

Abstract
Genetic adaptation to different environmental conditions is expected to lead to large differences between populations at selected loci, thus providing a signature of positive selection. Whereas balancing selection can maintain polymorphisms over long evolutionary periods and even geographic scale, thus leads to low levels of divergence between populations at selected loci. However, little is known about the relative importance of these two selective forces in shaping genomic diversity, partly due to difficulties in recognizing balancing selection in species showing low levels of differentiation. Here we address this problem by studying genomic diversity in the European common vole (Microtus arvalis) presenting high levels of differentiation between populations (average F ST = 0.31). We studied 3,839 Amplified Fragment Length Polymorphism (AFLP) markers genotyped in 444 individuals from 21 populations distributed across the European continent and hence over different environmental conditions. Our statistical approach to detect markers under selection is based on a Bayesian method specifically developed for AFLP markers, which treats AFLPs as a nearly codominant marker system, and therefore has increased power to detect selection. The high number of screened populations allowed us to detect the signature of balancing selection across a large geographic area. We detected 33 markers potentially under balancing selection, hence strong evidence of stabilizing selection in 21 populations across Europe. However, our analyses identified four-times more markers (138) being under positive selection, and geographical patterns suggest that some of these markers are probably associated with alpine regions, which seem to have environmental conditions that favour adaptation. We conclude that despite favourable conditions in this study for the detection of balancing selection, this evolutionary force seems to play a relatively minor role in shaping the genomic diversity of the common vole, which is more influenced by positive selection and neutral processes like drift and demographic history.
Introduction
Despite nearly six decades of genetic investigations, it remains unclear for most organisms to which extent the demographic history of populations, genetic drift or selection influences the pattern of genetic diversity of a species. Historically, the observation that many genes are genetically polymorphic within population was first explained by a selective advantage of heterozygotes [1]. This explanation was challenged by Kimura's neutral [2], [3] and nearly neutral [4] theory of molecular evolution, which provided a competing explanation for the high frequency of genetic polymorphism. Nowadays it is generally accepted that a majority of the genetic variations evolved nearly neutrally, but that natural selection plays a decisive role in evolution and leaves footprints in the genome. Natural selection acts in at least three forms, which are positive, purifying and balancing selection. Positive selection can lower genetic diversity locally but increase it globally, to a level depending on the spatial and environmental heterogeneity [5]–[7]. Balancing selection maintains genetic variation within populations [8] and leads to generally low levels of differentiation between populations, even though it can contribute to increase population differentiation if selective pressures are spatially heterogeneous [5]. Finally, purifying selection generally decrease levels of genetic diversity, even though strong background selection can promote increased difference between populations by lowering their effective size [9]. In the past, balancing selection played an important role in evolutionary genetics in explaining the high level of genomic polymorphism observed among species or populations [8], [10]. However, the effect of selection can be multifarious and the impact of each is still under debate [11], especially for balancing selection.
At least in humans a number of common genetic diseases have been proposed to be maintained in populations as a result of balancing selection, e.g. sickle-cell anaemia [12], [13], glucose-6-phosphate dehydrogenase deficiency [14], thalassemia [15] and cystic fibrosis [16]. Other examples are the ABO blood group [17], polymorphisms of beta-globin [18], the major histocompatibility complex (MHC; [19]) including the human HLA-G promoter [20], CCR5 in humans [21], the complementary sex determination locus in bees [22], response to pathogens [23], high diversity genes in Arabidopsis [24] or self-incompatibility and nuclear-cytoplasmic gynodioecy in plants (see e.g. [25]). However, all these examples were identified by a candidate gene approach and not genome-wide scans. Hence they do not give any information about the importance of balancing selection in shaping genomic diversity. In this context, there are only few genome-wide studies of balancing selection in humans [26], [27] or sticklebacks [28] and these studies remain inconclusive about the importance of balancing selection in shaping and maintaining genetic diversity, potentially due to methodological limitations (see below). Compared to balancing selection, the occurrence and influence of positive selection on an organism's genetic variation is much less questioned, as positive selection should allow the spread of advantageous traits and play a central role in the evolution of species (see e.g. [29], [30]).
The prevalence of balancing selection is still highly debated, mainly due to missing evidence in organisms other than humans, but also because methods developed specifically to detect balancing selection are still few (see e.g. [27], [31]–[34]). Moreover, the classical detection of balancing selection based on levels of differentiation between populations is difficult in organisms with low levels of differentiation (see [35]) like humans or Drosophila [36], [37] and a decent number of populations need to be investigated to have the statistical power to detect balancing selection [35].
In order to better detect signals of balancing selection, we focused in this study on an organism showing particularly high levels of differentiation, which is the common vole (Microtus arvalis). This species has a very wide distribution in Europe, and it is found in most open grassland and farmland habitats up to 2,000 m altitude [38]–[40]. It ranges from the Atlantic coast of France to Central Russia, as well as from the Orkney archipelago in the North to the Mediterranean coast in Spain (Figure 1). In previous studies it has been shown that the vole populations have an overall high levels of differentiation for both mtDNA (F ST = 0.7) and nuclear markers (STR, F ST = 0.17) [41]–[43]. The widespread distribution of this species in different habitats and environments, and its peculiar pattern of genetic diversity makes it particularly suitable for the detection of markers with high or low levels of differentiation, and by extension for the determination of the respective roles of positive or balancing selection over a large geographic scale.
Figure 1. Geographic location of the 21 Microtus arvalis populations analyzed.

The grey area corresponds to the European distribution of the species (after [98]). See Table 1 for sample abbreviations.
The aim of this study is to detect selective patterns in populations across the European mainland to disentangle the importance of balancing and positive selection in shaping the genetic diversity observed in the distribution range of the common vole. However, a major challenge in identifying genomic regions under selection is to separate the footprint of selection from that of population history and demography (e.g. [10], [44], [45]). Hence examining a large number of loci scattered throughout the genome is an effective way to tell apart the effect of selection from the confounding effects of population history and demography [10], [46], [47]. Cavalli-Sforza [48] and Lewontin and Krakauer [49] proposed that genetic drift and gene flow should affect all loci similarly, leading to some overall degree of differentiation between populations, but that selected loci would deviate significantly from this distribution. Indeed, positive selection acting on a given locus should increase population differentiation (and lead to high FST) whereas balancing selection should reduce it and lead to low FST (see e.g. [40], [47], [50], [51]).
For non-model organisms Amplified Fragment Length Polymorphisms (AFLPs) allow the screening of thousands of randomly distributed loci in a genome [52], [53]. To detect AFLP outliers, we used a recently developed extension of the Bayesian F ST-based approach [35], [54] based on the F-model [55]. BayeScan 2.1 [40] provides estimates of allele frequencies and F-statistics from AFLPs by incorporating for each individual the band intensity of a marker instead of simply using presence-absence patterns [56], [57]. This procedure implicitly allows one to distinguish between homo- and heterozygotes, and significantly improves the detection of selection with AFLP markers, which nearly reach the power obtained with single nucleotide polymorphism (SNP) data for which individual genotypes are known [40].
Materials and Methods
Sample and DNA extraction
We analysed 21 vole populations across most of the distribution range of M. arvalis in Europe, with a total of 444 individuals (see Figure 1 and Table 1). The populations were spread over 2,500 km from Spain (EAv) to Poland (PSr), and over a 750 km latitudinal gradient from Belgium (BSt) to Italy (INa).
Table 1. Location and properties of 444 M. arvalis samples from 21 populations across Europe.
| ID | Country | Location | N | Latitude | Longitude | F ST (CI) | FIS (CI) | Variable AFLPs % | AFLP diversity |
| BSt | Belgium | Stalhille | 21 | 51°13′N | 3°05′E | 0.304 (0.292–0.318) | 0.0010 (0.0000–0.0029) | 30.2 | 0.091 |
| BVe | Belgium | Veurne | 21 | 51°03′N | 2°40′E | 0.345 (0.331–0.359) | 0.0008 (0.0000–0.0024) | 25.3 | 0.078 |
| CZD | Czech Republic | Drnholec | 21 | 48°53′N | 16°27′E | 0.162 (0.151–0.172) | 0.0431 (0.0288–0.0576) | 46.3 | 0.132 |
| FAv | France | Avallon | 20 | 47°29′N | 3°54′E | 0.248 (0.236–0.261) | 0.0008 (0.0000–0.0023) | 37.3 | 0.115 |
| FCm | France | Clérmont-Ferrand | 21 | 45°47′N | 3°05′E | 0.262 (0.250–0.275) | 0.0006 (0.0000–0.0018) | 38.1 | 0.116 |
| FFr | France | Fressenneville | 22 | 50°05′N | 1°34′E | 0.302 (0.289–0.315) | 0.0020 (0.0001–0.0058) | 30.8 | 0.090 |
| FTh | France | Thaon | 21 | 49°16′N | 0°26′W | 0.294 (0.281–0.306) | 0.0058 (0.0004–0.0137) | 35.4 | 0.108 |
| DGo | Germany | Gotha | 20 | 50°57′N | 10°42′E | 0.224 (0.212–0.235) | 0.0238 (0.0118–0.0382) | 37.6 | 0.113 |
| DSc | Germany | Schiltach | 20 | 48°17′N | 8°21′E | 0.237 (0.225–0.250) | 0.0007 (0.0000–0.0022) | 33.8 | 0.107 |
| INa | Italy | Naturno | 21 | 46°39′N | 11°01′E | 0.349 (0.335–0.363) | 0.0030 (0.0002–0.0083) | 31.6 | 0.096 |
| PSr | Poland | Srodkowy | 21 | 53°35′N | 22°47′E | 0.297 (0.283–0.309) | 0.0055 (0.0000–0.0136) | 34.2 | 0.108 |
| EAv | Spain | Avila | 21 | 40°42′N | 4°48′W | 0.426 (0.411–0.440) | 0.0009 (0.0000–0.0026) | 27.8 | 0.084 |
| ESe | Spain | Segovia | 21 | 40°54′N | 4°06′W | 0.442 (0.429–0.457) | 0.0006 (0.0000–0.0019) | 26.1 | 0.078 |
| CHAP1 | Switzerland | Alp di Plaun | 20 | 46°47′N | 9°29′E | 0.224 (0.212–0.236) | 0.0129 (0.0001–0.0240) | 39.3 | 0.123 |
| CHBo1 | Switzerland | Bonaduz | 22 | 46°48′N | 9°24′E | 0.297 (0.285–0.310) | 0.0050 (0.0000–0.0125) | 32.3 | 0.098 |
| CHBw2 | Switzerland | Brienzwiler | 21 | 46°45′N | 8°07′E | 0.328 (0.315–0.343) | 0.0031 (0.0000–0.0085) | 27.7 | 0.082 |
| CHCa1 | Switzerland | Calandahütte | 22 | 46°53′N | 9°29′E | 0.257 (0.245–0.269) | 0.0124 (0.0003–0.0232) | 35.5 | 0.105 |
| CHDP1 | Switzerland | Domat/Ems | 22 | 46°50′N | 9°26′E | 0.278 (0.265–0.290) | 0.0061 (0.0000–0.0145) | 32.6 | 0.097 |
| CHGS2 | Switzerland | Grosse Scheidegg | 22 | 46°40′N | 8°06′E | 0.386 (0.372–0.400) | 0.0011 (0.0000–0.0034) | 22.6 | 0.068 |
| CHMe2 | Switzerland | Meiringen | 22 | 46°43′′N | 8°11′E | 0.411 (0.396–0.424) | 0.0007 (0.0000–0.0021) | 19.9 | 0.061 |
| CHSF2 | Switzerland | Schreck-Feld | 22 | 46°40′N | 8°04′E | 0.354 (0.341–0.368) | 0.0019 (0.0001–0.0055) | 25.1 | 0.076 |
We report the population ID, sampling site (country and location), number of individuals per population (N), coordinates, estimates of population specific F ST and F IS with 95% credibility intervals (CI), proportion of variable AFLP markers and AFLP marker diversity for each population.
AFLPs of six primer combinations have been published in Foll et al. [56].
AFLPs of 21 primer combinations have been published in Fischer et al. [40].
The samples for this study were obtained by strictly following the legislation on animal protection and experimentation of Switzerland and the other European countries involved. Microtus arvalis is not specifically protected by Swiss laws on animal protection (Tierschutzgesetz from December 16 2005) and hunting legislation (Verordnung zum Jagdschutzgesetz, February 29 1988) because of its role as an agricultural pest and general abundance. The use of snap traps for sampling M. arvalis is not a stress-inducing animal experiment (Schweregrad 0; Art. 137ff Swiss federal regulations on animal experimentation). However, Swiss samples analyzed in this study (some of them also covered in earlier studies; [40]–[43], [56], [58]–[62]) were obtained also under animal experimentation permits No. 55/02; 107/05; BE08/10; BE90/10 issued by the cantonal veterinary office of Bern according to federal law after ethical approval by the Bernese cantonal commission on animal experimentation. Additional samples were obtained from the researcher network on rodent-borne pathogens based at the German Federal Research Institute for Animal Health (FLI; http://www.fli.bund.de; GH is one of the coordinators) [63]–[65] and its international partners in the European projects EDEN and EDENext on biology and control of vector-borne infections in Europe (http://www.edenext.eu). Sample acquisition followed strictly the legislation of the relevant countries after approval by the according animal protection and ethics committees as required by the European Commission Seventh Framework Programme (FP7; http://cordis.europa.eu/fp7/home_en.html) [66], [67].
Total genomic DNA was extracted from foot, tail or liver tissue stored in absolute ethanol and later deep-frozen using a standard phenol-chloroform protocol [68]. The quality and quantity of the DNA was determined on 0.8% agarose gels and with a spectrophotometer (NanoDrop ND-1000 Spectrophotometer, NanoDrop Technologies, Inc., Wilmington, USA). The DNA concentration was standardized to 100 ng DNA/µL for all individuals to ensure similar PCR yield across samples [40].
AFLP analyses
AFLP analyses were performed according to standard protocols as established by Vos et al. [52] and modified by Fink et al. [69]. Selective amplifications were performed using 21 primer combinations (Table S1). These primer combinations were then named according to the last two selective bases of each primer, e.g. the combination E01-AAC/M02-cag is referred to as ACag. Special care was taken to guarantee the reproducibility of AFLP marker analyses: a liquid-handling robot (Microlab STAR, Hamilton Bonaduz AG, Bonaduz, Switzerland) was used for selective amplification, multiplexing of PCR products and loading of the 96-well sequencer plate, and 38 individuals (9%) were independently replicated for all 21 primer combinations (see [40] for more details).
AFLP fragment scoring and diversity
AFLP fragment scoring was performed with GeneMapper software version 3.7 (Applied Biosystems). Bin sets were created automatically and manually revised [40]. Two AFLP data matrices were produced, one with band intensity information and one with a standard binary presence-absence matrix. The AFLPs binary data matrix was used to estimate reproducibility, AFLP diversity estimates, and to run the first PCA analyses. A particular AFLP band intensity was scored as ‘present’ (1), if its value was larger than 10% of the 95% band intensity distribution quantile, or ‘absent’ (0), if its intensity was smaller than 10% of the 95% quantile value. AFLP marker frequencies, the number of variable markers per population and AFLP diversity were then calculated with the program AFLPDAT [70]. AFLP diversity was calculated as the average proportion of pairwise differences between individuals for each population, which is an index similar to Nei's gene diversity calculated from marker frequencies [71], [72].
Outlier detection
A Bayesian genome scan approach (BayeScan) was used to detect markers under selection. This procedure is more efficient than classical outlier detection methods (like DETSELD, modified version of [73] or DFDIST, modified version of [74]) in the discovery of true selected loci, as it results in a lower number of false positives [75]. BayeScan 2.1 was specifically developed for AFLP markers. The inclusion of band intensity information makes the BayeScan analysis of dominant AFLPs almost as powerful as an analysis of the same number of codominant markers (e.g. reaching 92% of the power of a SNP data set) to detect selection (for more details see [40]). Moreover, this additional information makes it possible to infer population-specific inbreeding coefficient (F IS) from AFLP data [56]. Band intensity information required by BayeScan 2.1 was obtained from the AFLP data matrix of marker band intensity provided by GeneMapper. Since markers with a low minor allele frequency systematically bias the F ST estimates downwards [76], only markers with band frequencies between 5% and 95% were used for subsequent analyses. This procedure prevents an artificial increase in the number of inferred outlier markers under positive selection [76]. Note that markers having band frequencies higher than 95% were still considered as polymorphic if the distribution of band intensity across all individuals was bimodal [40] and if they did not exceed three-times the 95% quantile of the band intensity distribution for that marker to avoid artefacts of the sequencing machine. These markers are probably informative to infer F IS, as they contain a high proportion of fixed and/or heterozygous individuals.
BayeScan assumes that allele frequencies within populations follow a multinomial-Dirichlet distribution [55], [77], [78] with F ST parameters being a function of population-specific components shared among all loci and of locus-specific components shared among all populations. For a given locus, departure from neutrality is assumed when the locus-specific component is required to explain the observed pattern of diversity. BayeScan directly infers the posterior probability of each locus to be under the effect of selection by defining and comparing two alternative models: one model includes the locus-specific component, while the other excludes it [35]. The ratio of the model posterior probabilities is used to calculate then the posterior odds (PO), which measures how much more likely the model with selection is compared to the model without selection (see [40]). We used a threshold of PO>10 for a marker to be considered under selection, which refers to “strong evidence” for the alternative model (in this case the model with selection) as defined by Jeffreys [79]. For the Markov chain Monte Carlo (MCMC) algorithm we used 20 pilot runs of 5,000 iterations to adjust the proposal distribution to acceptance rates between 0.25 and 0.45 for the runs. A burn-in of 50,000 iterations was used and visually checked for convergence of the MCMC chains, followed by 50,000 iterations for estimation using a thinning interval of 10. False Discovery Rate (FDR) was used to control for multiple testing [40], [80].
Inference of neutral genetic structure across Europe
We performed two principal component analyses (PCA) in R [81] to infer the patterns of neutral genetic structure in common voles across Europe. PCA analyses were performed on the neutral (excluding outlier loci) and evolutionary informative AFLP markers. Evolutionary informative AFLPs have band frequencies ranging between 5% and 95%, which excludes uninformative and rare markers [76]. One PCA analysis was done at the individual level using AFLP marker presence/absence data for all 444 individuals and the second analysis was done at the population level, on the basis of marker allele frequencies estimated by BayeScan [40], [56] using band intensity information.
Inference of balancing selection
Markers detected under balancing selection were investigated in more detail, as heterozygosity information can be gained from the population-specific band intensity distribution for a specific AFLP marker. A marker under balancing selection should indeed have evenly distributed allele frequencies across most populations and heterozygous individuals should be observed within populations, leading to a bimodal band intensity distribution for this AFLP marker [56]. The markers inferred as under balancing selection were thus carefully examined for bimodality of band intensities. However, sex-chromosome linked markers may also show bimodal distributions and low differentiation between populations in samples with equal sex ratios, as males only have one X-chromosome. A t-test implemented in R was thus used to check for association between band intensity and gender, using a threshold of p>0.05 without correction for multiple testing, to be conservative in the identification of marker under balancing selection. We have used the same approach to test for any amplification difference among different 96-well PCR plates of the same primer pair (batch effect).
Inference of positive selection patterns across Europe
To infer the patterns of positive selection in common voles across Europe we performed scaled PCA in R of the population allele frequencies of loci inferred under positive selection by BayeScan.
To identify the strongest geographic patterns of selection across Europe, we used a locus-by-locus SAMOVA approach [82] to separate for each marker populations into groups (k = 2) leading to the highest level of genetic differentiation (F CT). The three outlier loci showing the highest F CT were identified and plotted onto the European map using the R package plotrix to visualize the population-specific allele frequencies of these patterns of selection. To find loci showing similar geographic patterns of selection across Europe, which could be the cause of multi-genic adaptation due to similar selective pressures on different loci or genetic linkage of markers, we computed a pairwise Pearson's correlation between the population-specific allele frequencies of the outlier loci using the R package psych and Holm's correction for multiple testing [83].
Results
AFLP variation and neutral genetic structure across Europe
The AFLP analyses of the 21 European vole populations provided 3,839 markers. The majority of these AFLP markers were polymorphic (3,318; 86%) and 2,054 (54%) showed informative band frequencies between 5% and 95% overall. For each individual, we obtained on average 2,342 AFLPs (range: 2,169–2,418) across all primer combinations, and the mean length of the fragments was 239 bp. An average of 183 AFLP markers was scored per primer combination across all individuals (range: 86–256; Table 2). The average proportion of variable AFLP bands per population was 31%, with an average AFLP diversity of 9.6%. F IS estimates were low for all populations, ranging between 0.001 and 0.043 (Table 1). Average genetic differentiation among populations was globally high with an average population-specific F ST of 0.31. The population from the Czech Republic (CZD) had the highest number of variable AFLP bands per population (46%), and consequently the lowest population-specific F ST (0.16), whereas the lowest diversity was observed in a population of the Swiss Alps (CHMe), with only 20% of variable markers and hence a high population-specific F ST (0.41).
Table 2. AFLP markers detected under positive selection with BayeScan.
| Primers | Markers | # BayeScan | Marker ID of specific primer combination |
| ACag | 201 | 9 | 33; 77; 84; 85; 119; 136; 186; 187; 200 |
| ACtc | 175 | 2 | 21; 94 |
| ACtt | 157 | 5 | 40; 76; 79; 115; 142 |
| AGaa | 147 | 4 | 5; 56; 60; 101 |
| AGac | 200 | 7 | 3; 13; 84; 144; 161; 188; 190 |
| AGtg | 141 | 4 | 97; 103; 106; 126 |
| CAat | 210 | 5 | 102; 103; 145; 166; 181 |
| CAta | 185 | 8 | 73; 84; 116; 155; 157; 158; 165; 169 |
| CCac | 195 | 4 | 17; 129; 153; 167 |
| CCta | 219 | 3 | 88; 153; 213; |
| CCtt | 256 | 12 | 2; 34; 41; 42; 50; 67; 97; 158; 168; 169; 193; 208 |
| CGag | 101 | 4 | 67; 69; 70; 97 |
| CGtt | 86 | 6 | 24; 40; 50; 51; 79; 87 |
| CTaa | 239 | 11 | 3; 7; 78; 86; 166; 173; 184; 185; 187; 208; 232 |
| CTag | 208 | 9 | 64; 90; 102; 125; 132; 133; 147; 154; 176 |
| CTtg | 211 | 10 | 46; 93; 97; 104; 117; 133; 170; 174; 175; 187 |
| GCat | 224 | 12 | 15; 31; 60; 106; 169; 175; 180; 181; 192; 208; 212; 213 |
| GCta | 199 | 6 | 7; 20; 33; 82; 98; 187 |
| GCtc | 181 | 7 | 16; 22; 40; 43; 160; 166; 172 |
| GGac | 168 | 4 | 30; 31; 36; 164 |
| GGtc | 136 | 6 | 15; 18; 64; 85; 125; 129 |
| Total | 3839 | 138 |
Given are the 21 primer combinations investigated, the total number of scored AFLP markers per primer combination across all individuals, the number of markers with a posterior odds (PO)>10 and the primer combination specific IDs of the markers under selection.
The two “evolutionary neutral” PCAs were based on 1,843 neutral AFLP markers - these were the 2,054 evolutionary informative AFLP markers minus the 211 inferred outlier loci (see more details below). These neutral markers led to a clustering of individuals that approximately matches the geographic origin of the samples (Figure 1) except that the Swiss vole populations were somewhat farther apart than geography would suggest. The entire individual-based AFLP data set (Figure 2A) as well as the PCA from estimated population-specific allele frequencies (Figure 2B) show very similar patterns and allow a clear separation of the populations, which indicates the high information content of these AFLP markers.
Figure 2. Principal component analysis (PCA) of (A) the neutral binary AFLP data matrix of 444 individuals from 21 populations across Europe using 1,843 neutral and evolutionary informative AFLP markers (see Material and Methods).
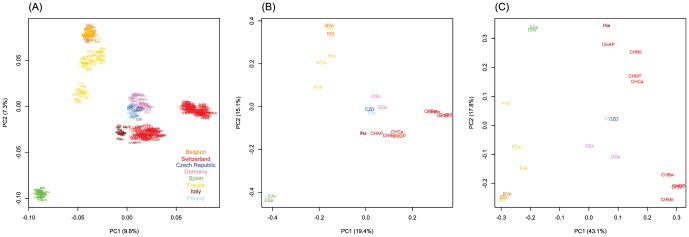
(B) PCA of the 21 population-specific allele frequency estimates of neutral AFLP markers by BayeScan. The distribution of the populations on the plot roughly follows the geographic origin of the samples. (C) PCA of the estimated population-based allele frequencies of the 138 outliers probably under positive selection. For population IDs see Table 1. Colours correspond to country affiliation (see Figure 1).
Genome scan
The BayeScan analysis of the 2,054 informative AFLP markers in 21 populations across Europe revealed 211 markers with a PO for selection larger than 10 with an associated FDR of less than 1.4%. Among these markers under selection, 138 (6.7%) had high F ST (mean F ST: 0.52) indicative of positive selection, and 73 were associated with very low F ST (mean F ST: 0.08) indicative of balancing selection (Figure 3; Table 2).
Figure 3. Results of BayeScan analysis for 2,054 informative AFLPs genotyped in 444 M. arvalis voles sampled in 21 populations.

The marker-specific F ST is plotted against the posterior odds (PO) of being under selection. The vertical line shows the critical PO of 10 used to identify outlier markers. Markers on the right side of the vertical line are outliers: 138 markers with high F ST indicative of positive selection and 73 markers with low F ST indicative of balancing selection were identified. Markers having a log10(PO)>4 were summarized in the category 4.
Inference of balancing selection
Bimodal band intensity information of AFLPs (for more details see Figure 4A and B, or [40], [56], [57]) was used to identify prime candidates for balancing selection and to exclude false positives among the 73 low F ST outliers. Among these, 40 markers were considered as unlikely to be under balancing selection, either because outliers showed significant band intensity differences between males and females (t-test, p<0.05) and were thus likely sex-chromosome linked (33 markers, Table 3, Figure 4C and D) or because of PCR amplification strength differences between 96-well plates (7 markers of the primer combination GGtc).
Figure 4. Bimodal band intensity distribution of two low F ST outlier markers.

(A) Bimodal distribution of a marker likely to be under balancing selection (CTaa125). The zero class of the distribution represents individuals not showing any band, the following first peak corresponds to heterozygous individuals and the second peak represents homozygous individuals. The black line represents a fitted density curve. (B) Comparison of band intensities in males and females for marker CTaa125 shown in (A) revealing no statistical difference and suggesting it is an autosomal marker. (C) Bimodal distribution for marker ACtt16. (D) Corresponding box plot of sex-specific band intensities for marker ACtt16 where females have band intensities about twice larger than males, hence suggesting it is an X-linked marker.
Table 3. List of the 73 markers identified by BayeScan as being potentially under balancing selection.
| Frequency | |||||
| ID | PO | F ST | Marker | Allele | Marker categorization |
| CCta185 | 226.3 | 0.105 | 0.991 | 0.843 | bimodal (balancing selection) |
| CGag21 | ∞ | 0.057 | 0.918 | 0.764 | bimodal (balancing selection) |
| CTaa125 | 63.1 | 0.136 | 0.927 | 0.699 | bimodal (balancing selection) |
| CTaa235 | 14.0 | 0.157 | 0.813 | 0.618 | bimodal (balancing selection) |
| GCat153 | ∞ | 0.094 | 0.723 | 0.476 | bimodal (balancing selection) |
| GCtc42 | 118.0 | 0.120 | 0.904 | 0.770 | bimodal (balancing selection) |
| CAta44 | ∞ | 0.032 | 1 | 0.738 | bimodal a |
| GCtc76 | ∞ | 0.029 | 0.984 | 0.847 | bimodal a |
| ACtc64 | 999.0 | 0.063 | 0.995 | 0.972 | multimodal b |
| AGaa48 | 554.6 | 0.084 | 1 | 0.844 | multimodal b |
| AGaa96 | 22.5 | 0.153 | 0.785 | 0.546 | unimodal b |
| AGtg79 | 130.6 | 0.117 | 0.941 | 0.728 | unimodal b |
| CAat185 | 249.0 | 0.078 | 0.969 | 0.812 | unimodal b |
| CAta9 | ∞ | 0.035 | 1 | 0.940 | unimodal b |
| CAta91 | 32.1 | 0.107 | 0.932 | 0.783 | unimodal b |
| CCta61 | 28.6 | 0.126 | 0.934 | 0.761 | unimodal b |
| GCat12 | ∞ | 0.033 | 1 | 0.982 | unimodal b |
| GCat95 | ∞ | 0.054 | 0.913 | 0.739 | unimodal b |
| GCat152 | 1249 | 0.075 | 0.93 | 0.740 | unimodal b |
| GCta26 | 91.6 | 0.124 | 0.888 | 0.696 | unimodal b |
| GCtc48 | 12.1 | 0.148 | 0.857 | 0.596 | unimodal b |
| ACag100 | 134.1 | 0.113 | 0.226 | 0.131 | low frequency c |
| CCac84 | 11.2 | 0.120 | 0.052 | 0.049 | low frequency c |
| CCta81 | 27.9 | 0.105 | 0.074 | 0.044 | low frequency c |
| CCta90 | 59.2 | 0.100 | 0.064 | 0.052 | low frequency c |
| CCta180 | 31.5 | 0.102 | 0.06 | 0.037 | low frequency c |
| CCta184 | 10.7 | 0.127 | 0.065 | 0.043 | low frequency c |
| CCtt32 | 10.1 | 0.132 | 0.07 | 0.054 | low frequency c |
| CCtt65 | ∞ | 0.078 | 0.33 | 0.206 | low frequency c |
| CCtt131 | 39.3 | 0.109 | 0.119 | 0.082 | low frequency c |
| CCtt145 | 383.6 | 0.068 | 0.075 | 0.074 | low frequency c |
| CGtt4 | 16.5 | 0.139 | 0.204 | 0.123 | low frequency c |
| CGtt6 | 40.0 | 0.109 | 0.098 | 0.069 | low frequency c |
| ACtc47 | ∞ | 0.066 | 0.991 | 0.812 | sex-linked d |
| ACtt16 | ∞ | 0.026 | 1 | 0.787 | sex-linked d |
| ACtt18 | 105.4 | 0.120 | 0.881 | 0.734 | sex-linked d |
| ACtt31 | ∞ | 0.024 | 0.998 | 0.762 | sex-linked d |
| ACtt46 | 276.8 | 0.121 | 0.938 | 0.713 | sex-linked d |
| AGac127 | ∞ | 0.079 | 0.998 | 0.829 | sex-linked d |
| AGtg131 | 160.3 | 0.135 | 0.78 | 0.596 | sex-linked d |
| CAat112 | 33.2 | 0.144 | 0.847 | 0.618 | sex-linked d |
| CAta114 | ∞ | 0.024 | 0.986 | 0.744 | sex-linked d |
| CAta142 | ∞ | 0.068 | 0.961 | 0.772 | sex-linked d |
| CCac46 | ∞ | 0.038 | 0.995 | 0.747 | sex-linked d |
| CCac57 | ∞ | 0.029 | 1 | 0.763 | sex-linked d |
| CCta11 | ∞ | 0.024 | 1 | 0.752 | sex-linked d |
| CCta29 | 34.7 | 0.148 | 0.66 | 0.477 | sex-linked d |
| CCta73 | 12.4 | 0.101 | 0.998 | 0.983 | sex-linked d |
| CCtt100 | ∞ | 0.031 | 0.991 | 0.710 | sex-linked d |
| CCtt150 | ∞ | 0.042 | 0.998 | 0.757 | sex-linked d |
| CCtt191 | ∞ | 0.029 | 0.995 | 0.712 | sex-linked d |
| CTaa27 | ∞ | 0.040 | 0.984 | 0.775 | sex-linked d |
| CTaa102 | ∞ | 0.040 | 0.934 | 0.662 | sex-linked d |
| CTaa113 | ∞ | 0.049 | 1 | 0.780 | sex-linked d |
| CTag19 | ∞ | 0.045 | 1 | 0.772 | sex-linked d |
| CTag94 | ∞ | 0.027 | 0.998 | 0.797 | sex-linked d |
| CTtg10 | ∞ | 0.076 | 0.896 | 0.738 | sex-linked d |
| CTtg105 | ∞ | 0.038 | 0.971 | 0.730 | sex-linked d |
| CTtg155 | ∞ | 0.059 | 0.943 | 0.721 | sex-linked d |
| GCat44 | ∞ | 0.068 | 0.946 | 0.778 | sex-linked d |
| GCat69 | ∞ | 0.023 | 0.995 | 0.742 | sex-linked d |
| GCat98 | ∞ | 0.044 | 0.995 | 0.792 | sex-linked d |
| GCta14 | ∞ | 0.060 | 1 | 0.817 | sex-linked d |
| GCta96 | 4999 | 0.090 | 0.895 | 0.632 | sex-linked d |
| ACag54 | 262.2 | 0.125 | 0.576 | 0.831 | sex-linked d |
| AGtg13 | 13.4 | 0.153 | 0.444 | 0.301 | sex-linked d |
| GGtc1 | ∞ | 0.033 | 0.998 | 0.866 | lab amplification difference e |
| GGtc9 | 45.3 | 0.122 | 0.988 | 0.748 | lab amplification difference e |
| GGtc10 | ∞ | 0.047 | 0.995 | 0.981 | lab amplification difference e |
| GGtc21 | 4999 | 0.097 | 0.846 | 0.714 | lab amplification difference e |
| GGtc34 | 16.2 | 0.089 | 0.995 | 0.968 | lab amplification difference e |
| GGtc37 | ∞ | 0.023 | 0.998 | 0.863 | lab amplification difference e |
| GGtc55 | ∞ | 0.057 | 0.998 | 0.817 | lab amplification difference e |
The top six markers are prime candidates for being under balancing selection (underlined markers) due to their clear bimodal intensity distribution in all populations. Thirty-three additional markers are sex-lined. Given are marker ID, posterior odds (PO) for the marker, marker frequency, estimated allele frequency and marker categorization.
clear bimodality was not found in single population.
unimodal or multimodal band intensity distribution.
low allele frequency.
sex-chromosome linkage.
bimodal distribution probably due to PCR amplification strength difference between 96-well plates of one specific primer pair (GGtc).
Among the remaining 33 markers with low F ST values, 27 showed distributions that could be compatible with other factors than just balancing selection. Two markers (CAta44 and GCtc76) had an overall bimodal distribution, but a clear bimodality was missing in individual populations. Thirteen markers had either a unimodal or multimodal band intensity distribution. Twelve markers had low allele frequencies (0.04–0.21) that could be a consequence of negative selection or frequency dependent selection, which is also form of balancing selection. Finally, six markers were identified as prime candidates for balancing selection (Table 3), as homozygous individuals had approximately twice the band intensity of heterozygous individuals (Figure 4A) and all populations showed intermediate allele frequencies across the European continent (see e.g. Figure 5A).
Figure 5. Allele frequency distributions of a locus potentially under balancing and three loci potentially under positive selection across Europe.
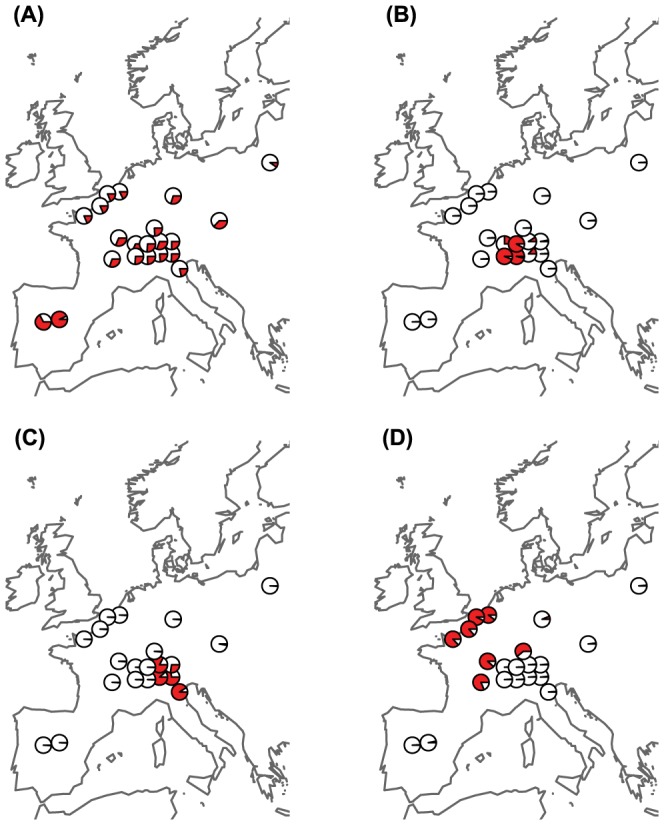
Pie charts indicate the minor allele frequency. (A) Potential patterns of continental balancing selection of the locus CTaa125, which shows very homogenous allele frequencies across Europe. (B–D) Three loci under potential positive selection, which produced the strongest splits (F CT) between two groups of populations across Europe identified by the locus-by-locus SAMOVA approach [82]. Shown are the loci (B) ACag119 with a F CT of 0.93, (C) CTaa3 with a F CT of 0.89, and (D) GGac31 with a F CT of 0.87.
Inference of positive selection across Europe
We detected a total of 138 markers potentially under positive selection across Europe, with an average of 6.6 outliers per tested primer combination (range: 2–12; Table 2). For these outliers, strong allele frequency differences were always identified in three or more populations compared to the rest, showing that selection was inferred independently in multiple populations (see e.g. Figure 5 B–D).
The PCA based on allele frequencies estimated for the 138 loci potentially under positive selection revealed a different pattern than the neutral markers (Figure 2C). Especially the populations within the Swiss Alps (CHAP, CHBo, CHCa, CHDP, CHBw, CHMe, CHGS and CHSF) and Italian Alps (INa) are much more separated from the other populations and show larger extent of differentiation among themselves compared to the PCA on neutral loci (Figure 2A and B), which is potentially indicative of strong selection pressures in the alpine area.
SAMOVA allowed us to identify the outlier loci that produced the strongest splits between two groups of populations across Europe leading to the highest level of genetic differentiation (F CT), which might be an indication of the strength of selection. The three loci that showed the strongest splits are ACag119 with a F CT of 0.93 (Figure 5B), CTaa3 with a F CT of 0.89 (Figure 5C) and GGac31 with a F CT of 0.87 (Figure 5D). The pairwise comparison of allele frequencies of outlier loci identified that ACag119 showed a significant correlation with only three other loci, CTaa3 with six and GGac31 with 16 loci. Among the 138 loci under positive selection the average number of associations was 6.1 with a range of 0 to 24 associations. Additional information for all 138 outlier loci can be found in Table S2.
Discussion
The current study illustrates the capacity of Bayesian F ST outlier approaches to identify the signature of positive and balancing selection in non-model organisms. The nearly 4,000 AFLP markers, of which 2,054 were evolutionarily informative, clearly allowed us to screen a representative part of the common vole genome for loci linked to recent adaptation on a continental scale in Europe.
Genetic structure across Europe inferred by AFLPs
The neutral AFLP markers allowed us to accurately resolve population genetic structure of the 21 vole populations across the European continent and the PCA led to a clustering of individuals and populations that corresponds approximately to the geographic origin of the samples (Figure 1 vs. 4A and B). Similar patterns were found in humans were genetic data also mirror geography in Europe [84]. This high resolution indicates the large information content present in this AFLP data set and is further supported by a very similar PCA-based clustering of populations inferred by 6,807 polymorphic SNPs (see Figure S2 in [60]), which were used to resolved the four evolutionary lineages present in Europe [43].
Pattern of selection across European continent
We scanned 21 vole populations across the European continent for evidence of selection. Overall slightly more than 8% of all markers were under positive or balancing selection. Despite the detection of some candidate loci for balancing selection (1.6%), more loci for local positive selection (6.7%) were identified. These results suggest that drift and the demographic history of vole populations have strongly influenced the observed genetic diversity, but that also positive selection plays an important role in shaping the genetic diversity of vole populations, while balancing selection is less common. Nevertheless, the detection of several markers with multiple evidence of balancing selection is remarkable, especially the signature of a stabilizing evolutionary process on such a large geographic scale.
Contrasting to our results, balancing selection played in the past an important role in evolutionary genetics in explaining the high level of genomic polymorphism observed among species or populations [8], [10]. Six decades ago Dobzhansky [1] suggested that genetic polymorphisms were maintained in populations by selection favouring heterozygotes, thus by balancing selection. Later Kimura [2], [85] has shown that most polymorphisms in the genome should be selectively neutral after the action of purifying selection. It follows that clear examples of balancing selection in any organism should be quite limited and mainly inferred by a candidate gene approach (see Introduction and e.g. [12]–[25]), but little is known about the prevalence of balancing selection on a genome wide scale [26]–[28]. In humans balancing selection is thought to have a limited role in preserving genome-wide polymorphisms [26], [86], as a specific survey of balancing selection in humans identified only 60 out of 13,400 genes [27]. In this study we identified 33 loci with significantly low levels of differentiation among populations, which represent slightly more than 1.5% of all informative markers and hence slightly more that the 0.5% inferred in humans [27]. Our findings, together with the human studies [27], indicate that large geographic scale balancing selection is probably not as frequent as previously suspected, and hence only plays a minor role in maintaining polymorphism in a population or in shaping the genetic diversity of a species.
The observation of evenly distributed allele frequencies across the whole European continent (e.g. see Figure 5A) despite extremely strong levels of differentiation among populations (average F ST = 0.31) is quite remarkable, especially for a species with limited dispersal ability [43], [87]. Such even allele frequencies across a large geographic range are difficult to explain in absence of strong stabilizing selection and hence good support for the presence of balancing selection.
This study used a conservative post-hoc evaluation of AFLP marker band intensity distributions to provide further support for the authenticity of the signature of balancing selection, which allowed us to prioritize prime candidate loci for balancing selection. Six markers were characterized by low F ST values, evenly distributed allele frequencies among populations (Figure 5A) and especially by the bimodal band intensity distribution, which clearly indicates the presence of heterozygous individuals in several populations (Figure 4A). Apart from these six loci, 27 markers showed peculiarities also compatible with other factors than only balancing selection. Twelve markers had low allele frequencies across Europe, maybe as a result of frequency-dependent selection, a selective mechanism that favours alleles when they are rare and might result in balanced genetic polymorphisms in populations [11]. But the observed low allele frequencies might also be explained by slightly negative selection [27]. For 15 markers no obvious bimodal band intensity distribution was observed, hence no clear signal of heterozygous individuals within populations could be identified, which might be explained by the stochasticity of slight technical variation in the sequencing machine that might have eroded the signal. However, especially the detection of 33 sex-chromosome linked markers (Figure 4B) clearly supports the use of AFLPs as a partially codominant marker system and indicates that heterozygous individuals or individuals carrying only one gene copy can reliably be estimated from the band intensity distribution in AFLP markers [56], [57].
Compared to balancing selection the inference of directional selection is less questioned, even though some confounding demographic factors (e.g. surfing during range expansions; [88], [89]) might produce some false positives. However, as we have used a quite stringent threshold for accepting a locus to be under selection (PO>10), our results suggest that we have here a very low false discovery rate of less than 1.4%. We detected that 6.7% of the informative markers probably evolved as a consequence of directional selection, which might be linked to adaptation to spatial heterogeneity of the environments of European vole populations. Given the wide distribution range and highly heterogeneous environments where these voles are found, it is indeed expected that different polymorphisms might be selected in different populations and habitats [5], [26]. The markers detected under positive selection in this study display a wide variety of allele frequency patterns across Europe. The PCA based on 138 markers under positive selection revealed a quite different structure (Figure 2C) than the PCA computed on 1,843 informative and neutral AFLPs (Figure 2A and B), indicating that selection acts differently on these loci than the interplay of drift and geographic separation. It is difficult to draw conclusions on the selection pressure from the allele frequency distribution of these markers; nevertheless there are some interesting patterns, which might be explained by environmental differences among populations. The two outlier loci that showed the strongest splits between two groups of populations across Europe (Figure 5B and C), were driven by populations from Alpine areas (some of the vole populations lived above 2,000 m asl). Hence they might be related to an adaptation to high elevation [40], [90] or just to the highly heterogeneous environment observed at a small geographic scale, which is specific to Alpine regions [91]. These Swiss and Italian Alpine populations are much more separated in the PCA on loci under selection (Figure 2A) than in the neutral PCAs, indicating that probably many loci are under selection in this region. However, there are also patterns that are more difficult to interpret in environmental or geographic context, e.g. Figure 5D, but biotic interactions can be also very important for local adaption and are much more difficult to infer.
Outlook
AFLP genome scans enable us to detect markers under recent selection in the common vole genome, but it is unfortunately impossible to determine their function and location in the absence of a sequenced genome for this species. New high-throughput technologies make full genomes more accessible than before (for review see [92]–[94]), but target-capture sequencing of hundreds of individuals is still prohibitive for most non-model organisms [57] and full genome re-sequencing studies of pooled population data (Pool-Seq) is only possible for rather small genomes (see e.g. [91], [95], [96]). An alternative strategy would be the investigation of candidate loci for selection by direct high-throughput sequencing of AFLP fragments [60], [97], which could be useful to further characterize candidate regions and genes linked with AFLP markers in this non-model organism.
Supporting Information
The 21 selective primer combinations and their fluorescent labels used in the AFLP assay.
(DOCX)
SAMOVA results of the 138 loci probably under positive selection ranked by F CT and the number of significant association with other loci having similar allele frequencies across Europe.
(DOCX)
Acknowledgments
We are grateful to L. L. Bischoff, T. Hofer, E. Kindler and Y. Liu for discussions and comments during the preparation of the manuscript. We thank S. Tellenbach for technical assistance and I. Dupanloup for support with SAMOVA. We are very grateful to the following people for providing access to samples: J.B. Searle, M. Thoma, J. Thiel, J. Bryja, M. Ratkiewicz, B. Thorenz, N. Martinkova, M. Pascal, E. Jones and T. Cucchi.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was supported by grants from the University of Bern Research Foundation and the Swiss National Science Foundation (grants 3100A0-126074 and 31003A-143393 to LE). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Dobzhansky T (1955) A review of some fundamental concepts and problems of population genetics. Cold Spring Harbor Symposia on Quantitative Biology 20: 1–15. [DOI] [PubMed] [Google Scholar]
- 2.Kimura M (1983) The neutral theory of molecular evolution. In: M.Nei, R.K.Koehn, editors.Evolution of Genes and Proteins.Sunderland, MA, USA: Sinauer. pp. 208–233.
- 3. Kimura M (1968) Evolutionary rate at the molecular level. Nature 217: 624–626. [DOI] [PubMed] [Google Scholar]
- 4. Ohta T (1973) Slightly deleterious mutant substitutions in evolution. Nature 246: 96–98. [DOI] [PubMed] [Google Scholar]
- 5. Huang Y, Wright SI, Agrawal AF (2014) Genome-wide patterns of genetic variation within and among alternative selective regimes. PLoS Genet 10: e1004527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levene H (1953) Genetic equilibrium when more than one ecological niche is available. The American Naturalist 87: 331–333. [Google Scholar]
- 7. Felsenstein J (1976) The theoretical population genetics of variable selection and migration. Annual Review of Genetics 10: 253–280. [DOI] [PubMed] [Google Scholar]
- 8. Charlesworth D (2006) Balancing selection and its effects on sequences in nearby genome regions. PLoS Genetics 2: 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maruki T, Kumar S, Kim Y (2012) Purifying selection modulates the estimates of population differentiation and confounds genome-wide comparisons across single-nucleotide polymorphisms. Molecular Biology and Evolution 29: 3617–3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nielsen R (2005) Molecular signatures of natural selection. Annual Review of Genetics 39: 197–218. [DOI] [PubMed] [Google Scholar]
- 11. Mitchell-Olds T, Willis JH, Goldstein DB (2007) Which evolutionary processes influence natural genetic variation for phenotypic traits? Nature Reviews Genetics 8: 845–856. [DOI] [PubMed] [Google Scholar]
- 12. Aidoo M, Terlouw DJ, Kolczak M, McElroy PD, ter Kuile FO, et al. (2002) Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet 359: 1311–1312. [DOI] [PubMed] [Google Scholar]
- 13. Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, et al. (2010) Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nature Communications 1: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verrelli BC, McDonald JH, Argyropoulos G, Destrol-Bisol G, Froment A, et al. (2002) Evidence for balancing selection from nucleotide sequence analyses of human G6PD . American Journal of Human Genetics 71: 1112–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Allen SJ, O'Donnell A, Alexander NDE, Alpers MP, Peto TEA, et al. (1997) Alpha(+)-thalassemia protects children against disease caused by other infections as well as malaria. Proceedings of the National Academy of Sciences of the United States of America 94: 14736–14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schroeder SA, Gaughan DM, Swift M (1995) Protection against bronchial asthma by CFTR delta F508 mutation: a heterozygote advantage in cystic fibrosis. Nature Medicine 1: 703–705. [DOI] [PubMed] [Google Scholar]
- 17. Calafell F, Roubinet F, Ramirez-Soriano A, Saitou N, Bertranpetit J, et al. (2008) Evolutionary dynamics of the human ABO gene. Human Genetics 124: 123–135. [DOI] [PubMed] [Google Scholar]
- 18. Baum J, Ward RH, Conway DJ (2002) Natural selection on the erythrocyte surface. Molecular Biology and Evolution 19: 223–229. [DOI] [PubMed] [Google Scholar]
- 19. Garrigan D, Hedrick PW (2003) Perspective: detecting adaptive molecular polymorphism: lessons from the MHC. Evolution 57: 1707–1722. [DOI] [PubMed] [Google Scholar]
- 20. Tan Z, Shon AM, Ober C (2005) Evidence of balancing selection at the HLA-G promoter region. Human Molecular Genetics 14: 3619–3628. [DOI] [PubMed] [Google Scholar]
- 21. Wooding S, Stone AC, Dunn DM, Mummidi S, Jorde LB, et al. (2005) Contrasting effects of natural selection on human and chimpanzee CC chemokine receptor 5. American Journal of Human Genetics 76: 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cho SC, Huang ZY, Green DR, Smith DR, Zhang JZ (2006) Evolution of the complementary sex-determination gene of honey bees: balancing selection and trans-species polymorphisms. Genome Research 16: 1366–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, et al. (2010) Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genetics 6: e1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cork JM, Purugganan MD (2005) High-diversity genes in the Arabidopsis genome. Genetics 170: 1897–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delph LF, Kelly JK (2014) On the importance of balancing selection in plants. New Phytologist 201: 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Asthana S, Schmidt S, Sunyaev S (2005) A limited role for balancing selection. Trends in Genetics 21: 30–32. [DOI] [PubMed] [Google Scholar]
- 27. Andres AM, Hubisz MJ, Indap A, Torgerson DG, Degenhardt JD, et al. (2009) Targets of balancing selection in the human genome. Molecular Biology and Evolution 26: 2755–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Makinen HS, Cano M, Merila J (2008) Identifying footprints of directional and balancing selection in marine and freshwater three-spined stickleback (Gasterosteus aculeatus) populations. Molecular Ecology 17: 3565–3582. [DOI] [PubMed] [Google Scholar]
- 29. Cutter AD, Payseur BA (2013) Genomic signatures of selection at linked sites: unifying the disparity among species. Nature Reviews Genetics 14: 262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barrett RDH, Schluter D (2008) Adaptation from standing genetic variation. Trends in Ecology and Evolution 23: 38–44. [DOI] [PubMed] [Google Scholar]
- 31. Weedall GD, Conway DJ (2010) Detecting signatures of balancing selection to identify targets of anti-parasite immunity. Trends in Parasitology 26: 363–369. [DOI] [PubMed] [Google Scholar]
- 32. Thomas JC, Godfrey PA, Feldgarden M, Robinson DA (2012) Candidate targets of balancing selection in the genome of Staphylococcus aureus . Molecular Biology and Evolution 29: 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Raj S, Pagani L, Gallego Romero I, Kivisild T, Amos W (2013) A general linear model-based approach for inferring selection to climate. BMC Genetics 14: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amambua-Ngwa A, Tetteh KKA, Manske M, Gomez-Escobar N, Stewart LB, et al. (2012) Population genomic scan for candidate signatures of balancing selection to guide antigen characterization in malaria parasites. PLoS Genet 8: e1002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180: 977–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, et al. (2002) Genetic structure of human populations. Science 298: 2381–2385. [DOI] [PubMed] [Google Scholar]
- 37. Caracristi G, Schlotterer C (2003) Genetic differentiation between American and European Drosophila melanogaster populations could be attributed to admixture of African alleles. Molecular Biology and Evolution 20: 792–799. [DOI] [PubMed] [Google Scholar]
- 38.Niethammer J, Krapp F (1982) Handbuch der Säugetiere Europas. Wiesbaden: Akademische Verlagsgesellschaft. 649 p.
- 39.Hausser J (1995) Säugetiere der Schweiz: Verbreitung, Biologie, Oekologie. Basel: Birkhäuser Verlag.
- 40. Fischer MC, Foll M, Excoffier L, Heckel G (2011) Enhanced AFLP genome scans detect local adaptation in high-altitude populations of a small rodent (Microtus arvalis). Molecular Ecology 20: 1450–1462. [DOI] [PubMed] [Google Scholar]
- 41. Braaker S, Heckel G (2009) Transalpine colonisation and partial phylogeographic erosion by dispersal in the common vole (Microtus arvalis). Molecular Ecology 18: 2518–2531. [DOI] [PubMed] [Google Scholar]
- 42. Fink S, Excoffier L, Heckel G (2004) Mitochondrial gene diversity in the common vole Microtus arvalis shaped by historical divergence and local adaptations. Molecular Ecology 13: 3501–3514. [DOI] [PubMed] [Google Scholar]
- 43. Heckel G, Burri R, Fink S, Desmet J-F, Excoffier L (2005) Genetic structure and colonization processes in European populations of the common vole Microtus arvalis . Evolution 59: 2231–2242. [PubMed] [Google Scholar]
- 44. Vitalis R, Dawson K, Boursot P (2001) Interpretation of variation across marker loci as evidence of selection. Genetics 158: 1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Teshima KM, Coop G, Przeworski M (2006) How reliable are empirical genomic scans for selective sweeps? Genome Research 16: 702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schlotterer C (2003) Hitchhiking mapping - functional genomics from the population genetics perspective. Trends in Genetics 19: 32–38. [DOI] [PubMed] [Google Scholar]
- 47. Storz JF (2005) Using genome scans of DNA polymorphism to infer adaptive population divergence. Molecular Ecology 14: 671–688. [DOI] [PubMed] [Google Scholar]
- 48. Cavalli-Sforza LL (1966) Population structure and human evolution. Proceedings of the National Academy of Sciences of the United States of America 164: 362–379. [DOI] [PubMed] [Google Scholar]
- 49. Lewontin RC, Krakauer J (1973) Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 74: 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beaumont MA (2005) Adaptation and speciation: what can Fst tell us? Trends in Ecology and Evolution 20: 435–440. [DOI] [PubMed] [Google Scholar]
- 51. Luikart G, England PR, Tallmon D, Jordan S, Taberlet P (2003) The power and promise of population genomics: from genotyping to genome typing. Nature Reviews Genetics 4: 981–994. [DOI] [PubMed] [Google Scholar]
- 52. Vos P, Hogers R, Bleeker M, Reijans M, Vandelee T, et al. (1995) AFLP - a new technique for DNA-fingerprinting. Nucleic Acids Research 23: 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Albertson RC, Markert JA, Danley PD, Kocher TD (1999) Phylogeny of a rapidly evolving clade: the cichlid fishes of Lake Malawi, East Africa. Proceedings of the National Academy of Sciences of the United States of America 96: 5107–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Beaumont MA, Balding DJ (2004) Identifying adaptive genetic divergence among populations from genome scans. Molecular Ecology 13: 969–980. [DOI] [PubMed] [Google Scholar]
- 55. Balding DJ, Nichols RA (1995) A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica 96: 3–12. [DOI] [PubMed] [Google Scholar]
- 56. Foll M, Fischer MC, Heckel G, Excoffier L (2010) Estimating population structure from AFLP amplification intensity. Molecular Ecology 19: 4638–4647. [DOI] [PubMed] [Google Scholar]
- 57. Gaggiotti OE (2010) Bayesian statistical treatment of the fluorescence of AFLP bands leads to accurate genetic structure inference. Molecular Ecology 19: 4586–4588. [DOI] [PubMed] [Google Scholar]
- 58. Beysard M, Heckel G (2014) Structure and dynamics of hybrid zones at different stages of speciation in the common vole (Microtus arvalis). Molecular Ecology 23: 673–687. [DOI] [PubMed] [Google Scholar]
- 59. Hamilton G, Ray N, Heckel G, Beaumont M, Excoffier L (2005) Bayesian estimation of recent migration rates after a spatial expansion. Genetics 170: 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lischer HEL, Excoffier L, Heckel G (2014) Ignoring heterozygous sites biases phylogenomic estimates of divergence times: implications for the evolutionary history of Microtus voles. Molecular Biology and Evolution 31: 817–831. [DOI] [PubMed] [Google Scholar]
- 61. Martínková N, Barnett R, Cucchi T, Struchen R, Pascal M, et al. (2013) Divergent evolutionary processes associated with colonization of offshore islands. Molecular Ecology 22: 5205–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sutter A, Beysard M, Heckel G (2013) Sex-specific clines support incipient speciation in a common European mammal. Heredity 110: 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schmidt-Chanasit J, Essbauer S, Petraityte R, Yoshimatsu K, Tackmann K, et al. (2010) Extensive host sharing of central European Tula virus. Journal of Virology 84: 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ulrich RG, Heckel G, Pelz HJ, Wieler LH, Nordhoff M, et al. (2009) Rodents and rodent associated disease pathogen. Bundesgesundheitsblatt-Gesundheitsforschung-Gesundheitsschutz 52: 352–369. [DOI] [PubMed] [Google Scholar]
- 65. Ulrich RG, Schmidt-Chanasit J, Schlegel M, Jacob J, Pelz HJ, et al. (2008) Network “Rodent-borne pathogens” in Germany: longitudinal studies on the geographical distribution and prevalence of hantavirus infections. Parasitology Research 103: S121–S129. [DOI] [PubMed] [Google Scholar]
- 66. Ali H, Drewes S, Sadowska E, Mikowska M, Groschup M, et al. (2014) First molecular evidence for Puumala hantavirus in Poland. Viruses 6: 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt S, Essbauer S, Mayer-Scholl A, Poppert S, Schmidt-Chanasit J, et al. (2014) Multiple infections of rodents with zoonotic pathogens in Austria. Vector-Borne and Zoonotic Diseases, in press. [DOI] [PMC free article] [PubMed]
- 68.Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- 69. Fink S, Fischer MC, Excoffier L, Heckel G (2010) Genomic scans support repetitive continental colonization events during the rapid radiation of voles (Rodentia: Microtus): the utility of AFLPs versus mitochondrial and nuclear sequence markers. Systematic Biology 59: 548–572. [DOI] [PubMed] [Google Scholar]
- 70. Ehrich D (2006) AFLPDAT: a collection of R functions for convenient handling of AFLP data. Molecular Ecology Notes 6: 603–604. [Google Scholar]
- 71.Nei M (1987) Molecular evolutionary genetics. New York: Columbia University Press.
- 72. Kosman E (2003) Nei's gene diversity and the index of average differences are identical measures of diversity within populations. Plant Pathology 52: 533–535. [Google Scholar]
- 73. Vitalis R, Dawson K, Boursot P, Belkhir K (2003) DetSel 1.0: a computer program to detect markers responding to selection. Journal of Heredity 94: 429–431. [DOI] [PubMed] [Google Scholar]
- 74. Beaumont MA, Nichols RA (1996) Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society B-Biological Sciences 363: 1619–1626. [Google Scholar]
- 75. Perez-Figueroa A, Garcia-Pereira MJ, Saura M, Rolan-Alvarez E, Caballero A (2010) Comparing three different methods to detect selective loci using dominant markers. Journal of Evolutionary Biology 23: 2267–2276. [DOI] [PubMed] [Google Scholar]
- 76. Roesti M, Salzburger W, Berner D (2012) Uninformative polymorphisms bias genome scans for signatures of selection. BMC Evolutionary Biology 12: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rannala B, Hartigan JA (1996) Estimating gene flow in island populations. Genetical Research 67: 147–158. [DOI] [PubMed] [Google Scholar]
- 78. Balding DJ (2003) Likelihood-based inference for genetic correlation coefficients. Theoretical Population Biology 63: 221–230. [DOI] [PubMed] [Google Scholar]
- 79.Jeffreys H (1961) The theory of probability: Oxford University Press.
- 80. Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B-Methodological 57: 289–300. [Google Scholar]
- 81.R Development Core Team (2011) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
- 82. Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Molecular Ecology 11: 2571–2581. [DOI] [PubMed] [Google Scholar]
- 83. Holm S (1979) A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics 6: 65–70. [Google Scholar]
- 84. Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, et al. (2008) Genes mirror geography within Europe. Nature 456: 98–U95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kimura M (1968) Genetic variability maintained in a finite population due to mutational production of neutral and nearly neutral isoalleles. Genetical Research 11: 247–269. [DOI] [PubMed] [Google Scholar]
- 86. Bubb KL, Bovee D, Buckley D, Haugen E, Kibukawa M, et al. (2006) Scan of human genome reveals no new loci under ancient balancing selection. Genetics 173: 2165–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schweizer M, Excoffier L, Heckel G (2007) Fine-scale genetic structure and dispersal patterns in the common vole Microtus arvalis . Molecular Ecology 16: 2463–2473. [DOI] [PubMed] [Google Scholar]
- 88. Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annual Review of Ecology Evolution and Systematics 40: 481–501. [Google Scholar]
- 89. Klopfstein S, Currat M, Excoffier L (2006) The fate of mutations surfing on the wave of a range expansion. Molecular Biology and Evolution 23: 482–490. [DOI] [PubMed] [Google Scholar]
- 90. Storz JF (2010) Genes for high altitudes. Science 329: 40–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Fischer MC, Rellstab C, Tedder A, Zoller S, Gugerli F, et al. (2013) Population genomic footprints of selection and associations with climate in natural populations of Arabidopsis halleri from the Alps. Molecular Ecology 22: 5594–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ellegren H (2008) Sequencing goes 454 and takes large-scale genomics into the wild. Molecular Ecology 17: 1629–1631. [DOI] [PubMed] [Google Scholar]
- 93. Tautz D, Ellegren H, Weigel D (2010) Next generation molecular ecology. Molecular Ecology 19: 1–3. [DOI] [PubMed] [Google Scholar]
- 94. Gibbons JG, Janson EM, Hittinger CT, Johnston M, Abbot P, et al. (2009) Benchmarking next-generation transcriptome sequencing for functional and evolutionary genomics. Molecular Biology and Evolution 26: 2731–2744. [DOI] [PubMed] [Google Scholar]
- 95. Turner TL, Bourne EC, Von Wettberg EJ, Hu TT, Nuzhdin SV (2010) Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nature Genetics 42: 260–263. [DOI] [PubMed] [Google Scholar]
- 96. Rellstab C, Zoller S, Tedder A, Gugerli F, Fischer MC (2013) Validation of SNP allele frequencies determined by pooled next-generation sequencing in natural populations of a non-model plant species. PLoS One 8: e80422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Paris M, Despres L (2012) Identifying insecticide resistance genes in mosquito by combining AFLP genome scans and 454 pyrosequencing. Molecular Ecology 21: 1672–1686. [DOI] [PubMed] [Google Scholar]
- 98.Mitchell-Jones AJ, Amori G, Bogdanowicz W, Krystufek B, Reijnders PJH, et al. (1999) The atlas of European mammals. London: T. Poyser, A. D. Poyser.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The 21 selective primer combinations and their fluorescent labels used in the AFLP assay.
(DOCX)
SAMOVA results of the 138 loci probably under positive selection ranked by F CT and the number of significant association with other loci having similar allele frequencies across Europe.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.