

Abstract
Abnormal expression or mutations in Ras proteins has been found in up to 30% of cancer cell types, making them excellent protein models to probe structure-function relationships of cell-signaling processes that mediate cell transformtion. Yet, there has been very little development of therapies to help tackle Ras-related diseased states. The development of small molecules to target Ras proteins to potentially inhibit abnormal Ras-stimulated cell signaling has been conceptualized and some progress has been made over the last 16 or so years. Here, we briefly review studies characterizing Ras protein-small molecule interactions to show the importance and potential that these small molecules may have for Ras-related drug discovery. We summarize recent results, highlighting small molecules that can be directly targeted to Ras using Structure-Based Drug Design (SBDD) and Fragment-Based Lead Discovery (FBLD) methods. The inactivation of Ras oncogenic signaling in vitro by small molecules is currently an attractive hurdle to try to and leap over in order to attack the oncogenic state. In this regard, important features of previously characterized properties of small molecule Ras targets, as well as a current understanding of conformational and dynamics changes seen for Ras-related mutants, relative to wild type, must be taken into account as newer small molecule design strategies towards Ras are developed.
Keywords: Ras [Rat Sarcoma], Small Molecule Target, Structure-Based Drug Design, Fragment-Based Drug Design, GTP Hydrolysis, Guanine Nucleotide Exchange Factors [GEF]
1. Introduction
Abnormal expression of Ras [Rat sarcoma] proteins plays a significant role in events leading to cellular proliferation, inhibition of cell death, and malignant transformation [1]. In fact, aberrant signaling mechanisms mediated by Ras proteins have been implicated in many cancers [2]. Although the characterization of Ras proteins, and their roles in these aberrant cell-signaling mechanisms have been studied for many years, these proteins still lack effective agents to attack their abnormal functioning states. In fact, Gysin et al. pointed out, as recently as 2011, there were no drugs that could directly target Ras proteins in a way that might facilitate the deregulation of Ras-stimulated oncogenic activity [3]. Therefore, approaches are still needed to develop strategies to inhibit oncogenic Ras signaling. Ras proteins are small GTPases that functions as nucleotide-dependent switches which are “on” when GTP-bound and “off” when GDP-bound. Ras proteins possess intrinsic GTPase activity and the nucleotide exchange is catalyzed by guanine nucleotide exchange factors [GEFs]. When bound to GTP, Ras proteins are active and capable of recruiting downstream effectors and influencing cell function. Effector binding essentially involves protein domains that have been designated as the Switch 1 and Switch 2 regions. GTP hydrolysis leads to a GDP-bound, inactive Ras protein. Both the intrinsic GTPase activity and the GTP binding activity of Ras proteins are fairly low and are accelerated by regulatory proteins that control Ras function. In addition to the GEFs, these regulatory proteins include the guanine activating proteins [GAPs]. Loss of GAP function, or inhibition of GTP hydrolysis [4], might raise the levels of GTP-bound Ras proteins and culminate in Ras over activity, thereby leading to oncogenic behavior. In the same vein, if interactions with GEFs are not properly controlled, then the levels of GTP-bound Ras can also increase and lead to Ras-stimulated overactivity. Accordingly, considerable effort has been spent on developing small molecules as possible “Direct” targets (Figure 1) to Ras proteins to potentially inhibit abnormal Ras-stimulated cell signaling. Here, we briefly review studies characterizing Ras protein-small molecule interactions. Indeed, studies to date show the rationale as well as the importance for continued development of small molecules targets to Ras proteins. We also summarize recent results on the development of small molecule inhibitors of Ras using Structure-Based Drug Design [SBDD] and Fragment-Based Lead Discovery [FBLD]. These techniques can be expected to provide a new perspective on how subsequent targeting of Ras proteins for therapeutic treatment may be approached.
Figure 1.

Illustration of a Ras-related protein interaction with an effector protein [right], and the potential effect [i.e. disturbance of protein interaction] that a small molecule, with direct binding capability to Ras, could have on the activation state of the Ras protein [left]. (a) A representation of a GTPase inhibitor effector interaction; and (b) A representation of a GEF effector interaction. The Ras-related protein model shown in blue is that of Cell division cycle 42 [Cdc42], PDB access code: [1aje.pdb].
2. Targeting Small Molecules to Ras
The development of small molecules that bind to Ras proteins has presented formidable challenges for quite some time. Nevertheless, the importance of this development is paramount to the overall goal of inactivating oncogenic Ras. Small molecules might be expected to help de-regulate “over activity” caused by the instability of a bound nucleotide that has been shown for some Ras-related mutant proteins through solution NMR studies [5] and biochemical studies [6]. The development of small molecules to target Ras proteins has been limited, which has hampered the development of approaches to tackle to the Ras-stimulated oncogenic state [3]. One consideration has been a perceived lack of binding regions for small molecules on Ras proteins [3] or on effector or regulatory proteins. The concept of targeting oncogenic Ras proteins with small molecules as a novel approach was first highlighted by Tavares, et al. [7] who used mass spectrometry, NMR spectroscopy and molecular modeling as tools to characterize a small molecule target that was not nucleotide based. The molecule did not displace the nucleotide, and it bound to Ras in the Switch 2 domain of Ras [7]. This small molecule, SCH 54292, when complexed to Ras, decreased the rate of Ras-stimulated nucleotide exchange [7]. The potential of this approach was further supported by studies of Ahmadian et al., who observed that oncogenic Ras mutants, with diminished capability to hydrolyze GTP, were rescued by a GTP analog that was modified to contain a catalytically active functional group, diaminobenzophe-none-phosphoroamidate [DABP-GTP] [8]. The compound SCH 54292 has not been further developed, most likely due to its insolubility in an aqueous environment; however, these studies illustrated the potential of small molecules as intracellular inhibitors of abnormal Ras function. This compound was used as the basis for the synthesis of a water-soluble ligand of Ras by Palmioli, et al. [9], who designed a glycosylated derivative of SCH 54292 that exhibited significant water solubility and could bind to Ras. Through multi-dimensional NMR spectroscopy, and biochemical characterization studies, this compound showed inhibitory activity towards nucleotide-exchange of GTP for GDP on Ras; however, the binding affinity for the compound was low [μM] [9]. While the low affinity of this ligand was disappointing, the generation of a water-soluble compound that was able to target Ras showed the promise of this approach.
Targeting small molecules to regulate protein-protein interactions has also proven to be challenging. However, Waldmann et al., found that some derivatives of the nonsteroidal anti-inflammatory drug, Sulindac, had an inhibitory effect on the Ras signaling pathway [10]. This class of compounds, as well as derivatives of MCP-1 reduced Ras-induced Raf stimulation to highlight the potential targeting of Ras down stream effector proteins [11,12]. Inhibiting Ras-effector protein interactions as a way to deregulate over active GTP-bound Ras was also shown for Cu2+ and Zn2+ cyclens based on studies conducted by Rosnizeck et al., [13]. It was hypothesized by these authors, based on NMR and crystal structure data analysis, that the presence of these cyclens caused a disruption of the nucleotide-binding site in the presence of GTP, facilitating weaker effector interactions and leading to the over-active state, but the binding sites were not located in the Ras-GDP bound state. However, the cyclens showed binding affinities with even lower [mM affinity] than that discussed above for the glycosylated derivative of SCH 54292 [10]. Nonetheless, the studies of Waldmann et al., and Rosnizeck et al., again emphasized the potential that small molecule targets might provide for disturbing potentially oncogenic Ras protein interactions. A recent review by Wang et al. discusses in great detail the potential binding site(s) on Ras, as well as functional activities of the SCH-54292 derivatives, the cyclens and the Sulindac-based compounds, as well as the MCP-1 compounds [14].
3. Considerations for Future Directions and Emerging Approaches to Target Ras
Recently, evidence has emerged showing the potential for small molecules to be targeted to small surface “pockets” on proteins, thus allowing these molecules to possibly block protein interactions leading to abnormal biological function [15,16]. Solution-based structure studies on a Switch 1 mutant of the Ras-related protein Cdc42 performed in our laboratory have outlined differences in conformational dynamics in the mutant protein construct, relative to wild type Cdc42, which were correlated to a reduced binding of an effector protein that inhibits GTP hydrolysis [17]. This finding suggested that conformational changes imparted by mutations in Ras-related proteins have the potential to alter effector protein interactions. As such, it should be considered that the development of small molecule targets directed towards Ras proteins could facilitate changes in local conformational dynamics, possibly without affecting the overall stability of the Ras protein. This type of approach might be a viable subsequent direction for drug development research efforts. Fragment Based Lead Discovery (FBLD) involves the screening of low molecular weight compounds (typically < 300 Da) at concentrations that allow for their characterization using biophysical techniques such as NMR spectroscopy and X-Ray crystallography (Figure 2). The use of these complementary approaches has facilitated the identification of fragments with properties that may make them more suitable ligands than fragments developed from the “High Throughput Screening” methods used more typically [18,19]. In addition, the FBLD approach has been used successfully to identify small molecule fragments that bind at the interface of protein-protein interaction sites [16]. To this end, using an FBLD approach, Maurer et al. identified three small molecules: benzamidine [BZDN], benzimidazole [BZIM] and 4,6-dichloro-2-methyl-3aminoethyl-indole (DCAI), which recognized the same binding pocket on Ras, near the Switch 1 and 2 regions, and characterized their potential as targets for Ras inactivation [20]. High-resolution co-crystal structures revealed a ligand-binding pocket on the Ras protein and it was observed that the binding pocket on Ras could be expanded for DCAI relative to its size when binding BZDN or BZIM to Ras [20]. Biochemical assays revealed that the binding of DCAI affected the ability of the GEF effector protein Son of Sevenless [SOS] to activate nucleotide exchange in Ras, and it was postulated that the inhibition of nucleotide exchange was due to the blocking of the Ras-SOS interaction by DCAI [20]. As the authors point out, the issue of binding strength for small molecule targets to Ras is still a challenge, as their studies with DCAI showed very low affinity for Ras, as has been also seen with other Ras small molecule targets. These FBLD studies, nevertheless, show promise for efforts to develop small molecule inhibitors of abnormal Ras-stimulated activity.
Figure 2.
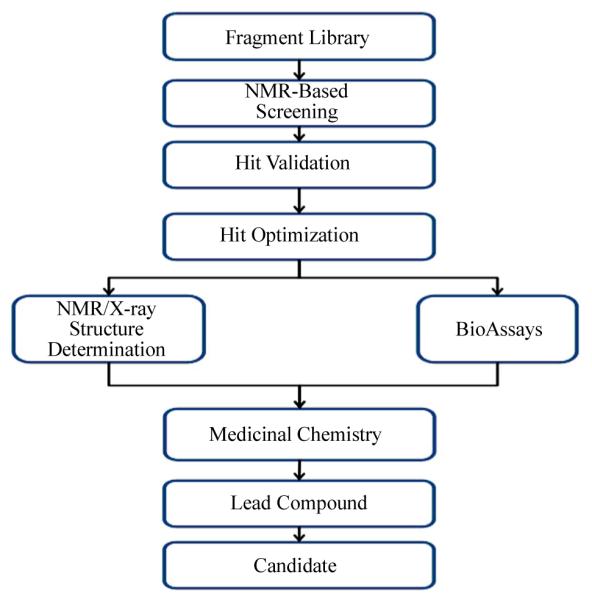
Schematic flow chart of a general approach of FBLD to lead candidate development towards a Ras-related protein.
In silico-based methods have also been used to identify and characterize novel small molecule targets to Ras and the Ras-related protein Cdc42. Friesland, et al., used high-throughput screening to identify ZCL278, which was docked to the surface of Cdc42 between the Switch 1 and Switch 2 regions, and mimicked an interaction between Cdc42 and the GEF effector intersectin [21]. Biochemical and cell-based assays showed that ZCL278 led to a significant reduction in cell-signaling activity, presumably due to an altered interaction with the GEF effector which helped to stabilize the inactive GDP-bound state of Cdc42 (see Figure 1(b)). These findings suggest that this small molecule could serve as a direct target to Cdc42. In another study, Shima et al. examined structures of H-Ras, M-Ras and several mutants and identified surface regions that they hypothesized could serve as binding sites for small compounds [22-24]. These sites were examined by in silico-based Structure Based Drug Design (SBDD) studies, and a group of small molecules. The Kobe0065 family, were identified to bind to Ras [25]. Subsequent biochemical assays showed the anti-tumor activity of members of the Kobe0065 family [25]. Although the authors point out that the inhibitory activity of these compounds is low, again suggesting weak binding, this study shows added promise for the identification of small molecule targets for Ras that may show clinical potential [25].
In summary, the inactivation of Ras oncogenic signaling still remains a daunting task for the scientific community. The scope of current efforts highlights the importance of the Ras proteins as targets for treatment of the oncogenic state. The detection and characterization of small molecule targets, not only to Ras proteins, but to proteins in general has been dependent upon sensitive biophysical technologies capable of detecting low affinity interactions of low molecular weight compounds. Over the last decade, approaches such as nuclear magnetic resonance [NMR], X-ray crystallography, and Surface Plasmon Resonance [SPR] have become core technologies in many pharma and biotech settings. Moreover, approaches such as FBLD and in silico-based structure design have become powerful and exciting tools for exploring the targeting of Ras GTPases. These novel approaches may contribute to the subsequent development of therapeutic strategies to tackle diseased states caused by Ras-related proteins. As small molecule targeting to Ras progresses, it will be imperative to consider the development of design strategies that will be relevant for in vivo study. Additionally, our increased knowledge of the conformational and dynamics changes seen for Ras-related mutant proteins should also be exploited in subsequent design strategies to target small molecules to adjacent regions on Ras proteins. Although the state of knowledge and the tangible applications are at an early stage, small molecule drug candidates nevertheless show promise for disturbing disease-causing Ras-related protein-protein interactions.
4. Acknowledgements
We thank Drs. Roger Koeppe II and Robert Oswald for providing critical feedback and suggestions on the content and scope of this manuscript, and Mrs. Kyla Morris and Mr. Hans Wang with Figure design. This publication was supported by Grant Number 1K-01-CA113753 to P.D.A. from the National Cancer Institute of the National Institutes of Health (NIH), Grant Number P30 GM1034-50 from the National Institute of General Medical Sciences of the NIH, and the Arkansas Biosciences Institute.
REFERENCES
- [1].Bos JL. Ras Oncogenes in Human Cancer: A Review. Cancer Research. 1989;Vol. 49(No. 17):4682–4689. [PubMed] [Google Scholar]
- [2].Baines AT, Xu D, Der CJ. Inhibition of Ras for Cancer Treatment: The Search Continues. Future Medicinal Chemistry. 2011;Vol. 3(No. 14):1787–1808. doi: 10.4155/fmc.11.121. http://dx.doi.org/10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for Targeting Ras Proteins. Genes & Cancer. 2011;Vol. 2(No. 3):359–372. doi: 10.1177/1947601911412376. http://dx.doi.org/10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guo W, Sutcliffe MJ, Cerione RA, Oswald RE. Identification of the Binding Surface on Cdc42Hs for p21-Activated Kinase. Biochemistry. 1998;Vol. 37(No. 40):14030–14037. doi: 10.1021/bi981352+. http://dx.doi.org/10.1021/bi981352+ [DOI] [PubMed] [Google Scholar]
- [5].Adams PD, Loh AP, Oswald RE. Backbone Dynamics of an Oncogenic Mutant of Cdc42Hs Shows Increased Flexibility at the Nucleotide-Binding Site. Biochemistry. 2004;Vol. 43(No. 31):9968–9977. doi: 10.1021/bi0490901. http://dx.doi.org/10.1021/bi0490901. [DOI] [PubMed] [Google Scholar]
- [6].Lin R, Bagrodia S, Cerione R, Manor D. A Novel Cdc42Hs Mutant Induces Cellular Transformation. Current Biology. 1997;Vol. 7(No. 10):794–797. doi: 10.1016/s0960-9822(06)00338-1. http://dx.doi.org/10.1016/S0960-9822(06)00338-1. [DOI] [PubMed] [Google Scholar]
- [7].Taveras AG, Remiszewski SW, Doll RJ, Cesarz D, Huang EC, Kirschmeier P, Pramanik BN, Snow ME, Wang YS, del Rosario JD, Vibulbhan B, Bauer BB, Brown JE, Carr D, Catino J, Evans CA, Girijavallabhan V, Heimark L, James L, Liberles S, Nash C, Perkins L, Senior MM, Tsarbopoulos A, Webber SE, et al. Ras Oncoprotein Inhibitors: The Discovery of Potent, Ras Nucleotide Exchange Inhibitors and the Structural Determination of a Drug-Protein Complex. Bioorganic & Medicinal Chemistry. 1997;Vol. 5(No. 1):125–133. doi: 10.1016/s0968-0896(96)00202-7. http://dx.doi.org/10.1016/S0968-0896(96)00202-7. [DOI] [PubMed] [Google Scholar]
- [8].Ahmadian MR, Zor T, Vogt D, Kabsch W, Selinger Z, Wittinghofer A, Scheffzek K. Guanosine Triphosphatase Stimulation of Oncogenic Ras Mutants. Proceedings of the National Academy Sciences of the USA. 1999;Vol. 96(No. 12):7065–7070. doi: 10.1073/pnas.96.12.7065. http://dx.doi.org/10.1073/pnas.96.12.7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Palmioli A, Sacco E, Abraham S, Thomas CJ, Di Domizio A, De Gioia L, Gaponenko V, Vanoni M, Peri F. First Experimental Identification of Ras-Inhibitor Binding Interface Using a Water-Soluble Ras Ligand. Bioorganic & Medicinal Chemistry Letters. 2009;Vol. 19(No. 15):4217–4222. doi: 10.1016/j.bmcl.2009.05.107. http://dx.doi.org/10.1016/j.bmcl.2009.05.107. [DOI] [PubMed] [Google Scholar]
- [10].Waldmann H, Karaguni IM, Carpintero M, Gourzoulidou E, Herrmann C, Brockmann C, Oschkinat H, Muller O. Sulindac-Derived Ras Pathway Inhibitors Target the Ras-Raf Interaction and Downstream Effectors in the Ras Pathway. Angewandte Chemie International Edition. 2004;Vol. 43(No. 4):454–458. doi: 10.1002/anie.200353089. http://dx.doi.org/10.1002/anie.200353089. [DOI] [PubMed] [Google Scholar]
- [11].Kato-Stankiewicz J, Hakimi I, Zhi G, Zhang J, Serebriiskii I, Guo L, Edamatsu H, Koide H, Menon S, Eckl R, Sakamuri S, Lu Y, Chen QZ, Agarwal S, Baumbach WR, Golemis EA, Tamanoi F, Khazak V. Inhibitors of Ras/Raf-1 Interaction Identified by Two-Hybrid Screening Revert Ras-Dependent Transformation Phenotypes in Human Cancer Cells. Proceedings of the National Academy Sciences of the USA. 2002;Vol. 99(No. 22):14398–14403. doi: 10.1073/pnas.222222699. http://dx.doi.org/10.1073/pnas.222222699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gonzalez-Perez V, Reiner DJ, Alan JK, Mitchell C, Edwards LJ, Khazak V, Der CJ, Cox AD. Genetic and Functional Characterization of Putative Ras/Raf Interaction Inhibitors in C. elegans and Mammalian Cells. Journal of Molecular Signaling. 2010;Vol. 5(No. 1):2. doi: 10.1186/1750-2187-5-2. http://dx.doi.org/10.1186/1750-2187-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rosnizeck IC, Graf T, Spoerner M, Trankle J, Filchtinski D, Herrmann C, Gremer L, Vetter IR, Wittinghofer A, Konig B, Kalbitzer HR. Stabilizing a Weak Binding State for Effectors in the Human Ras Protein by Cyclen Complexes. Angewandte Chemie International Edition. 2010;Vol. 49(No. 22):3830–3833. doi: 10.1002/anie.200907002. http://dx.doi.org/10.1002/anie.200907002. [DOI] [PubMed] [Google Scholar]
- [14].Wang W, Fang G, Rudolph J. Ras Inhibition via Direct Ras Binding—Is There a Path Forward? Bioorganic & Medicinal Chemistry Letters. 2012;Vol. 22(No. 18):5766–5776. doi: 10.1016/j.bmcl.2012.07.082. http://dx.doi.org/10.1016/j.bmcl.2012.07.082. [DOI] [PubMed] [Google Scholar]
- [15].Scott DE, Coyne AG, Hudson SA, Abell C. Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry. 2012;Vol. 51(No. 25):4990–5003. doi: 10.1021/bi3005126. http://dx.doi.org/10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- [16].Scott DE, Ehebauer MT, Pukala T, Marsh M, Blundell TL, Venkitaraman AR, Abell C, Hyvonen M. Using a Fragment-Based Approach to Target Protein-Protein Interactions. ChemBioChem. 2013;Vol. 14(No. 3):332–342. doi: 10.1002/cbic.201200521. http://dx.doi.org/10.1002/cbic.201200521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chandrashekar R, Salem O, Krizova H, McFeeters R, Adams PD. A Switch I Mutant of Cdc42 Exhibits Less Conformational Freedom. Biochemistry. 2011;Vol. 50(No. 28):6196–6207. doi: 10.1021/bi2004284. http://dx.doi.org/10.1021/bi2004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Baker M. Fragment-Based Lead Discovery Grows Up. Nature Reviews and Drug Discovery. 2013;Vol. 12(No. 1):5–7. doi: 10.1038/nrd3926. http://dx.doi.org/10.1038/nrd3926. [DOI] [PubMed] [Google Scholar]
- [19].Carr RA, Congreve M, Murray CW, Rees DC. Fragment-Based Lead Discovery: Leads by Design. Drug Discovery Today. 2005;Vol. 10(No. 14):987–992. doi: 10.1016/S1359-6446(05)03511-7. http://dx.doi.org/10.1016/S1359-6446(05)03511-7. [DOI] [PubMed] [Google Scholar]
- [20].Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, Ludlam MJ, Bowman KK, Wu J, Giannetti AM, Starovasnik MA, Mellman I, Jackson PK, Rudolph J, Wang W, Fang G. Small-Molecule Ligands Bind to a Distinct Pocket in Ras and Inhibit SOS-Mediated Nucleotide Exchange Activity. Proceedings of the National Academy Sciences of the USA. 2012;Vol. 109(No. 14):5299–5304. doi: 10.1073/pnas.1116510109. http://dx.doi.org/10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Friesland A, Zhao Y, Chen YH, Wang L, Zhou H, Lu Q. Small Molecule Targeting Cdc42-Intersectin Interaction Disrupts Golgi Organization and Suppresses Cell Motility. Proceedings of the National Academy Sciences of the USA. 2013;Vol. 110(No. 4):1261–1266. doi: 10.1073/pnas.1116051110. http://dx.doi.org/10.1073/pnas.1116051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Muraoka S, Shima F, Araki M, Inoue T, Yoshimoto A, Ijiri Y, Seki N, Tamura A, Kumasaka T, Yamamoto M, Kataoka T. Crystal Structures of the State 1 Conformations of the GTP-Bound H-Ras Protein and Its Oncogenic G12V and Q61L Mutants. FEBS Letters. 2012;Vol. 586(No.12):1715–1718. doi: 10.1016/j.febslet.2012.04.058. http://dx.doi.org/10.1016/j.febslet.2012.04.058. [DOI] [PubMed] [Google Scholar]
- [23].Shima F, Ijiri Y, Muraoka S, Liao J, Ye M, Araki M, Matsumoto K, Yamamoto N, Sugimoto T, Yoshikawa Y, Kumasaka T, Yamamoto M, Tamura A, Kataoka T. Structural Basis for Conformational Dynamics of GTP-Bound Ras Protein. Journal of Biological Chemistry. 2010;Vol. 285:22696–22705. doi: 10.1074/jbc.M110.125161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ye M, Shima F, Muraoka S, Liao J, Okamoto H, Yamamoto M, Tamura A, Yagi N, Ueki T, Kataoka T. Crystal Structure of M-Ras Reveals a GTP-Bound ‘Off’ State Conformation of Ras Family Small GTPases. Journal of Biological Chemistry. 2005;Vol. 280:31267–31275. doi: 10.1074/jbc.M505503200. http://dx.doi.org/10.1074/jbc.M505503200. [DOI] [PubMed] [Google Scholar]
- [25].Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, Nishiyama Y, Sato C, Muraoka S, Tamura A, Osoda T, Tsuda K, Miyakawa T, Fukunishi H, Shimada J, Kumasaka T, Yamamoto M, Kataoka T. In Silico Discovery of Small-Molecule Ras Inhibitors that Display Antitumor Activity by Blocking the Ras-Effector Interaction. Proceedings of the National Academy Sciences of the USA. 2013;Vol. 110(No. 20):8182–8187. doi: 10.1073/pnas.1217730110. http://dx.doi.org/10.1073/pnas.1217730110. [DOI] [PMC free article] [PubMed] [Google Scholar]