

Abstract
MHC class I molecules (HLA-I in humans) present peptides derived from endogenous proteins to CTLs. Whereas the peptide-binding specificities of HLA-A and -B molecules have been studied extensively, little is known about HLA-C specificities. Combining a positional scanning combinatorial peptide library approach with a peptide–HLA-I dissociation assay, in this study we present a general strategy to determine the peptide-binding specificity of any MHC class I molecule. We applied this novel strategy to 17 of the most common HLA-C molecules, and for 16 of these we successfully generated matrices representing their peptide-binding motifs. The motifs prominently shared a conserved C-terminal primary anchor with hydrophobic amino acid residues, as well as one or more diverse primary and auxiliary anchors at P1, P2, P3, and/or P7. Matrices were used to generate a large panel of HLA-C–specific peptide-binding data and update our pan-specific NetMHCpan predictor, whose predictive performance was considerably improved with respect to peptide binding to HLA-C. The updated predictor was used to assess the specificities of HLA-C molecules, which were found to cover a more limited sequence space than HLA-A and -B molecules. Assessing the functional significance of these new tools, HLA-C*07:01 transgenic mice were immunized with stable HLA-C*07:01 binders; six of six tested stable peptide binders were immunogenic. Finally, we generated HLA-C tetramers and labeled human CD8+ T cells and NK cells. These new resources should support future research on the biology of HLA-C molecules. The data are deposited at the Immune Epitope Database, and the updated NetMHCpan predictor is available at the Center for Biological Sequence Analysis and the Immune Epitope Database.
Introduction
The major histocompatibility gene complex is generically known as the MHC system and in humans as the HLA system. It encodes a highly diverse set of important immunological proteins, including the classical MHC (or HLA) class I (MHC-I) and II Ags. The major function of classical HLA class I (HLA-I) molecules is to sample peptides derived from cytosolic proteins and present them to CD8+ CTLs, thereby allowing CTLs to survey the ongoing protein metabolism of our cells and monitor it for the presence of intracellular pathogens. In a complementary function, some HLA-I molecules serve as ligands for killer Ig–related receptors (KIRs), thereby allowing NK cells to survey our cells for HLA-I expression and counteract any pathogen, which, in an attempt to reduce HLA-I expression and escape CTL-mediated immune reactions, might interfere with Ag processing and presentation. This enforces Ag presentation and assures that our protein metabolism is subject to CTL scrutiny at all times.
The HLA system is polygenic and extremely polymorphic. For HLA-I Ags, there are three classical isotypes corresponding to the three loci, HLA-A, -B, and -C. Each of these loci has a very large number of unique alleles encoding a great number of variants, or allotypes. Currently, the numbers of known HLA-A, -B, and -C alleles are 1833, 2459, and 1507, respectively (http://www.ebi.ac.uk/ipd/imgt/hla/stats.html; accessed: March 2014). The polymorphic residues are primarily concentrated at the lining of the peptide-binding site where they determine the shape and functionality (i.e., the specificity) of the site. Owing to the polygenic and polymorphic nature of the HLA system, each individual presents a unique selection of peptides derived from our cells’ protein metabolism. This individualizes the specificity of our cellular immune systems and reduces the risk of developing population-wide microbial escape mutants.
Peptide binding is a fundamental requirement for the proper folding, expression, and function of classical HLA molecules. Obviously, the final outcome depends on the ensemble of all these processes, but peptide binding is by far the dominant factor in determining which peptides are naturally presented. Thus, understanding the specificities of peptide interactions with HLA-I molecules is essential for our understanding of how the CTL and the NK systems operate. Currently, our knowledge of the peptide-binding specificity of HLA-I molecules differs considerably between the three HLA-I isotypes. A total of some 112,000 and 53,500 examples of HLA-A and -B interactions representing 48 and 66 different HLA-A and -B allotypes, respectively, are currently registered in the Immune Epitope Database (IEDB; www.iedb.org; accessed March 2014). These data have enabled the generation of accurate quantitative predictors of peptide binding to HLA-A and -B molecules (including but not limited to Refs. 1–3). With this coverage of allotypes, it has been possible to leverage the data from different HLA-I allotypes and generate pan-specific predictors such as the NetMHCpan, which is a reasonably accurate predictor even for allotypes and species that have not yet been described experimentally (4, 5). In contrast, much fewer data are available for HLA-C where only some 1200 examples of peptide interactions representing merely eight different HLA-C allotypes have been registered at the IEDB. Therefore, predictions of peptide binding to HLA-C are less accurate, and it is more difficult to extend pan-specific predictions to all allotype members of this isotype. This lack of information about the specificity of HLA-C molecules effectively limits our understanding of this isotype and its role in human immunity.
There is no doubt that HLA-C merits considerable interest. It appears to be particularly adapted for recognition by KIR molecules of the NK cells. Whereas fewer than half of the HLA-A and -B allotypes serve as KIR ligands, and this only in conjunction with a few peptides, all HLA-C allotypes express KIR ligands, either in the form of the C1 or C2 epitopes, and this in conjunction with many different peptides (6–9). In another function, it has been suggested that the interaction of uterine NK cells with HLA-C, which is uniquely expressed on fetal extravillous trophoblast, is important for successful placentation (10, 11). Finally, the polymorphism of the HLA-C isotype indicates that it is capable of binding a diverse repertoire of peptides in keeping with it also having an important role in Ag presentation and generation of CTL responses. Indeed, HLA-C molecules are known to bind peptides of microbial origin and present them to CTLs [e.g., HIV (12–14) and CMV (15)]. The recent discovery of the ability of HIV-1 Nef proteins to selectively downregulate HLA-A and HLA-B, but not HLA-C, expression (16) suggests that HLA-C may play a unique role in Ag presentation.
In this study, we have developed a general approach to address the specificity of any hitherto unknown MHC-I molecule. Generating a panel of highly active, recombinant HLA-C allotype molecules, we have determined the peptide-binding motifs of 16 HLA-C molecules. All molecules were found to have a primary anchor at the peptide C terminus with preferences for hydrophobic residues; N-terminally, strong anchors were generally observed in P2 and P3, and auxiliary anchors were sometimes seen in P1 and P7. We have applied this novel approach to identify and measure the binding of a panel of individual peptides to these HLA-C allotypes. The resulting data were used to update our pan-specific predictor, NetMHCpan, thereby significantly improving the accuracy of this predictor. The pan-specific predictor was used to analyze the specificity of all HLA-C allotypes, and the results indicate that the peptide-binding repertoire of HLA-C is more limited compared with the repertoires of HLA-A and -B. Finally, we validated these tools by identifying a panel of HLA-C*07:01–binding peptides and demonstrated that six of six stable peptides were immunogenic in an HLA-C*07:01 transgenic mouse model. The updated NetMHCpan predictor should support further investigations into the nature of HLA-C–mediated immunity, including both CTL and NK activity. It is available at the Web sites http://www.cbs.dtu.dk/services/NetMHCpan and http://www.iedb.org/.
Materials and Methods
Peptides
All peptides were purchased from Schafer-N (Copenhagen, Denmark). Briefly, peptides were synthesized using standard Fmoc (9-fluorenylmethyloxycarbonyl) chemistry. Synthesized peptides were purified by reverse-phase HPLC to >80% purity and validated by mass spectrometry. Positional scanning combinatorial peptide libraries (PSCPL) were synthesized as previously described (17). Briefly, eight of nine positions comprised an equimolar pool of 19 of the 20 natural amino acids (i.e., excluding cysteine), whereas the remaining position comprised 1 of the 20 natural amino acids (i.e., including cysteine), thereby interrogating the position-specific effect of this latter amino acid. In one synthesis, the amino acid pool was used in all nine positions. The library therefore consisted of (20 × 9) +1, or 181 individual peptide libraries (where X denotes the random incorporation of an amino acid from the mixture, and the fixed amino acid and its identity is indicated by the single letter amino acid abbreviation): 20 PSCPL sublibraries describing position 1, AX8, CX8, DX8, ….YX8; 20 PSCPL sublibraries describing position 2, XAX7, XCX7, XDX7, …. XYX7; and so forth to 20 PSCPL sublibraries describing position 9, X8A, X8C, X8D, …. X8Y; and finally a completely random peptide library, X9.
Protein production
Recombinant biotinylated MHC-I molecules were produced as previously described (18–20). Briefly, relevant HLA-encoding genes (α1α2α3, residues 1–275) were cloned into a pET28a expression vector already containing a biotin signal peptide and a histidine affinity tag. For HLA-C alleles encoding cysteine in residue 1, this was mutated to glycine to prevent unwanted disulfide formation. Constructs were validated by DNA sequencing and transformed into Escherichia coli BL21(DE3) expression strains carrying the pACYC vector encoding BirA for in vivo biotinylation. Transformed cells were grown in a 2.5-l Infors fermentor. When the cell density reached OD600 of 25, protein production was induced with isopropyl β-d-thiogalactoside. The cells were harvested after 3 h and lysed by high-pressure cell disruption. Inclusion bodies containing recombinant MHC-I were isolated by centrifugation and washed to remove cell debris. Inclusion bodies were extracted into Tris-buffered 8 M urea (pH 8.0), and HLA molecules were purified under denaturing and nonreducing conditions by successive immobilized metal affinity, hydrophobic interaction, and size exclusion chromatography. Isolated HLA molecules were stored at −24°C until use.
Native recombinant β2-microglobulin (β2m) was produced as described previously (21).
Radiolabeling
Recombinant β2m was radiolabeled with [125I] according to the chloramine-T labeling procedure (22). At room temperature, 20 μg β2m was mixed with 1 mCi [125I] (PerkinElmer, NEZ033A005MC) and 5 μg chloramine-T (Sigma-Aldrich, C9887) in a total volume of 65 μl. The reaction was stopped after 1 min by addition of 5 μl metabisulfite (1 mg/ml). Free iodine was removed by Sephadex G10 size exclusion chromatography using a 4-ml column equilibrated and eluted in PBS/2% ethanol/0.1% azide. Fractions (200 μl) were collected and peak fractions containing radioactivity were pooled. The radioactivity incorporated was measured on a gamma counter (Packard Cobra 5010). Radiolabeled β2m was stored at 4°C for ≤4 wk.
Determining peptide-binding motifs by a scintillation proximity-based dissociation assay
Nonameric peptide-binding motifs were determined for HLA-C*01:02, -C*02:02, -C*03:02, -C*03:03, -C*03:04, -C*04:01, -C*05:01, -C*06:02, -C*07:01, -C*07:02, -C*07:04, -C*08:01, -C*08:02, -C*12:03, -C*14:02, -C*15:02, and -C*16:01 using a recently developed scintillation proximity assay (SPA)–based peptide–MHC-I dissociation assay (23) in combination with nonameric PSCPL (17). In 384-well streptavidin-coated FlashPlate HTS Plus microplates (PerkinElmer, SMP410), recombinant biotinylated MHC-I H chain in 8 M urea was diluted at least 100 times (to 50 nM) into PBS/0.1% Lutrol F68 containing 78 μg/ml (≈78 μM) of each sublibrary with 2–10 nM [125I]-radiolabeled β2m in PBS, giving a total volume of 60 μl/well. The plates were sealed and incubated overnight at 18°C. Dissociation was started by adding 10 μl/well unlabeled β2m in PBS (1.4 μM, final concentration 200 nM) to prevent reassociation of any dissociated radiolabeled β2m, and transferring the plate to a TopCount NXT scintillation counter set to 37°C. The amount of labeled β2m still associated with HLA-C was measured continuously for 24 h as previously described (23).
Analysis of PSCPL data
The area under the dissociation curve (AUC) was calculated after background subtraction by summarizing the cpm from 0 to 24 h. The relative binding (RB), that is, the relative contribution of each sublibrary to peptide binding, was calculated according to the following equation: RB = AUCsublibrary/AUCX9. The RB values of each amino acid in a given position were summarized and normalized so the sum equals 20 (17). An anchor position (AP) value for each peptide position, defining the relative contribution of this position to peptide binding, was calculated as:
Sequence logos describing the amino acid binding specificity captured by the PSCPL were calculated using the Seq2Logo sequence logo generator (24). In short, the RB values at each peptide position were normalized by the sum of the RB values at the given position. This normalized PSCPL matrix was submitted to the Seq2Logo server, and binding motif was visualized as a p-weighted Kullback–Leibler logo generated using a flat amino acid background distribution (i.e., all amino acids are assumed to be found with an equal frequency). In this type of logo, anchor positions are characterized by a tall stack of letters, amino acids presented on the positive y-axis are enriched in peptide binders, and amino acids on the negative y-axis are depleted from peptide binders.
Peptide–MHC-I-binding assay
Peptide affinities of HLA-C molecules were determined using a luminescent oxygen channeling immunoassay (25). Briefly, peptides were dissolved in PBS/0.1% Lutrol F68 by sonication for 10 min. Peptides were titrated in 384-well microplates using a Microlab STAR liquid handling robot (Hamilton Robotics). Recombinant, denatured HLA-C H chain was diluted into PBS/0.1% Lutrol F68/100 mM Tris/maleate (pH 6.6) containing prefolded, recombinant β2m on ice. The H chain/β2m mix was added 1:1 to the peptide titrations and incubated for 48 h at 18°C to allow peptide–MHC-I complex folding. After complex folding, samples were transferred to 384-well OptiPlates, and streptavidin-coated donor beads (PerkinElmer, 6760002) and W6/32-conjugated acceptor beads (PerkinElmer, 6762001, in-house conjugated with W6/32) diluted in PBS/0.1% Lutrol F68 were added to a final concentration of 5 μg/ml each. The OptiPlates were incubated overnight and luminescence was measured in an EnVision 2103 multilabel reader.
Bioinformatics analysis of the peptide-binding specificity of HLA-C allotypes
A set of 121 frequent HLA-A, -B, and -C alleles present in the European population was identified using the dbMHC database (http://www.ncbi.nlm.nih.gov/gv/mhc/) using an allele frequency threshold of 0.05%. The set contains 34 HLA-A, 60 HLA-B, and 27 HLA-C allotypes. Next, binding affinity to a set of 50,000 random natural 9-mer peptides was predicted for each of these HLA molecules using NetMHCpan (version 2.8) (4, 5) that has been trained on data, including the HLA-C–binding data described in this work.
The functional distance between two HLA molecules was calculated as described for the MHCcluster method (24) (version 2.0, using NetMHCpan version 2.8 as prediction method). In short, the similarity between any two molecules is estimated from the correlation between the union of the top 10% strongest binding peptides for each allele. This similarity is 1 when the two molecules have a perfect binding specificity overlap and negative when the two molecules share no specificity overlap. The distance between two molecules is next defined as 1 − similarity. Using this distance measure, a distance matrix is calculated containing the distance between any two HLA molecules. The distance matrix is converted to an unweighted pair group method with arithmetic mean distance tree. To estimate the significance of the MHC distance tree, a large set of distance trees is generated using the bootstrap method, and a final tree is summarized in the form of a “greedy” consensus tree with corresponding branch bootstrap values.
Sequence logos describing the binding motif of each HLA molecule were generated using the top 1% highest scoring predictions as input to the Seq2Logo sequence logo generator (24) with default options.
CTL responses in HLA-C*07:01 transgenic mice
Ficoll-purified splenocytes from HLA-C*07:01 transgenic mice were assayed for CD8+ T cell responses following peptide and DNA immunization by an IFN-γ ELISPOT assay as described previously (26).
Donors
The study of donor immune responses was approved by the Committees on Biomedical Research Ethics of the Capital Region (De Videnskabsetiske Komiteer for Region Hovedstaden), and informed written consent was obtained. Blood samples were obtained from healthy yellow fever vaccinees 14–20 d after vaccination. PBMCs were isolated by density gradient centrifugation using Ficoll-Paque Plus (GE Healthcare Europe, Brøndby, Denmark) and stored until use at −150°C. Chromosomal DNA was isolated from and typed for HLA-A/B/C and HLA-DR/DQ/DP using sequence-based typing (Genome Diagnostics, Utrecht, The Netherlands).
Tetramer staining
Tetramers of HLA-C*03:04 were produced as previously described (20). Briefly, biotinylated recombinant HLA class I H chains were diluted into a reaction buffer containing 50 mM Tris-maleate (pH 6.6), 0.1% Lutrol F68 NF (BASF, a surfactant compatible with cellular use), and excess of β2m and peptide and incubated for 48 h at 18°C. To tetramerize the resulting peptide–HLA–I monomers, streptavidin-R–PE or streptavidin-allophycocyanin was sequentially added over 60 min at a 1:4 molar ratio of streptavidin to peptide–HLA-I monomers. PBMCs (106) were resuspended in 25 μl PE- and allophycocyanin-conjugated tetramer and incubated for 20 min at room temperature followed by 30 min incubation with a mixture of anti-CD3, anti-CD4, anti-CD8, and anti-CD56 Ab (BioLegend, San Diego, CA).
Data analysis
All data handling and nonlinear regression analyses were done using Microsoft Office Excel 2007 and GraphPad Prism. Flow cytometry data were analyzed by FACSDiva software.
Results
Determining MHC-I peptide-binding motifs by combining PSCPL with an SPA-based dissociation assay
We have recently published a MHC-I dissociation assay based on recombinant biotinylated MHC-I H chain and radiolabeled β2m (23). The strength of this assay lies in that it requires no labeling of the peptide in question, it is very robust and accurate, and it has a high throughput allowing the measurement of 384 distinct dissociation curves in parallel in a single experiment. We reasoned that the nature of this assay would make it suitable for PSCPL analysis (17) of the peptide-binding specificity of any MHC-I molecule. In a PSCPL-based specificity analysis of peptide–MHC-I interaction, all possible peptides of a given size (in this case 9 mers) are represented by a systematic set of sublibraries. For each sublibrary, one amino acid in one position is kept constant, whereas the remaining positions contain mixtures of amino acids. The PSCPL approach yields a complete representation of MHC-I specificity encompassing all positions (including primary and secondary anchors) and all possible residues (i.e., all 20 naturally occurring residues, including favored and disfavored amino acids). The resulting description of MHC specificity can be represented as a quantitative position-specific scoring matrix, which for every peptide position gives the RB of each of the 20 naturally occurring amino acids (as detailed in Materials and Methods). Originally, Falk et al. (27) defined an AP as a position that reveals “a strong signal for only one residue, or alternatively, if a position is occupied by a few residues with closely related side chains.” Operationally, the AP values are calculated as the sum of squared deviations of RB values for a given position. This readily indicates the location of anchor positions and their relative importance. Traditionally, affinity measurements of each PSCPL sublibrary have been used to determine PSCPL matrices (17, 28, 29). To examine whether dissociation measurements could be used to obtain identical or similar data, we initially compared affinity- versus dissociation-based PSCPL matrices representing the specificity of the well-characterized HLA-A*02:01 allotype (Table I). Predictably, the enormous diversity of peptides present in a PSCPL sublibrary led to a multiphase dissociation curve (data not shown), which complicates any analysis based on using the t1/2 as the parameter of dissociation. Therefore, we tested different parameters including 1) the signal at time 0 (Y0), 2) the half-life determined from the first 9 h of dissociation as fitted by GraphPad Prism, 3) integrating the counts over the first 24 h dissociation period (normalized for Y0), and 4) integrating the counts over the first 24 h dissociation period (without normalization). The latter measure gave (sublibrary by sublibrary, position by position) a nearly perfect match with the results obtained using affinity measurements (Table I). Thus, the well-known HLA-A*02:01 specificity with positions 2 and 9 as anchor positions (preferred residues Leu and Met in position 2; preferred residues Val, Leu, Ile, and Met in position 9) was revealed by the stability-driven PSCPL analysis using integrated counts without Y0 normalization. This is perhaps not surprising given that this read-off mode captures both complex formation and the subsequent dissociation phase. In contrast, both the integrated counts normalized for Y0 and the fitted half-life solely capture dissociation, and these stricter measures reduced the signals, making it much more difficult to discern motifs (data not shown).
Table I. Determining the peptide-binding motifs of MHC-I allotypes using PSCPL.
| Residue | Amino Acid Position in Peptide |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| Peptide–MHC-I affinity | |||||||||
| A | 1.2 | 0.5 | 1.0 | 0.2 | 1.1 | 1.1 | 0.8 | 1.0 | 1.9 |
| C | 0.7 | 0.1 | 0.2 | 1.0 | 0.6 | 0.6 | 0.9 | 1.0 | 0.2 |
| D | 0.0 | 0.0 | 1.1 | 2.8 | 1.4 | 1.4 | 0.5 | 0.8 | 0.0 |
| E | 0.1 | 0.0 | 0.4 | 2.6 | 1.4 | 0.7 | 1.0 | 1.6 | 0.0 |
| F | 3.8 | 0.2 | 2.9 | 0.0 | 1.5 | 1.4 | 3.2 | 1.2 | 1.0 |
| G | 1.1 | 0.3 | 0.6 | 1.1 | 1.9 | 0.7 | 0.1 | 0.9 | 0.5 |
| H | 0.9 | 0.1 | 0.2 | 0.9 | 1.1 | 0.7 | 0.4 | 0.7 | 0.0 |
| I | 0.6 | 1.8 | 1.1 | 0.5 | 0.9 | 1.5 | 1.7 | 0.4 | 2.5 |
| K | 1.2 | 0.1 | 0.2 | 0.8 | 0.5 | 0.0 | 0.2 | 0.5 | 0.0 |
| L | 0.8 | 5.0 | 1.2 | 0.5 | 0.8 | 1.7 | 0.9 | 1.1 | 2.7 |
| M | 1.4 | 6.9 | 2.4 | 0.9 | 0.9 | 1.0 | 1.5 | 0.9 | 2.1 |
| N | 0.4 | 0.1 | 1.0 | 0.6 | 0.9 | 1.5 | 0.6 | 0.9 | 0.1 |
| P | 0.1 | 0.0 | 0.6 | 1.9 | 0.6 | 1.8 | 1.2 | 2.2 | 0.1 |
| Q | 0.4 | 1.8 | 1.0 | 1.2 | 0.8 | 0.5 | 1.0 | 1.3 | 0.1 |
| R | 1.4 | 0.1 | 0.2 | 0.7 | 0.8 | 0.4 | 0.3 | 0.9 | 0.1 |
| S | 1.1 | 0.1 | 1.3 | 0.6 | 0.6 | 1.2 | 0.6 | 0.7 | 0.1 |
| T | 0.7 | 1.0 | 0.9 | 1.1 | 0.8 | 0.6 | 0.9 | 1.0 | 0.1 |
| V | 1.1 | 1.6 | 0.6 | 0.8 | 1.0 | 1.2 | 0.6 | 0.6 | 8.4 |
| W | 0.6 | 0.1 | 1.2 | 0.4 | 1.4 | 0.8 | 1.7 | 0.6 | 0.0 |
| Y | 2.5 | 0.2 | 1.8 | 1.3 | 0.8 | 1.0 | 1.9 | 1.4 | 0.1 |
| Sum | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| AP value | 14 | 64 | 10 | 10 | 2 | 4 | 10 | 3 | 74 |
| Peptide–MHC-I complex dissociation | |||||||||
| A | 1.01 | 0.7 | 1.7 | 1.2 | 1.1 | 1.2 | 1.5 | 1.2 | 2.2 |
| C | 0.5 | 0.5 | 1.6 | 1.1 | 0.9 | 0.7 | 1.4 | 1.2 | 0.5 |
| D | 0.0 | 0.0 | 1.3 | 2.7 | 0.8 | 0.7 | 0.4 | 0.5 | 0.0 |
| E | 0.0 | 0.0 | 0.2 | 1.9 | 0.7 | 0.8 | 0.8 | 1.7 | 0.0 |
| F | 2.3 | 0.2 | 2.5 | 0.8 | 1.3 | 0.9 | 2.7 | 1.5 | 0.4 |
| G | 0.8 | 0.2 | 0.7 | 0.0 | 1.3 | 0.5 | 0.3 | 1.0 | 0.2 |
| H | 0.7 | 0.0 | 0.7 | 1.3 | 1.2 | 1.0 | 1.3 | 0.9 | 0.0 |
| I | 0.7 | 1.9 | 0.7 | 0.3 | 0.8 | 1.6 | 0.4 | 0.4 | 2.2 |
| K | 1.8 | 0.1 | 0.1 | 0.7 | 1.0 | 0.6 | 0.1 | 0.5 | 0.1 |
| L | 1.1 | 6.4 | 1.7 | 0.2 | 0.8 | 1.4 | 0.9 | 0.5 | 4.2 |
| M | 1.5 | 5.6 | 2.7 | 0.4 | 0.8 | 1.0 | 1.5 | 0.3 | 2.4 |
| N | 0.4 | 0.1 | 1.0 | 1.1 | 0.9 | 1.6 | 0.8 | 0.5 | 0.0 |
| P | 0.0 | 0.1 | 0.4 | 1.3 | 0.6 | 1.7 | 2.3 | 2.6 | 0.1 |
| Q | 0.4 | 1.5 | 0.9 | 1.0 | 0.6 | 1.3 | 0.8 | 1.1 | 0.0 |
| R | 1.4 | 0.1 | 0.1 | 0.7 | 1.2 | 0.5 | 0.3 | 0.8 | 0.0 |
| S | 1.1 | 0.2 | 1.2 | 1.0 | 0.9 | 0.9 | 0.5 | 0.9 | 0.4 |
| T | 1.1 | 0.9 | 1.1 | 1.1 | 1.0 | 0.5 | 1.4 | 1.3 | 0.8 |
| V | 1.2 | 1.3 | 0.5 | 0.6 | 0.7 | 0.9 | 1.3 | 0.6 | 6.4 |
| W | 0.9 | 0.1 | 0.4 | 0.9 | 1.9 | 0.7 | 0.6 | 0.6 | 0.0 |
| Y | 2.9 | 0.1 | 0.7 | 1.5 | 1.5 | 1.5 | 0.7 | 1.9 | 0.0 |
| Sum | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| AP value | 11 | 62 | 10 | 7 | 2 | 3 | 9 | 7 | 56 |
The peptide-binding motif of HLA-A*02:01 was determined via nonameric PSCPL analysis either by peptide–MHC-I affinity measurement or peptide–MHC-I complex dissociation. Residues with RB values larger than 2 are in terms of peptide binding considered favored (bold numbers) in the given position, residues with RB values <0.5 are considered disfavored (italic numbers), and residues with 0.5 < RB < 2.0 are accepted (numbers in regular font). Both methods capture the well-described peptide-binding motif of the HLA-A*02:01 allotype, having strong anchors in peptide positions 2 and 9 with preference for Leu/Met and Leu/Val, respectively. RB values are shown.
Determining the peptide-binding motif of 16 HLA-C allotypes
Using the dissociation-driven PSCPL analysis, we analyzed the specificity of 17 recombinant HLA-C molecules: HLA-C*01:02, -C*02:02, -C*03:02, -C*03:03, -C*03:04, -C*04:01, -C*05:01, -C*06:02, -C*07:01, -C*07:02, -C*07:04, -C*08:01, -C*08:02, -C*12:03, -C*14:02, -C*15:02, and -C*16:01. For 16 of these 17 HLA-C molecules, distinct peptide-binding specificities were readily revealed, whereas low or inconsistent signals were obtained for one molecule, HLA-C*01:02. The resulting motifs are shown as LOGO representations in Fig. 1 and the full peptide-binding matrix representations are shown in Supplemental Table I). At first glance, the overall shape of the motifs of these HLA-C allotypes appeared very similar to those seen for HLA-A and -B allotypes. In terms of the number and distribution of anchor positions, all motifs featured a pronounced primary anchor position at the C-terminal, and most motifs also featured a more or less pronounced primary anchor position at P2 and/or P3 as well as some secondary anchor positions scattered in P1, P2, P3, and P7; the only exception appeared to be HLA-C*14:02, which almost exclusively exhibited C-terminal specificity. In terms of which amino acid residues were present in these primary anchor positions, the primary C-terminal anchor position of all these HLA-C allotypes contained a restricted repertoire of hydrophobic and aromatic residues such as Phe, Ile, Leu, Met, Val, and Tyr. In contrast, the primary P2/P3 anchor positions contained a much more variable repertoire of hydrophobic, polar, or charged (both positively and negatively) amino acids (summarized in Table II). Secondary anchors were observed at P1, P2, P3, and P7 (Table II).
FIGURE 1.
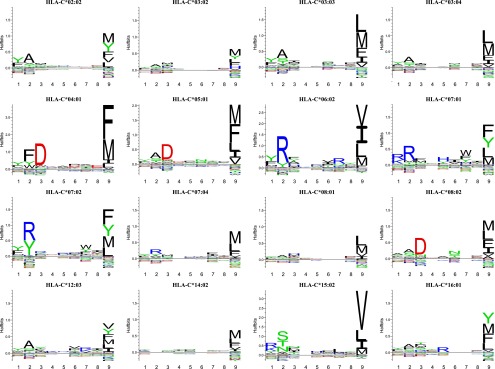
HLA-C allotypes exhibit classical MHC-I peptide-binding motifs. The peptide-binding motifs of 16 HLA-C allotypes were determined by SPA-driven PSCPL analysis using a nonameric peptide library. Binding motifs are represented as logos generated from peptide-binding matrices using the Seq2Logo server (24). The height of each letter indicates the information content, that is, larger letters indicate residues having more impact on binding in the given position, be it positive or negative. Amino acids plotted on the positive y-axis contribute positively to peptide binding, whereas amino acids plotted on the negative y-axis contribute negatively to peptide binding. The total height of a given position indicates the information content, and thus higher stacks represent amino acid position important for peptide binding. Each logo is an average of two independent experiments.
Table II. Peptide anchor positions and preferred anchor residues of HLA-C allotypes identified from nonameric PSCPL analysis.
| Allotype | Peptide Position |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | |
| HLA-C*02:02 | FY | A | F LMVY | ||||||
| HLA-C*03:02 | A | F LM Y | |||||||
| HLA-C*03:03 | FY | AS | FILMV | ||||||
| HLA-C*03:04 | Y | AS | FILM | ||||||
| HLA-C*04:01 | FY | FWY | D | FILM | |||||
| HLA-C*05:01 | Y | AS | D | FILMV | |||||
| HLA-C*06:02 | FY | RY | AFY | KQR | ILMV | ||||
| HLA-C*07:01 | KR | R | FM | H | I | FWY | F LM Y | ||
| HLA-C*07:02 | FY | RY | FWY | F LM Y | |||||
| HLA-C*07:04 | Y | R | F | F LM | |||||
| HLA-C*08:01 | FY | A | LMV | ||||||
| HLA-C*08:02 | AS | D | N | FILM | |||||
| HLA-C*12:03 | Y | A | Y | FILMVY | |||||
| HLA-C*14:02 | F LMV | ||||||||
| HLA-C*15:02 | KRY | NST | H | L | ILMV | ||||
| HLA-C*16:01 | AF | R | F LM Y | ||||||
Sixteen HLA-C molecules were analyzed for peptide-binding specificity by SPA-driven PSCPL analysis. Amino acid residue preferences are indicated in standard single-letter amino acid code. An RB value of >2 was used as a threshold for preferred binding. Boldface letters denote a primary anchor residue; italic letters denote an auxiliary anchor.
High-affinity peptide binders predicted by PSCPL matrices
A PSCPL-derived HLA-I motif represents the combined effect of all the peptides of the individual PSCPL sublibraries, which each encompass billions of different peptides. In reality, peptide binding to HLA-I involves components of correlated effects where one amino acid in one position may affect the specificity at other positions. Correlated effects cannot be captured by a PSCPL matrix approach, which inherently assumes that all amino acids in all positions are recognized independently of the amino acids present in all other positions (also known as “independent binding of side-chains”) (30). In contrast, neural networks (and other higher order data-mining methods) are ideally suited to capture and predict peptide–HLA-I binding including any correlated effects. However, the development of neural networks is dependent on the availability of a large body of data representing many different single peptide-binding events (31). To generate single peptide-binding data and support the development of neural networks, we used the PSCPL matrices to identify peptides that were predicted to bind to the different HLA-C allotypes. Over the years, we have collected ∼9000 nonamer peptides from various sources. Multiplying the relevant RB values of the different amino acids in the different positions, we predicted the binding of each peptide/HLA-C allotype combination. For each HLA-C allotype, we then ranked the peptides, selected the 100 highest scoring peptides, and measured the affinity of binding of each peptide using a previously published high-throughput peptide-binding assay (25). Eight HLA-C allotypes were included in this analysis (i.e., HLA-C*03:03, -C*04:01, -C*05:01, -C*06:02, -C*07:02, -C*12:03, -C*14:02 and -C*15:02). Overall, this selection method was very successful in identifying binding peptides (Supplemental Table IIA). Thus, the frequency of binders with a KD < 500 nM, which is commonly regarded as the threshold for a peptide–HLA-I combination being immunogenic, was 71–85% for all HLA-C allotypes studies, except for HLA-C*04:01 (18%). The fewer number of binders found for HLA-C*04:01 is likely due to a bias in our peptide repository, which has been built to match HLA-I specificities of interest over the years. These results are in agreement with observations by other groups using PSCPL-based peptide selection (17).
Additional peptide binders predicted by NetMHCpan
We reasoned that the pan-specific NetMHCpan predictor might identify peptides that have not been captured by, or do not conform to, PSCPL-generated motifs and therefore could expand our selection of peptides with some that are particularly valuable and information-rich. To this end, we selected an additional set of peptides from our nonamer peptide repository, but this time we searched for peptides with a high NetMHCpan but a low PSCPL ranking and measured the binding of these peptides. For HLA-C*04:01, C*05:01, -C*06:02, and -C*07:02, most peptides selected according to this stringent strategy were nonbinders, and the binders, which were identified, were only intermediary binders, suggesting that for these HLA-C allotypes the PSCPL matrices had already captured most of the information about peptide binders. For HLA-C03:03, -C12:03, -C*14:02, and -C*15:02, however, NetMHCpan managed to identify additional, non–matrix-conforming peptide binders, many of which were strong binders. This is particularly obvious for HLA-C*12:03 and HLA-C*14:02, where 75 and 84%, respectively, of the additional peptides identified by NetMHCpan were binders (Supplemental Table IIB). This demonstrates that for these latter HLA-C allotypes, NetMHCpan captured valuable information that had not been captured by the PSCPL matrices.
Improving NetMHCpan predictions of HLA-C binding
Overall, the combined strategy described above assured that both unbiased (PSCPL) and “experienced” (NetMHCpan) components were involved in the important selection of informative peptides needed for efficient neural network development. The new HLA-C–binding data were added to the data that originally were used to generate NetMHCpan (version 2.3, predominantly generated from HLA-A– and HLA-B–binding data), and the predictor was retrained. The ability to identify peptides known to bind to HLA-A, -B, and -C according to the SYFPEITHI database of natural HLA bound and/or immunogenic peptides was used to validate the updated predictor and to compare it with its predecessor. With respect to predicting peptide binding to HLA-C, the performance of the updated NetMHCpan method was significantly improved when the new HLA-C data were added to the training data (Fig. 2). In contrast, the prediction of peptide binding to HLA-A and -B was unaffected. For HLA-C, the predictive performance measured in terms of the AUC for a set of 77 HLA-C ligands described in the SYFPEITHI database (www.syfpeithi.de) increased from 0.87 to 0.97, improving the performance of HLA-C predictions to a level comparable to those of HLA-A and HLA-B (Fig. 2). This improvement is highly significant (p < 10−6, binomial test excluding ties).
FIGURE 2.

Improvement of HLA-C–binding prediction tools. The NetMHCpan prediction method was enriched with HLA-C binding data for eight HLA-C allotypes (HLA-C*03:03, -C*04:01, -C*05:01, -C*06:02, -C*07:02, -C*12:03, -C*14:02, and -C*15:02). Comparison of performance efficacy, measured by the AUC, of the previous version of the NetMHCpan predictor with a new version updated with HLA-C–binding data shows that enrichment with HLA-C–binding data improved the performance of peptide–HLA-C-binding prediction.
Evaluating HLA-C binding
The improved NetMHCpan predictor (version 2.8) was subsequently used to examine the peptide-binding specificities of the HLA-C isotype. Initially, the most frequent members of HLA-A, -B, and -C isotypes were compared. One hundred twenty-one different HLA-I allotypes with an allele frequency of >0.05% in the European population were extracted from the dbMHC database: 34 HLA-A, 60 HLA-B, and 27 HLA-C allotypes. The functional distance between any two HLA-I allotypes was calculated using the MHCcluster method (32) as described in Materials and Methods. This analysis revealed that the peptide-binding specificities of the HLA-C isotype are distinctly different from those of HLA-A or -B isotypes (colored red, green, and blue for HLA-A, -B, and -C, respectively, Fig. 3). Furthermore, isotype-specific histograms of the pairwise distances between the 34 HLA-A, 60 HLA-B, and 27 HLA-C allotypes (colored red, green, and blue for HLA-A, -B, and -C, respectively, Fig. 4) revealed that the HLA-C isotype has a more narrow functional diversity, that is, that it presents a less diverse peptide repertoire than do the HLA-A or HLA-B isotypes (Fig. 4). To further illustrate this, we estimated the repertoire of peptides presented by the HLA-A, -B, and -C isotypes. From a set of 50,000 random natural 9-mer peptides, we extracted the 1% highest scoring peptides for each of the 34, 60, and 27 prevalent members of the HLA-A, -B, and -C isotypes, respectively, and then identified the number of unique peptides per isotype, that is, 5491, 6376, and 2624, for the HLA-A, -B, and -C isotypes, respectively. Compared to the HLA-A and -B isotypes, it would appear that the HLA-C isotype binds a more distinct and limited part of the universe of peptides.
FIGURE 3.

In silico functional clustering of 121 HLA-A, -B, and -C molecules. Using the updated NetMHCpan predictor to cluster 121 common HLA-I allotypes (34 HLA-A, 60 HLA-B, and 27 HLA-C) according to predicted binding motif shows that allotypes of the HLA-C isotype have distinctly different peptide-binding specificities from those of the HLA-A and HLA-B isotypes. The clustering is calculated from the predicted binding of a set of 50,000 random natural 9-mer peptides using 100 bootstrapped distance trees. Sequence logos are added to the plot displaying the predicted binding motif for a representative allotype for each cluster in the tree. Tree and sequence logos are calculated as described in Materials and Methods.
FIGURE 4.

HLA-C allotypes bind a less diverse set of peptides compared with HLA-A and HLA-B allotypes. Normalized histograms of HLA intraloci functional similarity distances are shown. The figure displays the probability (p) of observing a given functional distance (d) between different HLA molecules within each of the three loci (HLA-A, HLA-B, and HLA-C). Functional distances between two HLA molecules were calculated as described in the text using the MHCcluster similarity measure. Distances were normalized so that the maximal distance for any HLA pair was 1, and the minimal distance was 0. The histograms were calculated from the 121 HLA molecules included in Fig. 3.
Next, we extracted a representative set of HLA-C allotypes and constructed a tree of the peptide-binding specificities of the HLA-C isotype. To obtain a nonredundant representation of HLA-C, we extracted representative allotypes by homology reduction ad modum Hobohm II (33). The resulting 86 HLA-C allotypes were analyzed as described above using the MHCcluster method and illustrated as an unweighted pair group method with arithmetic mean distance tree (Fig. 5, including LOGO representation of the peptide-binding specificity of allotypes representing the main branches). This analysis illustrates the limited functional (i.e., peptide-binding specificity) diversity of the HLA-C isotype. In particular, the strong preference for a hydrophobic residue at P9 is a shared feature of all allotype members of the HLA-C isotype, a requirement that can be viewed as a prerequisite for being an HLA-C–binding epitope. Once that universal requirement is met, the functional diversity is primarily seen in less influential positions of the motif such as the recognition of the P2/P3 anchor residues. As shown, the tree is largely divided into two major partitions: one partition that prefers Tyr and Phe or Arg at P2 (seen to the left of the tree, Fig. 5), and another partition that prefers Ala, Ser, and Thr at P2 or Asp and Glu at P3 (seen to the right part of the tree, Fig. 5).
FIGURE 5.
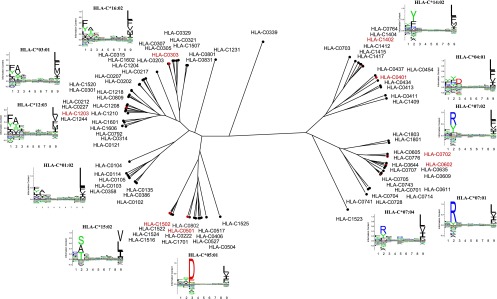
In silico functional clustering of 86 representative HLA-C allotypes. Eighty-six representative HLA-C allotypes were clustered according to predicted binding motif. The clustering was calculated from the predicted binding of a set of 50,000 random natural 9-mer peptides using 100 bootstrapped distance trees. Sequence logos are added to the plot displaying the binding motif for a representative molecule for each cluster in the tree. The eight HLA-C molecules characterized with binding data are highlighted in red. Tree and sequence logos are calculated as described in Materials and Methods.
The HLA-C isotype is unique in the sense that virtually all HLA-C allotypes express either the C1 or C2 KIR ligands, which are determined by the presence of Asn or Lys, respectively, in position 80 of the HLA-C (the only exception being HLA-C*07:115, which expresses an Asp in position 80). It is not known to what extent this essentially dimorphic position affects peptide binding and the subsequent generation of the C1/C2 epitopes (34). Being in close proximity to the bound peptide, position 80 may potentially affect the peptide-binding specificity directly. Indeed, position 80 is part of the HLA-I pseudosequence that is used to generate the NetMHCpan predictor (5). Because the predictor allows the submission of custom sequences, we reasoned that it might empower a systematic analysis of the effect of virtual Asn80Lys (i.e., C1 to C2 epitope) or Lys80Asn (i.e., C2 to C2 epitope) single-point mutations and thereby shed light on the possible contribution of the C1/C2 epitope dimorphism to the peptide-binding specificity of the corresponding HLA-C allotypes. The Asn80 (i.e., the C1 epitope) and Lys80 (i.e., the C2 epitope) are expressed in 47 and 39 of the 86 representative wild-type HLA-C allotypes included in this study, respectively. Similar to the MHCcluster approach described above, we extracted the 1% top predicted peptide binders from 50,000 random natural 9-mer peptides for the 47 wild-type C1-expressing HLA-C allotypes and the corresponding Asn80Lys single-point mutations, as well as for the 39 wild-type C2-expressing HLA-C allotypes and the corresponding Lys80Asn single-point mutations. Only 1.1 and 1.3% of the resulting peptides differed between the wild-type and the virtual single point-mutated molecules, suggesting that the C1/C2 dimorphism does not contribute much to the peptide-binding specificity of the HLA-C isotype.
Using the improved predictors to identify HLA-C–restricted peptides
In the past, the prediction of HLA-C–restricted epitopes has been hampered by the relative lack of data and accurate predictors relevant for HLA-C allotypes. We have recently generated an HLA-C*07:01 transgenic mouse strain expressing the HLA-C*07:01 α1α2 H chain domains fused to a mouse α3 domain and covalently linked to human β2m. Initially, we used published HLA-C*07:01–restricted epitopes (35–37) to test the HLA-C*07:01 restricting capacity of transgenic mice. These mice were immunized either 1) with peptide epitopes formulated with a helper epitope in IFA and injected s.c., or 2) with epitope-encoding DNA vaccines and injected i.m. In either event, we would detect epitope-specific CD8+ T cell responses using an IFN-γ ELISPOT assay. However, three of three previously published epitopes failed to induce CD8+ T cell responses whether immunized with peptide or through DNA vaccination. This raises the question of the sensitivity of this transgenic system. Using the improved predictor we noted that none of these peptides would be predicted to be binders to HLA-C*07:01 according to the work presented in this study (Table III). We have recently confirmed previous reports stating that peptide–HLA-I stability, rather than affinity, is indicative of immunogenicity (38). Therefore, we selected the six most stable HLA-C*07:01 binders from the panel of HLA-C*07:01–binding peptides identified in this study and used these to immunize HLA-C*07:01 transgenic animals using either the peptide or DNA immunization strategies. All six of these in silico predicted and biochemically verified high-stability binders were able to induce strong CD8+ T cell responses (Table III; note that two of these six epitopes have been reported previously; see Ref. 26). Thus, the HLA-C data and predictors presented in the present study appear to capture the peptide-binding activity and specificity of HLA-C allotypes, resulting in improved predictions of immunogenic epitopes (at least for HLA-C*07:01).
Table III. HLA-C*07:01 transgenic mice have CD8+ T cell responses against HLA-C*07:01–binding peptides of high stability.
| Peptide Sequence | HLA-C*07:01–Binding Prediction |
pHLA-I Stability, t1/2 (h) | References | ||||
|---|---|---|---|---|---|---|---|
| NetMHCpan |
PSCPL Scoreb | CD8+ T Cell IFN-γ Responses (INF-γ/106 Cells) |
|||||
| % Ranka | Binding Level | DNA Immunization | Peptide Immunization | ||||
| PLADLSPFA | 50.00 | Nonbinder | 0 | ND | 0/0/0/0 | 0/0/0/0 | (26, 35) |
| EGDCAPEEK | 50.00 | Nonbinder | 0 | ND | 0/0/0/0 | 0/0/0/0 | (26, 36) |
| RNGYRALMDKS | 50.00 | Nonbinder | N/Ac | ND | 0/0/0/0 | 0/0/0/0 | (26, 37) |
| RRRPVTRPL | 0.12 | Strong binder | 85 | 16.2 | 33/33/24/1 | 619/599/177/0 | (26) |
| RRARYWLTY | 0.30 | Strong binder | 81 | 27.2 | 273/246/42/9 | 643/640/133/97/92 | (26) |
| RRMATTFTF | 0.12 | Strong binder | 267 | 34.6 | 338/185/125/101 | 317/225/69/21/0 | |
| RRNDGVVQY | 0.25 | Strong binder | 99 | 16.3 | 17/0/0/0 | 308/289/189/50/34 | |
| RYSGFVRTL | 0.25 | Strong binder | 50 | 17.7 | 427/371/56/5 | 388/185/7/5 | |
| SRPLVSFSF | 1.50 | Weak binder | 40 | 22.4 | 329/318/20/3 | 221/123/49/38 | |
Predicted HLA-C*07:01–binding peptides of high stability and previously published HLA-C*07:01–restricted peptides were used to immunize HLA-C*07:01 transgenic mice. IFN-γ T cell responses were assayed by ELISPOT following peptide and DNA immunization. Peptide-binding predictions were done using the updated NetMHCpan server, and PSCPL scores were calculated by multiplication of the individual RB value from each peptide position using the PSCPL-derived HLA-C*07:01–binding matrix. The peptides reported were selected from a large repository of peptides collected for many different purposes. The pHLA-I complex stability was measured by an SPA-based pMHC-I dissociation assay. ND, not determined.
Values of <0.5 are considered high-affinity interactions, 0.5–2 medium-affinity interaction, and values of >2 are considered of low affinity or nonbinding.
PSCPL scores are calculated by multiplication of the PSCPL-derived RB value for each peptide position (see Materials and Methods); higher values predict higher affinity.
Not available. PSCPL prediction scores are based on a nonamer PSCPL and can thus only be calculated for nonameric peptides.
Using recombinant HLA-C molecules to generate tetramers and label lymphocytes
Finally, we wanted to examine whether our recombinant HLA-C preparations are suitable for tetramer generation. To this end, we generated HLA-C tetramers to evaluate a yellow fever NS2A4–13 (HAVPFGLVSM)–specific response, which have been observed in HLA-C*03:04+ yellow fever vaccinees (to be reported elsewhere). HLA-C*03:04 tetramers incorporating either the NS2A4–13 or a negative control peptide were generated. Both peptides could bind to HLA-C*03:04 and support tetramer formation (data not shown); however, only the NS2A4–13/HLA-C*03:04 tetramer could label cells in the lymphocyte gate as detected by flow cytometry. As shown in Fig. 6, the specific tetramer labeling did not only involve a CD8+ T cell population, but also a prominent CD3−, CD4−, CD8−, CD56+ population, suggesting a NK cell origin of the latter population. Indeed, gating on tetramer+ lymphocytes suggested the existence of at least two tetramer+ subpopulations: a CD8+/CD56− population and a CD8-/CD56int population accounting for 4 and 87% of the tetramer+ cells, respectively. We suggest that the former population consists of classical CD8+ CTLs whereas the latter consists of NK cells; however, a more detailed analysis of these phenotypic characteristics is beyond the scope of the present study. Suffice it to say that these HLA-C tetramers can label lymphocytes in a peptide-dependent manner.
FIGURE 6.

pHLA-C tetramer staining of CD8+ T lymphocytes. PBMCs from a yellow fever–vaccinated donor were stained with Abs against cell surface markers CD3, CD4, CD8, and CD56 and with two specific tetramers, that is, HLA-C*03:04-NS5477–485 (YMWLGARYL) (A and B) and HLA-C*03:04-NS2A4–13 (HAVPFGLVSM) (C–E). The results in (A)–(D) are gated for the total lymphocyte population whereas (E) is gated for all tetramer+ lymphocytes.
Discussion
Information about peptide binding to HLA-C is somewhat limited. Thus, as of March 2014, <1% of the HLA-I ligand data and <1% of the HLA-I allotypes hosted at the authoritative IEDB database originated from the HLA-C isotype; peptide-binding predictions perform poorer for the HLA-C isotype than for the HLA-A and -B isotypes; and only three published structures of HLA-C complexes exist in the PDB structure database (1QQD, 1IM9, and 1EFX) and they are all at a relatively low level of resolution (2.7–3.0 Å). In this study, we present a systematic analysis of the peptide-binding specificity of the HLA-C isotype. We have selected a panel of the most commonly expressed HLA-C allotypes and generated them as recombinant molecules, developed a general approach to uncover the peptide-binding specificity of these molecules, generated peptide-binding data for several different HLA-C allotypes, and updated the NetMHCpan method for MHC peptide-binding predictions. The updated predictor is improved considerably with respect to HLA-C predictions and is now at par with predictions for HLA-A and -B isotypes. Being pan-specific, this predictor can be applied to all HLA-C allotypes irrespective of whether experimental data exist for a specific allotype of interest. In other words, the improved NetMHCpan predictor can be used to describe the specificity of the entire HLA-C isotype.
The motifs identified in the present study largely match those previously described for HLA-C*03:04 (9), HLA-C*06:02 (39, 40), HLA-C*07:01 (40), and HLA-C*07:02 (39), supporting the notion that the approach taken in the present study yields an accurate representation of the peptide-binding properties of HLA-I molecules. For all HLA-C allotypes, whether tested experimentally or inferred bioinformatically, we observed the presence of a strong and dominant C-terminal primary anchor position, which encompassed a rather restricted repertoire of hydrophobic and aromatic amino acid residues (Phe, Ile, Leu, Met, Val, and Tyr). Whereas a dominant C-terminal anchor position is also commonly observed within the HLA-A and -B isotypes, for these later isotypes the amino acid composition of this anchor position is often more diverse. This explains, at least partially, the more limited peptide-binding repertoire of the HLA-C isotype compared with the HLA-A and -B isotypes. For most HLA-C allotypes, we also observed the presence of primary P2 and/or P3 anchor positions. Encompassing a greater repertoire of amino acids than the C-terminal anchor position, the P2/P3 anchor positions contribute with most of the differences in peptide-binding specificity observed within the HLA-C isotype. Using the P2/P3 specificity as a token, one can identify at least four major groups of HLA-C specificities: Ser/Ala/Thr in P2 (e.g., HLA-C*03:03), Tyr/Phe in P2 (e.g., HLA-C*04:01 and HLA-C*14:02), Asp/Glu in P3 (e.g., HLA-C*05:01), and Arg in P2 (e.g., HLA-C*07:01).
We have analyzed the motifs of HLA-C more systematically and in more detail, anchor position by anchor position. All HLA-C allotypes studied share a preference for aromatic and/or hydrophobic amino acids such as Phe, Ile, Leu, Met, Val, and Tyr at the C-terminal anchor position (Supplemental Table III). All molecules accept Phe, Ile, Leu, Met, and Val at the C-terminal position (RB values > 0.5), whereas Tyr is only accepted by selected allotypes (HLA-C*02:02, -C*03:02, -C*06:02, -C*07:01, -C*07:02, -C*12:03, -C*14:02, and -C*16:01). Of the polymorphic HLA residues interacting with the peptide C-terminal position (residues 74, 77, 80, 81, 84, 95, 97, 116, 118, 143, and 147 according to the NetMHCpan pseudosequence defined by a 4 Å threshold) (5), residues 77, 80, 95, 97, 116, and 147 are polymorphic among the HLA-C molecules. The allotypes accepting a C-terminal Tyr all express Ser at position 116 (Supplemental Table III), whereas allotypes having Tyr116 (HLA-C*03:03 and -C*03:04), Phe116 (HLA-C*04:01, -C*05:01, -C*07:04, -C*08:01, and -C*08:02), or Leu116 (HLA-C*15:02) disfavor the presence of Tyr at the peptide C-terminal position (RB values < 0.5). Residue 116 of the MHC-I molecule is placed at the bottom of the P9 pocket, greatly influencing the peptide-binding motif of the MHC-I molecule (41). In lieu of structural data for HLA-C, one can make a comparison with a similar observation that has been reported for HLA-B*35:01 (Ser116) and HLA-B*35:03 (Phe116), where HLA-B*35:01 allows binding of a C-terminal Tyr, whereas HLA-B*35:03 cannot accommodate a Tyr residue in the F pocket (41).
Strong anchors were also observed at the N-terminal end of the peptide, namely at P2 and/or P3 (Supplemental Table IV). This is a common positioning of a primary anchor in the HLA-C isotype, as well as in the HLA-A and -B isotypes. Concerning P2, both HLA-C*03:03, HLA-C*03:04, and HLA-C*12:03 have an anchor position at P2 featuring either Ala or Ser (Supplemental Tables IC, ID, IM, and IV). Of the polymorphic residues that interact with P2 according to the pseudosequence (5), HLA-C*03:03, HLA-C*03:04, and HLA-C*12:03 share a Tyr9, Ala24, Tyr99 motif (Supplemental Table IV), where the Tyr9 significantly restricts the size of the P2 pocket (42) favoring Ala (9, 39) and, as shown in the present study, the Ser in the P2 pocket. The Tyr9, Ala24, Tyr99 motif is also present in HLA-C*05:01 and HLA-C*08:02, which could account for their preference at P2 for Ala or Ser, although the concurrent presence of the strong Asp anchor residue in P3 seems to lessen the importance of P2, making P2 an auxiliary anchor in these latter molecules. Of the HLA-C molecules tested, HLA-C*04:01 has a unique P2 motif favoring aromatic residues Phe, Trpm and Tyr (Supplemental Table IV), and, although not confirmed by the PSCPL analysis, NetMHCpan prediction shows that HLA-C*14:02 shares this motif (Fig. 5, Supplemental Table IV). The crystal structure of the QYDDAVYKL peptide in complex with HLA-C*04:01 (PDB 1QQD) shows that the P2 Tyr fits in a pocket formed by the residues Tyr7, Phe22, Tyr67, and Phe99, creating a spacious pocket allowing the binding of tyrosine (43), but rotated differently than in most other HLA-I molecules. Among the HLA-C molecules studied, residues 7, 22, and 67 are monomorphic, whereas residue 99 is polymorphic. HLA-C*04:01 and HLA-C*14:02 share the Phe99 phenotype whereas the remaining molecules express either Ser99 (HLA-C*07:02 only) or Tyr99 (Supplemental Table IV), suggesting that this residue could be important for the P2 specificity of HLA-C molecules. In line with this, HLA-C*04:01 and HLA-C*14:02 also have Ser at position 9, whereas other molecules have Tyr9 or Asp9. Position 9 of the HLA-I molecules is situated at the bottom of the peptide-binding groove near the N-terminal end of the peptide, and expressing a bulky amino acid such as Phe or Tyr at residue 9 could restrict the size of the N-terminal–binding pockets.
Concerning P3, HLA-C*04:01, -C*05:01, and -C*08:02 share a strong preference for Asp (Fig. 1, Supplemental Tables IE, IF, IL, and IV). In the crystal structure of the QYDDAVYKL peptide in complex with HLA-C*04:01 (43), the Asp side chain of P3 cannot fit into the small D pocket of HLA-C*04:01, and thus hydrogen bonds with the peptide backbone and Arg156 (43). This Arg156 is shared between HLA-C*04:01, -C*05:01, -C*08:02, and -C*14:02, suggesting a contribution of this residue to the specificity at the P3 position of HLA-C molecules.
Auxiliary anchor positions were often observed in P1, P2, P3, and/or P7 (Supplemental Table IV). HLA-C*02:02, -C*03:03, -C*03:04, -C*04:01, -C*05:01, -C*06:02, -C*07:02, -C*07:04, -C*08:01, and -C*12:03 have an auxiliary anchor at P1 favoring Tyr or Phe/Tyr, whereas HLA-C*07:01 and -C*15:02 have an auxiliary anchor at P1 favoring the positively charged Lys and Arg residues. Whereas the P1 and P2 pockets of the latter group have a net negative charge, which confers a preference for positive amino acids at P1 and P2 (40), the former group has an Asn66 reducing the negative charge of their P1 and P2 pockets (Supplemental Table IV), which is likely to shift their preference from positive toward noncharged amino acids. As alluded to above, HLA-C*05:01 and -C*08:02 show auxiliary preference for Ala and Ser in position 2. This motif is shared with HLA-C*03:03, HLA-C*03:04, and HLA-C*12:03; however, the presence of the strong P3 anchor with preference for Asp likely lessens the significance of the residue in P2 for HLA-C*05:01 and HLA-C*08:02 (Supplemental Table IV). An auxiliary anchor was observed in P7 where HLA-C*07:01, HLA-C*07:02, and HLA-C*07:04 prefer aromatic amino acids Phe, Trp, and Tyr. These allotypes have a Leu147–Ala152 motif, compared with a Trp147–(Ala/Glu/Ser)152 motif for the remaining molecules studied. Residues 147 and 152 are located in the α2 helix of the MHC-I molecule facing in toward the peptide-binding groove, and the smaller size of the Leu147–Ala152 compared with the Trp147–(Ala/Glu/Ser)152 could indicate the presence of a P7 pocket along the α2 helix in HLA-C*07:01, HLA-C*07:02, and HLA-C*07:04.
Finally, our functional analysis shows that HLA-C*14:02 has a very unusual and promiscuous binding motif with no clear P2 or P3 anchor residues and only a weak P9 primary anchor (Supplemental Tables III, IV). The lack of a P2 anchor residue is not obvious because HLA-C*14:02 has a Ser9,indicating that an aromatic residue could accommodate the P2 pocket of molecule. The relative lack of anchors may explain the very high frequency of HLA-C*14:02 binders and may explain why the PSCPL predictions were less efficient for this allotype.
The HLA-C isotype is unique to hominoids where it has evolved as the most recent of the HLA-I isotypes (34, 44). The HLA-C is under two different evolutionary constraints serving both as TCR ligands for CD8+ T cells and as KIR ligands for NK cells. Both receptors interact with the outward-facing peptide–MHC complex and engage both the peptide and the MHC. Thus, there is considerable overlap between the ligands of the two receptors, although the KIR is more focused at the part of the peptide-binding cleft, which engages the C-terminal end of the peptide, including the C1/C2 dimorphism at position 80 of the HLA-C molecule, as well as the P8 and P7 residues of the bound peptide. This raises the question whether the peptide-binding repertoire of the HLA-C isotype is affected by the dual requirements of being a TCR and a KIR ligand. Our data clearly demonstrate that the peptide-binding repertoire of the HLA-C isotype is much more limited than are the repertoires of the HLA-A and -B isotypes, and that this is primarily caused by a restricted repertoire of amino acids at the C-terminal anchor. Being in the vicinity of the KIR recognition site, it is tempting to speculate that the KIR ligand requirement is behind this restriction of the peptide-binding repertoire of the HLA-C isotype. Another question is whether the C1/C2 dimorphism directly affects the peptide-binding repertoire of the HLA-C isotype. We have exploited the capability of the NetMHCpan predictor to handle custom MHC-I sequences to perform a systematic in silico study of the effect of this dimorphism on the peptide-binding specificity. This analysis showed no sign of gross alternations of the peptide-binding specificity caused directly by the C1/C2 dimorphism.
We present a novel method to characterize the HLA-I peptide-binding specificity based on SPA-driven PSCPL analysis. We show that an SPA-driven MHC-I dissociation assay can be used in combination with a nonameric PSCPL matrix to obtain an unbiased rendition of the peptide-binding motif for known as well as unknown HLA-I molecules. In theory, this approach could be extended to any MHC-I molecule from any species where the β2m sequence is a known, and it should be extremely robust because it does not depend on any preconceived information (e.g., knowledge of an indicator peptide sequence) or reagents (i.e., availability of a specific Ab) needed for detecting peptide interaction with the MHC-I in question. In fact, we have recently exploited this aspect of the approach to uncover the specificity of panels of bovine, swine, and bird MHC-I molecules (to be reported elsewhere). In the case of the HLA-C isotype, we have performed follow-up studies where we used the HLA-C peptide-binding matrices to assign a rank score to our panel of >10,000 nonameric peptides. For eight HLA-C molecules the 100 highest scoring peptides were tested for binding. In several cases, we found that ∼70% of these peptides bound with a KD of <500 nM. This underscores that PSCPL-driven matrix analysis is a fast and efficient method to identify binders to HLA molecules of unknown specificity. To extend the sampling of peptides, we also used the pan-specific NetMHCpan predictor to identify 100 peptides with high NetMHCpan scores but low PSCPL scores. It appears that NetMHCpan does capture additional peptide binders, particularly when the PSCPL has been less successful in identifying binders. Thus, the two selection methods complement each other, and we envision that this dual peptide selection principle could be used as a general method to acquire information-rich peptide–MHC-I interaction data and develop improved predictors.
Our overall conclusion is that HLA-C isotypes have peptide-binding features similar to those of a bona fide CTL restriction element. The suite of tools developed in the present study (recombinant HLA-C molecules and the accompanying binding assays, motif definitions, predictors, and tetramers) should support discovery of HLA-C–restricted T cell epitopes and NK targets and enable a more comprehensive analysis of the usage of HLA-C as CTL restriction element and as targets of NK cells.
Supplementary Material
Acknowledgments
We acknowledge the expert technical assistance of Anne Bregnballe Kristensen, Iben Sara Pedersen, and Anne Schmiegelow. We thank David Ostrov for valuable discussions on HLA-C structure.
This work was supported by National Institutes of Health/National Institute of Allergy and Infectious Diseases Grant N01-AI-2008032, HHSN272200900045C, the University of Copenhagen, Faculty of Health and Medical Sciences, and by Agence Nationale de la Recherche Grant ANR 2010 BIOT 008 01.
The online version of this article contains supplemental material.
- AP
- anchor position
- AUC
- area under the curve
- HLA-I
- HLA class I
- IEDB
- Immune Epitope Database
- KIR
- killer Ig–related receptor
- β2m
- β2-microglobulin
- MHC-I
- MHC class I
- PSCPL
- positional scanning combinatorial peptide library
- RB
- relative binding
- SPA
- scintillation proximity assay.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Lundegaard C., Lamberth K., Harndahl M., Buus S., Lund O., Nielsen M. 2008. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 36: W509–W512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karosiene E., Lundegaard C., Lund O., Nielsen M. 2012. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 64: 177–186. [DOI] [PubMed] [Google Scholar]
- 3.Kim Y., Sidney J., Pinilla C., Sette A., Peters B. 2009. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics 10: 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoof I., Peters B., Sidney J., Pedersen L. E., Sette A., Lund O., Buus S., Nielsen M. 2009. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielsen M., Lundegaard C., Blicher T., Lamberth K., Harndahl M., Justesen S., Røder G., Peters B., Sette A., Lund O., Buus S. 2007. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2: e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fadda L., Borhis G., Ahmed P., Cheent K., Pageon S. V., Cazaly A., Stathopoulos S., Middleton D., Mulder A., Claas F. H., et al. 2010. Peptide antagonism as a mechanism for NK cell activation. Proc. Natl. Acad. Sci. USA 107: 10160–10165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mandelboim O., Wilson S. B., Valés-Gómez M., Reyburn H. T., Strominger J. L. 1997. Self and viral peptides can initiate lysis by autologous natural killer cells. Proc. Natl. Acad. Sci. USA 94: 4604–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajagopalan S., Long E. O. 1997. The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J. Exp. Med. 185: 1523–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zappacosta F., Borrego F., Brooks A. G., Parker K. C., Coligan J. E. 1997. Peptides isolated from HLA-Cw*0304 confer different degrees of protection from natural killer cell-mediated lysis. Proc. Natl. Acad. Sci. USA 94: 6313–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lash G. E., Robson S. C., Bulmer J. N. 2010. Review: functional role of uterine natural killer (uNK) cells in human early pregnancy decidua. Placenta 31(Suppl.): S87–S92. [DOI] [PubMed] [Google Scholar]
- 11.Parham P., Norman P. J., Abi-Rached L., Hilton H. G., Guethlein L. A. 2012. Review: immunogenetics of human placentation. Placenta 33(Suppl): S71–S80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulpa D. A., Collins K. L. 2011. The emerging role of HLA-C in HIV-1 infection. Immunology 134: 116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alter G., Altfeld M. 2011. Mutiny or scrutiny: NK cell modulation of DC function in HIV-1 infection. Trends Immunol. 32: 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zipeto D., Beretta A. 2012. HLA-C and HIV-1: friends or foes? Retrovirology 9: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braendstrup P., Mortensen B. K., Justesen S., Osterby T., Rasmussen M., Hansen A. M., Christiansen C. B., Hansen M. B., Nielsen M., Vindeløv L., et al. 2014. Identification and HLA-tetramer-validation of human CD4+ and CD8+ T cell responses against HCMV proteins IE1 and IE2. PLoS ONE 9: e94892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins K. L., Chen B. K., Kalams S. A., Walker B. D., Baltimore D. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391: 397–401. [DOI] [PubMed] [Google Scholar]
- 17.Stryhn A., Pedersen L. O., Romme T., Holm C. B., Holm A., Buus S. 1996. Peptide binding specificity of major histocompatibility complex class I resolved into an array of apparently independent subspecificities: quantitation by peptide libraries and improved prediction of binding. Eur. J. Immunol. 26: 1911–1918. [DOI] [PubMed] [Google Scholar]
- 18.Ostergaard Pedersen L., Nissen M. H., Hansen N. J., Nielsen L. L., Lauenmøller S. L., Blicher T., Nansen A., Sylvester-Hvid C., Thromsen A. R., Buus S. 2001. Efficient assembly of recombinant major histocompatibility complex class I molecules with preformed disulfide bonds. Eur. J. Immunol. 31: 2986–2996. [DOI] [PubMed] [Google Scholar]
- 19.Ferré H., Ruffet E., Blicher T., Sylvester-Hvid C., Nielsen L. L., Hobley T. J., Thomas O. R., Buus S. 2003. Purification of correctly oxidized MHC class I heavy-chain molecules under denaturing conditions: a novel strategy exploiting disulfide assisted protein folding. Protein Sci. 12: 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leisner C., Loeth N., Lamberth K., Justesen S., Sylvester-Hvid C., Schmidt E. G., Claesson M., Buus S., Stryhn A. 2008. One-pot, mix-and-read peptide-MHC tetramers. PLoS One 3: e1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferré H., Ruffet E., Nielsen L. L., Nissen M. H., Hobley T. J., Thomas O. R., Buus S. 2005. A novel system for continuous protein refolding and on-line capture by expanded bed adsorption. Protein Sci. 14: 2141–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hunter W. M., Greenwood F. C. 1962. Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature 194: 495–496. [DOI] [PubMed] [Google Scholar]
- 23.Harndahl M., Rasmussen M., Roder G., Buus S. 2011. Real-time, high-throughput measurements of peptide-MHC-I dissociation using a scintillation proximity assay. J. Immunol. Methods 374: 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomsen M. C., Nielsen M. 2012. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 40: W281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harndahl M., Justesen S., Lamberth K., Røder G., Nielsen M., Buus S. 2009. Peptide binding to HLA class I molecules: homogenous, high-throughput screening, and affinity assays. J. Biomol. Screen. 14: 173–180. [DOI] [PubMed] [Google Scholar]
- 26.Boucherma R., Kridane-Miledi H., Bouziat R., Rasmussen M., Gatard T., Langa-Vives F., Lemercier B., Lim A., Bérard M., Benmohamed L., et al. 2013. HLA-A*01:03, HLA-A*24:02, HLA-B*08:01, HLA-B*27:05, HLA-B*35:01, HLA-B*44:02, and HLA-C*07:01 monochain transgenic/H-2 class I null mice: novel versatile preclinical models of human T cell responses. J. Immunol. 191: 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falk K., Rötzschke O., Stevanović S., Jung G., Rammensee H. G. 1991. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351: 290–296. [DOI] [PubMed] [Google Scholar]
- 28.Sidney J., Assarsson E., Moore C., Ngo S., Pinilla C., Sette A., Peters B. 2008. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 4: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamberth K., Røder G., Harndahl M., Nielsen M., Lundegaard C., Schafer-Nielsen C., Lund O., Buus S. 2008. The peptide-binding specificity of HLA-A*3001 demonstrates membership of the HLA-A3 supertype. Immunogenetics 60: 633–643. [DOI] [PubMed] [Google Scholar]
- 30.Parker K. C., Bednarek M. A., Coligan J. E. 1994. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 152: 163–175. [PubMed] [Google Scholar]
- 31.Buus S., Lauemøller S. L., Worning P., Kesmir C., Frimurer T., Corbet S., Fomsgaard A., Hilden J., Holm A., Brunak S. 2003. Sensitive quantitative predictions of peptide-MHC binding by a “Query by Committee” artificial neural network approach. Tissue Antigens 62: 378–384. [DOI] [PubMed] [Google Scholar]
- 32.Thomsen M., Lundegaard C., Buus S., Lund O., Nielsen M. 2013. MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics 65: 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hobohm U., Scharf M., Schneider R., Sander C. 1992. Selection of representative protein data sets. Protein Sci. 1: 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parham P., Norman P. J., Abi-Rached L., Guethlein L. A. 2012. Human-specific evolution of killer cell immunoglobulin-like receptor recognition of major histocompatibility complex class I molecules. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367: 800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Redchenko I., Harrop R., Ryan M. G., Hawkins R. E., Carroll M. W. 2006. Identification of a major histocompatibility complex class I-restricted T-cell epitope in the tumour-associated antigen, 5T4. Immunology 118: 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Breckpot K., Heirman C., De Greef C., van der Bruggen P., Thielemans K. 2004. Identification of new antigenic peptide presented by HLA-Cw7 and encoded by several MAGE genes using dendritic cells transduced with lentiviruses. J. Immunol. 172: 2232–2237. [DOI] [PubMed] [Google Scholar]
- 37.Larrieu P., Renaud V., Godet Y., Jotereau F., Fonteneau J. F. 2008. A HLA-Cw*0701 restricted Melan-A/MART1 epitope presented by melanoma tumor cells to CD8+ tumor infiltrating lymphocytes. Cancer Immunol. Immunother. 57: 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harndahl M., Rasmussen M., Roder G., Dalgaard Pedersen I., Sørensen M., Nielsen M., Buus S. 2012. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur. J. Immunol. 42: 1405–1416. [DOI] [PubMed] [Google Scholar]
- 39.Falk K., Rötzschke O., Grahovac B., Schendel D., Stevanović S., Gnau V., Jung G., Strominger J. L., Rammensee H. G. 1993. Allele-specific peptide ligand motifs of HLA-C molecules. Proc. Natl. Acad. Sci. USA 90: 12005–12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dionne S. O., Lake D. F., Grimes W. J., Smith M. H. 2004. Identification of HLA-Cw6.02 and HLA-Cw7.01 allele-specific binding motifs by screening synthetic peptide libraries. Immunogenetics 56: 391–398. [DOI] [PubMed] [Google Scholar]
- 41.Gao X., Nelson G. W., Karacki P., Martin M. P., Phair J., Kaslow R., Goedert J. J., Buchbinder S., Hoots K., Vlahov D., et al. 2001. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N. Engl. J. Med. 344: 1668–1675. [DOI] [PubMed] [Google Scholar]
- 42.Boyington J. C., Motyka S. A., Schuck P., Brooks A. G., Sun P. D. 2000. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature 405: 537–543. [DOI] [PubMed] [Google Scholar]
- 43.Fan Q. R., Wiley D. C. 1999. Structure of human histocompatibility leukocyte antigen (HLA)-Cw4, a ligand for the KIR2D natural killer cell inhibitory receptor. J. Exp. Med. 190: 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moesta A. K., Abi-Rached L., Norman P. J., Parham P. 2009. Chimpanzees use more varied receptors and ligands than humans for inhibitory killer cell Ig-like receptor recognition of the MHC-C1 and MHC-C2 epitopes. J. Immunol. 182: 3628–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.