

Abstract
Pre-mRNA splicing by the spliceosome is an essential step in the maturation of nearly all human mRNAs. Mutations in six spliceosomal proteins, PRPF3, PRPF4, PRPF6, PRPF8, PRPF31 and SNRNP200, cause retinitis pigmentosa (RP), a disease characterized by progressive photoreceptor degeneration. All splicing factors linked to RP are constituents of the U4/U6.U5 tri-snRNP subunit of the spliceosome, suggesting that the compromised function of this particle may lead to RP. Here, we report the identification of the p.R192H variant of the tri-snRNP factor PRPF4 in a patient with RP. The mutation affects a highly conserved arginine residue that is crucial for PRPF4 function. Introduction of a corresponding mutation into the zebrafish homolog of PRPF4 resulted in a complete loss of function in vivo. A series of biochemical experiments suggested that p.R192H disrupts the binding interface between PRPF4 and its interactor PRPF3. This interferes with the ability of PRPF4 to integrate into the tri-snRNP, as shown in a human cell line and in zebrafish embryos. These data suggest that the p.R192H variant of PRPF4 represents a functional null allele. The resulting haploinsufficiency of PRPF4 compromises the function of the tri-snRNP, reinforcing the notion that this spliceosomal particle is of crucial importance in the physiology of the retina.
Introduction
Mutations that interfere with splicing are a frequent cause of human disease [1], [2]. The majority of these mutations affect pre-mRNA splice sites or regulatory elements in cis [3]. However, also mutations that interfere with the trans-acting machinery that catalyzes the splicing reaction – the spliceosome – can lead to disease [4]–[7]. Surprisingly, despite the essential role of pre-mRNA splicing for gene expression on a transcriptome-wide scale, mutations in general splicing factors are often associated with specific phenotypes that are restricted to a particular tissue or even cell type [2]. A prime example for such a tissue specificity paradox is the hereditary eye disease retinitis pigmentosa (RP), which is characterized by a selective loss of photoreceptor cells in the retina. As a consequence, patients suffer from a severe visual handicap [8]. Although this phenotype is highly selective and affects primarily rod photoreceptors, a substantial fraction of RP- cases have been linked to mutations in the general splicing factors PRPF3, PRPF4, PRPF6, PRPF8, PRPF31 and SNRNP200 [9]–[14].
The aforementioned proteins are part of the spliceosome, a highly dynamic molecular machine that consists of five small nuclear ribonucleoprotein particles (snRNPs; termed according to their RNA content U1, U2, U4/U6 and U5) and a large set of non-snRNP factors [15]. It is formed in a stepwise manner on the cis-acting sequence elements of introns, including the branch point and the 5′ and 3′ splice sites, which define the boundaries to exons. In a first step, U1 and U2 snRNPs bind to the 5′-splice site and the branch point, respectively. The resulting pre-spliceosomal complex then recruits U4/U6 and U5 snRNPs, which form prior to their integration into the spliceosome the so-called U4/U6.U5-tri-snRNP. Structural rearrangements then lead to the release of U1 and U4 snRNPs, and the formation of the catalytically active spliceosome. After splicing, the spliceosome is disassembled and its individual subunits are recycled so that they can re-assemble on another intron. While U1 and U2 snRNPs are recycled as “mono-snRNPs”, the tri-snRNP is rebuilt by the joining of the snRNPs U4 and U6, which generates a U4/U6 di-snRNP that is subsequently bound by the U5 snRNP [16].
Intriguingly, every splicing factor linked to RP thus far is a component of the U4/U6.U5 tri-snRNP. The tri-snRNP is essential for the formation of the active center of the spliceosome, participates in splice site selection and has been recently proposed to recruit the nuclear RNA surveillance machinery to mRNAs [17]–[19]. It has been hypothesized that defects in RP-linked splicing factors affect gene expression in a manner to which the retina is uniquely sensitive [20], [21]. In line with this hypothesis, we have recently shown in a zebrafish model for RP that the deficiency of PRPF31 predominantly affects the expression of retina-specific transcripts [22]. Many of the affected transcripts were also associated with retinal disease, thereby providing a direct link between defects in general splicing factors and a retinal phenotype. Of note, the knockdown of another tri-snRNP protein, PRPF4, resulted in a photoreceptor-specific phenotype that was indistinguishable from that of the PRPF31 model [22]. Therefore, we reasoned that PRPF4 could be similarly involved in the pathogenesis of RP and screened patients for mutations in the PRPF4 gene.
Here, we characterize the molecular and physiological effects of a missense mutation (p.R192H) in the tri-snRNP factor, PRPF4, which was identified in a sporadic RP patient. Several lines of evidence suggest that this variant represents a loss-of-function allele. First, a corresponding mutation in the zebrafish PRPF4 homolog rendered it physiologically inactive. Second, binding of PRPF4 to its interactor PRPF3 was abrogated by the p.R192H amino acid exchange. Third, variant PRPF4 failed to integrate into the tri-snRNP in human cell lines and zebrafish embryos. Together, these findings indicate that PRPF4 haploinsufficiency may contribute to RP. Furthermore, they underline the role of tri-snRNP splicing factors as mutational hotspots in RP and demonstrate the feasibility of a direct analysis of these candidate genes in RP patients.
Results
Identification of a loss-of-function variant of the splicing factor PRPF4 in a patient with RP
We screened DNA samples from 85 patients with sporadic or autosomal dominant RP (adRP) for mutations in PRPF4. This revealed a heterozygous non-synonymous nucleotide exchange, c.575G>A, in the sporadic female patient RP-106 (Fig. 1A). At the age of 39 years, RP-106 had stopped driving because of night blindness. Constriction of the visual field was noted at the age of approximately 45 years. With 55 years of age, early cataracts were found. Funduscopy showed pale papillae, narrowing of retinal vessels, and bone spicule-like pigmentation in the periphery. The scotopic electroretinogram (ERG) was extinguished, the photopic ERG was almost extinguished. RP-106 has two daughters (III:2, 54 years, and III:4, 52 years). III:4 was found to carry the c.575G>A variant. III:2 and III:4 underwent detailed ophthalmological investigation including assessment of visual acuity, funduscopy, perimetry, dark adaptation and ERG and did not show any signs of retinal degeneration. As shown in a recent study by Chen et al. [10], the onset of PRPF4-associated RP is variable (between 15 and 27 years of age). Both inter- and intrafamilial clinical variability are common in RP. The reported onset of visual complaints (20 years of age) in our patient corresponds to the range reported by Chen et al [10].
Figure 1. RP family described herein.
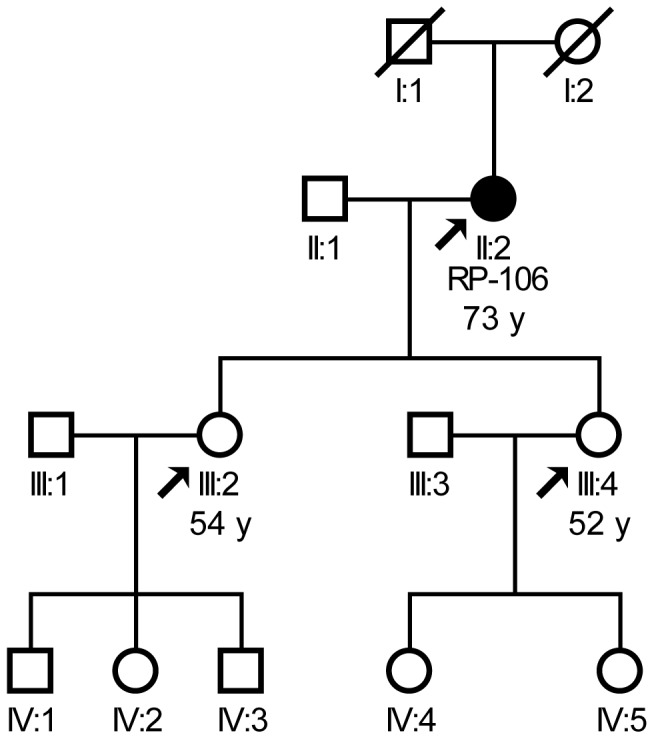
II:2, index patient and heterozygous carrier of the PRPF4 variant p.R192H. Arrows, individuals who were available for genetic and clinical characterization. Circles, female individuals; Squares, male individuals. Filled symbols indicate affected persons. White symbols, unaffected persons by detailed ophthalmological investigation (III:2, III:4) or medical history. III:4, healthy carrier of the PRPF4 variant. Crossed symbols, deceased persons. Age at the time of examination is given below the pedigree symbols.
Although c.575G>A was annotated as a single nucleotide polymorphism (SNP) in the dbSNP database (rs41296057; http://www.ncbi.nlm.nih.gov/SNP/), this entry did not contain information about allele frequency or validation status. Furthermore, c.575G>A was not present in 13,006 control chromosomes deposited in the Exome Variant Server (http://evs.gs.washington.edu/EVS/) and 2,188 chromosomes deposited in the 1000genomes database (http://www.1000genomes.org/). These data suggested that c.575G>A is a very rare PRPF4 variant. The predicted amino acid exchange affects a highly conserved arginine residue at position 192 (p.R192H) and was predicted by the PROVEAN, PolyPhen-2 and SIFT algorithms to have a deleterious effect [23]–[25]. Hence, we set out to analyze the impact of the p.R192H variant on PRPF4 protein function.
First, we wanted to investigate whether the mutant allele acts in a dominant negative manner. For this, zebrafish embryos were injected with mRNAs transcribed in vitro encoding for a fusion protein of an N-terminal hemagglutinin epitope (HA-tag) and zebrafish Prpf4, either in wildtype form or with an amino acid exchange that corresponds to p.R192H. The HA-tag allowed for the simultaneous and semi-quantitative detection of exogenous and endogenous protein by Western blotting using a Prpf4-specific antibody. This revealed an overexpression of exogenous HA-prpf4 (wildtype and mutant) in fish that were injected with the corresponding mRNAs (Figure 2A, upper panel). Yet, neither of the proteins interfered with normal embryonic development (Fig.2 A, lower panel), suggesting that p.R192H does not have a strong dominant negative effect.
Figure 2. The p.R192H variant leads to a loss of function in vivo.

(A) Expression of zebrafish Prpf4 carrying a mutation corresponding to p.R192H does not have a dominant negative effect. Upper panel: Western blot detection of endogenous Prpf4 and exogenous HA-tagged Prpf4 in embryos injected with RNAs indicated above. Lower panel: No defects in embryonic development were observed upon overexpression of wildtype or p.R192H Prpf4. (B) Western blot showing Prpf4 protein levels in zebrafish injected with the indicated combinations of prpf4 morpholino (MO) and RNA. (C–F) The p.R192H mutant fails to rescue a lethal prpf4 deficiency. Representative selections of 3 days post fertilization (dpf) larvae from embryos injected with the indicated combinations of prpf4 morpholino (MO) and RNA. (G–J) Quantification of normal, deformed and dead animals from four independent rescue experiments. The rescue effect (H vs. I) and the loss of function effect of the p.R192H mutation (I vs. J) were highly significant (Pearson χ2 test; n is the total number of injected animals). Please note that the control experiments (uninjected, MO and MO + wt RNA), but not the characterization of p.R192H, were previously published as part of a larger knockdown study [22]. As the characterization of p.R192H has been performed in the context of this study, we reproduce here the control data for clarity (with permission from Oxford University Press).
Next, we addressed the possibility that p.R192H results in a loss of function. For this, we included the p.R192H mutant in our rescue analysis of severe Prpf4 deficiency [22]. In this assay, zebrafish embryos were injected with a dose of antisense morpholino that strongly reduces Prpf4 expression (Fig. 2B, compare lanes 1 and 2 with lanes 3 and 4) and leads to an embryonic lethal phenotype (Fig. 2, compare C and D). Although the co-injection of in vitro transcribed, morpholino-insensitive mRNAs encoding HA-Prpf4 in either wildtype or mutant form led to the expression of comparable amounts of protein (Fig. 2B, compare lanes 5 and 6 with lanes 7 and 8), only the injection of wildtype mRNA resulted in an improvement of the phenotype (Fig. 2, compare E and F). Quantification of these results (Fig. 2 G–J) confirmed the significantly lower rescue effect of mutant Prpf4 compared to wildtype protein (Pearson χ2 test, p = 0.00001), suggesting that p.R192H results in a loss of function.
The p.R192H variant disrupts binding of PRPF4 to PRPF3
Phylogenetic alignment of the amino acid sequence of PRPF4 homologs revealed that arginine 192 of human PRPF4 is highly conserved in evolution (Fig. 3A) and locates in a 34 amino acids stretch essential for Prp4 function in yeast [26]. Since this basic region contains a putative nuclear localization signal [27], it was first tested whether p.R192H interferes with nuclear import and/or subnuclear localization of PRPF4. The localization of human, HA-tagged wildtype and p.R192H PRPF4 was analyzed in transfected HeLa cells by indirect immunofluorescence. Both proteins were found in the nucleus in a speckled pattern that is characteristic for splicing factors and co-localized with the U5 snRNP associated splicing factor EFTUD2 (Fig. 3B–I). These results indicate that the missense mutation p.R192H does not affect the sub-cellular localization of PRPF4.
Figure 3. Characterization of the PRPF4 missense variant p.R192H identified in an RP patient.

(A) Domain organization of PRPF4 (upper panel). The p.R192H amino acid exchange is located between the splicing factor motif (SFM) and the WD40 repeat domain (WD). Amino acid sequence alignment revealed a strong conservation of the affected arginine residue across species (lower panel). Identical residues are shown in red; similarity greater than 70% (BLOSUM62) is indicated by a grey box. See table S1 for sequence accession numbers. (B–I) The subcellular localization of PRPF4 is not affected by the p.R192H mutant. Wildtype (B–E) and the p.R192H form of PRPF4 (F–I) were detected in the nuclei of transfected HeLa cells. Both proteins co-localized with EFTUD2 protein in a speckled pattern typical for splicing factors (B, F). Nuclei were control-stained with DAPI (E, I). The scale bar in K is 20 µm and accounts for all images. (J) The p.R192H missense mutant impairs binding to PRPF3. Immunoprecipitation analysis of in vitro translated [35S]-labeled PRPF4-PRPF3 complex (upper panel) using an anti-PRPF3 antibody revealed a significant reduction of co-precipitated p.R192H-PRPF4 (compare lanes 3 and 4). The interaction of PRPF4 to PPIH in contrast, was not affected (compare lanes 9 and 10). Lanes 1, 2, 7 and 8 show 10% of the inputs, lanes 5, 6, 11 and 12 are mock immunoprecipitations.
PRPF4 is a constituent of the U4/U6 di-snRNP and as such becomes also integrated into the U4/U6.U5 tri-snRNP. In the context of these snRNPs, PRPF4 directly interacts with PRPF3 (the product of the RP disease gene PRPF3), and the cyclophilin PPIH [28], [29]. To analyze whether p.R192H interferes with these interactions, we generated [35S]-labeled, HA-tagged wildtype and mutant PRPF4 by translation in rabbit reticulocyte lysates in vitro, along with their potential interacting proteins. The proteins of interest were then immunoprecipitated by anti-PRPF3 antibodies and detected by autoradiography. As shown in Figure 3 J, anti-PRPF3 antibodies efficiently co-precipitated wildtype PRPF4 but not the p.R192H mutant (upper panel, compare lanes 3 and 4). The interaction of PRPF4 with PPIH was not affected, suggesting that PRPF4 p.R192H is not globally misfolded (Fig. 3J, lower panel). Thus, the p.R192H amino acid exchange results in a specific disruption of the binding of PRPF4 to PRPF3.
The p.R192H variant leads to a snRNP integration defect of PRPF4
The data presented above pointed to a crucial function of the residue 192 in the interaction of PRPF4 with PRPF3. The missense mutation identified may disrupt this interaction by local unfolding, or by preventing a defined interaction of arginine 192 with the corresponding interaction surface in PRPF3. To distinguish between both possibilities and to assess the importance of the physicochemical properties of this residue, we produced a series of mutants where arginine 192 was replaced with lysine, tryptophan or glutamate. These mutants were co-translated with PRPF3 and assayed for binding by co-immunoprecipitation with a PRPF3 antibody. Although the p.R192H mutant showed the strongest effect, a decrease in PRPF3 binding was also observed for all other variants tested (Fig. 4A). These data suggest that the arginine at position 192 engages in a specific contact with the PRPF3 binding interface that is prevented in the p.R192H variant.
Figure 4. The arginine residue at position 192 is essential for integration of PRPF4 into snRNPs.
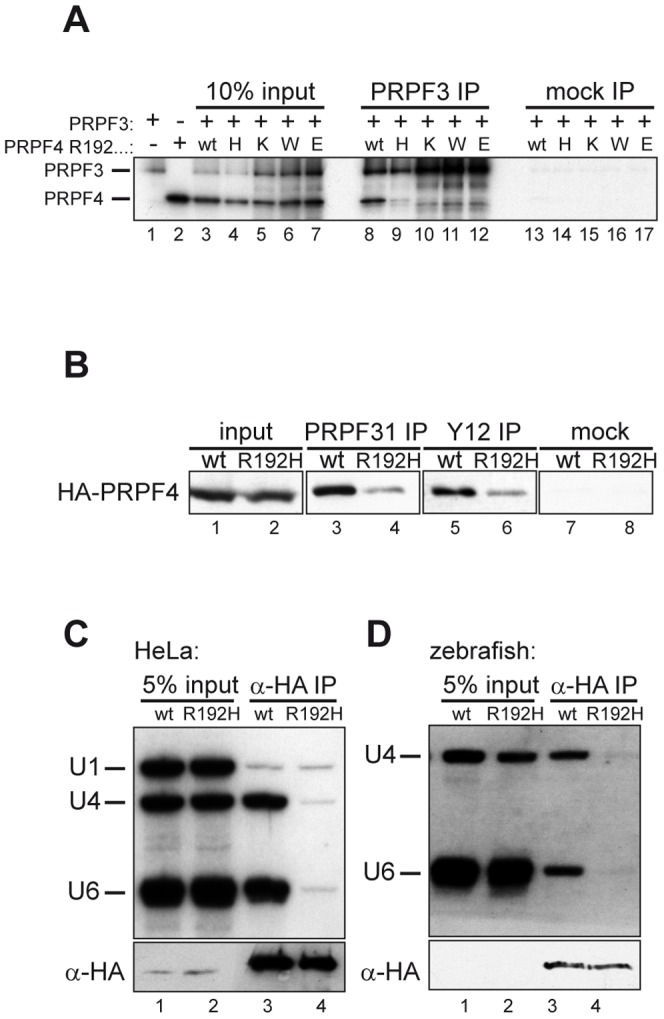
(A) A series of mutants at position 192 was analyzed for binding to PRPF3. Neither lysine (positively charged) nor tryptophan (aromatic) nor glutamate (negatively charged) were tolerated at this position. (B) Reduced co-precipitation of p.R192H PRPF4 with snRNPs. HEK293 cells were transfected with wildtype and mutant HA-PRPF4 and lysates were immunoprecipitated using an anti-PRPF31 antibody (lanes 3 and 4), anti-Sm-antibody Y12 (lanes 5 and 6) or mock precipitated (lanes 7 and 8). Lanes 1 and 2 show 10% of the input used for the immunoprecipitations. (C) PRPF4 p.R192H fails to associate with U4 and U6 snRNA. HEK293 cells were transfected with HA-tagged PRPF4 in either wildtype or p.R192H mutant form. Anti-HA immunoprecipitation was controlled by Western blot (lower panel) and co-precipitated di-snRNP RNAs U4 and U6 were analyzed by Northern blotting (upper panel). U1 snRNA served as a specificity control. (D) snRNA co-immunoprecipitation experiments using zebrafish embryo lysates. Embryos were co-injected with 50 pg wildtype or p.R192H HA-prpf4 mRNA and 0.5 ng prpf4-MO to facilitate snRNP integration of exogenous protein. At 10 hpf, extracts were prepared and immunoprecipitations were analyzed as described in (C).
Mutational studies in yeast have suggested that Prp4 is integrated into the U4/U6 snRNP via its binding to Prp3 [27]. It was hence a possibility that the p.R192H mutant of PRPF4 would not only disrupt the interaction with PRPF3 but also interfere with its integration into spliceosomal complexes. Two independent experiments were performed to test this possibility. In a first approach, HEK293 cells were transfected with HA-tagged versions of either wildtype or p.R192H PRPF4. Cellular lysates were then immunoprecipitated with antibodies directed against either PRPF31 (a U4/U6-snRNP specific component that does not directly bind to PRPF4) or the Sm proteins common to spliceosomal snRNPs (Fig. 4 B). A robust co-precipitation of wildtype protein by both antibodies was observed, whereas the p.R192H mutant PRPF4 was poorly co-precipitated (compare lanes 3 with 4 and 5 with 6, respectively; see also lanes 1 and 2 for inputs). In a second approach, wildtype and p.R192H mutant HA-PRPF4 were immunopecipitated with anti-HA antibodies from transfected HeLa cells and analyzed for co-precipitation of the tri-snRNP snRNAs U4 and U6 as well as snRNA U1 as a control by Northern blotting (Fig. 4 C). Although wildtype and mutant proteins were precipitated with similar efficiency (lower panel), co-precipitation of the snRNAs U4 and U6 was markedly reduced for mutant PRPF4 (compare lanes 3 and 4). Thus, the p.R192H PRPF4 variant is not stably incorporated into snRNPs.
We next considered the possibility that the uncovered biochemical defect might also underlie the inability of p.R192H mutant prpf4 mRNA to rescue the lethal Prpf4 knockdown in zebrafish. This was addressed by analyzing the U snRNP integration of wild type and mutant Prpf4 in zebrafish embryos. Co-injections of morpholino against endogenous prpf4 together with morpholino-insensitive in vitro transcripts encoding either wildtype or p.R192H HA-Prpf4 were performed in an analogous manner as described for the rescue experiments. At 10 hours post fertilization (hpf), lysates of the embryos were prepared and subjected to anti-HA immunoprecipitation. Co-precipitated snRNAs were then analyzed by northern blotting using probes specific for zebrafish U4 and U6 snRNAs. Similar levels of HA-Prpf4 were recovered from animals injected with wildtype or p.R192H RNA (Fig. 4D, lower panel), but the amount of associated di-snRNP snRNAs was severely reduced for p.R192H HA-Prpf4 (Fig. 4D, upper panel). Thus, the p.R192H mutation interferes with the integration of PRPF4 into spliceosomal complexes in vivo. Given the essential nature of this step in pre-mRNA processing, this defect is a likely cause for the inability of p.R192H RNA to rescue Prpf4 deficiency.
Discussion
In this study, we describe the identification of an isolated RP patient that carries a heterozygous missense variant (p.R192H) in the gene encoding the tri-snRNP splicing factor PRPF4. We show that this variant disrupts the binding of PRPF4 to its interactor PRPF3 in vitro. This leads to an impaired integration into spliceosomal particles and a loss of physiological function in vivo.
PRPF4 has been shown previously to form a very stable ternary complex with PRPF3 and the peptidyl-prolyl isomerase PPIH. Within this ternary complex, PRPF4 acts as a bridging factor by contacting PPIH via its N-terminal part and PRPF3 via its central and C-terminal part [28], [29]. Consistent with this mapping is our observation that the p.R192H mutation affected PRPF3 binding but not the association with PPIH. This mode of binding suggests that p.R192H not only causes a defective integration of PRPF4 into the tri-snRNP, but that it might also lead to the concomitant loss of PPIH, a protein necessary for efficient splicing [30]. Remarkably, a similar mechanism has been proposed for an RP-mutant form of PRPF31 that is thought to sequester its interactor PRPF6 out of the tri-snRNP [31], and an altered composition of the tri-snRNP has also been described for RP-causing missense mutations in PRPF8 [21].
Our biochemical data strongly indicate that the p.R192H variant leads to the production of non-functional protein and thus represents a functional null allele. However, patient RP-106 has two daughters, one of which was found to have inherited the c.575G>A variant (Fig. 1). Both daughters underwent detailed ophthalmological investigation and did not show signs of retinal degeneration. While the genetic study presented here thus does not allow to conclude whether PRPF4 is causally linked to RP, additional evidence indicates that this is indeed the case: Our data obtained in zebrafish showed that the sub-lethal knockdown of Prpf4 leads to a retina-specific phenotype similar to that of RP [22]. The data presented here shows that wildtype Prpf4, but not a variant corresponding to p.R192H can rescue such Prpf4 deficiency (Fig. 2). Further support for a link between PRPF4 deficiency and RP comes from a recent study where PRPF4 mutations were found to cause adRP in a Chinese cohort [10]. Also, a potentially pathogenic missense variant of PRPF4 (p.P187A) was reported in two German siblings with RP [32]. Although this latter study failed to demonstrate a clear association between PRPF4 and RP in North American and European populations [32], these data indicate that rare defects in PRPF4 might cause RP in a limited subset of patients.
The absence of RP symptoms in the daughter who carries the p.R192H variant of PRPF4 might be due to incomplete penetrance or largely variable disease expressivity, both being phenomena frequently observed for RP-associated mutations in splicing factors [33]–[35]. For the PRPF31-linked autosomal dominant RP, this has been explained by a modifier gene that modulates PRPF31 transcription. Low expression of the modifier increases wild-type PRPF31 levels to an extent that are sufficient to prevent the development of disease symptoms in heterozygous mutation carriers [36]. It has been proposed that the vulnerability of retinal cells to splicing factor defects arises from an unmet requirement for the production of sufficient amounts of mature retinal mRNAs [20]. Thus, defects in different splicing factors might act together to decrease the level of functional tri-snRNPs below the threshold required in retinal cells. This does not necessarily require a complete loss of the mutant protein from the tri-snRNP, as demonstrated by an RP-causing missense mutation in SNRNP200 which still allows integration of the mutant protein into the tri-snRNP but causes a decrease in splicing efficiency and fidelity [37]. It is possible that a hypomorphic variant in another splicing factor is present in the patient, but not in her daughter, and leads to the manifestation of RP. Moreover, it can not be excluded that the RP of the patient is caused by biallelic mutations in a recessive RP gene, and that p.R192H represents a rare non-pathogenic variant.
Support for the notion that a decrease in overall tri-snRNP activity rather than individual splicing factor defects lead to photoreceptor damage comes from a zebrafish mutant of the tri-snRNP recycling factor SART3: Although SART3 is an assembly factor that is not itself part of the tri-snRNP [38], its mutation leads to the reduced expression of photoreceptor mRNAs [39]. Taken together, our data are consistent with a model in which rare mutations of tri-snRNP splicing factors act together to decrease the level of functional tri-snRNPs below the threshold required in retinal cells.
Materials and Methods
Plasmids and antibodies
Zebrafish prpf4 was amplified from 14 hpf embryonic oligo-dT cDNA using gene-specific primers (see table S1 for sequences). Human PRPF4 was amplified using the “Marathon human fetal brain” cDNA library (Clontech) as a template and gene-specific primers. cDNAs were inserted into a modified version of the pcDNA3 vector that adds an N-terminal hemagglutinin- (HA-) tag for the transfection of HeLa cells, into the pET21a, pET28a and the pGEX-6P1 vector for in vitro translation and the production of recombinant proteins and into an HA-tagged version of the pCS2+ vector for in vitro transcription. To generate the morpholino-insensitive prpf4, synonymous mutations were introduced in the ATG region by site directed mutagenesis, resulting in 6 mismatches to the antisense morpholino sequence (see table S1 for sequences of mutagenic primers). The same approach was applied to introduce the p.R177H mutation that corresponds to the human p.R192H exchange. Antibodies against PRPF3, PRPF4 and PRPF31 were described previously [22]. Antibodies against EFTUD2 were a gift from R. Lührmann. The anti-HA antibody (MMS-101R) was purchased from HiSS Diagnostics.
Microinjection and Western blot analysis of zebrafish embryos
Microinjection of zebrafish embryos and Western blot analysis of embryo lysates was performed as described previously [22]. Briefly, one- or two-cell stage embryos were injected with a morpholino oligo directed against the start codon region of prpf4 (prpf4-MO: TGGAGCTTCATCTTCATCTGACATC; Gene Tools, LLC). Lysates for Western blots were prepared by homogenization of single larvae in 20 µl of a 1∶1 mixture of Laemmli sample buffer and buffer B (100 mM NaH2PO4, 10 mM Tris pH 8.0, 8 M Urea).
Rescue experiments
Single clutches of embryos were divided into four groups that (1) were left uninjected, (2) were injected with a lethal dose of prpf4-MO (2 µg/µl), (3) were co-injected with prpf4-MO and wildtype prpf4 mRNA or (4) were co-injected with prpf4-MO and p.R192H mutant prpf4 mRNA. The co-injected mRNAs were transcribed in vitro, capped and purified as described previously [22]. Successful injection (knockdown of endogenous Prpf4 and expression of exogenous HA-Prpf4) was monitored by Western blotting at 12 hpf. After 4 dpf, larvae were scored as described [22] as either lethal, severely deformed or slightly deformed/not affected. At least three independent experiments were performed for each analysis.
Immunocytochemistry
HeLa cells were grown on coverslips in DMEM (PAA) containing 10% fetal calf serum (PAA) at 37°C and 5% CO2. HA-PRPF4 inserted into the pcDNA3 vector was transfected using Nanofectin (PAA) and two days after transfection cells were fixed and immunocytochemistry was performed as described previously [40].
In vitro translation, co-immunoprecipitations and Northern blotting
Proteins were translated from pET21a and pET28a vectors using the T7 TNT coupled transcription and translation kit (Promega) in the presence of [35S]-L-methionine.
Extracts form HEK293 cells were obtained by sonication in IPP300 (20 mM Tris pH 7.0, 300 mM NaCl, 0.05% NP40) containing 1.5 mM MgCl2, 0.5 mM DTT and protease inhibitors. Zebrafish embryo lysates were prepared from ∼100 embryos (10hpf) by sonication in 200 µl of IPP300 containing 0.5% NP40. Crude lysates were extracted using 100 µl N-heptane and centrifuged at 16.000 g and 4°C for 30 minutes.
For immunoprecipitations, antibodies were coupled to either protein A- or protein G-Sepharose and incubated with translated proteins/cellular extract in IPP300. After excessive washing with IPP300, bound proteins were eluted using Laemmli sample buffer and analyzed by Western blotting or autoradiography.
For Northern blot analyses, PK-buffer (2x: 200 mM Tris-HCl pH 7.5, 25 mM EDTA, 300 mM NaCl, 2% SDS) and phenol were added to the immunoprecipitates. After phase separation, the RNA was ethanol-precipitated, subjected to PAGE (10%, 8 M Urea) and electroblotted onto Hybond N+ Nylon membrane (GE Healthcare). Hybridization of [32P]-labeled oligonucleotides specific for U1, U4 and U6 snRNAs (see table S1 for sequences) was carried out at 42°C in RapidHyb solution (GE Healthcare).
Patient screening and analysis
Samples were obtained with written informed consent. Clinical investigations were conducted according to the Declaration of Helsinki, and the study was approved by the institutional review board of the Ethics Committee of the University Hospital of Cologne. 85 patients with sporadic or autosomal dominant RP were screened for mutations in the PRPF4 gene. All 14 coding exons were PCR-amplified and the products were subjected to SSCP (single strand conformation polymorphism) analysis. Products with band shifts were directly sequenced. All 85 patients with no previously identified RP-mutations and 100 healthy control individuals were genotyped for the presence of p.R192H by direct sequencing.
Supporting Information
Oligonucleotide sequences and accession numbers used.
(PDF)
Acknowledgments
We thank R. Lührmann for reagents, E. Kunkel for excellent technical assistance and T. Meitinger for critically reading the Manuscript.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by German Research Foundation (DFG) grants to U. Fischer and an Academic Research Fund (AcRF) grant (T207B3107) of the Ministry of Education, Singapore to C. Winkler. B. Linder and A. Hirmer received fellowships from the German ProRetina Foundation and the Graduiertenkolleg GK 1048, respectively. This publication was funded by the German Research Foundation (DFG) and the University of Wuerzburg in the funding programme Open Access Publishing. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Faustino NA, Cooper TA (2003) Pre-mRNA splicing and human disease. Genes Dev 17: 419–437. [DOI] [PubMed] [Google Scholar]
- 2. Cooper TA, Wan L, Dreyfuss G (2009) RNA and disease. Cell 136: 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cartegni L, Chew SL, Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3: 285–298. [DOI] [PubMed] [Google Scholar]
- 4. Edery P, Marcaillou C, Sahbatou M, Labalme A, Chastang J, et al. (2011) Association of TALS Developmental Disorder with Defect in Minor Splicing Component U4atac snRNA. Science 332: 240–243. [DOI] [PubMed] [Google Scholar]
- 5. He H, Liyanarachchi S, Akagi K, Nagy R, Li J, et al. (2011) Mutations in U4atac snRNA, a Component of the Minor Spliceosome, in the Developmental Disorder MOPD I. Science 332: 238–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jia Y, Mu JC, Ackerman SL (2012) Mutation of a U2 snRNA Gene Causes Global Disruption of Alternative Splicing and Neurodegeneration. Cell 148: 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pasternack SM, Refke M, Paknia E, Hennies HC, Franz T, et al. (2013) Mutations in SNRPE, which Encodes a Core Protein of the Spliceosome, Cause Autosomal-Dominant Hypotrichosis Simplex. Am J Hum Genet 92: 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hartong DT, Berson EL, Dryja TP (2006) Retinitis pigmentosa. Lancet 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- 9. Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, et al. (2001) A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol Cell 8: 375–381. [DOI] [PubMed] [Google Scholar]
- 10. Chen X, Liu Y, Sheng X, Tam POS, Zhao K, et al. (2014) PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum Mol Genet 23: 2926–2939. [DOI] [PubMed] [Google Scholar]
- 11. Chakarova CF, Hims MM, Bolz H, Abu-Safieh L, Patel RJ, et al. (2002) Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet 11: 87–92. [DOI] [PubMed] [Google Scholar]
- 12. Zhao C, Bellur DL, Lu S, Zhao F, Grassi MA, et al. (2009) Autosomal-Dominant Retinitis Pigmentosa Caused by a Mutation in SNRNP200, a Gene Required for Unwinding of U4/U6 snRNAs. Am J Hum Genet 85: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, et al. (2001) Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum Mol Genet 10: 1555–1562. [DOI] [PubMed] [Google Scholar]
- 14. Tanackovic G, Ransijn A, Ayuso C, Harper S, Berson EL, et al. (2011) A Missense Mutation in PRPF6 Causes Impairment of pre-mRNA Splicing and Autosomal-Dominant Retinitis Pigmentosa. Am J Hum Genet 88: 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wahl MC, Will CL, Lührmann R (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- 16. Raghunathan PL, Guthrie C (1998) A Spliceosomal Recycling Factor That Reanneals U4 and U6 Small Nuclear Ribonucleoprotein Particles. Science 279: 857–860. [DOI] [PubMed] [Google Scholar]
- 17.Galej WP, Oubridge C, Newman AJ, Nagai K (2013) Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature advance online publication. [DOI] [PMC free article] [PubMed]
- 18. Bonnal S, Martínez C, Förch P, Bachi A, Wilm M, et al. (2008) RBM5/Luca-15/H37 Regulates Fas Alternative Splice Site Pairing after Exon Definition. Mol Cell 32: 81–95. [DOI] [PubMed] [Google Scholar]
- 19. Nag A, Steitz JA (2012) Tri-snRNP-associated proteins interact with subunits of the TRAMP and nuclear exosome complexes, linking RNA decay and pre-mRNA splicing. RNA Biol 9: 0–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mordes D, Luo X, Kar A, Kuo D, Xu L, et al. (2006) Pre-mRNA splicing and retinitis pigmentosa. Mol Vis 12: 1259–1271. [PMC free article] [PubMed] [Google Scholar]
- 21. Tanackovic G, Ransijn A, Thibault P, Abou Elela S, Klinck R, et al. (2011) PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum Mol Genet 20: 2116–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Linder B, Dill H, Hirmer A, Brocher J, Lee GP, et al. (2011) Systemic splicing factor deficiency causes tissue-specific defects: a zebrafish model for retinitis pigmentosa. Hum Mol Genet 20: 368–377. [DOI] [PubMed] [Google Scholar]
- 23. Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 24. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 7: e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu J, Xu Y, Schappert K, Harrington T, Wang A, et al. (1994) Mutational analysis of the PRP4 protein of Saccharomyces cerevisiae suggests domain structure and snRNP interactions. Nucleic Acids Res 22: 1724–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ayadi L, Callebaut I, Saguez C, Villa T, Mornon JP, et al. (1998) Functional and structural characterization of the prp3 binding domain of the yeast prp4 splicing factor. J Mol Biol 284: 673–687. [DOI] [PubMed] [Google Scholar]
- 28. Horowitz DS, Kobayashi R, Krainer AR (1997) A new cyclophilin and the human homologues of yeast Prp3 and Prp4 form a complex associated with U4/U6 snRNPs. RNA 3: 1374–1387. [PMC free article] [PubMed] [Google Scholar]
- 29. Teigelkamp S, Achsel T, Mundt C, Gothel SF, Cronshagen U, et al. (1998) The 20 kD protein of human [U4/U6.U5] tri-snRNPs is a novel cyclophilin that forms a complex with the U4/U6-specific 60 kD and 90 kD proteins. RNA 4: 127–141. [PMC free article] [PubMed] [Google Scholar]
- 30. Horowitz DS, Lee EJ, Mabon SA, Misteli T (2002) A cyclophilin functions in pre-mRNA splicing. EMBO J 21: 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huranova M, Hnilicova J, Fleischer B, Cvackova Z, Stanek D (2009) A mutation linked to retinitis pigmentosa in HPRP31 causes protein instability and impairs its interactions with spliceosomal snRNPs. Hum Mol Genet 18: 2014–2023. [DOI] [PubMed] [Google Scholar]
- 32. Benaglio P, San Jose PF, Avila-Fernandez A, Ascari G, Harper S, et al. (2014) Mutational screening of splicing factor genes in cases with autosomal dominant retinitis pigmentosa. Mol Vis 20: 843–851. [PMC free article] [PubMed] [Google Scholar]
- 33. Evans K, al-Maghtheh M, Fitzke FW, Moore AT, Jay M, et al. (1995) Bimodal expressivity in dominant retinitis pigmentosa genetically linked to chromosome 19q. Br J Ophthalmol 79: 841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaclavik V, Gaillard M-C, Tiab L, Schorderet DF, Munier FL (2010) Variable phenotypic expressivity in a Swiss family with autosomal dominant retinitis pigmentosa due to a T494M mutation in the PRPF3 gene. Mol Vis 16: 467–475. [PMC free article] [PubMed] [Google Scholar]
- 35. Maubaret CG, Vaclavik V, Mukhopadhyay R, Waseem NH, Churchill A, et al. (2011) Autosomal Dominant Retinitis Pigmentosa with Intrafamilial Variability and Incomplete Penetrance in Two Families Carrying Mutations in PRPF8. Invest Ophthalmol Vis Sci 52: 9304–9309. [DOI] [PubMed] [Google Scholar]
- 36. Venturini G, Rose AM, Shah AZ, Bhattacharya SS, Rivolta C (2012) CNOT3 Is a Modifier of PRPF31 Mutations in Retinitis Pigmentosa with Incomplete Penetrance. PLoS Genet 8: e1003040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cvačková Z, Matějů D, Staněk D (2014) Retinitis Pigmentosa Mutations of SNRNP200 Enhance Cryptic Splice-Site Recognition. Hum Mutat 35: 308–317. [DOI] [PubMed] [Google Scholar]
- 38. Bell M, Schreiner S, Damianov A, Reddy R, Bindereif A (2002) p110, a novel human U6 snRNP protein and U4/U6 snRNP recycling factor. EMBO J 21: 2724–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trede NS, Medenbach J, Damianov A, Hung L-H, Weber GJ, et al. (2007) Network of coregulated spliceosome components revealed by zebrafish mutant in recycling factor p110. Proc Natl Acad Sci U A 104: 6608–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Linder B, Plottner O, Kroiss M, Hartmann E, Laggerbauer B, et al. (2008) Tdrd3 is a novel stress granule-associated protein interacting with the Fragile-X syndrome protein FMRP. Hum Mol Genet 17: 3236–3246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotide sequences and accession numbers used.
(PDF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.