

Abstract
This chapter focuses on the aggregation of glutamine containing peptides and proteins with an emphasis on huntingtin protein, whose aggregation leads to the development of Huntington’s disease. The kinetics that leads to the formation of amyloids, the structure of aggregates of various types and the morphological mechanical properties of amyloid fibrils are described. The kinetics of amyloid fibril formation has been proposed to follow a nucleation dependent polymerization model, dependent upon the size of the nucleus. This model and the effect of the polyglutamine length on the nucleus size are reviewed. Aggregate structure is characterized at two different levels. The atomic-scale resolution structure of fibrillar and crystalline aggregates of polyglutamine containing proteins and peptides was determined by X-ray crystallography and solid-state nuclear magnetic resonance (NMR). The chapter outlines the results obtained by both these techniques. Atomic force microscopy (AFM) was instrumental in elucidating the morphology of fibrils, their organization and assembly. The chapter also discusses the high stability of amyloid fibrils, including their mechanical properties as revealed by AFM.
Keywords: Huntington’s disease, Amyloids, Self-assembly, Fibrillogenesis, Atomic Force Microscopy, AFM, Solid-state NMR
10.1 Introduction
Misfolding and aggregation of proteins is a common thread linking a number of important human health problems associated with protein deposition diseases, including neurodegenerative disorders such as Parkinson’s disease, Down’s syndrome, Alzheimer’s and Huntington’s diseases, systemic and localized amyloidoses and transmissible encephalopathies (Dobson 2004b). Each of these diseases is associated with misfolding and aggregation of one or two specific proteins. Altogether, the accumulation of abnormal protein aggregates exerts toxicity by disrupting intracellular transport, overwhelming protein degradation pathways, and/or disturbing vital cellular functions. Since protein refolding is frequently accompanied by transient association of partially folded intermediates, the propensity to aggregate is considered a general characteristic of the majority of partially folded proteins (review Straub and Thirumalai 2011 and references therein). Thus, protein folding abnormalities and subsequent events underlie a multitude of pathologies and can lead to difficulties with protein therapeutic applications. Current demographic trends indicate the need for therapeutics targeting age-related degenerative disorders and macromolecule therapeutics will be at the forefront of future medical developments. The field of medicine therefore can be dramatically advanced by establishing a fundamental understanding of key factors leading to the misfolding and self-aggregation of proteins involved in various protein folding pathologies. This chapter is focused primarily on misfolding and self-assembly studies of proteins containing glutamine repeat (polyQ) of various lengths, which define the aggregation ability of the protein.
10.2 Aggregation Kinetics of Amyloids
10.2.1 Glutamine Repeats and Huntington’s Disease
The origin of Huntington’s disease (HD) is an extension of polyQ in the huntingtin (Htt) protein sequence. Normally Htt contains 5–35 Q’s, but when the number of Q’s exceeds 36, pathology develops (MacDonald et al. 1993). The extension of the polyQ sequence results from mutation of the gene, leading to multiplication of a trinucleotide CAG repeat (Walker 2007), resulting in polyglutamine stretches in the protein. There are 66 human proteins containing homopolymeric stretches of five or more glutamines. Many of them are non-disease related proteins that contain polyQ repeats, which are intrinsically prone to expansion at the genetic level (Robertson et al. 2011). In addition to Huntington’s diseases, polyQ repeats are responsible for the development of spinobulbar muscular atrophy, dentatorubral-pallidoluysian atrophy and a number of spinocerebellar ataxias (Zoghbi and Orr 2000). The critical threshold length of polyQ varies for different proteins. For most disease related proteins, the glutamine repeat threshold is approximately 35. There are only a few exceptions; for example, the critical threshold for spinocerebrallar ataxia 3 is beyond 50, while the polyQ length varies between 18 and 20 residues for spinocerebrallar ataxia 6 (Gillian 2003). These diseases have little in common at the genetic level except for the presence of long CAG repeats. However, for all of these diseases, there is a strong correlation between the length of the extended repeats, propensity toward aggregation and disease onset (Gatchel and Zoghbi 2005; Gusella and MacDonald 2000). It has also been observed that the proteolytic cleavage of the polyQ containing segment of Htt exon 1 in vitro initiates aggregation (Graham et al. 2006). The neurotoxicity of polyQ aggregation was demonstrated with direct experiments in which the intranuclear injection of polyQ aggregates caused cell death (Yang et al. 2002). These and other observations led to the hypothesis that length–dependent aggregation of polyQ may be the primary trigger in the expanded CAG repeat diseases (Thakur et al. 2009). Therefore, simple polyQ sequences have been used as model systems to reveal the common principles that trigger pathology of the expanded CAG disease family.
10.2.2 Nucleation Dependent Polymerization Model of Amyloid Aggregation
The kinetics of amyloid aggregation has a specific sigmoidal shape with an extended lag period, as illustrated in Fig. 10.1a. This figure shows the time-dependent aggregation of a short fragment of prion protein from yeast, detected by probing aliquots taken from the reaction mixture with Thioflavin T (ThT) fluorescence (Lyubchenko et al. 2010). The kinetics depends on various conditions, such as the acidity of the solvent. For example, the lag is 195 h at pH 2.0, but it is 11 h if the aggregation experiment is performed at pH 5.6.
Figure 10.1.
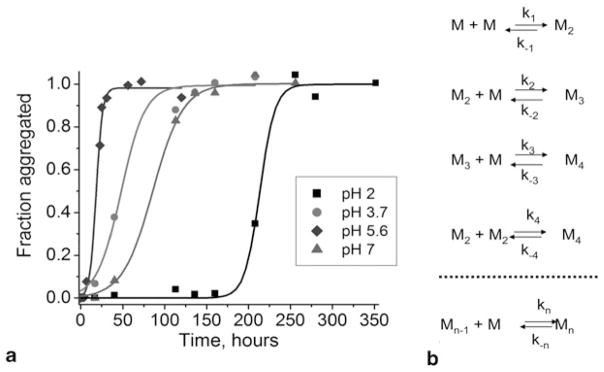
a Normalized kinetic curves of short peptide aggregation from Sup35 yeast prion protein at various pH values: pH 7.0 (triangles, tlag = 49 h), pH 5.6 (diamonds, tlag = 11 h), pH 3.7 (circles, tlag = 19 h), and pH 2.0 (squares, tlag = 195 h). b Schematic for the aggregation kinetics of misfolded monomers (M) with formation of the dimer (M2), trimer (M3), tetramer (M4), and so on up to the n-mer (Mn)
Historically, self-assembly kinetics were observed initially in the earlier work of Hofrichter et al. (Hofrichter et al. 1974), in which the gelation phenomenon of purified deoxyhemoglobin was investigated. The authors proposed a model that dissected the growth kinetics of fibrils into two phases. The first phase is the nucleation process. During this phase, a critical oligomer of a particular size (nucleus) is formed. The nucleus undergoes a thermodynamically favorable elongation process in which monomers are added via consecutive steps (Fig. 10.1b). This model applied to the aggregation of deoxyhemoglobin suggested that the size of a nucleus could be as large as 30 monomeric units (Hofrichter et al. 1974). Later, Jarrett and Lansbury applied this model to analyze the aggregation of amyloids that followed a similar kinetic profile (Jarrett and Lansbury 1993). It was recently shown that the growth of amyloid plaques in vivo follows the same model (Meyer-Luehmann et al. 2008). Both in vivo and in vitro studies indicate a considerably long lag period during which the formation of stable nuclei occurs. This period is considered a key step in the process of amyloid growth. However, a number of important questions arise. What are these nuclei? How large are they? How long do they live? The analysis of experimental aggregation kinetics data similar to the one shown in (Fig. 10.1) is inconclusive. For example, the analysis performed in (Ferrone 1999) led to the conclusion that nuclei for the aggregation of polyQ peptides are monomers that are in equilibrium with the rest of the polyQ peptide samples (Chen et al. 2002a). At the same time, a similar theoretical model predicted nuclei as large as hexamers for Sup35p, but only trimers for Sup35 NMp species (Krzewska et al. 2007). Bernacki and Murphy have addressed this controversy by evaluating the ability of available kinetic models to predict mechanisms of fibril growth (Bernacki and Murphy 2009). The major conclusion of this analysis was that the ThT kinetic curves do not provide sufficient information for developing a mechanistic model of the aggregation process; thus, additional information such as fibril size over time is required. Therefore, there is a need for methods capable of unambiguously detecting and characterizing all oligomeric species formed during the entire aggregation process.
10.2.3 PolyQ Peptides—Aggregation and Structure
An extensive study of a series of simple polyQ peptides with varying lengths has established that the peptides follow a nucleated growth polymerization mechanism (Chen et al. 2002b; Kar et al. 2011). One of the most critical parameters determining how fast polyQ peptide aggregates is the length of the polyQ stretch. Increasing the length of polyQ shortens the lag phase, thus accelerating the overall aggregation rate (Chen et al. 2002b; Perutz et al. 2002; Perutz and Windle 2001; Scherzinger et al. 1999). It has also been suggested that the critical nucleus of polyQ aggregation is a rarely populated form of a monomer for peptides with pathological repeat lengths (Chen et al. 2002b; Kar et al. 2011). Therefore, the stable nucleus of aggregation for pathologically long polyQ is a monomer, i.e., n = 1 (Chen et al. 2002a). However, the size of the nucleus is dependent on the repeat length. A very sharp transition from n = 1 to n = 4 over a short range of repeat lengths, from Q26 to Q23, was observed (Kar et al. 2011). Within this short increase in sequence, there is a tight transition of the size of the critical nucleus, from monomeric through dimeric, to a tetrameric nucleus. Regardless of the length, however, polyQ peptides form aggregates of similar structure (Thakur et al. 2009).
10.2.4 Solution Structure of PolyQ
Circular dichroism (CD) analysis of freshly disaggregated monomeric Q42 peptide showed that when incubated at 37 °C for long periods, the peptide undergoes a conformational transition to a β-sheet (Chen et al. 2002b). The acquisition of a β-sheet structure in the polyQ was found to be intimately associated with the development of aggregate structure, with no evidence of significant development of β-structure within the polyQ peptide before its aggregation. Under normal conditions, monomeric polyQ peptides lack any regular defined secondary structure (Walters and Murphy 2009). Recent experimental and computational studies have demonstrated that polyQ chains in aqueous solutions form collapsed, disordered globules (Vitalis et al. 2007). The formation of such compact structures by polyQ peptides suggests that water is a poor solvent for these peptides. Interestingly, the compactness of the polyQ depends on its length, in which peptides are extended at short lengths (Q < 10) and become increasingly more compact at Q lengths between 10 and 16 (Walters and Murphy 2009). The critical turning point of such a transition takes place at Q16, where the peptides are as compact as many folded proteins (Digambaranath et al. 2011). According to (Walters and Murphy 2009), the following model can explain why polyQ peptides become more collapsed as the length of polyQ increases. Glutamine is an amino acid that contributes hydrophilic side chains to a polypeptide chain. The side chain of glutamine, especially its amide group, participates with water as a donor as well as an acceptor. Hydrogen bonding with water dominates for short polyQ, so that the peptide is well solvated. With increased length of polyQ, the probability of forming various intramolecular hydrogen contacts increases. Multiple side chain-side chain, side chain-backbone, or backbone-backbone hydrogen bonds are formed as the number of Q residues increases. The network of hydrogen bonds stabilizes the collapsed conformation of the peptide, which does not have any regular secondary structure.
This model was also supported by a recent single molecule force spectroscopy study that directly probed the mechanical properties of single polyQ chains (Dougan et al. 2009). Single polyQ chains of varying lengths have been pulled by application of an external force onto constructs where polyQ expansions were flanked by the I27 titin module. Interestingly, no extension has been observed for any polyQ under the application of force, suggesting that polyQ chains form mechanically stable collapsed structures. Disruption of the polyQ sequence with proline residues resulted in their easier extensibility under an applied force. Moreover, such mechanical extensibility appeared to be sensitive to the position of the proline interruption. Therefore, several experimental and theoretical studies demonstrated that polyQ chains collapse to form a heterogeneous ensemble of conformations. Interestingly, the degree of collapse was found to be strongly correlated with the aggregation properties of polyQ (Walters and Murphy 2009).
10.2.5 Effect of Flanking Sequences on PolyQ Aggregation
Huntingtin (Htt) and other polyQ containing proteins differ profoundly in the amino acids flanking the polyQ region, and the role of these flanking sequences in the aggregation of such proteins has been studied. Htt contains proline motifs (polyP) flanking polyQ at the C-terminus, and the incorporation of polyP sequence to the C terminal side of a simple polyQ reduces aggregation kinetics and aggregate stability (Bhattacharyya et al. 2006). The effect of the polyP expansion was found to be directional: the addition of polyP on the N terminal end of polyQ40 did not have any impact on its aggregation (Bhattacharyya et al. 2006). However, although the addition of a polyP sequence at the C-terminus dramatically changes kinetics of aggregation, it does not alter the aggregation mechanism that essentially follows the nucleation model (Bhattacharyya et al. 2005). The inhibitory effect of proline motifs depends on their length, with a minimum of five proline residues required for efficient deceleration of aggregation kinetics (Kar et al. 2011). It was proposed that polyP induces an altered structure in the conformationally fluctuating monomer ensemble. This conformational heterogeneity was confirmed by the crystallographic studies of polyQ (Kim et al. 2009).
10.2.6 Mechanism of PolyQ Aggregation
The aggregation of synthetic peptides with pathologic lengths of polyQ stretches follows the nucleation polymerization model described above. The analysis of kinetic curves for such peptides led to the conclusion that the nucleus of aggregation is a monomeric peptide; therefore, the rate-limiting nucleation event for polyQ peptides is folding within a monomer, rather than unfavorable assembly of monomers into a stable nucleus of finite size (Chen et al. 2002b; Kar et al. 2011). Such a folding event within the monomer requires a conformational transition with the formation of β-sheets (Fig. 10.2a), proposed in the early work of Perutz (Perutz et al. 1994). The β-strands in such structures are held together by hydrogen bonds between both main-chain and side-chain amides. The structures were called “polar zippers” explaining the stabilizing nature of bonds holding them together. Such a “folding within the monomer” mechanism has been tested using mutational analysis of polyQ stretches (Thakur and Wetzel 2002). Prolineglycine (PG) pairs were introduced at different intervals within the polyQ sequence in order to establish the length of the basic fold. It has been found that when PG separate stretches of nine glutamines, the aggregation kinetics is not altered significantly, in contrast to Q8 or Q7 (Thakur and Wetzel 2002). On the other hand, when proline was introduced in the middle of Q9, aggregation was completely abolished. Based on these observations and the fact that PG favors β-turns (Venkatraman et al. 2001), it has been proposed that an antiparallel β-sheet folding motif is the fundamental unit critical for aggregation of polyQ (Thakur and Wetzel 2002). This hypothesis is further supported by the observation that DPG, instead of PG, enhances the aggregation efficiency of the peptide (Thakur and Wetzel 2002) (Walters and Murphy 2011), consistent with a stronger preference for β-turns of DPG (Venkatraman et al. 2001). However, shortening of the polyQ peptides, from 26–23 residues, changes the aggregation mechanism from a monomeric mechanism to the multimeric nucleus model.
Figure 10.2.

Proposed mechanism for polyQ peptide aggregation from an unfolded monomer to an insoluble fibrillar aggregate. The right-hand side shows the nucleation–elongation mechanism (steps (a) to (c)). a A monomer is in rapid equilibrium with a thermodynamically unfavorable β-sheet nucleus, b The β-Sheet nucleus serves as a template for the addition of a monomer. c Fibrils elongate in repeated rounds of monomer addition. The left-hand side outlines the association–conformational conversion mechanism (steps (d) to (f)). d Monomers lacking regular secondary structure rapidly associate into large soluble oligomers, driven by hydrophobic interactions, e Conformational rearrangement within the large oligomers leads to the formation of β-sheet nodes (indicated by the small dashed box), f β-Sheet formation propagates throughout the oligomers, producing insoluble fibrillar aggregates
A more complex aggregation pathway was found for a polyQ containing the N-terminal 17 amino acids of Htt exon 1 (NT17). The formation of intermediate structures was observed during aggregation of this peptide (Thakur et al. 2009). These first 17 amino acids were shown to play a critical role in altering the aggregation pathway of the peptide. Thakur et al. (Thakur et al. 2009) observed the formation of oligomers with the first 17 amino acids of the protein, comprising the core of oligomers and polyQ sequences exposed on the surface. Disordered monomers self-associate into soluble oligomers using hydrophobic interactions of NT17. It has been suggested that N-terminal interactions facilitate intermolecular contacts within the polyQ stretch (Fig. 10.2d). Conformational rearrangement within these oligomers results in aggregation-prone β-sheet “nodes” (Fig. 10.2e) (Tam et al. 2009; Thakur et al. 2009). The β-sheet conformation then induces conformational conversion of peptides within oligomers leading to the formation of insoluble aggregates and ultimately to fibrillar structures with a high β-sheet content and a low amount of water (Fig. 10.2f). The mechanism resembles the nucleated conformational conversion model, proposed to describe amyloid formation by the yeast prion protein (Serio et al. 2000). According to this model, a rapid assembly of monomers into oligomeric structures is followed by conformational conversion within these complexes into pathologic aggregation prone conformations, which induces conformational changes in oncoming monomers and fuels the ongoing fibril-elongation reaction (Serio et al. 2000).
10.3 The Structure of Aggregates Formed by Glutamine Repeats
The quest for the structure of glutamine repeats started when Perutz was investigating the equilibrium constant of the reaction of hemoglobin with oxygen. The equilibrium constant varied over five orders of magnitude among species and the authors identified the cause in a novel protein structural element formed by the sequence Glu-Glu-His-Lys repeated four times; this structural element shown in Fig. 10.3 was coined polar zippers (De Baere et al. 1992; Sherman et al. 1992). Realizing that a variety of polar zipper sequences might exist, Perutz subsequently identified long repeats of only glutamines or serines that could “readily be tipped together into pleated antiparallel β-sheets, with optimal hydrogen bonding distances and angles between their side chains, which are placed symmetrically above and below the plane of the sheet, such that the amino group of each glutamine side chain on one strand donates a hydrogen bond to the carbonyl of each glutamine side chain on the opposite strand” (Perutz et al. 1993). These initial studies inspired many subsequent structural studies of glutamine-rich peptides and proteins (Perutz 1995), providing a structural basis to understand the molecular mechanisms of HD development.
Figure 10.3.

The first cartoon depiction of polar zippers formed by glutamate repeats by Perutz. Reproduced with permission from (De Baere et al. 1992)
10.3.1 Glutamine-repeat Peptides
The X-ray fiber diffraction data for D2Q15K2 led to the water-filled β-helix model of fibrils, in which individual β-sheets are arranged around a 31.0 Å cylinder with 20 amino acid residues per turn and with a water-filled inner pore that is 11.8 Å (Perutz et al. 2002). The original diagram of the model is shown in Fig. 10.4. In this arrangement, alternating side chains point into and out of the cylinder, and stabilizing hydrogen bonds are formed parallel to the cylinder axis between both the backbone and side chains.
Figure 10.4.
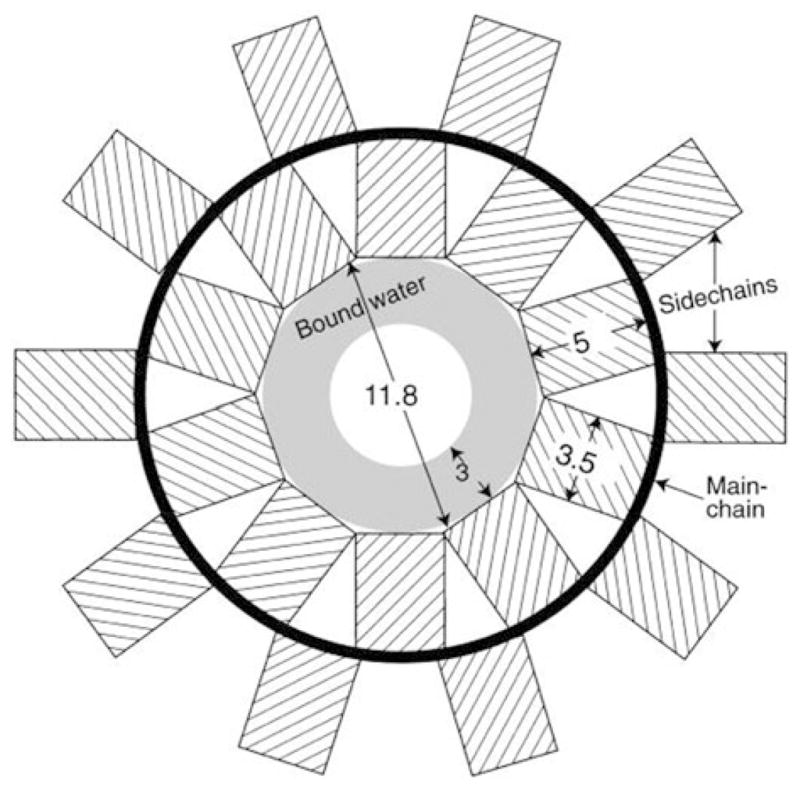
Diagram representation of the β-helical water-filled nanotube model proposed by Perutz for the amyloid structure. β-strands become a cylinder that is 31 Å in diameter (thick black line) with side chains alternatively pointing into and out of the cylinder (hashed rectangles). The inner channel is 11.8 Å and is filled with water, and the 3 Å layer is a structural part of the model. Reproduced with permission from (Perutz et al. 2002)
This model was extended to fit existing x-ray diffraction data on fibrils formed by amyloid domains of Htt and yeast prion Sup35 proteins, as well as α-synuclein and amyloid β fragments (Perutz et al. 2002). However, recently an alternative interpretation of the same data was suggested (Sikorski and Atkins 2005). In this model, β-hairpins are tightly stacked on top of each other, 8.3 Å apart, to form the cross-β structure—the superpleated model. Subsequent molecular modeling studies revealed that the proposed β-helix model is unstable (Zanuy et al. 2006), and suggested that triangular β-helices with a dry fibril core might be a more suitable model (Stork et al. 2005). However, more recent simulations suggest that the β-helix model can be stable if the glutamine repeat is larger than 30 residues (Ogawa et al. 2008).
A recent solid-state NMR study provided the fibril structure formed by the D2Q15K2, GK2Q38K2, and GK2Q54K2 peptides with glutamine repeats below, at, and respectively above the 38 polyQ threshold necessary for triggering HD (Schneider et al. 2011). Lateral packing of protofilaments, with approximate widths of 70–80 Å as observed by EM and NMR, favors structural variability in all three fibril preparations. Additionally, solid-state NMR 1H-13C and 1H-15N correlations suggest extensive hydrogen bonding involving glutamine side chains, as well as tight packing of β-sheets, resulting in the close proximity of side chain termini of one β-sheet and the backbone of an adjacent β-sheet. These data validates the steric zipper model (Sawaya et al. 2007) (see subsection 10.3.2 below). Water-edited solid-state NMR spectroscopy employs transfer of proton magnetization from water to protein, to probe solvent accessibility at the amino acid residue level. When applied to these fibrils, the results suggest that the fibrils possess a dry, water-inaccessible core of at least 70–80 Å in diameter, which is incompatible with the water-filled β-helix model. This study also demonstrates that two equally populated conformations for the glutamine side chains exist in all three peptides. It is further shown through 13C/15N-13C correlations that these populations do not belong to two distinct fibril conformations present within the same sample, but rather occur within individual fibrils and even peptide monomers. The authors suggest a model in which peptides comprising 15 sequential glutamine residues form fibrils with single extended β-strands, while longer constructs most likely form a superpleated fibril structure in which each molecule contributes two or more antiparallel β-strands. Figure 10.5 illustrates this model.
Figure 10.5.

Sketches of the proposed polyglutamine fibril model derived from solid-state NMR data. An approximate scale bar is indicated at the top. Minimal repeat units of (a) polyQ15, (b) polyQ38, and (c) polyQ54 fibrils, assuming similar β-strand lengths, viewed down the fibril axis. (d) Illustration of the superpleated antiparallel cross-β arrangement of monomers in polyQ38 fibrils, viewed down the fibril axis. The fibril repeat unit consists of one GK2Q38K2 molecule that forms two β-strands and contributes to two stacked β-sheets. (Reproduced with permission from Schneider et al. 2011)
10.3.2 Atomic Structure of the GNNQQNY Peptide of the Prion-protein Sup35
One of the most extensive studied glutamine/asparagine rich peptide aggregates to date are those formed by the segment 7–13 of the prion protein Ure35, for which the first high-resolution atomic structure of an amyloid-like fibril was determined by the Eisenberg group (Nelson et al. 2005). This work on the crystal structure of GNNQQNY was later extended to crystallographic studies of other small peptide domains from amyloid-forming proteins, including insulin, islet amyloid polypep-tide, and amyloid-β (Sawaya et al. 2007). The common feature of all structures is the self-complementarity in their binding within the crystal. This is denoted by “dry interfaces” in Fig. 10.6 and suggests that polar zippers are an example of a more general steric zipper motif, for which a total of eight structural classes have been assigned (Sawaya et al. 2007). These classes are defined by the parallel or anti-parallel arrangement of adjacent β-sheets as well as the parallel arrangement of adjacent strands within β-sheets, and by the inter-packing of adjacent sheets with the same or opposed surfaces.
Figure 10.6.
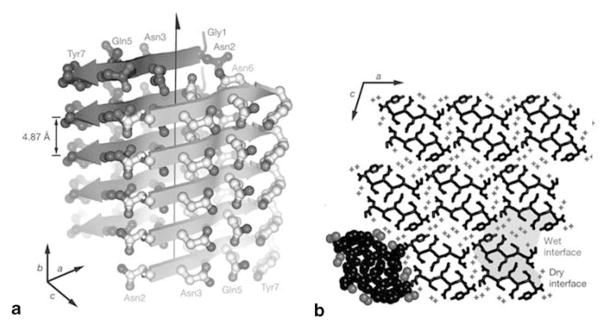
The X-ray structure of monoclinic GNNQQNY crystals according to (Nelson et al. 2005). In (a) the fibril structure is depicted with individual β-strands represented as pointed arrows and side chains represented as balls and sticks. The polar zipper with stacked asparagines (Asn2 and Asn3) and glutamines (Gln5) can be observed. In (b) a transversal section through the crystal is shown and the “wet” and “dry” interfaces are highlighted. (Reproduced with permission from Nelson et al. 2005)
At the same time, it is not known to what extent the crystal structures of these small peptides relate to the amyloid fibrils that they form. This gap in knowledge was filled by solid-state NMR studies performed by the group of Griffin. In an initial study (van der Wel et al. 2007), both monoclinic and orthorhombic crystals and three different fiber morphologies were prepared and analyzed by solid-state NMR. Interestingly, by determining the 13C and 15N chemical shifts for isotopically enriched crystalline and fibrillar preparations, the authors found that all five structures were different in small, but distinct details. In particular, all structures consist of parallel and in-register extended β-sheets, but the mobility of the aromatic tyrosine ring differs among the structures. This initial study was further expanded (van der Wel et al. 2010) to determine the complete assignment of 13C and 15N chemical shifts within the three fibril morphologies, and the precise backbone torsion angles ψ and φ, as well as the determination of the intermolecular contacts. All three GNNQQNY fibrils were found to consist of parallel and in-register β-sheets, with one form exhibiting a highly localized backbone distortion.
A third study by the Griffin group analyzed polymorphism of GNNQQNY fibrils (Lewandowski et al. 2011). Several fibrilization conditions were developed (involving temperature variations and/or changes in solution pH), and the resulting fibrils were investigated by electron microscopy (EM). EM images, supported by solid-state NMR experiments with samples of selectively [13C,15N]-labeled GN-NQQNY peptides, found that the protofilaments within the fibril were tightly associated with each other in specific ways to form mature fibrils, greatly increasing the structural complexity of the amyloid fibrils. It seems counterintuitive that these assemblies have no apparent dependence on fibrilization conditions, which is in contrast with other amyloid fibrils. For example, Aβ fibrils formed under quiescent and agitated conditions are very different (Petkova et al. 2005), and furthermore, the fibrils formed by de novo designed peptides at different pH also exhibit different polymorphisms.
10.3.3 Fibrils Formed by Prion Domains of Ure2p
The structure determination of amyloids formed by large glutamine- and asparagine-rich proteins of biological relevance is difficult. These fibrils typically exhibit structural heterogeneity which make crystallization virtually impossible and also negatively affects the quality of solid-state NMR spectra. The large number of glutamines and asparagines that directly participate in β-sheet formation also results in severe spectral overlap, further impeding solid-state NMR studies. Selective labeling of individual amino acids is not possible, as these proteins cannot be produced by solid phase peptide synthesis (the yields are impractical for large proteins); but rather, the proteins have to be recombinantly expressed in bacteria. Electron paramagnetic resonance (EPR) studies are more appropriate in this case because cysteine mutants required for nitroxyl spin labeling can be easily produced during protein expression in bacteria. The majority of structural studies on large glutamine/asparagine rich proteins to date have been performed on the yeast prion protein Ure2p. The results described below qualitatively identify and describe the Ure2p amyloid core, but do not provide atomic level detail about its structure.
Guo (Ngo et al. 2011) recently performed a detailed EPR investigation of the Ure2p amyloid core, involving residues 1–89 (Ure2p(1–89)). This protein fragment was expressed as a fusion with the non-amyloidogenic M domain of the yeast prion protein Sup35 (residues 124–253) to increase solubility. The resulting construct forms typical amyloid fibrils that are 12 nm wide, and EPR spectroscopy demonstrated that the entire sequence is involved in the formation of an amyloid core with a parallel in-register β structure. In particular, 15 samples with a nitroxyl spin label at every fifth residue, from position 5 to 75, exhibit a single-line EPR spectra that are typical for parallel in-register amyloid fibrils (Margittai and Langen 2008).
In addition, EPR data shows that the Ure2(30–65) segment is more compact and less exposed to water than the rest of the fibril core. For example, careful quantitative analysis of spin exchange frequencies obtained from spectral simulations reveals that the typical 4.7 Å separation between the β-strands is smaller by 0.1 Å in this region. The associated increase in the stability of the Ure2(30–65) segment is further supported by measurements of reduced side chain local mobility in the presence of 3M of the denaturant guanidine hydrochloride. Finally, reduced solvent accessibility was also found for the Ure2(30–65) domain by measuring the spin exchange frequency between the spin label and a large paramagnetic reagent present in solution.
Solid-state NMR experiments have been performed on the Ure2p amyloid by Tycko (Baxa et al. 2007; Chan et al. 2005). An initial study involved the Ure2p(10–39) segment (Chan et al. 2005)-the most highly conserved sequence in the prion domain of Ure2p, which was previously shown to form amyloid like fibrils (Baxa et al. 2005; Kajava et al. 2004). The parallel and in-register arrangement of β-strands within these fibrils was demonstrated by solid-state NMR experiments that precisely measured the 4.7 Å inter-strand separation in samples selectively labeled with 13C at the methyl group of alanine 15 and the carbonyl group of phenylalanine 37. This study (Chan et al. 2005) also experimentally demonstrated for the first time (in parallel with the x-ray analysis of the GNNQQNY crystals (Nelson et al. 2005)) the existence of hydrogen bonds between the glutamine side chains, which represents a key structural element of polar zippers (Perutz et al. 1993). For this purpose, a novel solid-state NMR technique to measure inter-molecular side chain contacts between the 13C-carbonyl and 15N-amine of glutamine 18 was devised.
Finally, a quantitative analysis of solid-state NMR 13C spectra was performed for samples of Ure2p(10–39) fibrils in the following states: (1) hydrated, centrifuged fibril pellet, (2) dry, lyophilized fibrils, and (3) lyophilized and subsequently rehydrated fibrils. In all cases, both the frequency and the line width of the 13C signals were preserved, except for a small increase in the line width for the lyophilized sample, due to the inhomogeneous broadening induced by local static structural disorder. This strongly supports the idea that hydration does not affect the structure of the Ure2p(10–39) fibrils and is incompatible with the model of water-filled nanotubes (Perutz et al. 2002), which would collapse upon water removal.
This work was later extended to fibrils formed by Ure2p(1–89)—the entire prion domain of Ure2p (Baxa et al. 2007). These fibrils were shown to also adopt an in-register, parallel structure that permits polar zipper interactions between the glutamine and asparagine side chains. An extensive analysis of 13C and 15N line widths in uniformly labeled and fully hydrated Ure2p(1–89) fibrils strongly supports a higher degree of structural variability when compared to fibrils formed by residues 218–289 of the HET-s prion protein (Van Melckebeke et al. 2010; Wasmer et al. 2008). The authors suggest that HET-s has biologically evolved to form amyloid fibrils, and consequently, these fibrils exhibit an unusually high degree of structural order among amyloids. Quantitative analysis of asparagine-specific NMR signals also reveals that the asparagine-rich domain, Ure2p(44–76), may not participate in the formation of a compact β-sheet structure. A molecular explanation may reside in the shorter side chain length of asparagine versus glutamine: only polyQ segments may be able to form networks of polar zippers with optimal geometry in cross-β structures (Baxa et al. 2007).
Subsequent studies involving full-length Ure2p also established a partial degree of structural disorder in fibrils formed under physiological-like conditions (Kryndushkin et al. 2011; Loquet et al. 2009). In addition, these studies demonstrate that the Ure2p functional C-terminal domain remains very well-structured and unchanged within the fibrils and is not part of the Ure2p amyloid core. This is supported by the fact that solid-state NMR spectra of the fibrillar full-length Ure2p and the crystalline Ure2p(70–354) C-terminal domain share a very high degree of similarity (Loquet et al. 2009). In addition, since solid-state NMR experiments only reveal signals from the remaining β-sheet fibril core, the Ure2p C-terminal domain was shown to be completely accessible to and removed by proteases in fibrillar samples of full-length Ure2p (Kryndushkin et al. 2011).
All experimental data on the atomic structure of Ure2p can be summarized into a model schematically shown in Fig. 10.7. In sharp contrast to the β-helix model (Perutz et al. 2002), the amyloid core is formed by compact, dry, parallel and in-register β-sheets of the N-terminal prion domain that is folded into a β-serpentine. The functional C-terminus is not part of the amyloid core and maintains the same structure as in the non-amyloid form of Ure2p. The exact number and distribution of the β-strands within the Ure2p prion domain is currently not known. As a result, several models for Ure2p amyloid were actually proposed. One model proposes that the β-serpentine fold of Ure2p prion domain contains nine β-strands (Kajava et al. 2004), while another model suggests the presence of five β-strands (Kryndushkin et al. 2011). Additional experiments are required to improve our current understanding of the Ure2p amyloid structure.
Figure 10.7.
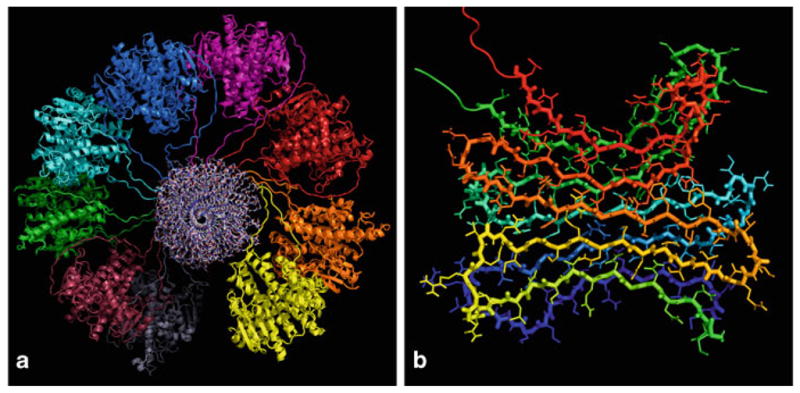
a Proposed structural model for full-length Ure2p fibrils. In this view along the fibril axis, the amyloid core formed by the prion domain is in the center (residues 1–81 shown in the all-atom representation) and the C-terminal domains are folded as dimers and decorate the fibril laterally (residues 82–354 shown in cartoon representation). b A detailed view of the β-serpentine fold of the Ure2p prion domain: two molecules in a parallel, in-register β-sheet arrangement are shown, each monomer encompassing five β-strands. (Reproduced with permission from Kryndushkin et al. 2011)
10.4 Morphology, Structure and Mechanical Properties of Amyloid Fibrils
The interest in large amyloid aggregates, particularly nanofibrils, remains high for several reasons. First, the plaque deposits consist of primarily fibrils; therefore structural analysis of these aggregates is needed to understand their formation. Second, elucidating the mechanism of fibril growth aids in the understanding of the mechanism of protein self-assembly into nanoaggregates. Finally, amyloid fibrils are natural nanomaterials, with a number of interesting physico-chemical properties, such as a relatively high mechanical stability, which make fibrils an attractive resource for a number of practical applications, such as biomaterials. The recent findings that fibrils are non-toxic to cells relieves the concern that such applications could be biohazardous. The subsections below outline specifics for each of these features of amyloid fibrils.
10.4.1 Morphology of Amyloid Fibrils
Structural organization of amyloid fibrils is predominantly studied by nanoimaging techniques, such as electron microscopy (EM) and AFM (reviewed in (Lyubchenko et al. 2006; Stromer and Serpell 2005)). Figure 10.8 shows AFM images of fibrils formed by spontaneous aggregation of α-synuclein protein in an aqueous solution. The sample contains both long fibrils (indicated with 2), and short, thin fibrils (indicated with 1). These thin fibrils are protofibrils of the protein, which associate to form mature thick and straight fibrils. The association is evidenced by the section indicated by the arrow in the long fibril 2, on the top of the figure. Additional evidence comes from fibrils, indicated by 3 that have a clear twisted morphology with frayed ends. The smooth and twisted morphology of the fibrils suggests that different assembly pathways exist for the fibrils formation, and/or it indicates the difference in the structure of the protofibrils themselves.
Figure 10.8.

AFM images of α-synuclein fibrils. Figures 1, 2, and 3 indicate protofibrils, thick smooth fibrils and twisted fibrils respectively. Arrow points to the bulge region of the thick fibril. Images were taken in air with Nanoscope IIIa AFM operating in Tapping mode
A rather straight shape is a common feature of amyloid fibrils, however fibrils formed by Htt Exon 1 (Q67) have an alternate morphology with extensive branching (Dahlgren et al. 2005). A gallery of four aggregates with different shapes is shown in Fig. 10.9. It was suggested that the fibril branching was likely caused by structural features close to the polyQ region, but not to the C-terminus of Htt. Branched morphology is a distinct feature of the Htt fibrils that differ them dramatically from other filamentous aggregates, such as those formed by amyloid β peptide and α-synuclein. The majority of models for the fibrils formation fall into a category of in-register, parallel intermolecular alignment of monomers, when strands form two or more parallel β-sheets through intermolecular hydrogen bonding (e.g. Ma and Nussinov 2002; Petkova et al. 2002; Tycko 2003). These models interpret the amyloid-type fibril structure to be long and straight with an un-branched morphology (see Fig. 10.8), but they do not explain branch morphology and thickness variations observed for the huntingtin fibrils. The explanation provided in (Dahlgren et al. 2005) suggests that Htt fibrils can grow in two directions—perpendicular and parallel to the fibrils structure proposed in (Der-Sarkissian et al. 2003). The parallel orientation of flat β-sheet structures allows for greater variations in the number of stacked sheets, and thus predicts the irregularity in the fiber thickness. The bulges, which are formed by the parallel orientation of the fibril axis, function as buds for growing new fibrils. In the majority of amyloids, such as fibrils formed by amyloid β peptides and α-synuclein, the lateral interaction between β-sheets is relatively weak, therefore the stacked arrangement of the planes is most likely the predominant conformation of the fibril. The formation of branched structures suggests that in the case of Htt protein, both lateral and stacked interactions are comparable; therefore, lateral interactions leads to the arrangement of the planes along the fibril’s axis, in parallel with the stacking arrangement of the planes, which leads to branching.
Figure 10.9.
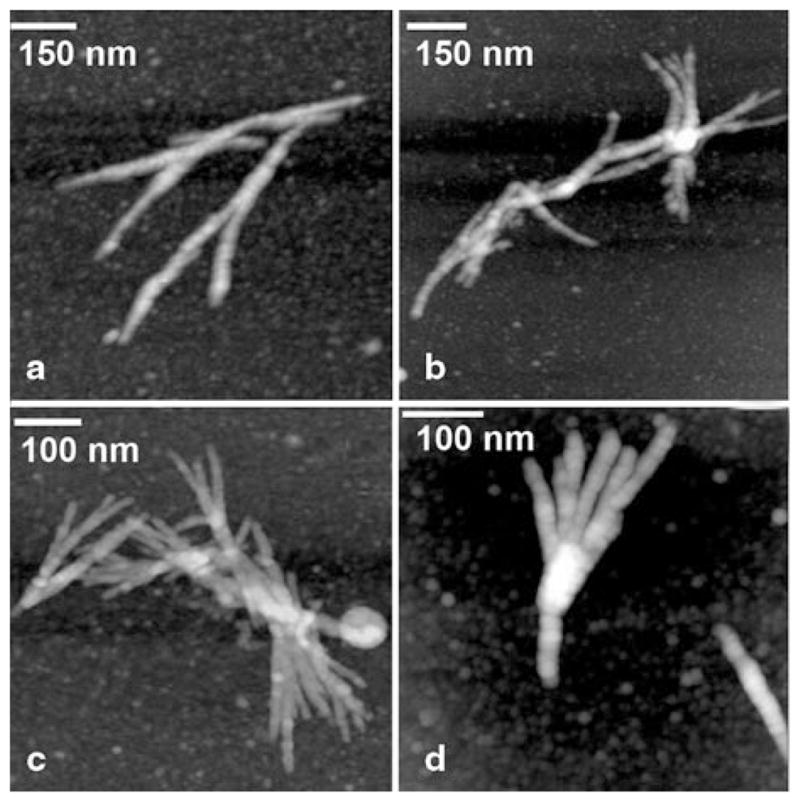
AFM images of Htt (Q7) aggregates. Images were taken in air with Nanoscope IIIa AFM operating in Tapping mode
10.4.2 Mechanics and Stability of Amyloid Fibrils
This and the following subsections outlines the results obtained on various amyloid fibrils, and although the mechanical properties of Htt and other polyglutamine fibrils have not been studied to date, some of the mechanical properties can be translated to Htt fibrils.
There is growing evidence supporting the conclusion that the ability to form fibrils is a generic property of the polypeptide chain; i.e., many proteins, perhaps all, are potentially capable of forming amyloid fibrils under appropriate conditions (Dobson 1999, 2004a). This means that amyloidogenic polypeptides are unrelated in terms of sequence or structure. The experiments which probed aggregated amyloids with antibodies demonstrated that there is a class of structure specific antibodies that recognize protein aggregates (e.g. fibrils), but do not bind to a monomeric form of the same protein (O’Nuallain and Wetzel 2002). Importantly, among those structure-specific antibodies are those that distinguish different aggregated morphologies of the same protein (Glabe 2004; Kayed et al. 2003). At the same time, it was also established that some antibodies were able to recognize aggregates formed by different proteins. The striking conclusion based on these experiments was that the same structural morphologies formed by different proteins are recognized by the same structure-specific antibody. These studies lead to the intriguing conclusion that aggregated proteins might have common structural motifs, even if they are not structurally similar prior to fibrillation: being rich in β-sheet, α-helix, β-helix, contain both α-helices and β-sheets, globular proteins with rigid 3D-structure or belong to the realm of natively unfolded (or intrinsically unstructured) proteins (Uversky and Fink 2004). Despite these differences, amyloid fibrils have similar structural features, such as fibril straightness.
Estimates of fibril stiffness can be made based on their morphology and the use of general principles of polymer statistics. Such an approach was applied initially in (Lyubchenko et al. 2006). According to the polymer statistics, the flexibility of linear polymers is characterized by the persistence length: the larger the persistence length, the stiffer the polymer (Flory 1953). For example, the persistence length of the DNA double helix (diameter of 2 nm) is ca. 45 nm (Lu et al. 2001), which appears on AFM images as a flexible polymer with straight regions, in the range of 10–20 nm. Amyloid β protofilaments appear with a similar morphology (Lyubchenko et al. 2006). RecA-DNA fibrils are 15 times stiffer, with a persistence length of approximately 700 nm and straight regions as long as 200–300 nm (Lyubchenko et al. 1995). The amyloid fibrils have a comparable width, but they are much straighter and some of them appear as straight as carbon nanofibrils, one of the stiffest fibrils.
The quantitative analysis based on a similar idea for fifty individual amyloid fibrils was performed in (Knowles et al. 2007) and revealed that the stiffness of amyloid fibrils measured by the bending rigidity varied by two orders of magnitude. The stiffest fibrils were made by the short peptide of transthyretin, while the most flexible were fibrils of α-lactabumin. The data for the main amyloids, such as amyloid β and prion peptides of different lengths, were also converted into Young’s modulus and varied between 2 and 14 GPa. Note for comparison that Young’s modulus for such biological proteinaceous materials as dragline silk, collagen and keratin is in the range of 10 GPa, approaching the stiffness of bones and concrete (Knowles and Buehler 2011). The combination of AFM imaging with vibrational spectroscopy (vibrational sum-frequency generation, VSFG) revealed an interesting correlation between the stiffness of amyloid fibrils formed by β-lactoglobulin and the secondary structure of the fibril sample (van den Akker et al. 2011). It was found that straight fibrils have a 100 % β-sheet conformation, whereas softer fibrils have a substantial α-helical content.
Experimental approaches for measuring fibril stiffness are primarily based on direct probing of isolated fibrils with the AFM tip, in which the AFM cantilever is used as a sensor to measure the sample stiffness. Young’s modulus measurement for individual amyloid fibrils formed by insulin was 3.3 ± 0.4 GPa, which is in the range of the above-mentioned estimates (Smith et al. 2006). Similar approaches applied to α-synuclein fibrils, produced Young’s moduli values within the range of 1.3–2.1 GPa (Sweers et al. 2011). Note that these measurements were perfromed with the use of three different single-point nanoindentation approaches, which resulted in close values. Altogether, the theoretical analysis and experimental studies lead to the conclusion that amyloid fibrils have a high mechanical stiffness. These characteristics are important for potential biotechnological applications, including the use of amyloids as biomaterials. This issue is discussed in the recent review and references therein (Sweers et al. 2011).
10.4.3 Interaction within Amyloid Fibrils
The studies described above provide firm evidence of the high stiffness of amyloid fibrils, but they do not answer the question of how strong the interactions within the fibrils are. The AFM approach schematically shown in Fig. 10.10 was proposed to measure the strength of interprotein interactions within fibrils (Kellermayer et al. 2005).
Figure 10.10.
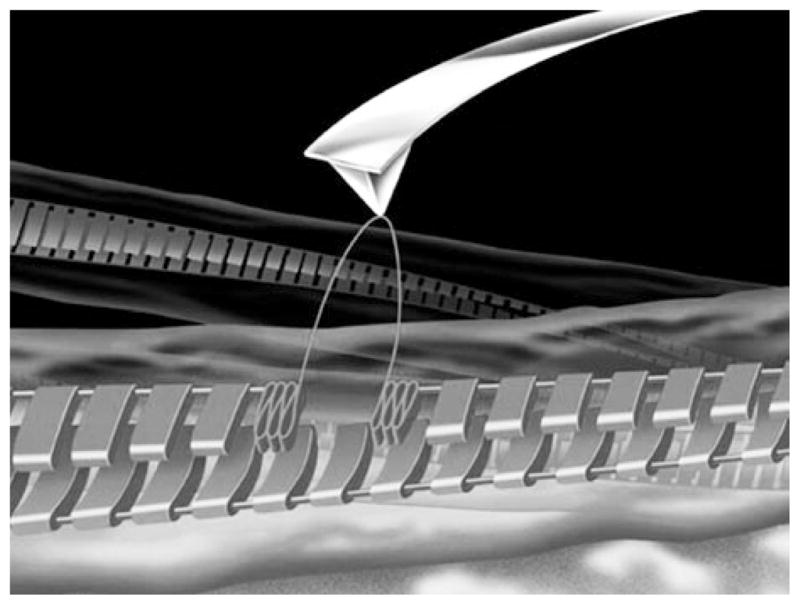
Schematic for the AFM probing experiments on measuring interactions within the fibril. The AFM tip picks a protein monomer inside the fibril shown as a periodic ladder and unfolds it upon the pulling step. (Reproduced with permission from (Mostaert et al. 2006)
In these experiments, the AFM tip was pressed against the fibril selected within the previously imaged area to create a strong interaction between the tip and the selected area on the fibril. The tip was pulled away from the fibril to measure the forces stabilizing the structure of the fibril and/or the unzipping of the protofibril within the fibril.
Initial experiments performed with fibrils formed by amyloid β peptides 1–40 and 25–35 showed that this approach is capable of probing the mechanics of amyloid fibrils (Kellermayer et al. 2005). Different sets of force spectroscopy data obtained for these peptides can be explained by different unzipping mechanisms of β-sheets for both peptides. Acetylation of the peptide led to a dramatic decrease of the rupture forces, although both native and acetylated peptides form morphologically undistinguishable fibrils (Karsai et al. 2005). However, pulling experiments performed with fibrils formed by α-synuclein protein revealed problems with the data interpretation and the experimental setup (Lyubchenko et al. 2006). First, the fibril has to be strongly attached to the surface; otherwise sliding of the filament can be interpreted as a long unwrapping event. Second, performing AFM imaging both prior to and after probing, enables one to identify the damaged area of the fibrils, which can be used for structural interpretation of the pulling experiments (Mostaert et al. 2006). These conclusions are illustrated in Fig. 10.11 in which pulling experiments for α-synuclein fibrils at six selected points were performed; they are numbered in Fig. 10.11a and the probing points are indicated with crosses and arrows. Note that the fibrils were covalently immobilized on the surface. The AFM image in Fig. 10.11b shows the results of a pulling experiment. Unambiguously identified gaps on fibrils are indicated in image b with white arrows and numbered according to the previous image (a). The filaments were removed from the fibril from points 4 to 6. The force measurements (Fig. 10.11c) revealed a characteristic extension-rupture pattern, which is an indication of fibril unzipping (extension part of the force curve), followed by the rupture event. In contrast to the results of (Kellermayer et al. 2005), extended plateaus on the force curves were not observed. However, such force curves were detected in the case when the fibrils were deposited on the functionalized APS-surface without cross-linking (Fig. 10.11d). The values of rupture forces in these experiments range between 100 pN and 1.5 nN, suggesting that in addition to unwrapping of individual proteins, multiple rupture events can occur. The use of low forces during the approach steps is beneficial for pulling individual proteins, enabling the authors to narrow the range of rupture forces to 108 ± 35 pN (Mostaert et al. 2009).
Figure 10.11.

AFM pulling experiments of α-synuclein fibrils. Pulling points (1–6) are indicated with arrows in image (a) obtained before pulling. The fibrils were covalently immobilized on the surface. Inset (i) illustrates the pulling approach. Image (b) was taken after pulling. Damaged sections of fibrils in image (b) are indicated with white arrows and numbered according to image (a). (c) Force curves for pulling the fibrils cross-linked to the APS-mica surface (trigger—100pN, dwell time—2s). The tip spring constant, k = 67.31 pN/nm. (d) Force curves for pulling of the non-covalently bound fibrils (trigger 500 pN, dwell time 2 s). The tip spring constant, k = 51.69 pN/nm. Pulling was done in PBS buffer. (Reproduced with permission from Lyubchenko et al. 2006)
The approach proposed in (Dong et al. 2010) allows one to measure the strength of interaction within the individual fibril. In this approach, the monomeric form of the yeast prion protein was immobilized on the surface. The fibril assembled by the same protein was attached to the monomers at the end of the fibril and the strength of the construct was probed by pulling the fibril at another end with the use of an optical tweezers puller. The strength of the noncovalent monomer-fibril interaction was more than 250 pN, the limit of the puller, but the use of guanidine hydrochloride allowed the authors to stretch and rupture the fibrils. The main assumption for the proposed methodology was that the monomeric protein is capable of strong binding to the preformed fibrils. This assumption has recently been confirmed by detailed atomistic calculations in which the interaction of a monomeric amyloid β peptide with fibrils was modeled (Straub and Thirumalai 2011). According to these computer simulations, a non-structured monomer undergoes a structural transition to fit the conformation of monomers within the oligomer, leading to the elongation of the oligomer. Recent molecular dynamic simulations showed that the conformational fitting mechanism also works when monomers interact with dimers, the shortest oligomer (Rojas et al. 2011).
These recent advances in computer simulation highlighted an important property of amyloid protein—the inducible conformational transition enabling the proteins to adopt the conformation favorable for the aggregates’ growth. In fact, this property of amyloid proteins was hypothesized earlier (McAllister et al. 2005) and resulted in a novel technique for probing misfolded states of proteins (see review (Lyubchenko et al. 2010) and references therein). In this approach, protein monomers are covalently immobilized on the AFM tip and substrate, and the interaction between the proteins is measured by multiple approach-retraction cycles of the AFM force spectroscopy operation mode. It was shown that at conditions facilitating protein aggregation, strong interactions between the proteins are detected. Moreover, this technique allows one to estimate the lifetime of the transient dimers, resulting in measurements as long as seconds. Compared to the conformational dynamics of monomeric amyloid peptides and protein that occurs in the nanosecond time scale, the formation of dimers leads to an enormous stabilization of the protein state in which they strongly interact. These findings suggest that the conformational fit of amyloid proteins occurs at the monomeric level, leading to the formation of mis-folded dimers with elevated stability. These studies suggest that the formation of stable dimers is a key property of amyloid proteins and that their formation is a trigger for further aggregation processes.
10.5 Conclusions
Structural studies performed with various techniques provided a wealth of information on the structure of amyloid fibrils at different levels. They revealed numerous similarities in the structure of the fibrils, but also revealed differences that may reflect their function. The high mechanical stability of amyloids is a property that attracted attention to these fibrils, as potential biomaterials with various applications. The use of novel nanoimaging approaches was instrumental in understanding the mechanism of fibril formation and their mechanical properties. The progress in computer simulation provided atomistic views of fibril assembly and the role of weak bonds in the formation of mechanically strong periodic filaments. Undoubtedly, further experimental studies of amyloid protein self-assembly, with the use of novel nanotools, will aid in understanding the mechanisms of the development of diseases triggered by protein aggregation, and will eventually lead to effective preventative and therapeutic treatments of amyloid diseases.
Acknowledgments
The work was supported by National Institutes of Health Grants (1P01GM091743–01A1 and 1 R01 GM096039-01A1), U.S. Department of Energy Grant DE-FG02-08ER64579, National Science Foundation (EPS—1004094) and the Nebraska Research Initiative grant to Y.L.L.
Abbreviations
- Htt
Huntingtin protein
- Aβ
Amyloid-beta
- AFM
Atomic Force Microscopy
- NMR
Nuclear magnetic resonance
- EPR
Electron paramagnetic resonance
Contributor Information
Yuri L. Lyubchenko, Email: ylyubchenko@unmc.edu, Department of Pharmaceutical Sciences, Nanoimaging Core Facility College of Pharmacy, University of Nebraska Medical Center, COP 1012, 986025 Nebraska Medical Center, Omaha, NE 68198-6025, USA
Alexey V. Krasnoslobodtsev, Email: akrasnos@unmc.edu, Department of Pharmaceutical Sciences, Nanoimaging Core Facility College of Pharmacy, University of Nebraska Medical Center, COP 1012, 986025 Nebraska Medical Center, Omaha, NE 68198-6025, USA
Sorin Luca, Email: sluca@unmc.edu, Department of Pharmaceutical Sciences, Nanoimaging Core Facility College of Pharmacy, University of Nebraska Medical Center, COP 1012, 986025 Nebraska Medical Center, Omaha, NE 68198-6025, USA.
References
- Baxa U, Cheng N, Winkler DC, Chiu TK, Davies DR, Sharma D, Inouye H, Kirschner DA, Wickner RB, Steven AC. Filaments of the Ure2p prion protein have a cross-beta core structure. J Struct Biol. 2005;150:170–179. doi: 10.1016/j.jsb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Baxa U, Wickner RB, Steven AC, Anderson DE, Marekov LN, Yau W-M, Tycko R. Characterization of beta-sheet structure in Ure2p1–89 yeast prion fibrils by solid-state nuclear magnetic resonance. Biochemistry. 2007;46:13149–13162. doi: 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- Bernacki JP, Murphy RM. Model discrimination and mechanistic interpretation of kinetic data in protein aggregation studies. Biophys J. 2009;96:2871–2887. doi: 10.1016/j.bpj.2008.12.3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Thakur AK, Chellgren VM, Thiagarajan G, Williams AD, Chellgren BW, Creamer TP, Wetzel R. Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol. 2006;355:524–535. doi: 10.1016/j.jmb.2005.10.053. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya AM, Thakur AK, Wetzel R. Polyglutamine aggregation nucleation: thermodynamics of a highly unfavorable protein folding reaction. Proc Natl Acad Sci USA. 2005;102:15400–15405. doi: 10.1073/pnas.0501651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JCC, Oyler NA, Yau W-M, Tycko R. Parallel beta-sheets and polar zippers in amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p. Biochemistry. 2005;44:10669–10680. doi: 10.1021/bi050724t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Berthelier V, Hamilton JB, O’Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002a;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- Chen S, Ferrone FA, Wetzel R. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci USA. 2002b;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren PR, Karymov MA, Bankston J, Holden T, Thumfort P, Ingram VM, Lyubchenko YL. Atomic force microscopy analysis of the Huntington protein nanofibril formation. Nanomedicine. 2005;1:52–57. doi: 10.1016/j.nano.2004.11.004. [DOI] [PubMed] [Google Scholar]
- De Baere I, Liu L, Moens L, Van Beeumen J, Gielens C, Richelle J, Trotman C, Finch J, Gerstein M, Perutz M. Polar zipper sequence in the high-affinity hemoglobin of Ascaris suum: amino acid sequence and structural interpretation. Proc Natl Acad Sci USA. 1992;89:4638–4642. doi: 10.1073/pnas.89.10.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der-Sarkissian A, Jao CC, Chen J, Langen R. Structural organization of alpha-synuclein fibrils studied by site-directed spin labeling. J Biol Chem. 2003;278:37530–37535. doi: 10.1074/jbc.M305266200. [DOI] [PubMed] [Google Scholar]
- Digambaranath JL, Campbell TV, Chung A, McPhail MJ, Stevenson KE, Zohdy MA, Finke JM. An accurate model of polyglutamine. Proteins. 2011;79:1427–1440. doi: 10.1002/prot.22970. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Experimental investigation of protein folding and misfolding. Methods. 2004a;34:4–14. doi: 10.1016/j.ymeth.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004b;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Dong J, Castro CE, Boyce MC, Lang MJ, Lindquist S. Optical trapping with high forces reveals unexpected behaviors of prion fibrils. Nat Struct Mol Biol. 2010;17:1422–1430. doi: 10.1038/nsmb.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan L, Li J, Badilla CL, Berne BJ, Fernandez JM. Single homopolypeptide chains collapse into mechanically rigid conformations. Proc Natl Acad Sci USA. 2009;106:12605–12610. doi: 10.1073/pnas.0900678106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- Flory P. Principles of Polymer Chemistry. Cornell University Press; New York: 1953. [Google Scholar]
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genetics. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- Gillian B. Huntingtin aggregation and toxicity in Huntington’s disease. The Lancet. 2003;361:1642–1644. doi: 10.1016/S0140-6736(03)13304-1. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem Sci. 2004;29:542–547. doi: 10.1016/j.tibs.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat rev Neurosci. 2000;1:109–115. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- Hofrichter J, Ross PD, Eaton WA. Kinetics and mechanism of deoxyhemoglobin S gelation: a new approach to understanding sickle cell disease. Proc Natl Acad Sci USA. 1974;71:4864–4868. doi: 10.1073/pnas.71.12.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- Kajava AV, Baxa U, Wickner RB, Steven AC. A model for Ure2p prion filaments and other amyloids: the parallel superpleated beta-structure. Proc Natl Acad Sci USA. 2004;101:7885–7890. doi: 10.1073/pnas.0402427101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat Struct Mol Biol. 2011;18:328–336. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsai A, Nagy A, Kengyel A, Martonfalvi Z, Grama L, Penke B, Kellermayer MS. Effect of lysine-28 side-chain acetylation on the nanomechanical behavior of alzheimer amyloid beta25–35 fibrils. J Chem Inf Model. 2005;45:1641–1646. doi: 10.1021/ci0501701. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kellermayer MS, Grama L, Karsai A, Nagy A, Kahn A, Datki ZL, Penke B. Reversible mechanical unzipping of amyloid beta-fibrils. J Biol Chem. 2005;280:8464–8470. doi: 10.1074/jbc.M411556200. [DOI] [PubMed] [Google Scholar]
- Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary structure of Huntingtin amino-terminal region. Structure. 2009;17:1205–1212. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles TP, Buehler MJ. Nanomechanics of functional and pathological amyloid materials. Nature nanotechnology. 2011;6:469–479. doi: 10.1038/nnano.2011.102. [DOI] [PubMed] [Google Scholar]
- Knowles TP, Fitzpatrick AW, Meehan S, Mott HR, Vendruscolo M, Dobson CM, Welland ME. Role of intermolecular forces in defining material properties of protein nanofibrils. Science. 2007;318:1900–1903. doi: 10.1126/science.1150057. [DOI] [PubMed] [Google Scholar]
- Kryndushkin DS, Wickner RB, Tycko R. The core of Ure2p prion fibrils is formed by the N-terminal segment in a parallel cross-β structure: evidence from solid-state NMR. J Mol Biol. 2011;409:263–277. doi: 10.1016/j.jmb.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzewska J, Tanaka M, Burston SG, Melki R. Biochemical and functional analysis of the assembly of full-length Sup35p and its prion-forming domain. J Biol Chem. 2007;282:1679–1686. doi: 10.1074/jbc.M608110200. [DOI] [PubMed] [Google Scholar]
- Lewandowski JzR, Van Der Wel PCA, Rigney M, Grigorieff N, Griffin RG. Structural Complexity of a Composite Amyloid Fibril. J Am Chem Soc. 2011;133:14686–14698. doi: 10.1021/ja203736z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loquet A, Bousset L, Gardiennet C, Sourigues Y, Wasmer C, Habenstein B, Schütz A, Meier BH, Melki R, Böckmann A. Prion fibrils of Ure2p assembled under physiological conditions contain highly ordered, natively folded modules. Journal of Molecular Biology. 2009;394:108–118. doi: 10.1016/j.jmb.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Lu Y, Weers B, Stellwagen NC. DNA persistence length revisited. Biopolymers. 2001;61:261–275. doi: 10.1002/bip.10151. [DOI] [PubMed] [Google Scholar]
- Lyubchenko YL, Jacobs BL, Lindsay SM, Stasiak A. Atomic force microscopy of nucleoprotein complexes. Scanning Microsc. 1995;9:705–724. discussion 724–707. [PubMed] [Google Scholar]
- Lyubchenko YL, Sherman S, Shlyakhtenko LS, Uversky VN. Nanoimaging for protein misfolding and related diseases. J Cell Biochem. 2006;99:52–70. doi: 10.1002/jcb.20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubchenko YL, Kim BH, Krasnoslobodtsev AV, Yu J. Nanoimaging for protein misfolding diseases. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2:526–543. doi: 10.1002/wnan.102. [DOI] [PubMed] [Google Scholar]
- Ma B, Nussinov R. Stabilities and conformations of Alzheimer’s beta-amyloid peptide oligomers (Abeta 16–22, Abeta 16–35, and Abeta 10–35): Sequence effects. Proc Natl Acad Sci USA. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, MacFarlane H, Jenkins B, Anderson MA, Wexler NS, Gusella JF, Bates GP, Baxendale S, Hummerich H, Kirby S, North M, Youngman S, Mott R, Zehetner G, Sedlacek Z, Poustka A, Frischauf A-M, Lehrach H, Buckler AJ, Church D, Doucette-Stamm L, O’Donovan MC, Riba-Ramirez L, Shah M, Stanton VP, Strobel SA, Draths KM, Wales JL, Dervan P, Housman DE, Altherr M, Shiang R, Thompson L, Fielder T, Wasmuth JJ, Tagle D, Valdes J, Elmer L, Allard M, Castilla L, Swaroop M, Blanchard K, Collins FS, Snell R, Holloway T, Gillespie K, Datson N, Shaw D, Harper PS. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Margittai M, Langen R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Q Rev Biophys. 2008;41:265–297. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- McAllister C, Karymov MA, Kawano Y, Lushnikov AY, Mikheikin A, Uversky VN, Lyubchenko YL. Protein interactions and misfolding analyzed by AFM force spectroscopy. J Mol Biol. 2005;354:1028–1042. doi: 10.1016/j.jmb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostaert A, Crockett R, Kearn G, Cherny I, Gazit E, Serpell L, Jarvis S. Mechanically functional amyloid fibrils in the adhesive of a marine invertebrate as revealed by Raman spectroscopy and atomic force microscopy. Arch Histol Cytol. 2009;72:199–207. doi: 10.1679/aohc.72.199. [DOI] [PubMed] [Google Scholar]
- Mostaert AS, Higgins MJ, Fukuma T, Rindi F, Jarvis SP. Nanoscale mechanical characterisation of amyloid fibrils discovered in a natural adhesive. J biol Phys. 2006;32:393–401. doi: 10.1007/s10867-006-9023-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo S, Gu L, Guo Z. Hierarchical organization in the amyloid core of yeast prion protein ure2. J Biol Chem. 2011;286:29691–29699. doi: 10.1074/jbc.M111.269092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci USA. 2002;99:1485–1490. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H, Nakano M, Watanabe H, Starikov EB, Rothstein SM, Tanaka S. Molecular dynamics simulation study on the structural stabilities of polyglutamine peptides. Comput Biol Chem. 2008;32:102–110. doi: 10.1016/j.compbiolchem.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Perutz MF. Glutamine repeats as polar zippers: their role in inherited neurodegenerative disease. Mol Med. 1995;1:718–721. [PMC free article] [PubMed] [Google Scholar]
- Perutz MF, Staden R, Moens L, De Baere I. Polar zippers. Curr Biol. 1993;3:249–253. doi: 10.1016/0960-9822(93)90174-m. [DOI] [PubMed] [Google Scholar]
- Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci USA. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz MF, Windle AH. Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature. 2001;412:143–144. doi: 10.1038/35084141. [DOI] [PubMed] [Google Scholar]
- Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci USA. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau W-M, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Robertson AL, Bate MA, Androulakis SG, Bottomley SP, Buckle AM. PolyQ: a database describing the sequence and domain context of polyglutamine repeats in proteins. Nucleic Acids Res. 2011;39:D272–D276. doi: 10.1093/nar/gkq1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas AV, Liwo A, Scheraga HA. A Study of the alpha-Helical Intermediate Preceding the Aggregation of the Amino-Terminal Fragment of the beta Amyloid Peptide (Abeta(1–28)) J Phys Chem B. 2011;115:12978–12983. doi: 10.1021/jp2050993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen AØ, Riekel C, Eisenberg D. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci USA. 1999;96:4604–4609. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Schumacher MC, Mueller H, Nand D, Klaukien V, Heise H, Riedel D, Wolf G, Behrmann E, Raunser S, Seidel R, Engelhard M, Baldus M. Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy. J Mol Biol. 2011;412:121–136. doi: 10.1016/j.jmb.2011.06.045. [DOI] [PubMed] [Google Scholar]
- Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- Sherman DR, Kloek aP, Krishnan BR, Guinn B, Goldberg DE. Ascaris hemoglobin gene: plant-like structure reflects the ancestral globin gene. Proc Natl Acad Sci USA. 1992;89:11696–11700. doi: 10.1073/pnas.89.24.11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski P, Atkins E. New model for crystalline polyglutamine assemblies and their connection with amyloid fibrils. Biomacromolecules. 2005;6:425–432. doi: 10.1021/bm0494388. [DOI] [PubMed] [Google Scholar]
- Smith JF, Knowles TP, Dobson CM, Macphee CE, Welland ME. Characterization of the nanoscale properties of individual amyloid fibrils. Proc Natl Acad Sci USA. 2006;103:15806–15811. doi: 10.1073/pnas.0604035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork M, Giese A, Kretzschmar HA, Tavan P. Molecular dynamics simulations indicate a possible role of parallel beta-helices in seeded aggregation of poly-Gln. Biophys J. 2005;88:2442–2451. doi: 10.1529/biophysj.104.052415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub JE, Thirumalai D. Toward a molecular theory of early and late events in monomer to amyloid fibril formation. Ann Rev Phys Chem. 2011;62:437–463. doi: 10.1146/annurev-physchem-032210-103526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromer T, Serpell LC. Structure and morphology of the Alzheimer’s amyloid fibril. Microsc Res Tech. 2005;67:210–217. doi: 10.1002/jemt.20190. [DOI] [PubMed] [Google Scholar]
- Sweers K, Van Der Werf K, Bennink M, Subramaniam V. Nanomechanical properties of alpha-synuclein amyloid fibrils: a comparative study by nanoindentation, harmonic force microscopy, and Peakforce QNM. Nanoscale Res Lett. 2011;6:270. doi: 10.1186/1556-276X-6-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S, Spiess C, Auyeung W, Joachimiak L, Chen B, Poirier MA, Frydman J. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat Struct Mol Biol. 2009;16:1279–1285. doi: 10.1038/nsmb.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur AK, Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci USA. 2002;99:17014–17019. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nature structural & molecular biology. 2009;16:380–389. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R. Insights into the amyloid folding problem from solid-state NMR. Biochemistry. 2003;42:3151–3159. doi: 10.1021/bi027378p. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Fink AL. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Van DerWel PCA, Lewandowski JR, Griffin RG. Solid-state NMR study of amyloid nanocrystals and fibrils formed by the peptide GNNQQNY from yeast prion protein Sup35p. J Am Chem Soc. 2007;129:5117–5130. doi: 10.1021/ja068633m. [DOI] [PubMed] [Google Scholar]
- Van Der Wel PCA, Lewandowski JR, Griffin RG. Structural characterization of GNNQQNY amyloid fibrils by magic angle spinning NMR. Biochemistry. 2010;49:9457–9469. doi: 10.1021/bi100077x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Melckebeke H, Wasmer C, Lange A, Ab E, Loquet A, Böckmann A, Meier BH. Atomic-resolution three-dimensional structure of HET-s(218–289) amyloid fibrils by solid-state NMR spectroscopy. J Am Chem Soc. 2010;132:13765–13775. doi: 10.1021/ja104213j. [DOI] [PubMed] [Google Scholar]
- Van Den Akker CC, Engel MFM, Velikov KP, Bonn M, Koenderink GH. Morphology and Persistence Length of Amyloid Fibrils Are Correlated to Peptide Molecular Structure. J Am Chem Soc. 2011;133:18030–18033. doi: 10.1021/ja206513r. [DOI] [PubMed] [Google Scholar]
- Venkatraman J, Shankaramma SC, Balaram P. Design of folded peptides. Chemical reviews. 2001;101:3131–3152. doi: 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]
- Vitalis A, Wang X, Pappu RV. Quantitative characterization of intrinsic disorder in polyglutamine: insights from analysis based on polymer theories. Biophysical journal. 2007;93:1923–1937. doi: 10.1529/biophysj.107.110080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FO. Huntington’s disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Walters RH, Murphy RM. Examining polyglutamine peptide length: a connection between collapsed conformations and increased aggregation. Journal of molecular biology. 2009;393:978–992. doi: 10.1016/j.jmb.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RH, Murphy RM. Aggregation kinetics of interrupted polyglutamine peptides. J Mol Biol. 2011;412:505–519. doi: 10.1016/j.jmb.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- Yang L, He HY, Zhang XJ. Increased expression of intranuclear AChE involved in apoptosis of SK-N-SH cells. Neurosci Res. 2002;42:261–268. doi: 10.1016/s0168-0102(02)00005-6. [DOI] [PubMed] [Google Scholar]
- Zanuy D, Gunasekaran K, Lesk AM, Nussinov R. Computational study of the fibril organization of polyglutamine repeats reveals a common motif identified in beta-helices. J Mol Biol. 2006;358:330–345. doi: 10.1016/j.jmb.2006.01.070. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]