

Abstract
The potential of MUNE as a unique electrophysiological tool to detect early motor unit abnormalities during a clinically silent period was investigated in the plasma membrane calcium ATPase 2 (PMCA2)-heterozygous mice. There was a significant reduction in MUNE in the PMCA2-heterozygous mice as compared to the wild type littermates at two months of age. In contrast, the compound motor action potential (CMAP) was not altered. The conduction velocity (CV) of the sensory nerve and sensory nerve action potentials (SNAP) were not modified indicating lack of major sensory deficits. Interestingly, despite a decline in MUNE at this age, no changes were detected in choline acetyl transferase (ChAT) positive motor neuron number in the ventral horn of the lumbar spinal cord. Hindlimb grip strength, a test that evaluates clinical dysfunction, was also similar to that of the wild type controls. However, motor neuron number significantly decreased by five months suggesting that a drop in MUNE preceded motor neuron loss. In the two-month-old PMCA2-null mice, reduced MUNE measurements coincided with lower motor neuron number and decreased hindlimb grip strength. The fall in motor neuron number was already detectable at three weeks, the earliest time studied, and became more pronounced by five months. Our results show that even partial reductions in PMCA2 levels are sufficient to cause delayed death of motor neurons and that MUNE may be a reliable and sensitive approach to detect pathology prior to cell loss and in the absence of overt clinical signs.
Keywords: Neurodegeneration, calcium signaling, ATPb2b, amyotrophic lateral sclerosis, spinal cord, MUNE
Introduction
The quantification of the number of motor units innervating a muscle or group of muscles is increasingly used in clinical neurophysiology (Felice, 1997, Shefner, et al., 2006, Shefner, et al., 2002). Progressive loss of motor units is seen in several peripheral nervous system diseases and is the defining characteristic of most motor neuron disorders (Rowland and Shneider, 2001). Although traditional electrodiagnostic tests including nerve conduction studies and needle electromyography (EMG) provide qualitative estimates of motor units critical for the diagnosis of neurological disorders such as amyotrophic lateral sclerosis (ALS), they have limited success in assessing disease progression and mild axonal loss mainly because of physiological compensatory mechanisms of peripheral reinnervation (Brown, et al., 2006, Brown, 1973). MUNE is a unique electrophysiological tool that might offer the advantage of early quantitative detection of motor unit abnormalities during a period where both the clinical and traditional electrodiagnostic tests are normal (Bromberg, 2007, Cozzolino, et al., 2008, McComas, et al., 1971, Shefner, 2001, Shefner, et al., 1999). The current studies were undertaken in order to investigate whether MUNE detects pathology even in the absence of apparent clinical signs and prior to motor neuron loss.
To address this issue we evaluated MUNE and traditional nerve conduction in the PMCA2-heterozygous and null mice. PMCAs are P-type ATPases that play a major role in calcium extrusion. Four isoforms, PMCA1-4, encoded by different genes, have been described (Strehler and Zacharias, 2001). Alternative RNA splicing generates multiple variants with different functions (Strehler, et al., 2007). PMCA isoform 2 is enriched in the brain and heart and is predominantly expressed in neurons including motor neurons of the spinal cord (Burette, et al., 2003, Filoteo, et al., 1997, Lehotsky, et al., 1999, Nicot, et al., 2003, Stahl, et al., 1992, Stauffer, et al., 1995, Stauffer, et al., 1997).
PMCA2 appears to play a unique role in the function of certain neuronal subpopulations as indicated by the phenotype of the PMCA2-null mice which exhibit both hearing deficits and motor abnormalities including unsteady gait and balance (Kozel, et al., 1998, Shull, et al., 2003, Street, et al., 1998). Some of the motor deficits might be the consequence of both vestibular dysfunction and cerebellar abnormalities (Empson, et al., 2007, Kozel, et al., 1998, Kurnellas, et al., 2007, Shull, et al., 2003). Although, the motor phenotype of the PMCA2-heterozygous mice is reminiscent of the wild type, with no noticeable gait or balance abnormalities, we found that the performance of PMCA2-heterozygous mice on the accelerating rotarod is inferior to that of the wild type (unpublished results) raising the possibility of subtler motor dysfunction detectable only when mice are presented with a challenging task. These findings are in agreement with reports on the deafwaddler mice with different mutations in the gene encoding the PMCA2 protein (McCullough and Tempel, 2004). The poorer performance on the rotarod might partly stem from dysfunction of the vestibular system although pathology in other CNS regions including the cerebellum and the spinal cord could also be a contributing factor. Therefore, we initiated studies to examine the spinal cord of the heterozygous mice and especially focused on motor neurons because of our earlier studies showing a decrease in motor neuron number in the lumbar spinal cord of the PMCA2-null mice at two months of age (Kurnellas, et al., 2005). However, it was not known whether a partial decrease in PMCA2 levels in the heterozygous mice could affect motor neuron function and survival. As a first step we used MUNE measurements because we hypothesized that the sensitivity of this electrophysiological approach would facilitate detection of subtle motor neuron pathology, which is not demonstrable by other methods.
Methods
Animals
PMCA2+/+, +/− and −/− mice have been previously described (Kozel, et al., 1998, Kurnellas, et al., 2005). All animal protocols were performed according to institutional guidelines.
MUNE
MUNE was assessed by an experimenter blinded to the phenotype of the mice. Although PMCA2-null mice are easily recognizable due to their severe gait, balance and posture abnormalities, the PMCA2-heterozygous mice cannot be distinguished from the wild-type based on these criteria. Mice were anesthetized with sodium pentobarbital (50 mg/kg; i.p.) and immobilized on a board. The stimulating electrodes were 0.7 mm needles insulated with Teflon (Dantec sensory needle; Dantec, Skovlunde, Denmark). The cathode was placed close to the sciatic nerve at the proximal thigh, and the anode was placed subcutaneously 1 cm proximal to the anode. Motor responses were recorded from a ring electrode (Hush™ micro digital rings. Alpine Biomed, Fountain Valley, CA) which was placed circumferentially around the animal's hindlimb at 1 to 1.5 cm distace from the stimulating electrode. Activity was recorded in both flexor and extensor compartments. The reference ring was placed circumferentially around the hindlimb, 2 cm distal to the recording electrode. Recordings were obtained from both hindlimbs.
Stimuli were given with monophasic pulses from a Medtronic Keypoint through a constant current stimulator and delivered with a fine intensity control. Recordings were taken through Medtronic Keypoint EMG amplifiers. Data were collected on a computer and stored digitally for on-line (real-time) analysis. Filter settings were 500 Hz/5 KHz. For all studies, the position of the stimulating electrode was optimized by establishing a threshold for evoking a motor response at less than 0.7 mA. Stimulus intensity was increased until the CMAP was maximized. Potential amplitude (peak-peak) and distal latency (to negative onset) were then recorded. For sensory nerve conduction studies, orthodromic recording was used. Sensory stimulation was induced by ring electrodes placed around the tail (Hush™ micro digital rings, Alpine Biomed). The cathode was placed at 2 cm from the anode. Recordings were obtained by two monopolar needle electrodes, placed in the base of the tail at 1–2 cm from each other. The distance between the stimulating and the recording electrode was 3 cm. A ground electrode was placed in one hindlimb.
Incremental MUNE was performed at a standard amplifier gain using a modification of the technique described previously by McComas and colleagues (McComas, et al., 1971) and used by Shefner el al in mice.(Shefner, et al., 1999). Using a repetition rate of 1/s, the stimulus intensity was slowly increased from subthreshold levels until a small, all-or-none response was evoked. The response was digitally recorded after its stability was established by 3 to 4 identical repeats. The intensity was slowly augmented until the response increased in a quantal fashion. The increased response was again monitored for stability before a tracing was obtained for analysis. This process was repeated for a total of 10 increments. Before performing 10 increments, a supramaximal response was obtained and was used to calculate maximum CMAP area. Individual motor unit area was determined by subtracting the CMAP area of each response from that of the prior response. The average of individual values yielded an estimate of average single motor unit action potential (SMUAP) area. This value was divided by the area of the maximum CMAP to yield the MUNE. Motor unit size was calculated by dividing maximum CMAP area by the corresponding MUNE value.
Immunolabeling and quantification of alpha-motor neurons
PMCA2+/+, +/− and −/− mice were anesthetized with a mixture of ketamine/xylazine. They were perfused with 0.9% saline followed by 4% paraformaldehyde in phosphate buffer, pH 7.5. The spinal cords were dissected, post-fixed and cryoprotected in sucrose. They were then frozen and the lumbar spinal cords were sectioned on a cryostat. Ten μm sections of each PMCA2 wild type, heterozygous and null mouse lumbar spinal cord were mounted on slides, side by side. The beginning of the lumbar enlargement at L1 was used as a landmark for the first section obtained and seven to ten sections collected at 100 μm intervals were stained by immunocytochemistry using an antibody against ChAT (Chemicon, Temecula, CA 1:2000 dilution). This ensured that each cell was counted only once. After overnight incubation with the primary antibody, the sections were washed and incubated with biotinylated rabbit anti-goat secondary antibody (1:200, Vector Laboratories, Burlingame, CA) followed by avidinbiotin complex (Vectastain, Vector). Immunopositive cells were visualized using 3, 3'-diaminobenzidine (DAB, Sigma), and counted with a 40X objective employing an Olympus BX41 microscope. Alpha motor neurons were identified by immunoreactivity to ChAT, morphology and location in the anterior horn. Results were analyzed by ANOVA followed by post-hoc tests.
Grip strength analysis
Forelimb and hindlimb grip strength were measured from 2-3 month-old PMCA2+/+, +/− and −/− mice using the Chatillon CE digital force gauge (Columbus Instruments, Columbus, OH). Readings were taken in T-peak mode and measured in kilograms of force. To measure forelimb strength, the mice were positioned above a pull bar attached to the force gauge and lowered until they grasped the bar with their forelimb. The mice were slowly pulled back from the force gauge until the forelimbs released the bar. The maximum force was recorded and three individual trials were performed for each mouse. To measure hindlimb grip strength, the hindlimbs were placed on the thin bar attached to the force gauge and the forelimbs were stabilized on a rod. The mice were pulled back until the hindlimbs released the thin bar and the force was recorded as explained above.
Results
MUNE is decreased in the PMCA2-heterozygous and null mice
MUNE was assessed in two-month-old PMCA2 heterozygous and null mice as compared to wild type littermates. In heterozygous mice, there was a significant decrease in MUNE as recorded from the right and left leg (~29 and 25 % of wild type, respectively) whereas in the PMCA2-null mouse the reduction reached 65 and 60 %, respectively (Fig. 1A and B). In contrast, evaluation of CAMP failed to detect significant changes in the heterozygous or null mice as compared to wild type controls (Fig. 1C and D). Motor unit size analysis indicated a significant increase in the PMCA2-null mice as compared to wild type controls despite extensive variability between individual animals (Fig. 1E and F). Motor unit size showed a tendency to increase about 1.6-fold in heterozygous mice as compared to the wild type but the difference did not reach statistical significance. The CV of the sensory nerve and SNAP were not modified in the PMCA2-heterozygous and null mice indicating a lack of sensory deficits (Fig. 1G and H).
Figure 1. MUNE measurements in the PMCA2-heterozygous and null mice.

MUNE obtained from the right (A) and left (B) hindlimbs were decreased in two-month-old PMCA2-heterozygous (HET) and null (knockout, KO) mice as compared to wild types (WT). In contrast, CMAP recorded from the right (C) and left (D) hindlimbs was not altered. Calculation of the motor unit size (μV × msec) from data derived from right (E) and left hindlimb (F) recordings indicated a significant difference between WT and KO. Conduction velocity-CV (G) and sensory nerve action potential-SNAP (H) were not significantly different in PMCA2 wild type as compared to heterozygous and null mice. Graphs represent mean ± S.E.M. Statistical analysis was performed by ANOVA, followed by Fisher post-hoc test. * Significantly different from wild type, p< 0.03 and, ** significantly different from wild type and heterozygous, p<0.005. + Significantly different from wild type p< 0.02 and ++ significantly different from wild type and heterozygous, p<0.02. N=8–10 for wild type and heterozygous and N=6 for PMCA2-null mice.
Motor neuron loss in the lumbar spinal cord of PMCA2-heterozygous and null mice
We previously reported that motor neuron number is decreased in the lumbar spinal cord of two-month-old PMCA2-null mice (Kurnellas, et al., 2005). However, it was not known whether this was due to abnormalities in motor neuron development or loss of motor neurons. One way of addressing this issue was to examine the number of motor neurons at various ages in order to determine whether there is a progressive decrease. Therefore, we evaluated motor neuron number at 3 weeks, 2-3 and 5 months of age (Figure 2). At three weeks postnatal, the number of ChAT positive cells in the lumbar spinal cord of PMCA2−/− mice was 35.9 ± 2.5% lower than the wild type. At 2-3 months the reduction was 39.5 ± 2.3% confirming previous results (Kurnellas, et al., 2005). However, by 5 months, PMCA2-null mice had 58.1± 7.6% less ChAT positive neurons as compared to the wild type (Figures 2 and 3). These results indicate that the further reduction in ChAT immunoreactive cell number from 2-3 to 5 months of age is likely due to motor neuron loss.
Figure 2. Quantification of ChAT positive motor neuron numbers in the lumbar spinal cord of PMCA2- wild type (WT), heterozygous (HET) and null (KO) mice.

A) Western blots showing the reduction in PMCA2 levels in the lumbar spinal cord of the PMCA2-HET and KO mice (upper panel). α-Tubulin was used to correct for experimental variations (lower panel). Lanes 1–3, 4–6 and 7–9 are proteins obtained from different two-month-old WT, HET and KO mice. B) Graphic representation of the data shown in A. All PMCA2 band intensities were normalized to α-tubulin. Values are expressed as mean ± S.E.M. ¶ Significantly different from WT, p<0.008 by Student's t-test. C) Three-week-old mice; 92–118 motor neurons were counted in 8–10 sections from each wild type control. N=4. D) Two to three-month-old mice; 152 ± 9.07 motor neurons were counted in 10 sections from each wild type control. N=5. E) Five-month-old mice; 133–198 motor neurons were counted in 7–10 sections from each wild type control. N=3. Values are mean ± S.E.M. ** significantly different from wild type (p<0.002) and heterozygous (p< 0.02), *** significantly different from wild type (p< 0.0001) and heterozygous (p< 0.005). * significantly different from wild type (p< 0.02). All statistical analyses were performed by ANOVA followed by Fisher's post-hoc test.
Figure 3. ChAT positive motor neurons in the ventral horn of the five-month-old PMCA2 wild type (A), heterozygous (B) and null (C) mice.
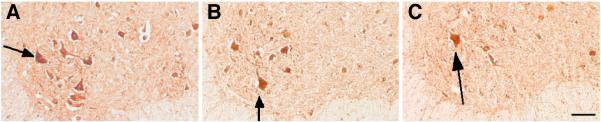
Arrows point at labeled neurons. Note the decrease in immunoreactive neurons in the heterozygous and null mouse. Bar=300 μm.
In contrast, there were no significant changes in the number of ChAT immunoreactive cell number in the lumbar spinal cord of the PMCA2-heterozygous mice at 3 weeks and 2-3 months of age despite lower PMCA2 levels in the spinal cord. However, by five months, there was about 25% reduction in the motor neuron number of the PMCA2-heterozygous mice as compared to the wild type controls. Thus, loss of motor neurons is delayed in the PMCA2-heterozygous mice and the extent of cell loss is less pronounced than that observed in the PMCA2-null mouse, at least at the ages analyzed.
Grip strength in PMCA2 heterozygous and null mice
To examine functional deficits in the PMCA2-heterozygous and null mice, we measured hindlimb and forelimb grip strength. Despite a reduction in MUNE in the two-month-old heterozygous mice, there were no alterations in the hindlimb grip strength indicating that MUNE can detect subtle abnormalities even in the absence of quantifiable functional deficits (Fig. 4). In contrast, a significant decrease in hindlimb grip strength is observed in the PMCA2-null mouse. Forelimb grip strength is not modified suggesting that the functional deficits are mostly attributable to pathology in the lumbar region. In accordance with these findings, motor neuron number at C7-T1 junction in the PMCA2 null mice were not significantly different from the wild type (results not shown) at 2-3 months indicating that loss of motor neurons is more pronounced in the lumbar region of the spine.
Figure 4. Forelimb (A) and hindlimb (B) grip strength in the two-month-old PMCA2 wild type, heterozygous (HET) and null (KO) mice.
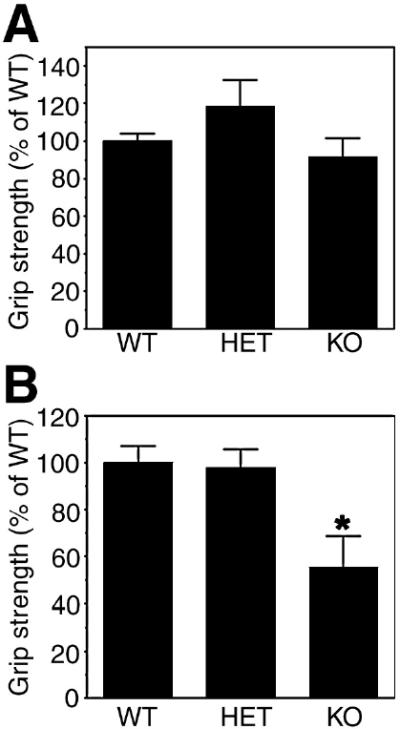
Values are mean ± S.E.M. * Significantly different from wild type by ANOVA, Tukey's post-hoc test; p< 0.03. N=3–4.
Discussion
The findings of this investigation suggest that MUNE can detect motor unit abnormalities even in the absence of noticeable clinical deficits or significant motor neuron loss in the PMCA2-heterozygous mice at 2-3 months of age. The early detection of anomalies by MUNE is important because it suggests that molecular changes associated with pathology and leading to neurodegeneration might already be occurring at this age. Therefore, trials on interventions to prevent motor neuron loss in the PMCA2-heterozygous mice should probably be performed at 2-3 months of age, the period prior to neuronal degeneration. Abnormalities in MUNE before detectable alterations in behavior and motor neuron density have also been reported in the mSOD1 (mutant superoxide dismutase 1) mouse, an animal model of ALS(Shefner, et al., 1999). Moreover, MUNE declines more rapidly than strength in ALS patients (Dantes and McComas, 1991, Felice, 1997, Shefner, et al., 2006). The decline in MUNE in the absence of a decrease in grip strength and motor neuron number in the two-month old PMCA2-heterozygous mice might be due to the dysfunction of distal axons and relative sparing of proximal axons and neurons, as previously suggested (Shefner, et al., 1999). Alternatively, functional remodeling of motor units in the absence of changes in motor neuron survival could also explain this divergence (Shefner, et al., 1999).
In contrast to MUNE, CAMP failed to reveal pathology not only in the PMCA2-heterozygous but also in the PMCA2-null mice. This is not surprising as CAMP amplitude, which measures the combined effect of denervation and compensatory reinnervation, is relatively insensitive to motor neuron loss until a substantial number of surviving motor units declines (Hansen and Ballantyne, 1978, McComas, et al., 1971).
In our studies, we used the incremental MUNE method which assumes that each response increment resulting from graded stimulation, represents the addition of a single motor unit. However, as the threshold for a given motor unit is probabilistic rather than absolute, a constant repetitive stimulus may evoke responses from different motor units at different times (Brown and Milner-Brown, 1976, Milner-Brown and Brown, 1976, Shefner, 2001). Another assumption is that the motor units measured at low stimulus intensity and used in the calculation of the average motor unit size, are representative of the total population of motor units in the entire muscle (Shefner, 2001). Finally, motor neuron instability in response to repeated electrical stimulation may add another source of bias to the estimation of the number of motor units in pathological conditions (Jillapalli and Shefner, 2004). Despite these technical limitations, the incremental MUNE method has been successfully used in several neuromuscular disorders and has provided insights into pathogenesis (Ballantyne and Hansen, 1974, Eisen, et al., 1974, Feeney, et al., 2001, Shefner, 2001, Shefner, et al., 2001, Shefner, et al., 1999). In particular, MUNE has been effectively employed in determining the progression of ALS and has detected motor neuron loss in animal models of ALS including the ALS2/alsin mouse model carrying a disrupted gene responsible for a number of juvenile recessive motor neuron diseases in humans (Azzouz, et al., 1997, Feeney, et al., 2001, Hadano, et al., 2006, Reaume, et al., 1996, Shefner, et al., 1999).
The current investigations highlight the importance of PMCA2 in the survival of motor neurons. Even partial reductions in PMCA2 expression are sufficient to decrease motor neuron number in the lumbar spinal cord of the adult PMCA2-heterozygous mice, albeit at a slower rate as compared to the null mice. In agreement with these results, we found that silencing of PMCA2 expression by siRNA causes death of spinal cord neurons even if the levels of PMCA2 protein are only partly reduced (unpublished results). It is not yet clear why motor neuron loss is considerably delayed in the heterozygous mice as compared to the PMCA2-null mice. It is possible to hypothesize that aberrance in calcium extrusion leads to continuous elevations in intracellular calcium levels which interferes with the normal signaling of the neuron and its communication with other cells. This might gradually weaken the motor neuron leading finally to death. In the PMCA2-null mouse, anomalies in developmental processes such as proliferation, differentiation and programmed cell death of motor neurons may also contribute to the drop in ChAT positive neuron number in the ventral horn since the decrease is already present at three week postnatal. However, the further fall in motor neuron number from 2-3 months to 5 months of age indicates that some of the decrease is due to motor neuron death.
In summary, our results indicate that MUNE measurements might detect early pathology even when clinical signs are weak. Furthermore, the studies described here suggest that PMCA2-heterozygous mice might be used as a model to study delayed and progressive lower motor neuron loss during adulthood, a pathological condition relevant to motor neuron diseases such as ALS.
Acknowledgments
This study was supported by NIH/NINDS grants NS 046363 to SE and NS 054336 to MPK. We thank Debrah J. Ehrlich for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Azzouz M, Leclerc N, Gurney M, Warter JM, Poindron P, Borg J. Progressive motor neuron impairment in an animal model of familial amyotrophic lateral sclerosis. Muscle Nerve. 1997;20:45–51. doi: 10.1002/(sici)1097-4598(199701)20:1<45::aid-mus6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 2.Ballantyne JP, Hansen S. New method for the estimation of the number of motor units in a muscle. 2. Duchenne, limb-girdle and facioscapulohumeral, and myotonic muscular dystrophies. J Neurol Neurosurg Psychiatry. 1974;37:1195–1201. doi: 10.1136/jnnp.37.11.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bromberg MB. Updating motor unit number estimation (MUNE) Clin Neurophysiol. 2007;118:1–8. doi: 10.1016/j.clinph.2006.07.304. [DOI] [PubMed] [Google Scholar]
- 4.Brown RH, Swash M, Pasinelli P. Informa Healthcare. North America by Taylor & Francis; Abingdon [England] Boca Raton, FL: 2006. Amyotrophic lateral sclerosis. [Google Scholar]
- 5.Brown WF. Functional compensation of human motor units in health and disease. J Neurol Sci. 1973;20:199–209. doi: 10.1016/0022-510x(73)90030-0. [DOI] [PubMed] [Google Scholar]
- 6.Brown WF, Milner-Brown HS. Some electrical properties of motor units and their effects on the methods of estimating motor unit numbers. J Neurol Neurosurg Psychiatry. 1976;39:249–257. doi: 10.1136/jnnp.39.3.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burette A, Rockwood JM, Strehler EE, Weinberg RJ. Isoform-specific distribution of the plasma membrane Ca2+ ATPase in the rat brain. J Comp Neurol. 2003;467:464–476. doi: 10.1002/cne.10933. [DOI] [PubMed] [Google Scholar]
- 8.Cozzolino M, Ferri A, Carri MT. Amyotrophic lateral sclerosis: from current developments in the laboratory to clinical implications. Antioxid Redox Signal. 2008;10:405–443. doi: 10.1089/ars.2007.1760. [DOI] [PubMed] [Google Scholar]
- 9.Dantes M, McComas A. The extent and time course of motoneuron involvement in amyotrophic lateral sclerosis. Muscle Nerve. 1991;14:416–421. doi: 10.1002/mus.880140506. [DOI] [PubMed] [Google Scholar]
- 10.Eisen A, Karpati G, Carpenter S, Danon J. The motor unit profile of the rat soleus in experimental myopathy and reinnervation. Neurology. 1974;24:878–884. doi: 10.1212/wnl.24.9.878. [DOI] [PubMed] [Google Scholar]
- 11.Empson RM, Garside ML, Knopfel T. Plasma membrane Ca2+ ATPase 2 contributes to short-term synapse plasticity at the parallel fiber to Purkinje neuron synapse. J Neurosci. 2007;27:3753–3758. doi: 10.1523/JNEUROSCI.0069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feeney SJ, McKelvie PA, Austin L, Jean-Francois MJ, Kapsa R, Tombs SM, Byrne E. Presymptomatic motor neuron loss and reactive astrocytosis in the SOD1 mouse model of amyotrophic lateral sclerosis. Muscle Nerve. 2001;24:1510–1519. doi: 10.1002/mus.1176. [DOI] [PubMed] [Google Scholar]
- 13.Felice KJ. A longitudinal study comparing thenar motor unit number estimates to other quantitative tests in patients with amyotrophic lateral sclerosis. Muscle Nerve. 1997;20:179–185. doi: 10.1002/(sici)1097-4598(199702)20:2<179::aid-mus7>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 14.Filoteo AG, Elwess NL, Enyedi A, Caride A, Aung HH, Penniston JT. Plasma membrane Ca2+ pump in rat brain. Patterns of alternative splices seen by isoform-specific antibodies. J Biol Chem. 1997;272:23741–23747. doi: 10.1074/jbc.272.38.23741. [DOI] [PubMed] [Google Scholar]
- 15.Hadano S, Benn SC, Kakuta S, Otomo A, Sudo K, Kunita R, Suzuki-Utsunomiya K, Mizumura H, Shefner JM, Cox GA, Iwakura Y, Brown RH, Jr., Ikeda JE. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum Mol Genet. 2006;15:233–250. doi: 10.1093/hmg/ddi440. [DOI] [PubMed] [Google Scholar]
- 16.Hansen S, Ballantyne JP. A quantitative electrophysiological study of motor neurone disease. J Neurol Neurosurg Psychiatry. 1978;41:773–783. doi: 10.1136/jnnp.41.9.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jillapalli D, Shefner JM. Single motor unit variability with threshold stimulation in patients with amyotrophic lateral sclerosis and normal subjects. Muscle Nerve. 2004;30:578–584. doi: 10.1002/mus.20147. [DOI] [PubMed] [Google Scholar]
- 18.Kozel PJ, Friedman RA, Erway LC, Yamoah EN, Liu LH, Riddle T, Duffy JJ, Doetschman T, Miller ML, Cardell EL, Shull GE. Balance and hearing deficits in mice with a null mutation in the gene encoding plasma membrane Ca2+-ATPase isoform 2. J Biol Chem. 1998;273:18693–18696. doi: 10.1074/jbc.273.30.18693. [DOI] [PubMed] [Google Scholar]
- 19.Kurnellas MP, Lee AK, Li H, Deng L, Ehrlich DJ, Elkabes S. Molecular alterations in the cerebellum of the plasma membrane calcium ATPase 2 (PMCA2)-null mouse indicate abnormalities in Purkinje neurons. Mol Cell Neurosci. 2007;34:178–188. doi: 10.1016/j.mcn.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurnellas MP, Nicot A, Shull GE, Elkabes S. Plasma membrane calcium ATPase deficiency causes neuronal pathology in the spinal cord: a potential mechanism for neurodegeneration in multiple sclerosis and spinal cord injury. FASEB J. 2005;19:298–300. doi: 10.1096/fj.04-2549fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehotsky J, Kaplan P, Racay P, Mezesova V, Raeymaekers L. Distribution of plasma membrane Ca2+ pump (PMCA) isoforms in the gerbil brain: effect of ischemia-reperfusion injury. Neurochem Int. 1999;35:221–227. doi: 10.1016/s0197-0186(99)00062-5. [DOI] [PubMed] [Google Scholar]
- 22.McComas AJ, Fawcett PR, Campbell MJ, Sica RE. Electrophysiological estimation of the number of motor units within a human muscle. J Neurol Neurosurg Psychiatry. 1971;34:121–131. doi: 10.1136/jnnp.34.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCullough BJ, Tempel BL. Haplo-insufficiency revealed in deafwaddler mice when tested for hearing loss and ataxia. Hear Res. 2004;195:90–102. doi: 10.1016/j.heares.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Milner-Brown HS, Brown WF. New methods of estimating the number of motor units in a muscle. J Neurol Neurosurg Psychiatry. 1976;39:258–265. doi: 10.1136/jnnp.39.3.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicot A, Ratnakar PV, Ron Y, Chen CC, Elkabes S. Regulation of gene expression in experimental autoimmune encephalomyelitis indicates early neuronal dysfunction. Brain. 2003;126:398–412. doi: 10.1093/brain/awg041. [DOI] [PubMed] [Google Scholar]
- 26.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr., Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 27.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 28.Shefner JM. Motor unit number estimation in human neurological diseases and animal models. Clin Neurophysiol. 2001;112:955–964. doi: 10.1016/s1388-2457(01)00520-x. [DOI] [PubMed] [Google Scholar]
- 29.Shefner JM, Brown RH, Jr., Cole D, Chaturvedi P, Schoenfeld D, Pastuszak K, Matthews R, Upton-Rice M, Cudkowicz ME. Effect of neurophilin ligands on motor units in mice with SOD1 ALS mutations. Neurology. 2001;57:1857–1861. doi: 10.1212/wnl.57.10.1857. [DOI] [PubMed] [Google Scholar]
- 30.Shefner JM, Cudkowicz M, Brown RH., Jr. Motor unit number estimation predicts disease onset and survival in a transgenic mouse model of amyotrophic lateral sclerosis. Muscle Nerve. 2006;34:603–607. doi: 10.1002/mus.20628. [DOI] [PubMed] [Google Scholar]
- 31.Shefner JM, Cudkowicz ME, Brown RH., Jr. Comparison of incremental with multipoint MUNE methods in transgenic ALS mice. Muscle Nerve. 2002;25:39–42. doi: 10.1002/mus.10000. [DOI] [PubMed] [Google Scholar]
- 32.Shefner JM, Reaume AG, Flood DG, Scott RW, Kowall NW, Ferrante RJ, Siwek DF, Upton-Rice M, Brown RH., Jr. Mice lacking cytosolic copper/zinc superoxide dismutase display a distinctive motor axonopathy. Neurology. 1999;53:1239–1246. doi: 10.1212/wnl.53.6.1239. [DOI] [PubMed] [Google Scholar]
- 33.Shull GE, Okunade G, Liu LH, Kozel P, Periasamy M, Lorenz JN, Prasad V. Physiological functions of plasma membrane and intracellular Ca2+ pumps revealed by analysis of null mutants. Ann N Y Acad Sci. 2003;986:453–460. doi: 10.1111/j.1749-6632.2003.tb07229.x. [DOI] [PubMed] [Google Scholar]
- 34.Stahl WL, Eakin TJ, Owens JW, Jr., Breininger JF, Filuk PE, Anderson WR. Plasma membrane Ca(2+)-ATPase isoforms: distribution of mRNAs in rat brain by in situ hybridization. Brain Res Mol Brain Res. 1992;16:223–231. doi: 10.1016/0169-328x(92)90229-5. [DOI] [PubMed] [Google Scholar]
- 35.Stauffer TP, Guerini D, Carafoli E. Tissue distribution of the four gene products of the plasma membrane Ca2+ pump. A study using specific antibodies. J Biol Chem. 1995;270:12184–12190. doi: 10.1074/jbc.270.20.12184. [DOI] [PubMed] [Google Scholar]
- 36.Stauffer TP, Guerini D, Celio MR, Carafoli E. Immunolocalization of the plasma membrane Ca2+ pump isoforms in the rat brain. Brain Res. 1997;748:21–29. doi: 10.1016/s0006-8993(96)01282-6. [DOI] [PubMed] [Google Scholar]
- 37.Street VA, McKee-Johnson JW, Fonseca RC, Tempel BL, Noben-Trauth K. Mutations in a plasma membrane Ca2+-ATPase gene cause deafness in deafwaddler mice. Nat Genet. 1998;19:390–394. doi: 10.1038/1284. [DOI] [PubMed] [Google Scholar]
- 38.Strehler EE, Filoteo AG, Penniston JT, Caride AJ. Plasma-membrane Ca(2+) pumps: structural diversity as the basis for functional versatility. Biochem Soc Trans. 2007;35:919–922. doi: 10.1042/BST0350919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strehler EE, Zacharias DA. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol Rev. 2001;81:21–50. doi: 10.1152/physrev.2001.81.1.21. [DOI] [PubMed] [Google Scholar]