

Abstract
Object
Chordoma cells can generate solid-like tumors in xenograft models that express some molecular characteristics of the parent tumor, including positivity for brachyury and cytokeratins. However, there is a dearth of molecular markers that relate to chordoma tumor growth, as well as the cell lines needed to advance treatment. The objective in this study was to isolate a novel primary chordoma cell source and analyze the characteristics of tumor growth in a mouse xenograft model for comparison with the established U-CH1 and U-CH2b cell lines.
Methods
Primary cells from a sacral chordoma, called “DVC-4,” were cultured alongside U-CH1 and U-CH2b cells for more than 20 passages and characterized for expression of CD24 and brachyury. While brachyury is believed essential for driving tumor formation, CD24 is associated with healthy nucleus pulposus cells. Each cell type was subcutaneously implanted in NOD/SCID/IL2Rγnull mice. The percentage of solid tumors formed, time to maximum tumor size, and immunostaining scores for CD24 and brachyury (intensity scores of 0–3, heterogeneity scores of 0–1) were reported and evaluated to test differences across groups.
Results
The DVC-4 cells retained chordoma-like morphology in culture and exhibited CD24 and brachyury expression profiles in vitro that were similar to those for U-CH1 and U-CH2b. Both U-CH1 and DVC-4 cells grew tumors at rates that were faster than those for U-CH2b cells. Gross tumor developed at nearly every site (95%) injected with U-CH1 and at most sites (75%) injected with DVC-4. In contrast, U-CH2b cells produced grossly visible tumors in less than 50% of injected sites. Brachyury staining was similar among tumors derived from all 3 cell types and was intensely positive (scores of 2–3) in a majority of tissue sections. In contrast, differences in the pattern and intensity of staining for CD24 were noted among the 3 types of cell-derived tumors (p < 0.05, chi-square test), with evidence of intense and uniform staining in a majority of U-CH1 tumor sections (score of 3) and more than half of the DVC-4 tumor sections (scores of 2–3). In contrast, a majority of sections from U-CH2b cells stained modestly for CD24 (scores of 1–2) with a predominantly heterogeneous staining pattern.
Conclusions
This is the first report on xenografts generated from U-CH2b cells in which a low tumorigenicity was discovered despite evidence of chordoma-like characteristics in vitro. For tumors derived from a primary chordoma cell and U-CH1 cell line, similarly intense staining for CD24 was observed, which may correspond to their similar potential to grow tumors. In contrast, U-CH2b tumors stained less intensely for CD24. These results emphasize that many markers, including CD24, may be useful in distinguishing among chordoma cell types and their tumorigenicity in vivo.
Keywords: chordoma, xenograft, U-CH1, U-CH2b, CD24, brachyury, oncology
Chordomas are rare, slow-growing but clinically aggressive tumors predominantly found in the sacrum, spine, and clivus.19 They are thought to be derived from vestiges of the notochord, a mesoderm-derived structure that is involved in the process of neurulation and embryonic development.7,16,21 Chordomas compose approximately 1%–4% of all primary bone tumors10 with an age-adjusted incidence rate in the general population of 0.8 per 1 million people.13,19 Their locally destructive behavior precludes gross-total resection in 50% of cases, contributing to its dismal prognosis with 5-, 10-, and 20-year survival rates of 67%, 40%, and 13%, respectively. A major contributor to the lack of efficacious therapy lies in the relative paucity of knowledge about in vivo chordoma biology and growth characteristics.3 Chordomas are diagnosed by their molecular presentation of S100 protein, epithelial membrane antigen, select cytokeratins (cytokeratin 5/8, 18, or 19), and Brachyury (T),14,22 a T-box transcription factor whose expression is limited to the notochord during development. Brachyury expression in adult tissue has been used as a specific and sensitive diagnostic aid for chordoma. Additional markers of chordoma have also been suggested, including elevated protein expression of alpha enolase (ENO1), pyruvate kinase M2 (PKM2), gp96,23 and CD24, a glycoprotein expressed on many B lymphocytes and mature granulocytes that is known to be specifically expressed in chordoma18 and notochordal nucleus pulposus cells.8 How these molecular markers relate to tumor growth or presentation in chordoma remains poorly understood.
Only recently have investigators begun to develop animal models of chordoma as a means to advance understanding of chordoma biology.11,12,14,20 To date, very few chordoma cell lines have been reported, namely U-CH117 and U-CH2b,2 along with many more putative chordoma cell lines. Only a few of these have been shown to generate tumors in established mouse xenograft models and to retain molecular characteristics of the parent tumor and expanded cells. Presneau and coworkers used an established murine model in the NOD/SCID/IL2Rγnull mouse to establish tumor formation using the human U-CH1 cell line.14 At 10 weeks after subcutaneous injection of U-CH1 cells in a laminin-rich Matrigel carrier, xenografts were generated with morphological and molecular profiles (that is, brachyury expression) typical of conventional chordomas. Subsequently, Hsu et al.11 and Siu et al.20 detailed an ability to generate chordoma xenografts from patient-derived primary human tumor cells (called “JHC7” and “JHH-2009–011,” from sacral and clival chordomas, respectively). A majority of the studies documenting successful xenograft growth from primary chordoma cells have used positive staining for brachyury, as well as cytokeratin, to confirm the existence of chordoma. These results have demonstrated that patient cell sources from primary tumor can generate solid tumors ex vivo that have some of the molecular characteristics and features of the parent tumor. However, information is still lacking about which molecular markers of chordoma cell lines and primary chordoma cells are associated with stable and successful xenograft growth of chordoma tissues.
The objective of this study was to isolate and expand chordoma cells from a primary tumor using modifications of methods previously developed for isolating notochordal cells of the nucleus pulposus.5,6,9 This work relied on enzymatic digestion protocols and plating on laminin-rich substrates to retain physaliphorous cells from the parent tumor that retain the CD24+ and T+ molecular profile of the parent tumor in cell culture and in xenografts generated in NOD/SCID/IL2Rγnull mice. Studies of isolated cells in culture and xenografts derived from these cells were compared with the U-CH1 and U-CH2b cell lines.
Methods
Tissue Harvest
Chordoma tissue (> 7 cm) was resected en bloc from the sacrum of a 74-year-old male patient and sent for pathology review. The tumor was described as mesenchymal with physaliphorous cells that were immunoreactive for keratins (monoclonal mouse anti–human cytokeratin, clones AE1/AE3 and MNF116, DakoCytomation; clone CAM5.2, Becton Dickinson) and epithelial membrane antigen and irregularly reactive for S100 protein. Tumor samples were collected in accordance with protocols approved by the Duke University Institutional Review Board, and written informed consent to remove and study the tumor tissues for research purposes was obtained from the patient before surgery. Tumor samples were placed in sealed tissue containers and stored at 4°C for less than 4 hours prior to transportation to the laboratory. Tumor samples were separated into 2 sections for cell isolation and histological examination.
Tissue Characterization by Immunostaining
Primary tumor tissue from the patient was embedded in optimal cutting temperature medium, immediately flash frozen in liquid nitrogen, and stored at −80°C for cryosectioning. Sections (7 μm) were fixed with formaldehyde (10 minutes, room temperature) and incubated with blocking serum (5% goat serum and 3.75% bovine serum albumin [BSA], Zymed). Sections were then incubated with primary antibodies against cytokeratin 5/8 primary antibody (MAB3228, mouse monoclonal anti-human antibody, Millipore), brachyury (Ab20109, polyclonal anti-human antibody, Santa Cruz Biotechnology Inc.), or CD24 (555426, mouse monoclonal anti-human antibody, BD Biosciences) for 2 hours. Sections were then incubated with appropriate secondary antibody (30 minutes, Alexa 488 goat anti-rabbit antibody or anti-mouse antibody, Molecular Probes) and counterstained with propidium iodide (0.2 mg/ml, 15 minutes, Sigma). Prior to incubation with the primary antibody, sections labeled for brachyury were first permeabilized with 0.2% Triton for 10 minutes at room temperature. For all sections, negative controls were incubated with rabbit IgG for polyclonal antibody and appropriate isotype controls (mouse IgG1 or IgG2a) for monoclonal antibodies to confirm specificity of the antibody.
Cell Isolation
For cell isolation, tumor tissues were digested using a sequential pronase-collagenase digestion previously developed for nucleus pulposus cells of the intervertebral disc.4 Cells were then plated on tissue culture plastic coated with various proteins to best promote retention of the chordoma morphology, as described here. To coat surfaces for cell culture, T25 flasks were first incubated with 2 ml of a 0.1% gelatin solution (2% stock solution diluted in H2O, G1393, Sigma) for approximately 30 minutes or with 2 ml of medium conditioned with 804G mouse chondrosarcoma cells1 at 37°C. This substrate was chosen based on prior studies suggesting that this matrix rich in laminin-5 (LM-332) was able to promote attachment and retention of larger vacuolated cells of the immature nucleus pulposus.9 Solution was then aspirated, and flasks were allowed to dry in an upright position at room temperature. Tumor tissues were first rinsed several times in wash medium (DMEM, high glucose, Gibco) supplemented with Fungizone (1 μg/ml, Gibco), kanamycin (diluted 1:100, K0129, Sigma), and gentamycin (0.165 mg/ml, Gibco), very lightly minced, and incubated 14–16 hours at 37°C in wash medium supplemented with 0.06% collagenase Type 2 (Worthington Biochemicals) and 0.04% pronase (Roche Applied Science). The remaining cell suspension was spun at 400g to collect the cell pellet and twice washed with wash medium. Isolated cells were resuspended in culture medium (4:1 ratio of Iscove’s modified Dulbecco’s medium [IMDM, 12440, Invitrogen]/RPMI 1640 [R8758, Sigma] with 10% fetal bovine serum [FBS, Hyclone]) supplemented with 100 U/ml penicillin-streptomycin (14140–122, Invitrogen/Gibco) and plated on T25 tissue culture flasks coated with 804G conditioned medium as described above (0.5–1 × 106 cells per flask).
Cells were incubated in culture at 37°C with infrequent medium changes (1–2 weeks) and were passaged when at 75% confluence. After the first passage, a subset of cells were split (1:2) and cultured on gelatin-coated as well as 804G-coated flasks. Cells were passaged at approximately 1- to 2-week intervals until Passages 12–15, after which doubling times appeared to slow for some populations. A subset of cells on both the 804G- or gelatin-coated flasks continued to grow with doubling times of approximately 10 days for more than 1 year. These cells were called “DVC-4 cells” (Duke-Veterans Affairs Chordoma-4), as they were from the fourth chordoma tumor resected for cell culture and expansion at that facility. Cells were periodically imaged or harvested for flow cytometry or immunohistochemical analyses.
Cell Culture
Two chordoma cell lines were acquired for comparative purposes through a materials transfer agreement with the Chordoma Foundation: human chordoma cell lines U-CH1 and U-CH2b (University Hospital of Ulm, Germany). Each cell line was separately cultured as follows.17 In brief, T75 flasks were coated with 3 ml of a 0.1% gelatin solution as described for the cell isolation protocol above. The U-CH1 or U-CH2b cells were plated on flasks at 106 cells/ml and cultured in a 4:1 ratio of IMDM (12440, Invitrogen)/RPMI 1640 (R8757, Sigma) supplemented with 10% FBS and 100 U/ml penicillin-streptomycin (15140–122, Invitrogen/Gibco).
Flow Cytometry
Primary cells obtained from culture and U-CH1 cell lines were periodically analyzed via flow cytometry to quantify expression levels of the cell surface marker CD24. Freshly isolated (Passage 0) and passaged cells were released from culture via trypsin and resuspended in phosphate-buffered saline (PBS, Gibco) at a concentration of 106 cells/ml. Cells were then incubated for 30 minutes at 4°C with CD24–fluorescein isothiocyanate (FITC) mouse monoclonal antibody (10 μg/ml, MCA1379FT, AbD Serotec) or mouse IgG1-FITC negative control (MCA928F, AbD Serotec), washed 2 times with PBS, and analyzed on an Accuri C6 flow cytometer (Becton Dickinson) to measure the (geometric) mean fluorescence intensity (MFI) and percentage of cells positive for surface proteins (%). Immunoglobulin G control values were recorded and subtracted from experimental values of fluorescence. A minimum of 10,000 cells were analyzed for each condition (CD24, control) for each time point.
To label cells for brachyury, cells were fixed with 4% formaldehyde, washed with PBS containing 0.1% saponin (558255, EMD Millipore Chemicals Inc.), and incubated with a polyclonal anti–human antibody (20109, Santa Cruz Biotechnology Inc.). Cells were washed twice with PBS containing 0.1% saponin and incubated with appropriate secondary antibody (10 μg/ml Alexa Fluor 488 goat anti-rabbit IgG, Molecular Probes) for 30 minutes at 4°C. Cells were analyzed on a flow cytometer to measure MFI and percentage of T+ cells as described above.
Immunostaining of Cultured Cells
Primary cells in culture and U-CH1 and U-CH2b cell lines were periodically examined for the presence of brachyury, as for the native tissue. Cells were released from substrates with trypsin and transferred to tissue chamber slides for labeling and visualization. Cells were fixed with 4% formaldehyde (10 minutes, room temperature), permeabilized with 0.2% Triton X-100 in PBS (2 minutes, Sigma), incubated with blocking serum (5% goat serum and 3.75% BSA), and incubated with primary and secondary antibodies, as for the tissue sections described above (polyclonal anti–human brachyury, Ab20109, Santa Cruz Biotechnology Inc.). Secondary antibody alone was used as a negative control.
Molecular Characterizations of Primary Cells
As the primary tumor cell isolates (DVC-4) were previously unstudied, the mRNA and genomic DNA for brachyury were evaluated as follows. Total mRNA and genomic DNA from DVC-4 cells (Passage 14) were isolated using standard techniques. Complementary DNA was generated from mRNA by random priming, and approximately 40 ng of RNA equivalents were used for quantitative polymerase chain reaction (PCR; Applied Biosystems 7900 PCR system) using primers for brachyury, CD24, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), as described previously.2 Genomic copy number was determined via multiplex quantitative PCR using 10 ng of genomic DNA and brachyury gene-specific primers and/or probe, with normalization for input DNA using commercially available telomerase reverse transcriptase–specific primers (4401633, Applied Biosystems).22 All assays were performed in quadruplicate.
Subcutaneous Xenograft Model
For this study, we used a strain of mice previously shown to be supportive of chordoma xenografts,14 the NOD/SCID/IL2Rγnull (NSG) mouse (25–30 g, Jackson Laboratory). Mice (8 mice per U-CH1, U-CH2b; 2 mice per DVC-4) underwent sedation for subcutaneous injection of cells (1–5 × 106 cells in 200 μl of Matrigel, Becton Dickinson) into 2 locations per mouse: the interscapular region and a region overlying the right or left iliac crest. The contralateral iliac crest site was used to deliver 200 μl of Matrigel alone as a control. Primary tumor cells (DVC-4) of Passage 2 were used for this procedure. Mice were returned to normal cage activity and monitored daily for evidence of tumor growth and adverse effects. All protocols were performed with approval from the Duke University Institutional Animal Care and Use Committee.
Gross measures of tumor size were obtained from photographs of each mouse to estimate a total tumor burden. All mice were euthanized when the tumor burden exceeded approximately 2000 mm3. The parameters of time to euthanization and percentage of tumors formed per injection site were recorded for all mice. When the mice were euthanized, subcutaneous tumors were excised, immediately flash frozen in liquid nitrogen, and stored at −80°C until cryosectioning.
Immunohistochemical Staining of Xenograft Tissue
For histological evaluation, solid tumor was prepared from mice representative of each xenograft model (5 tumor samples each for U-CH1 and U-CH2b; 2 tumor samples for DVC-4). Frozen tissue sections (7 μm, 3 sections per tumor, with 30-μm spacing) were prepared from excised tumor, fixed in 4% formaldehyde (10 minutes at room temperature), and incubated with primary anti–human brachyury, CD24, and appropriate secondary antibodies as described above. For each tumor section, the intensity of staining was recorded as 0 = no staining, 1 = diffuse and light intensity of staining (cell associated), 2 = moderate and moderately distributed intensity of staining (cell associated), and 3 = intense and widely distributed intensity of stain. As some regions in the xenograft tissues were very heterogeneous with evidence of different tissue types, we also provided a score for uniformity of staining: 0 = heterogeneous or nonuniform staining and 1 = homogeneous or uniform staining throughout the section. Pearson’s chi-square test was used to test for the significance of differences in intensity and uniformity.
Results
Histological Appearance of Primary Tumor and Isolated Cells
Sections of the patient tumor stained positively for cytokeratin, brachyury, and CD24 (Fig. 1). The sections studied were very homogeneous in appearance and contained physaliphorous cells in “stringy” arrays, as described previously. Primary cells isolated from the patient tumor attached to tissue culture plastic coated with either gelatin or the laminin-rich 804G-conditioned medium (Fig. 2). Cells maintained a rounded appearance on both substrates out to Passage 10, with many cells appearing very large in diameter and containing vacuoles typical of notochordal and chordoma cells. These cells were slightly slower to passage, however, with initial doubling times of approximately 10 days rather than the 7 days for U-CH1 cells. There was some evidence that cells began to spread out and elongate on the gelatin substrates, distinct from the chordoma-specific appearance.
FIG. 1.
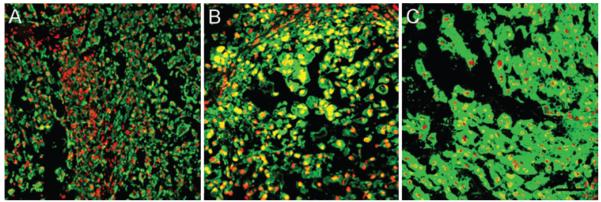
Patient tumor tissue stained positively for cytokeratin (A), brachyury (B), and CD24 (C). All sections were counter-stained with propidium iodide, and images were acquired after adjusting for the negative control (rabbit IgG). Bar = 100 μm.
FIG. 2.

Light microscopy images of cells plated on laminin-rich or gelatin-coated substrates after initial plating and after 6 (p6) or 10 (p10) passages (p). Bar = 100 μm.
Biomarkers of Chordoma Cells in Primary Cells and Tumor Cell Lines
Primary DVC-4 cells stained positively for brachyury immediately upon isolation. As shown in Fig. 3, brachyury staining was also present in virtually all cells and was clearly nucleus associated at Day 2 after isolation, again out to Passage 10 (Fig. 3A and B). This brachyury expression pattern in primary DVC-4 cells was very similar to that in U-CH1 cells lines plated on gelatin as shown (Fig. 3C). Messenger RNA expression levels of brachyury in DVC-4 cells were approximately 2-fold lower than those for U-CH1 according to quantitative PCR (0.50 ± 0.07–fold relative to U-CH1). In contrast, mRNA expression levels of CD24 in DVC-4 were 1.6-fold higher than in U-CH1 according to quantitative PCR (1.62 ± 0.46–fold relative to U-CH1). The brachyury gene (T) from genomic DNA of the DVC-4 tumor was not amplified compared with normal DNAs, as determined by quantitative PCR (data not shown).
FIG. 3.
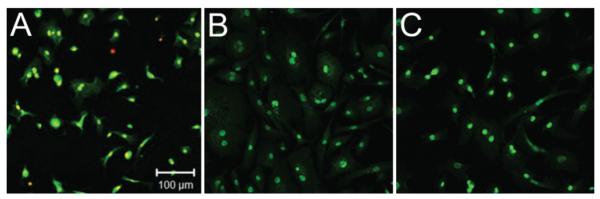
Cells were isolated from primary sacral tumor and expanded in culture for comparison with U-CH1 cells in monolayer. Cells here are labeled for anti–human brachyury antibody at the initial isolation and plating (A, Day 2 after isolation) and at Passage 10 (B). The U-CH1 cells are shown for comparison (C).
Primary DVC-4 cells also positively labeled for CD24 immediately upon isolation. Note that CD24 expression was assayed in DVC-4 cells via flow cytometry. At the time of isolation, 49% of all cells labeled positively for CD24 (Fig. 4), suggesting enrichment with chordoma cells during continued culture. With successive passages, the percent of cells labeling positive for CD24 increased to more than 95% of all cells by Passage 21.
FIG. 4.
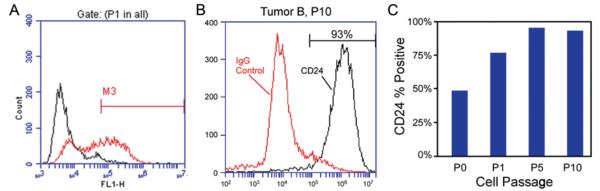
Primary human chordoma cells isolated from a sacral tumor obtained via resection. At the time of isolation and initial plating, expression of CD24 was detected in 49% of all cells via flow cytometry (A, anti–human CD24). CD24 expression continued to increase in cells with successive passages, as shown by the flow cytometry traces (B) and the graph (C).
In Vivo Tumorigenicity of Primary Cells in Mouse Xenograft Model
In this study, mice injected subcutaneously with primary cells (DVC-4) or with U-CH1 or U-CH2b cells were monitored via visual inspection and tumor dimensional measurement (Fig. 5). No tumors developed at the control sites (Matrigel alone) in any mouse. Growth kinetics of the DVC-4 tumors were much slower than those of the U-CH1 cells, with no tumors grossly evident in DVC-4–injected mice at 4 months after injection. In contrast, U-CH1 cells grew tumors very rapidly in NSG mice, as reported previously, with euthanization times of 15 weeks on average (Table 1) as compared with 23 weeks for mice injected with the DVC-4 cells. Tumor developed at nearly every site injected with U-CH1, whereas gross tumors arose at only 75% of the sites injected with the DVC-4 cells. Note that tumors formed from DVC-4 cells were solid, white, and distinct from the surrounding connective tissue.
FIG. 5.
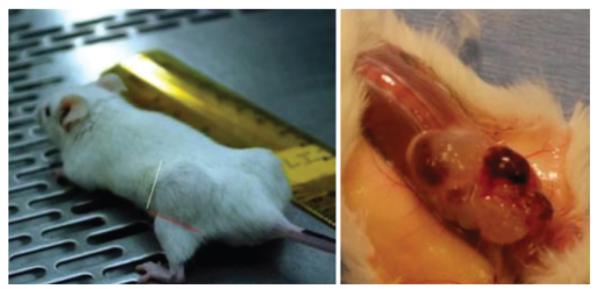
Method for estimating tumor xenograft dimensions in the flank after delivery of U-CH1 cells in a Matrigel carrier. Tumor appears on the mouse flank (left). Large tumor at time of excision exhibits the physaliphorous appearance of the native tumor (right).
TABLE 1.
Distribution of tumor xenografts generated in NSG mice
| Cells | No. of Mice Injected |
Mean Time to Death (wks)* |
Fraction of Injection Sites w/ Tumor |
|---|---|---|---|
| U-CH1 | 8 | 15.0 ± 0.89 | 93.75% |
| U-CH2b | 8 | 27.1 ± 0.89 | 43.75% |
| DVC-4 | 2 | 23 | 75% |
Values expressed as means ± standard deviation.
The U-CH2b cells contributed to the slowest growing tumors of all three cell types (Table 1). Tumors developed in less than 50% of injected sites as late as 27 weeks after injection. While some sites had not yet developed tumors, mice injected with U-CH2b cells were still euthanized at 27 weeks. It is noteworthy that tumors from U-CH2b cells were often gelatinous and stringy and appeared to be very confluent with the surrounding connective tissue.
Histological Features of the Xenograft-Derived Tumors
At least some sections of all xenografts stained positively for both brachyury and CD24, as shown in Table 2 and Fig. 6. There was little variation in the pattern of staining for brachyury among xenografts prepared from all three cell types, with approximately half of all examined sections showing some evidence of spatial heterogeneity in the pattern. A majority of tissue sections were intensely positive for brachyury staining, however, with virtually all examined sections scored at 2 or 3 for intensity of stain.
TABLE 2.
Uniformity and intensity of labeling for tumor xenografts generated in NSG mice*
| Labeling Pattern |
Labeling Intensity Score† |
|||||
|---|---|---|---|---|---|---|
| Marker & Cell | Heterogeneous | Homogeneous | 0 | 1 | 2 | 3 |
| brachyury | ||||||
| U-CH1 | 5 | 10 | 0 | 1 | 3 | 11 |
| U-CH2b | 10 | 5 | 0 | 1 | 8 | 6 |
| DVC-4 | 4 | 2 | 0 | 0 | 0 | 6 |
| CD24‡ | ||||||
| U-CH1 | 5 | 10 | 0 | 0 | 3 | 12 |
| U-CH2b | 11 | 4 | 0 | 9 | 5 | 1 |
| DVC-4 | 0 | 6 | 0 | 3 | 1 | 2 |
Values represent number of tissue sections.
Score 0–3: no stain to intense cell-associated stain.
Significant differences in scores among cell types for both uniformity and intensity of staining (p < 0.05, Pearson’s chi-square test).
FIG. 6.

Representative staining of mouse tumor xenografts harvested from NSG mice injected with cells from a primary tumor (DVC-4), U-CH1, or U-CH2b. Bar = 100 μm.
In contrast, differences in the pattern and intensity of staining for CD24 were noted among xenografts prepared from all 3 cell types (p < 0.05, chi-square test). All sections of U-CH1 xenografts stained intensely for CD24 (score of 3) with a predominantly uniform pattern, while a majority of those from U-CH2b xenografts had a low intensity of CD24 staining (score of 1) with a predominantly nonuniform or heterogeneous staining pattern. Xenografts from DVC-4 cells stained for CD24 with a consistent intensity of staining and with a highly homogeneous or spatially uniform pattern. These observations suggest that the heterogeneous staining for CD24, but not brachyury, was related to the morphology and growth characteristics of the cell-derived chordoma xenograft. Staining with cell-associated brachyury was common to all tumors and xenografts, whereas the pattern for CD24 positivity was distinct among the three different tumor sources.
Discussion
Animal models are important for studying cancer biology and assessing the preclinical efficacy of proposed therapeutic agents.3 In this study, we documented and compared the potential to generate chordoma xenografts across 2 cell lines and a primary chordoma cell called “DVC-4.” We showed that all 3 sources of chordoma tumor cells were able to form tumors in the immunocom-promised NSG mouse, as has already been shown for the U-CH1 cells.14 Importantly, this report is the first available on xenografts generated from U-CH2b cells, for which we discovered a relatively low tumorigenicity, with less than 50% of injected sites developing tumors at the latest time point. Our report of this behavior is both novel and significant, as prior studies of brachyury positivity and copy number aberrations in cultured U-CH2b cells2 were not predictive of the potential to generate tumor xenografts in vivo, pointing to the need for additional molecular markers to understand tumor progression. Several prior studies have confirmed that morphological appearance, cytoskeletal protein staining (vimentin, cytokeratins), and positivity for S100 and brachyury are essential for characterizing a chordoma xenograft;11,12,14,20 however, our results suggest that other molecular markers may be needed to distinguish tumorigenic potential and growth kinetics.
Brachyury is a transcription factor expressed in embryonic notochord, and prior studies have shown evidence that gene duplication can confer susceptibility to chordoma.3,22 Of interest here is our observation that the novel primary chordoma cells, DVC-4, had molecular staining for brachyury for cells in vitro and partly for tumor xenografts in vivo, which was similar to the pattern for U-CH1 cells. A majority of xenografts from U-CH1 or DVC-4 were intensely positive for brachyury, and both xenografts were morphologically solid and intact tissues. Moreover, mRNA levels for brachyury in DVC-4 were only modestly lower (2-fold) than those for U-CH1. However, there was no evidence of increased copy numbers for the brachyury gene (T) in DVC-4 genomic DNA, as compared with the U-CH1 cell line. Nevertheless, tumors formed at 75% of all sites injected with DVC-4 cells, as compared with a 95% success rate for U-CH1 cells. These observations suggest that DVC-4 may be an important cell line for additional studies of chordoma cell biology, providing the potential to study different mechanisms, yet similar propensities, to drive tumor formation. Additional work to more fully characterize a broader panel of molecular markers for DVC-4 would be important for confirming the utility of this novel chordoma cell line.
We did observe that the slow-growing U-CH2b cells, which were associated with infrequent tumor generation, were also linked to lower staining-intensity scores and more heterogeneity in the pattern of staining for CD24. The results shown for CD24 positivity suggest that this molecular marker may be useful for tracking relations to heterogeneity in growth potential and kinetics for chordoma. CD24 has already been documented as uniquely associated with nucleus pulposus cells8 and more recently has been suggested as a molecular marker of progenitor cells in the nucleus pulposus.15 The presentation of CD24 in chordoma emphasizes the link to the nucleus pulposus cell lineage and suggests that other molecular markers of nucleus pulposus cells may be useful for identifying chordoma cells and their relationship to xenograft growth potential.
Studies of chordoma xenografts in mouse models remain an important step toward understanding relations between the in vitro and in vivo biology of these complex tumors. Chordoma xenografts are very slow growing and challenging to support in vivo, and few reports are available for direct comparisons across multiple chordoma cell lines. Nevertheless, characterizations of xenografts in this study were limited by our knowledge of chordoma pathobiology so that we were only able to study the expression of a few molecular markers at the time of mouse termination. Future studies that make use of the transient expression of brachyury and other molecular markers would be important for tracking tumor growth and cell localization in real time in a living model.
Conclusions
This is the first report of xenografts generated from U-CH2b cells in which we discovered a low tumorigenicity for U-CH2b as compared with U-CH1, despite evidence of chordoma-like characteristics in vitro. For tumors derived from a primary chordoma cell and U-CH1, we observed heterogeneous and intense staining for CD24, which corresponded to their similar tumorigenic potential and growth kinetics. In contrast, U-CH2b tumors stained less intensely for CD24 in a manner that may relate to their low tumor growth potential in vivo. These results emphasize that many molecular markers, including CD24, may be important to distinguish chordoma cell types and their tumorigenicity in vivo.
Acknowledgment
We gratefully acknowledge the Chordoma Foundation for sharing their materials and expertise in performing this study.
This work was supported in part by a Young Investigator Grant from AO Spine North America (C.A.G.), NIH R01AR047442 (L.A.S.), and funds from the NIH R01EB002263 (I.O.K., L.A.S.) and NIH R01AR057410 (J.C.). The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.
Abbreviations used in this paper
- BSA
bovine serum albumin
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- IMDM
Iscove’s modified Dulbecco’s medium
- MFI
mean fluorescence intensity
- NSG
NOD/SCID/IL2Rγnull
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
Footnotes
Disclosure Author contributions to the study and manuscript preparation include the following. Conception and design: Setton, Karikari, Gilchrist, Jing, Alcorta, Chen, Richardson, Kelley. Acquisition of data: Karikari, Gilchrist, Jing, Alcorta, Gabr. Analysis and interpretation of data: Setton, Karikari, Gilchrist, Jing, Alcorta, Chen, Richardson, Gabr, Bell, Kelley. Drafting the article: Setton, Karikari, Gilchrist, Jing, Alcorta, Kelley. Critically revising the article: all authors. Reviewed submitted version of manuscript: all authors. Approved the final version of the manuscript on behalf of all authors: Setton. Statistical analysis: Setton, Gilchrist, Bell. Administrative/technical/material support: Jing, Alcorta, Gabr, Bell. Study supervision: Setton, Chen, Richardson, Kelley, Bagley. Financial support of the study: Bagley, Setton, Chen.
References
- 1.Baker SE, Hopkinson SB, Fitchmun M, Andreason GL, Frasier F, Plopper G, et al. Laminin-5 and hemidesmosomes: role of the alpha 3 chain subunit in hemidesmosome stability and assembly. J Cell Sci. 1996;109:2509–2520. doi: 10.1242/jcs.109.10.2509. [DOI] [PubMed] [Google Scholar]
- 2.Brüderlein S, Sommer JB, Meltzer PS, Li S, Osada T, Ng D, et al. Molecular characterization of putative chordoma cell lines. Sarcoma. 2010;630129:2010. doi: 10.1155/2010/630129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bydon M, Papadimitriou K, Witham T, Wolinsky JP, Bydon A, Sciubba D, et al. Novel therapeutic targets in chordoma. Expert Opin Ther Targets. 2012;16:1139–1143. doi: 10.1517/14728222.2012.714772. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Baer AE, Paik PY, Yan W, Setton LA. Matrix protein gene expression in intervertebral disc cells subjected to altered osmolarity. Biochem Biophys Res Commun. 2002;293:932–938. doi: 10.1016/S0006-291X(02)00314-5. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Jing L, Gilchrist CL, Richardson WJ, Fitch RD, Setton LA. Expression of laminin isoforms, receptors, and binding proteins unique to nucleus pulposus cells of immature inter-vertebral disc. Connect Tissue Res. 2009;50:294–306. [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Yan W, Setton LA. Molecular phenotypes of notochordal cells purified from immature nucleus pulposus. Eur Spine J. 2006;15(Suppl 3):S303–S311. doi: 10.1007/s00586-006-0088-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi KS, Cohn MJ, Harfe BD. Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: implications for disk degeneration and chordoma formation. Dev Dyn. 2008;237:3953–3958. doi: 10.1002/dvdy.21805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujita N, Miyamoto T, Imai J, Hosogane N, Suzuki T, Yagi M, et al. CD24 is expressed specifically in the nucleus pulposus of intervertebral discs. Biochem Biophys Res Commun. 2005;338:1890–1896. doi: 10.1016/j.bbrc.2005.10.166. [DOI] [PubMed] [Google Scholar]
- 9.Gilchrist CL, Francisco AT, Plopper GE, Chen J, Setton LA. Nucleus pulposus cell-matrix interactions with laminins. Eur Cell Mater. 2011;21:523–532. doi: 10.22203/ecm.v021a39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Healey JH, Lane JM. Chordoma: a critical review of diagnosis and treatment. Orthop Clin North Am. 1989;20:417–426. [PubMed] [Google Scholar]
- 11.Hsu W, Mohyeldin A, Shah SR, ap Rhys CM, Johnson LF, Se-dora-Roman NI, et al. Generation of chordoma cell line JHC7 and the identification of Brachyury as a novel molecular target. Laboratory investigation. J Neurosurg. 2011;115:760–769. doi: 10.3171/2011.5.JNS11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, Nielsen GP, Rosenberg AE, Waterman PR, Yang W, Choy E, et al. Establishment and characterization of a novel chordoma cell line: CH22. J Orthop Res. 2012;30:1666–1673. doi: 10.1002/jor.22113. [DOI] [PubMed] [Google Scholar]
- 13.McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control. 2001;12:1–11. doi: 10.1023/a:1008947301735. [DOI] [PubMed] [Google Scholar]
- 14.Presneau N, Shalaby A, Ye H, Pillay N, Halai D, Idowu B, et al. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: a genetic and functional-based study. J Pathol. 2011;223:327–335. doi: 10.1002/path.2816. [DOI] [PubMed] [Google Scholar]
- 15.Sakai D, Nakamura Y, Nakai T, Mishima T, Kato S, Grad S, et al. Exhaustion of nucleus pulposus progenitor cells with ageing and degeneration of the intervertebral disc. Nat Commun. 2012;3:1264. doi: 10.1038/ncomms2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salisbury JR. The pathology of the human notochord. J Pathol. 1993;171:253–255. doi: 10.1002/path.1711710404. [DOI] [PubMed] [Google Scholar]
- 17.Scheil S, Brüderlein S, Liehr T, Starke H, Herms J, Schulte M, et al. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer. 2001;32:203–211. doi: 10.1002/gcc.1184. [DOI] [PubMed] [Google Scholar]
- 18.Schwab JH, Boland PJ, Agaram NP, Socci ND, Guo T, O’Toole GC, et al. Chordoma and chondrosarcoma gene profile: implications for immunotherapy. Cancer Immunol Immunother. 2009;58:339–349. doi: 10.1007/s00262-008-0557-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sciubba DM, Chi JH, Rhines LD, Gokaslan ZL. Chordoma of the spinal column. Neurosurg Clin N Am. 2008;19:5–15. doi: 10.1016/j.nec.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Siu IM, Salmasi V, Orr BA, Zhao Q, Binder ZA, Tran C, et al. Establishment and characterization of a primary human chordoma xenograft model. Laboratory investigation. J Neurosurg. 2012;116:801–809. doi: 10.3171/2011.12.JNS111123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi T, Iwata J, Sugihara S, McCarthy EF, Jr, Karita M, Murakami H, et al. Distinguishing benign notochordal cell tumors from vertebral chordoma. Skeletal Radiol. 2008;37:291–299. doi: 10.1007/s00256-007-0435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang XR, Ng D, Alcorta DA, Liebsch NJ, Sheridan E, Li SF, et al. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat Genet. 2009;41:1176–1178. doi: 10.1038/ng.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou H, Chen CB, Lan J, Liu C, Liu XG, Jiang L, et al. Differential proteomic profiling of chordomas and analysis of prognostic factors. J Surg Oncol. 2010;102:720–727. doi: 10.1002/jso.21674. [DOI] [PubMed] [Google Scholar]