it detects alterations in nontumor cells. With further validation this marker combination (EVX1 and FGF1) has the potential to decrease the need for repeat prostate biopsies, a procedure associated with cost and complications.
Keywords: prostate, prostatic neoplasms, epigenomics, biopsy, diagnosis
The concept of the field defect or field cancerization, which has been used to explain the multifocality of some cancers, suggests that preneoplastic molecular alterations may exist even in histologically normal tissues associated with tumor.1 A field defect has been best characterized in head and neck, colorectal, bladder, cervical and other cancers.2 Given the multifocality of PCa and its association with aging, interest has increased in defining a PCa field defect.
The diagnosis of PCa typically uses PSA, a test with high sensitivity but low specificity.3 Patients with persistently increased PSA readings may undergo repeat prostate biopsies, which are associated with increased cost and morbidity.4,5 Efforts have been made to develop other forms of testing that decrease the risk of unrecognized, under sampled cancer in negative biopsy patients, including the free-to-total PSA ratio and the PCA3 urine test.6 We hypothesized negative biopsy tissues from histologically normal areas of a prostate biopsy represent a unique opportunity for improved diagnosis.
Epigenetic changes, particularly DNA methylation, are a superb source of biomarkers that can be detected by polymerase chain reaction based assays, likely occur early and persist throughout tumorigenesis.7 In a recent study we noted a widespread DNA methylation field defect in histologically normal prostate tissues adjacent to and distant from tumor foci.8 Using an unbiased methylation microarray approach, we screened 385,000 loci and validated several candidate loci in additional tissue cohorts. Regions in the EVX1, CAV1 and FGF1 genes developed aberrant methylation in TA tissues but not in prostate tissues from men without cancer. These alterations occurred in widespread fashion and did not depend on distance from the tumor. In contrast, other single gene studies of RASSF1A and other genes suggest that DNA methylation alterations occur only in immediately adjacent benign prostate tissue.1,9-13 Field defect alterations in gene expression14 and telomere length15 were also previously described in PCa.
In this study we developed a multiplex model to predict PCa in histologically normal tissues. We then externally validated these models using biopsy negative tissue cores obtained from prostate biopsies done elsewhere. A marker combination incorporating EVX1 and FGF1 differentiated patients with and without PCa using histologically normal biopsy cores with a predictive AUC of 0.774 on external validation and an NPV of 0.909. This multiplex model identifies patients without cancer in the setting of increased PSA and negative biopsy, and has the potential to decrease the need for repeat biopsies.
MATERIALS AND METHODS
Training Set Tissues
Freshly frozen samples termed NTA from patients with a mean age of 63 years (range 55 to 81) were obtained from organ donation at University of Pittsburgh and University of Wisconsin-Madison. They were extensively evaluated histologically to rule out associated PCa. A total of 24 TA prostate tissues were obtained from patients with a mean age of 61 years (range 57 to 64) who underwent radical prostatectomy for PCa. Microdissection was performed to obtain TA tissue greater than 10 mm from tumor foci. In addition, a set of 15 low volume (less than 5% involvement) and 15 high volume (greater than 25% involvement) samples were obtained from men with a mean age of 59 (range 48 to 67) and 64.1 years (range 48 to 74), respectively. Table 1 lists training set clinicopathological features. Study and tissue collection was approved by the University of Wisconsin-Madison and University of Pittsburgh institutional review boards.
Table 1.
Training set clinicopathological features
| NTA | TA Vol
|
TA Greater Than 10 mm from Tumor | Totals | ||
|---|---|---|---|---|---|
| Low | High | ||||
| No. cores | 11 | 15 | 15 | 24 | 65 |
| Mean age | 63 | 59 | 64.1 | 57.6 | — |
| Mean PSA (ng/ml) | — | 6.3 | 12.2 | 6.9 | — |
| Gleason: | |||||
| 6 | — | 5 | 2 | 4 | 11 |
| 7 | — | 7 | 5 | 11 | 23 |
| 8 | — | 2 | 3 | 3 | 8 |
| 9 | — | 0 | 5 | 5 | 10 |
| Unknown | — | 1 | 0 | 1 | 2 |
| Pathological stage: | |||||
| T2a | — | 3 | 2 | 1 | 6 |
| T2b | — | 0 | 0 | 1 | 1 |
| T2c | — | 11 | 5 | 13 | 29 |
| T3a | — | 0 | 3 | 2 | 5 |
| T3b | — | 0 | 4 | 3 | 7 |
| T4 | — | 0 | 1 | 0 | 1 |
| Unknown | — | 1 | 0 | 4 | 5 |
Validation Set Tissues
A total of 47 fresh frozen biopsy negative cores were obtained from University of Washington under an institutional review board approved protocol. All biopsies were taken from the peripheral zone using a standard 12-core biopsy template. Of the biopsies 24 were TA and contained histologically normal core tissue from a biopsy set in which cancer was found in at least 1 other core. Mean patient age was 62.3 years (range 52 to 74). Another 23 biopsies were NTA and contained normal core tissue from a biopsy set in which no cancer was found. Mean patient age was 61.7 years (range 52 to 75). To minimize the possibility of occult disease, NTA tissues were obtained from patients who had at least 1 other negative biopsy set from biopsies done before or after the current set. These patients were followed an average of 6.9 years (range 4.2 to 9.2) without cancer detected on subsequent biopsies.
To definitively rule out PCa in the TA and NTA biopsy specimens analyzed, cores were re-reviewed by a genitourinary pathologist at University of Wisconsin before analysis. No extensive high grade PIN was present in the samples. Table 2 lists clinicopathological data on these patients.
Table 2.
Validation set clinicopathological features
| NTA | TA | |
|---|---|---|
| No. cores | 23 | 24 |
| Mean age | 61.7 | 62.3 |
| Mean PSA (ng/ml) | 4.5 | 6.6 |
| Gleason: | ||
| 6 | — | 14 |
| 7 | — | 9 |
| 8 | — | 1 |
| Clinical stage: | ||
| T1c | — | 6 |
| T2 | — | 2 |
| T2a | — | 2 |
| T2b | — | 4 |
| T2c | — | 4 |
| T3a | — | 1 |
| T3b | — | 1 |
| T4 | — | 1 |
| Unknown | — | 3 |
Quantitative Pyrosequencing
Sodium bisulfite modification of genomic DNA was performed and amplified using quantitative Pyrosequencing with appropriate controls, as previously reported.12 Primer sequences were described previously.8 Briefly, biotinylated polymerase chain reaction products were captured with streptavidin sepharose beads, denatured to single strand and annealed to the sequencing primer for the Pyrosequencing assay. Methylation was quantified with the PyroMark™ TMMD Pyrosequencing System. Two cores (biological replicates) were analyzed per patient. Percent methylation values at all CGs for each gene were almost identical (SE less than 5%) in cores from the same patient. All samples were analyzed in at least 2 independent experiments using duplicate DNA samples (technical replicates). Six, 10 and 4 CpG sites were assayed for EVX1, CAV1 and FGF1, respectively.
Statistical Analysis
The t and Wilcoxon rank sum tests were used to compare methylation differences between NTA specimens and TA prostate tissues at individual CpGs. We assessed the collinearity of individual CpG sites using correlation matrices for each gene. Since CG sites correlated highly, only 1 CG per locus was selected to enter multivariate logistic regression models to prevent over fitting. The strongest predicting CG at each locus was entered into a multivariate logistic regression model using a forward selection method with a model entry criterion of p <0.05 to drop insignificant terms from the model. After final model generation, the predictive probability of each sample was used as input to generate a ROC curve and the AUC was calculated. We externally validated the model using 47 biopsy specimens obtained from University of Washington (fig. 1). All statistical analyses were performed with SPSS®, version 20.0. All tests were 2 tailed with significance considered at p <0.05.
Figure 1.
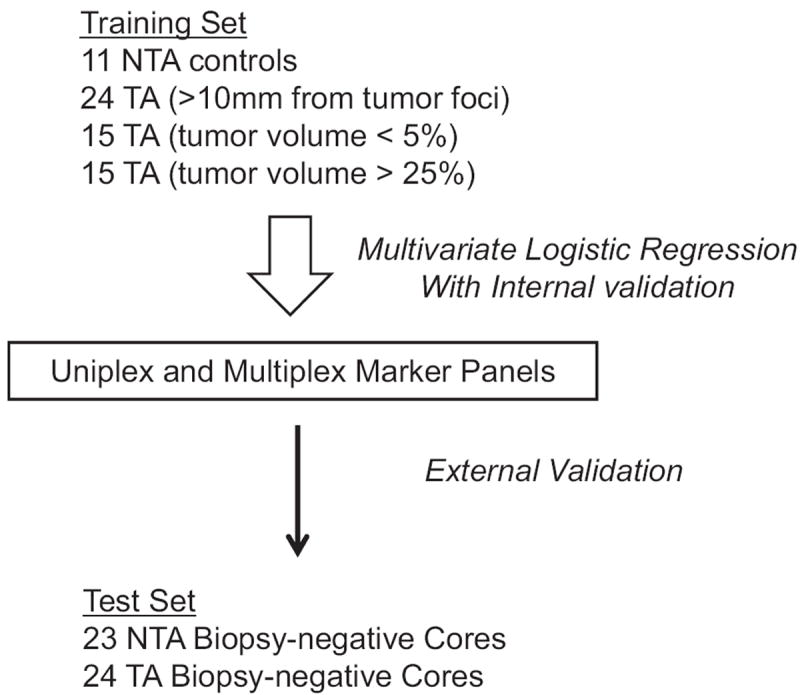
Regression model development and validation. To maximize model generalizability, model training sets included TA tissues from prostatectomy specimens of tumors of various volumes and at various distances from tumor foci. After internal validation, models were externally validated in 47 TA and NTA biopsy negative cores.
RESULTS
Field Defect Methylation Differences at EVX1, CAV1 and FGF1
An epigenetic field defect was initially defined by comparing methylation in NTA vs TA tissues at 385,000 loci using a methylation array with differences confirmed at a subset of loci.8 Methylation of 65 samples in the training set was first analyzed using quantitative Pyrosequencing at each locus. The t and nonparametric Wilcoxon rank sum tests showed highly significant differences between normal TA tissues and NTA benign prostate specimens at all loci (EVX1, CAV1 and FGF1) (fig. 2). EVX1 and CAV1 were hypermethylated, while FGF1 was hypomethylated at all CpGs assayed. The supplementary table http://jurology.com/ lists complete methylation data at each site with the SD and p value.
Figure 2.
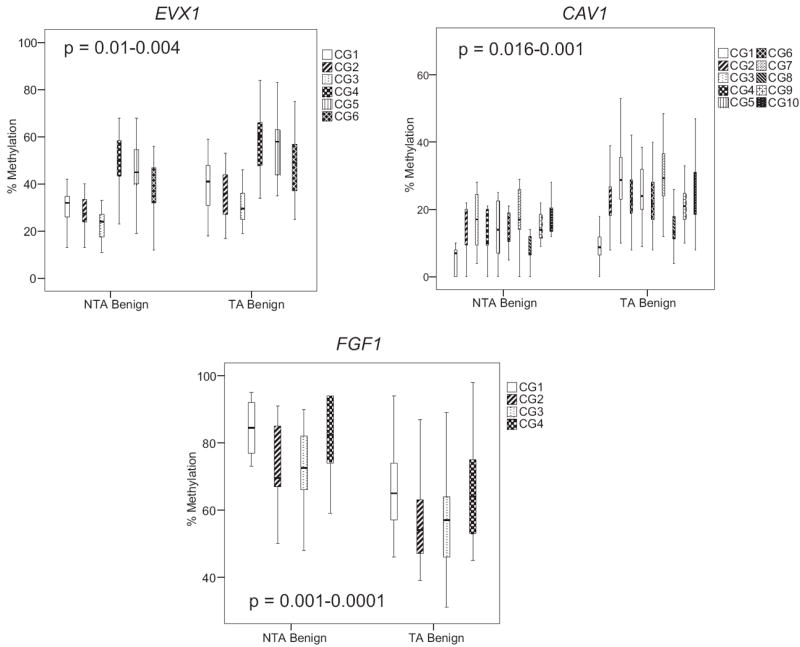
Quantitative Pyrosequencing revealed NTA and TA tissue EVX1, CAV1 and FGF1 methylation levels. EVX1 and CAV1 were hypermethylated in TA compared to NTA tissue. FGF1 was hypomethylated gene in TA tissue. All p values were highly statistically significant at all CpG sites (see supplementary table, http://jurology.com/). In all experiments SssI methylase treated, bisulfite converted DNA from HPEC and PPC1 cells served as positive controls. Water was substituted for DNA as negative control. Boxes represent 75th and 25th percentiles. Whiskers represent minimum and maximum. Bold lines represent median.
Effects of Tumor Volume on Field Defect Methylation
Methylation was compared between 15 low volume (5% or less tumor involvement) and 15 high volume (greater than 25% tumor involvement) TA tissues. Normal tissues obtained from low and high volume cancers were significantly different compared to NTA tissues (see supplementary figure, http://jurology.com/). There was a trend toward greater methylation differences in higher volume tumors but this was not statistically significant at all CG sites. An exception was FGF1 at 1 of 4 CpG sites (p = 0.04, see supplementary figure, http://.com/jurology.com/). At 19 other CpGs assayed there was no difference between low and higher volume cancer, suggesting that tumor volume has a minimal impact on the extent of the methylation field defect in these 3 regions. We subsequently selected 15 low volume (less than 5% involvement) and 15 high volume (greater than 25% involvement) specimens for inclusion in the training set to maximize the generalizability of these models for various clinical scenarios.
Regression Model Internal Validation
The predictive accuracy of these genes was assessed using regression models using EVX1, CAV1 and FGF1 alone (uniplex) or in combination (multiplex). Since tumor volume had a small effect on methylation levels, we used samples with low and high volume specimens in the training set (fig. 1). When used in uniplex models, each gene (EVX1, CAV1 and FGF1) had excellent predictive accuracy in the training sets (table 3). On multivariate analysis only EVX1 and FGF1 entered the model. When EVX1 and FGF1 were used in combination, the predictive accuracy for discriminating TA from NTA tissues was 0.898 in the training set (p <0.0001, table 3).
Table 3.
Uniplex and multiplex logistic regression model training set performance
| Model No. (gene) | Variable | Coefficient | Constant | AUC (95% CI) | p Value |
|---|---|---|---|---|---|
| Uniplex: | |||||
| 1 (EVX1) | CG3 | 0.197 | −3.8751 | 0.824 (0.686–0.963) | 0.001 |
| 2 (CAV1) | CG4 | 0.211 | −2.441 | 0.796 (0.669–0.923) | 0.002 |
| 3 (FGF1) | CG2 | −0.111 | 8.646 | 0.838 (0.711–0.965) | 0.001 |
| Multiplex 4: | |||||
| EVX1 | CG3 | 0.157 | 5.692 | 0.898 (0.795–1.00) | <0.0001 |
| FGF1 | CG2 | −0.131 |
Regression Model External Validation Predicted Cancer in Biopsy Specimens
Each logistic regression model generated was validated in a set of 47 biopsy negative cores from patients with a positive biopsy or with multiple negative biopsies. A cancer diagnosis was based on other cores sent for formal pathological analysis. Histopathology excluded cancer in the tested cores before use in the methylation assay. Biopsy samples were obtained from elsewhere and handled in blinded fashion. When used alone, EVX1, CAV1 and FGF1 discriminated between patients with and without known cancer (AUC 0.702, 0.696 and 0.658, respectively, p <0.05). A multiplex model incorporating EVX1 and FGF1 performed with higher predictive accuracy (AUC 0.774, p = 0.001, fig. 3, A). CAV1 did not enter the multivariate model (p >0.05). Using a predicted probability cutoff of 0.954, the sensitivity and specificity of the multiplex model incorporating EVX1 and FGF1 was 95.8% and 43.5%, respectively. At this cutoff the NPV was 0.909. Using a predicted probability cutoff of 0.995, the sensitivity and specificity of the multiplex model was 50% and 91.3%, respectively (fig. 3, B).
Figure 3.
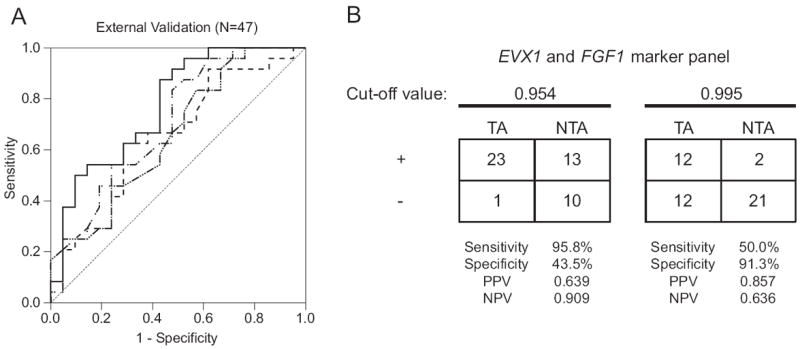
A, ROC curves were generated to externally validate predictive accuracy of uniplex and multiplex regression models for discriminating TA and NTA biopsy negative cores. When used alone, EVX1 (dashed and dotted curve), CAV1 (dashed curve with multiple dots) and FGF1 (dashed curve) discriminated between patients with and without known cancer (AUC 0.702, 0.696 and 0.658, respectively, p <0.05). Multiplex model incorporating EVX1 and FGF1 (solid curve) had highest predictive accuracy (AUC 0.774, 95% CI 0.64-0.908, p = 0.001) for discriminating TA vs NTA benign cores (47) from transrectal ultrasound guided biopsy. B, EVX1 and FGF1 multiplex model sensitivity, specificity, positive predictive value (PPV) and NPV. Two optimal cutoffs for predicted probabilities were used to define positive (+) and negative (−) test results.
DISCUSSION
Accumulating data suggest that a field defect occurs in patients with prostate, breast and other cancers.8,16 Epigenetic field defects were reported in premalignant and malignant conditions for many cancers, while for colorectal tumors they can even be detected earlier than cancerous histological changes.17 To our knowledge markers representing a widespread field defect that occurs not only adjacent to but also distant from tumors have not been used to date to detect cancer in biopsy specimens. In this first externally validated analysis using loci recently identified from a methylation array8 a multiplex model incorporating EVX1 and FGF1 had sensitivity that attained 96% on external validation. This study has important implications for the early detection of PCa since previous methylation studies relied on cancer cells to detect methylation differences.18
The genes evaluated in this study have biological importance in human PCa. Although the role of EVX1 in human tumorigenesis is unclear, EVX1 was recently found to be a frequently hypermethylated gene during PCa development and it independently predicted PSA failure after prostatectomy.19 CAV1 is also aberrantly methylated in PCa.20 We previously reported that EVX1 and CAV1 hypermethylation occurs not only in tumor specimens but also in normal TA tissues.8 Hypermethylation appears to spread from the edge of CpG islands and this early change in histologically normal-appearing cells appears to be detected by our assay.21 FGF1 is a paracrine factor that promotes PCa proliferation, resistance to cell death and invasiveness.22 We found that FGF1 is hypomethylated in histologically normal TA prostate tissues. In our previous study we did not observed altered expression of EVX1, FGF1 and CAV1, although these methylation differences were consistent with the location of these changes at a distance from the promoter.8 The effects of these field alterations in PCa development are not completely understood at this point but they may represent changes in nuclear structure.
Analysis of the effects of tumor volume on methylation at these 3 gene loci revealed that tumor volume has no significant effect at the CpG sites analyzed in EVX1 or CAV1. One of 4 sites showed a statistical trend for FGF1. This lack of an association with volume may be due to the initial methylation array analysis, which used primarily smaller volume cancers (6.3% average tumor involvement). However, there was a trend at most CG sites toward greater methylation in higher volume tumors (see supplementary figure, http://jurology.com/). Further data may support the use of this model in helping exclude small volume tumors, which typically have a more indolent clinical course.
Using GSTP1, APC and RASSF1, groups recently analyzed methylation changes involving adjacent abnormal tissue (peritumor or halo effect) that are induced by tumors.23-25 In these studies methylation results varied among biopsy cores from different locations in the same patient. Our approach is to determine alterations using an unbiased methylation array to define field defect changes in associated TA tissues and confirm that these changes are associated with the peritumor response as well as with widespread changes in the prostate.
Previous analysis of the markers used in this assay revealed that the distance from the tumor to the normal analyzed sample did not alter the methylation change.8 Remarkably, our assay demonstrated almost identical methylation values among cores taken from the same patient at all CGs for EVX1, CAV1 and FGF1. We believe that this highlights the sensitivity of the assay, a common problem for clinicians, since the assay does not rely on distance from the tumor or the presence of cancer cells. In addition, the data suggest that fewer cores are required for analysis.
Other groups identified RNA based changes when comparing TA and NTA tissues.14,26 We believe that DNA based changes have a number of inherent benefits, including relative stability. Combining them with clinicopathological features may further improve the accuracy of these field defect markers in subsequent studies. However, in this early study PSA did not improve the AUC of our methylation assay when used in combination (AUC 0.775). Recently, magnetic resonance imaging informed sampling of the prostate improved the detection rate of clinically significant cancers.27 Although magnetic resonance imaging is promising, unlike biological markers it has a limited role in improving the diagnosis of microscopic disease and causes significant expense.
In this study a multiplex model incorporating EVX1 and FGF1 revealed a high predictive accuracy of 0.774 on external validation. CAV1 was not ultimately incorporated into the multivariate model (p >0.05), possibly due to collinearity or lack of independence of CAV1 in relation to the other markers. The cutoff value for the multiplex model can be adjusted to serve the intended purpose of the clinician. If the intent is to rule out any possibility of cancer, using a predicted probability cutoff of 0.995 yields 91.3% specificity and 50.0% sensitivity. Conversely, if the intent is to detect more tumors, using a cutoff of 0.954 yields 95.8% sensitivity and 43.5% specificity.
Each individual gene performed robustly alone and a combination of markers allowed for even stronger predictive accuracy. Notably, each point along the ROC curve of the multiplex model performed better than individual gene markers (fig. 3). Our models do not rely on the combination of these markers with established markers, an approach that often results in falsely low p values for the proposed biomarker.28 Furthermore, the high negative predictive value of 0.909 indicates that the multiplex model will infrequently misclassify patients with cancer as normal.
Studies have shown that transrectal ultrasound guided prostate biopsy has a false-negative rate of 20% or higher.29 The performance of our multiplex model may be underestimated, given that some NTA biopsy tissues from the validation set may represent false-negative results and actually contain cancer. We used carefully analyzed donor or cystoprostatectomy samples for the training set to minimize this potential error. A theoretical limitation of the current approach is that in the training set NTA specimens were obtained from cystoprostatectomy, while TA specimens were obtained from radical prostatectomy. Differences in tissue handling might influence the field defect but this concern was mitigated by the validation in biopsy specimens in this study. Another potential limitation is that we used academic and Veterans Affairs populations. Assay sensitivity may be higher or lower in larger, more diverse populations with a different disease frequency.
This study shows the use of the field defect to accurately rule out cancer in the setting of increased PSA and negative biopsy. We did not test the ability of this assay to detect high grade disease since most validation set specimens were of intermediate grade, which is the most common grade diagnosed. In our previous series genes (NCR2 and WNT2) were associated with a field defect in high grade PCa.8 In future studies we will apply NCR2 and WNT2 using this approach to detect high grade disease in biopsy negative tissue. Whether this methylation field defect can predict individuals who will subsequently have a positive biopsy is also of interest.
CONCLUSIONS
We describe one of the first externally validated methylation assays to detect PCa using histologically normal biopsy tissue. Given the challenges of treating the patient with increased PSA and negative biopsy, this methylation assay may ultimately aid in the decision of whether to perform additional biopsies.
Supplementary Material
Acknowledgments
Supported by Institute for Clinical and Translational Research Training Grant NIH KL2 RR02012, the University of Wisconsin School of Medicine Shapiro Grant (MT), NIH 5RO1CA 097131, the Livesey Endowment (BY and DFJ), NIH 5R01CA 131255 and the Jim Allchin Prostate Cancer Research Fund (DWL).
Study received University of Wisconsin-Madison, University of Pittsburgh and University of Washington institutional review board approval.
Abbreviations and Acronyms
- CAV1
caveolin-1
- EVX1
even-skipped homeobox-1
- FGF1
fibroblast growth factor-1
- NPV
negative predictive value
- NTA
nonTA
- PCa
prostate cancer
- PSA
prostate specific antigen
- TA
tumor associated
References
- 1.Mehrotra J, Varde S, Wang H, et al. Quantitative, spatial resolution of the epigenetic field effect in prostate cancer. Prostate. 2008;68:152. doi: 10.1002/pros.20675. [DOI] [PubMed] [Google Scholar]
- 2.Dakubo GD, Jakupciak JP, Birch-Machin MA, et al. Clinical implications and utility of field cancerization. Cancer Cell Int. 2007;7:2. doi: 10.1186/1475-2867-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson IM, Pauler DK, Goodman PJ, et al. Prevalence of prostate cancer among men with a prostate-specific antigen level < or = 4.0 ng per milliliter. N Engl J Med. 2004;350:2239. doi: 10.1056/NEJMoa031918. [DOI] [PubMed] [Google Scholar]
- 4.Rosario DJ, Lane JA, Metcalfe C, et al. Short term outcomes of prostate biopsy in men tested for cancer by prostate specific antigen: prospective evaluation within ProtecT study. BMJ. 2012;344:d7894. doi: 10.1136/bmj.d7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gann PH, Fought A, Deaton R, et al. Risk factors for prostate cancer detection after a negative biopsy: a novel multivariable longitudinal approach. J Clin Oncol. 2010;28:1714. doi: 10.1200/JCO.2008.20.3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee GL, Dobi A, Srivastava S. Prostate cancer: diagnostic performance of the PCA3 urine test. Nat Rev Urol. 2011;8:123. doi: 10.1038/nrurol.2011.10. [DOI] [PubMed] [Google Scholar]
- 7.Nelson WG, De Marzo AM, Yegnasubramanian S. Epigenetic alterations in human prostate cancers. Endocrinology. 2009;150:3991. doi: 10.1210/en.2009-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang BB, Kueck J, Weeratunga P, et al. Methylation profiling defines a widespread field defect in histologically normal prostate tissues associated with prostate cancer. doi: 10.1593/neo.13280. Unpublished data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aitchison A, Warren A, Neal D, et al. RASSF1A promoter methylation is frequently detected in both pre-malignant and non-malignant microdissected prostatic epithelial tissues. Prostate. 2007;67:638. doi: 10.1002/pros.20475. [DOI] [PubMed] [Google Scholar]
- 10.Steiner I, Jung K, Schatz P, et al. Gene promoter methylation and its potential relevance in early prostate cancer diagnosis. Pathobiology. 2010;77:260. doi: 10.1159/000318017. [DOI] [PubMed] [Google Scholar]
- 11.Hanson JA, Gillespie JW, Grover A, et al. Gene promoter methylation in prostate tumor-associated stromal cells. J Natl Cancer Inst. 2006;98:255. doi: 10.1093/jnci/djj051. [DOI] [PubMed] [Google Scholar]
- 12.Bhusari S, Yang B, Kueck J, et al. Insulin-like growth factor-2 (IGF2) loss of imprinting marks a field defect within human prostates containing cancer. Prostate. 2011;71:1621. doi: 10.1002/pros.21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nonn L, Ananthanarayanan V, Gann PH. Evidence for field cancerization of the prostate. Prostate. 2009;69:1470. doi: 10.1002/pros.20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Risk MC, Knudsen BS, Coleman I, et al. Differential gene expression in benign prostate epithelium of men with and without prostate cancer: evidence for a prostate cancer field effect. Clin Cancer Res. 2010;16:5414. doi: 10.1158/1078-0432.CCR-10-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heaphy CM, Fleet TM, Treat EG, et al. Organ-wide telomeric status in diseased and disease-free prostatic tissues. Prostate. 2010;70:1471. doi: 10.1002/pros.21182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones TD, Wang M, Eble JN, et al. Molecular evidence supporting field effect in urothelial carcinogenesis. Clin Cancer Res. 2005;11:6512. doi: 10.1158/1078-0432.CCR-05-0891. [DOI] [PubMed] [Google Scholar]
- 17.Giovannucci E, Ogino S. DNA methylation, field effects, and colorectal cancer. J Natl Cancer Inst. 2005;97:1317. doi: 10.1093/jnci/dji305. [DOI] [PubMed] [Google Scholar]
- 18.Yegnasubramanian S, Kowalski J, Gonzalgo ML, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004;64:1975. doi: 10.1158/0008-5472.can-03-3972. [DOI] [PubMed] [Google Scholar]
- 19.Truong M, Yang B, Wagner J, et al. Even-skipped homeobox 1 is frequently hypermethylated in prostate cancer and predicts PSA recurrence. Br J Cancer. 2012;107:100. doi: 10.1038/bjc.2012.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui J, Rohr LR, Swanson G, et al. Hypermethylation of the caveolin-1 gene promoter in prostate cancer. Prostate. 2001;46:249. doi: 10.1002/1097-0045(20010215)46:3<249::aid-pros1030>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 21.Graff JR, Herman JG, Myöhänen S, et al. Mapping patterns of CpG island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation. J Biol Chem. 1997;272:22322. doi: 10.1074/jbc.272.35.22322. [DOI] [PubMed] [Google Scholar]
- 22.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 23.Trock BJ, Brotzman MJ, Mangold LA, et al. Evaluation of GSTP1 and APC methylation as indicators for repeat biopsy in a high-risk cohort of men with negative initial prostate biopsies. BJU Int. 2012;110:56. doi: 10.1111/j.1464-410X.2011.10718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Troyer DA, Lucia MS, de Bruïne AP, et al. Prostate cancer detected by methylated gene markers in histopathologically cancer-negative tissues from men with subsequent positive biopsies. Cancer Epidemiol Biomarkers Prev. 2009;18:2717. doi: 10.1158/1055-9965.EPI-09-0068. [DOI] [PubMed] [Google Scholar]
- 25.Stewart GD, Van Neste L, Delvenne P, et al. Clinical utility of an epigenetic assay to detect occult prostate cancer in histopathologically negative biopsies: results of the MATLOC study. J Urol. 2013;189:1110. doi: 10.1016/j.juro.2012.08.219. [DOI] [PubMed] [Google Scholar]
- 26.Yu YP, Landsittel D, Jing L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 27.Moore CM, Robertson NL, Arsanious N, et al. Image-guided prostate biopsy using magnetic resonance imaging-derived targets: a systematic review. Eur Urol. 2013;63:125. doi: 10.1016/j.eururo.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Taylor JM, Ankerst DP, Andridge RR. Validation of biomarker-based risk prediction models. Clin Cancer Res. 2008;14:5977. doi: 10.1158/1078-0432.CCR-07-4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Djavan B, Ravery V, Zlotta A, et al. Prospective evaluation of prostate cancer detected on biopsies 1, 2, 3 and 4: when should we stop? J Urol. 2001;166:1679. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.