

Abstract
Although primary biliary cirrhosis (PBC) is considered a model autoimmune disease, it has not responded therapeutically to traditional immunosuppressive agents. In addition, PBC may recur following liver transplantation, despite the absence of major histocompatibility complex (MHC) matching, in sharp contrast to the well-known paradigm of MHC restriction. We have suggested previously that invariant natural killer T (iNK T) cells are critical to the initiation of PBC. In this study we have taken advantage of our ability to induce autoimmune cholangitis with 2-octynoic acid, a common component of cosmetics, conjugated to bovine serum albumin (2-OA–BSA), and studied the natural history of pathology in mice genetically deleted for CD4 or CD8 following immunization with 2-OA–BSA in the presence or absence of α-galactosylceramide (α-GalCer). In particular, we address whether autoimmune cholangitis can be induced in the absence of traditional CD4 and CD8 responses. We report herein that CD4 and CD8 knock-out mice immunized with 2-OA–BSA/PBS or 2-OA–BSA/α-GalCer develop anti-mitochondrial antibodies (AMAs), portal infiltrates and fibrosis. Indeed, our data suggest that the innate immunity is critical for immunopathology and that the pathology is exacerbated in the presence of α-GalCer. In conclusion, these data provide not only an explanation for the recurrence of PBC following liver transplantation in the absence of MHC compatibility, but also suggest that effective therapies for PBC must include blocking of both innate and adaptive pathways.
Keywords: CD4 knock-out mice, CD8 knock-out mice, primary biliary cirrhosis, xenobiotics
Introduction
A paradox in our understanding of primary biliary cirrhosis (PBC) has been the recurrence of disease following liver transplantation despite the absence of major histocompatibility complex (MHC) matching. This observation seems in sharp contrast to the well-known paradigm of MHC restriction [1,2]. Furthermore, although PBC is considered to be a model autoimmune disease with the most highly directed and focused immune response against mitochondrial antigens, there is still a gap in our understanding of the relationships between the development of anti-mitochondrial antibody (AMA) and disease development and, more importantly, the relative failure of PBC to respond to conventional immunosuppressive agents [3,4]. The importance of adaptive immunity is highlighted by the targeting of the autoimmune response to liver, exemplified by the finding of a 15–100-fold enrichment of E2 subunits of the pyruvate dehydrogenase complex (PDC-E2)-specific CD8+ and CD4+ T cells in liver when compared with peripheral blood, as determined by immunohistochemical and tetramer analysis, respectively [5–7]. An important role for CD8+ T cells was noted by results from adoptive transfer studies in which CD8+ T cells from dominant negative transforming growth factor (TGF)-β receptor II (dnTGF-βRII) mice when injected into Rag−/− mice led to liver histopathology, remarkably similar to human PBC, suggesting a critical role of CD8+ T cells in biliary destruction [8]. Further, the increased expression of interferon (IFN)-γ and interleukin (IL)-17 in the liver was shown to be critical to the development of PBC [9–14].
A detailed understanding of the initiating events leading to the breaking of self-tolerance is key for future development of therapeutic measures [15–19]. With this goal in mind, our laboratory has demonstrated that in patients with PBC, both CD1d expression and the frequency of invariant natural killer T (iNK T) cells in liver are increased compared to controls [20–22]. In addition, in the dnTGF-βRII mouse model of human PBC, we demonstrated that mice exhibit hyperactive hepatic iNK T cells. The role of iNK T cells was further established by the demonstration that CD1d-deficient dnTGF-βRII mice exhibit significantly decreased hepatic lymphoid cell infiltrates and milder cholangitis [23]. We further found that iNK T cell activation by α-galactosylceramide (α-GalCer) led to a profound exacerbation of portal inflammation, granuloma formation, bile duct damage and hepatic fibrosis in our xenobiotic immunized mouse model of PBC [24]. In this study, we have continued our thesis that innate immunity is critical by taking advantage of our ability to induce autoimmune cholangitis with 2-octynoic acid conjugated to bovine serum albumin (2-OA–BSA) and studied the natural history of disease in two well-defined gene-deleted murine systems, mice with gene knock-outs for CD4 and CD8.
Materials and methods
Experimental mice
Female C57BL/6 mice aged 8–10 weeks were obtained from the National Laboratory Animal Center, Taipei, Taiwan. B6.129S6-Cd4tm1knw/J (CD4−/−) and B6.129S2-Cd8atm1Mak/J (CD8−/−) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All mice were maintained in the Animal Center of the College of Medicine, National Taiwan University. All experiments were performed following approval of The Institutional Animal Care and Use Committee (IACUC) of National Taiwan University College of Medicine and College of Public Health.
Experimental protocol
The protocol for induction of autoimmune cholangitis was modified from our previous report [24]. Briefly, mice were immunized intraperitoneally with 2-OA–BSA (100 μg) incorporated with complete Freund's adjuvant (CFA; Sigma-Aldrich, St Louis, MO, USA) and subsequently boosted at weeks 2, 4 and 8 with 2-OA–BSA incorporated in incomplete Freund's adjuvant (IFA; Sigma-Aldrich). Two μg of α-GalCer (KRN7000; Funakoshi, Tokyo, Japan) or phosphate-buffered saline (PBS) as a control were injected simultaneously with the first, second and third 2-OA–BSA immunizations. Sera were collected at 12 weeks post-immunization and titres of immunoglobulin (Ig)M and IgG anti-PDC-E2 autoantibodies were measured by enzyme-linked immunosorbent assay (ELISA). In all assays, positive and negative controls were included. Mice were killed at 12 weeks post-immunization for liver histopathology, including phenotypic analysis of mononuclear cell infiltrates. The methodology for each of the surrogate readouts are described below.
Determination of anti-PDC-E2 antibodies
Serological IgM and IgG anti-PDC-E2 were measured by ELISA using recombinant mouse PDC-E2. Briefly, purified recombinant mouse PDC-E2 at 1 μg/ml in carbonate buffer (pH 9·6) was coated onto ELISA plates at 4°C overnight. After blocking with 1% casein (Sigma-Aldrich) for 1 h, diluted sera were added for 2 h at room temperature. The ELISA plates were washed with PBS-Tween 20 followed by the addition of horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:5000; Zymed Laboratories, Carlsbad, CA, USA) or IgM (1:5000; Invitrogen, Camarillo, CA, USA). The plates were incubated for another 1 h and immunoreactivity was detected by measuring the optical density (OD) at 450 nm after exposure for 20 min to tetramethylbenzidine (TMB) substrate (R&D Systems, Minneapolis, MN, USA). Appropriate positive and negative controls were included with each assay.
Mononuclear cell preparation
Livers were perfused with PBS containing 0·2% BSA (PBS/0·2% BSA) (Sigma-Aldrich), passed through a 100 μm nylon mesh, and resuspended in PBS/0·2% BSA. The parenchymal cells were removed as pellets after centrifugation at 50 g for 5 min and the non-parenchymal cells were then isolated using Histopaque-1077 (Sigma-Aldrich). After centrifugation, the collected cells were washed with PBS/0·2% BSA and the viability of cells was confirmed to be >95% by trypan blue dye exclusion. Cell numbers were determined by an automated haemacytometer (XS-800i; Sysmex, Kobe, Japan).
Flow cytometry
Subsets of liver mononuclear cells were measured by flow cytometry. In all cases, we used a previously optimally defined dilution of monoclonal antibodies. Before staining, all cells were preincubated with anti-CD16/32 (clone 93) to block non-specific FcγR binding. The following antibodies were used in this study: anti-CD3, anti-CD4, anti-CD8a, anti-CD19, anti-TCR-β and anti-TCR-δ (Biolegend, San Diego, CA, USA) and anti-NK1.1 (eBioscience, San Diego, CA, USA). Stained cells were analysed using a fluorescence activated cell sorter (FACS)Calibur (BD Biosciences) and the data obtained analysed using FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Histopathology
Portions of the liver were excised and fixed immediately with 10% buffered formalin solution for 2 days at room temperature. Paraffin-embedded tissue sections were then cut into 4-μm slices for routine haematoxylin and eosin (H&E) and Masson's trichrome staining. Liver inflammation was evaluated under a microscope.
Statistical analysis
Results are expressed as the mean ± standard error of the mean (s.e.m.). Statistical analyses were performed using Prism (GraphPad Software, San Diego, CA, USA). P-values were calculated using a two-tailed unpaired Mann–Whitney U-test. Significance levels were defined as P-values ≤ 0·05.
Results
CD8 knock-out mice develop autoimmune cholangitis following immunization with 2-OA–BSA
Both IgM and IgG autoantibodies to PDC-E2 in 2-OA–BSA/α-GalCer-immunized CD8−/− mice were significantly higher when compared to 2-OA–BSA/PBS-immunized CD8−/− mice (Table 1, P < 0·05). Histologically, there was enhanced portal inflammation in 2-OA–BSA/α-GalCer-immunized CD8−/− mice compared to 2-OA–BSA/PBS-immunized CD8−/− mice (Table 2). There was also evidence for fibrosis in all (14 of 14) of the 2-OA–BSA/α-GalCer-immunized CD8−/− mice, but none of the 2-OA–BSA/PBS-immunized CD8−/− mice (Table 2). The total numbers of liver mononuclear cell infiltrates were significantly higher in 2-OA–BSA/α-GalCer-immunized CD8−/− mice than in 2-OA–BSA/PBS-immunized CD8−/− mice (Fig. 1a, P < 0·05). However, there were no differences in the number of total mononuclear cell infiltrates in the livers of CD8−/− mice immunized with 2-OA–BSA/PBS or 2-OA–BSA/α-GalCer compared to CD8+/+ mice with the same immunogen (Fig. 1a). In both CD8+/+ and CD8−/− mice the total numbers of T (CD3+ NK1.1−) cells, excluding CD8+ T cells and B cells, were also increased significantly in 2-OA–BSA/α-GalCer-immunized mice when compared to 2-OA–BSA/PBS-immunized mice (P < 0·01 for T cells and P < 0·05 for B cells). However, the numbers of CD3+ T cells without CD8+ T cells in the 2-OA–BSA/α-GalCer-immunized CD8−/− mice were significantly higher than those of CD8+/+ controls (Fig. 1b, P < 0·05). There was an increased T cell frequency in the 2-OA–BSA/α-GalCer-immunized CD8−/− mice consisted of double-negative T cells (Fig. 1c). Significantly increased CD4−CD8− double-negative T cells were also observed in 2-OA–BSA/PBS-immunized CD8−/− mice compared to 2-OA–BSA/PBS-immunized CD8+/+ mice (Fig. 1c, P < 0·01). In addition, there was a significant increase of double-negative T cells in CD8−/− mice immunized with 2-OA–BSA/α-GalCer compared to CD8−/− mice immunized with 2-OA–BSA/PBS (Fig. 1c, P < 0·005). Finally we note that γδ T cells were increased significantly in CD8−/− mice immunized with 2-OA–BSA/α-GalCer and 2-OA–BSA/PBS compared to control mice with the same immunogen (Fig. 1d, P < 0·01). The numbers of liver γδ T cells were significantly higher in CD8−/− mice immunized with 2-OA–BSA/α-GalCer than CD8−/− mice immunized with 2-OA–BSA/PBS (Fig. 1d, P < 0·05). Of note, the numbers of liver γδ T cells in naive CD8−/− and CD8+/+ mice were not different (Fig. 1d). Collectively, similar to CD8+/+ mice, CD8−/− mice develop autoimmune cholangitis following immunization with 2-OA–BSA and the administration of α-GalCer exacerbates disease in 2-OA–BSA-immunized CD8−/− mice. Significant increases of γδ T cells in immunized CD8−/− mice suggest that γδ T cells in CD8−/− mice may mediate effector functions that lead to liver damage.
Table 1.
Antibodies to PDC-E2 in immunized CD8+/+ and CD8−/− mice
| Group | 2-OA–BSA | α-GalCer | IgM anti-PDC-E2† | IgG anti-PDC-E2† |
|---|---|---|---|---|
| CD8+/+ | – | – | 0·121 ± 0·006 | 0·024 ± 0·006 |
| CD8+/+ | + | – | 0·622 ± 0·092 | 0·797 ± 0·105 |
| CD8+/+ | + | + | 0·928 ± 0·105** | 1·136 ± 0·097** |
| CD8−/− | – | – | 0·147 ± 0·016 | 0·029 ± 0·006 |
| CD8−/− | + | – | 0·680 ± 0·064 | 0·736 ± 0·114 |
| CD8−/− | + | + | 0·966 ± 0·095** | 1·126 ± 0·084** |
P < 0·05 compared to CD8−/− 2-octynoic acid conjugated bovine serum albumin (2-OA–BSA/PBS) controls.
O.D. ± s.e.m. α-GalCer = α-galactosylceramide; Ig = immunoglobulin; PDC-E2 = E2 subunits of the pyruvate dehydrogenase complex.
Table 2.
Histopathology of CD8+/+ and CD8−/− mice
| Group | 2-OA–BSA | α-GalCer | Portal inflammation | Fibrosis |
|---|---|---|---|---|
| CD8+/+ | – | – | 0/5 (0%) | 0/5 (0%) |
| CD8+/+ | + | – | 4/6 (66·7%) | 1/6 (16·67%) |
| CD8+/+ | + | + | 10/11 (90·91%) | 10/11 (90·91%) |
| CD8−/− | – | – | 0/5 (0%) | 0/5 (%) |
| CD8−/− | + | – | 6/8 (75%) | 0/8 (0%) |
| CD8−/− | + | + | 14/14 (100%) | 14/14 (100%) |
α-GalCer = α-galactosylceramide; 2-OA–BSA/PBS = 2-octynoic acid conjugated bovine serum albumin.
Fig. 1.
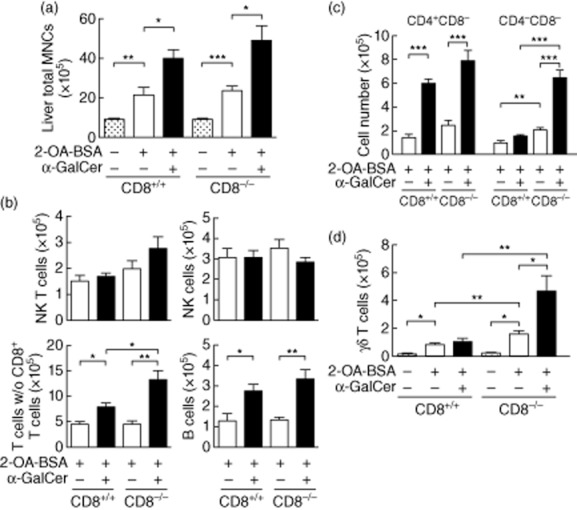
CD8 knock-out mice develop autoimmune cholangitis following immunization with 2-octynoic acid conjugated bovine serum albumin (2-OA–BSA). CD8−/− and CD8+/+ mice were immunized with either 2-OA–BSA/phosphate-buffered saline (PBS) or 2-OA–BSA/α-galactosylceramide (α-GalCer) killed at week 12. (a) Liver total mononuclear cells (MNCs). (b) Number of natural killer (NK) T (CD3+NK1.1+), NK (CD3−NK1.1+), T (CD3+ NK1.1−) excluding CD8+ T and B (CD19+) cells; (c) number of CD4+CD8− and CD4-CD8− T cells; (d) number of γδ T cells were determined by flow cytometry. Results are expressed as mean ± standard error of the mean (s.e.m.); n = five to 14 per group. *P < 0·05; **P < 0·01; ***P < 0·005.
CD4 knock-out mice develop autoimmune cholangitis following immunization with 2-OA–BSA
We subsequently investigated the role of CD4+ T cells in this murine model and accordingly immunized CD4−/− mice with 2-OA–BSA/PBS or 2-OA–BSA/α-GalCer and, as above, monitored for autoantibodies to PDC-E2 and liver histology. At 12 weeks, serum IgM antibodies to PDC-E2 were significantly higher in 2-OA–BSA/α-GalCer-immunized CD4−/− mice compared to those of 2-OA–BSA/PBS-immunized CD4−/− mice (Table 3, P < 0·01). However, sera from 2-OA–BSA/α-GalCer-immunized CD4−/− mice contained similar levels of IgM anti-PDC-E2 antibodies as CD4+/+ controls. The level of serum IgM anti-PDC-E2 antibodies was significantly lower in immunized CD4−/− mice when compared with 2-OA–BSA/PBS CD4+/+ controls (P < 0·01) (Table 3). Importantly, however, as expected there was no significant production of IgG anti-PDC-E2 in CD4−/− mice immunized with 2-OA–BSA/PBS or 2-OA–BSA/α-GalCer (Table 3). A higher frequency of liver or portal inflammation was found in 2-OA–BSA/α-GalCer-immunized CD4−/− mice than 2-OA–BSA/PBS-immunized CD4−/− mice (Table 4 and Fig. 3). There was also evidence for fibrosis in 14 of 16 2-OA–BSA/α-GalCer-immunized CD4−/− mice but none of 2-OA–BSA/PBS-immunized CD4−/− mice (Table 4 and Fig. 3). The total number of liver mononuclear cell infiltrates were significantly higher in 2-OA–BSA/α-GalCer-immunized CD4−/− mice than the 2-OA–BSA/PBS-immunized CD4−/− mice (Fig. 2a, P < 0·005). Indeed, there were no differences in the number of total mononuclear cell infiltrates in the liver of the CD4−/− mice immunized with 2-OA–BSA/PBS or 2-OA–BSA/α-GalCer compared to CD4+/+ controls with the same immunization (Fig. 2a). The total numbers of NK cells, T cells excluding CD4+ T cells and B cells, were also increased significantly in 2-OA–BSA/α-GalCer-immunized CD4−/− mice compared to 2-OA–BSA/PBS-immunized CD4−/− mice (Fig. 2b, NK cells, P < 0·05; T cells excluding CD4+ T cells, P < 0·005; B cells, P < 0·05). However, the numbers of NK T and NK cells in 2-OA–BSA/PBS-immunized CD4−/− mice were significantly lower than those of CD4+/+ controls (Fig. 2b, P < 0·01 for NK T cells and P < 0·005 for NK cells) and the number of NK cells in the 2-OA–BSA/α-GalCer-immunized CD4−/− mice was significantly lower than that of CD4+/+ controls (Fig. 2b, P < 0·05). Double-negative T cells were increased in CD4−/− mice immunized with 2-OA–BSA/α-GalCer compared to mice immunized with 2-OA–BSA/PBS (Fig. 2c, P < 0·05). Of note, significantly increased double-negative T cells were also observed in CD4−/− mice immunized with 2-OA–BSA/PBS or 2-OA–BSA/α-GalCer compared to CD4+/+ controls with the same immunization (Fig. 2c, P < 0·05 and P < 0·005). However, the numbers of γδ T cells were similar between CD4−/− and control mice (Fig. 2d). We should note that the increased numbers of double-negative T cells in 2-OA–BSA/α-GalCer immunized CD4−/− mice consisted of double-negative αβ T cells (data not shown). Collectively, similar to CD4+/+ mice, CD4−/− mice develop autoimmune cholangitis following immunization with 2-OA-BSA and α-GalCer exacerbates disease in 2-OA–BSA-immunized CD4−/− mice. Significant increases of double-negative αβ T cells in immunized CD4−/− mice indicate that double-negative T cells are also involved in mediating effector functions that lead to biliary damage.
Table 3.
Autoantibodies to PDC-E2 in immunized CD4+/+ and CD4−/− mice
| Group | 2-OA–BSA | α-GalCer | IgM anti-PDC-E2† | IgG anti-PDC-E2† |
|---|---|---|---|---|
| CD4+/+ | – | – | 0·098 ± 0·112 | 0·058 ± 0·009 |
| CD4+/+ | + | – | 0·606 ± 0·069 | 0·813 ± 0·122 |
| CD4+/+ | + | + | 0·878 ± 0·123** | 1·131 ± 0·096** |
| CD4−/− | – | – | 0·105 ± 0·013 | 0·084 ± 0·037 |
| CD4−/− | + | – | 0·236 ± 0·039*** | 0·138 ± 0·002*** |
| CD4−/− | + | + | 0·764 ± 0·169** | 0·102 ± 0·017*** |
**P < 0·05 and **P < 0·01 compared to CD8−/− 2-octynoic acid conjugated bovine serum albumin (2-OA–BSA/PBS) controls. ***P < 0·05 compared to CD4+/+ 2-OA–BSA-primary biliary cirrhosis (PBC) controls.
Optical density ± standard error of the mean. α-GalCer = α-galactosylceramide; PDC-E2 = E2 subunits of the pyruvate dehydrogenase complex; Ig = immunoglobulin.
Table 4.
Histopathology of CD4+/+ and CD4−/− mice
| Group | 2-OA–BSA | α-GalCer | Portal inflammation | Fibrosis |
|---|---|---|---|---|
| CD4+/+ | – | – | 0/4 (0%) | 0/4 (0%) |
| CD4+/+ | + | – | 6/8 (75%) | 0/8 (0%) |
| CD4+/+ | + | + | 13/13 (100%) | 12/13 (92·31%) |
| CD4−/− | – | – | 0/7 (0%) | 0/7 (0%) |
| CD4−/− | + | – | 5/8 (62·5%) | 0/8 (0%) |
| CD4−/− | + | + | 15/16 (93·75%) | 14/16 (87·5%) |
2-OA–BSA/PBS = 2-octynoic acid conjugated bovine serum albumin; α-GalCer = α-galactosylceramide.
Fig. 3.
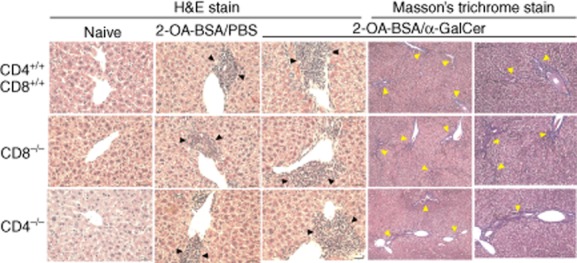
Representative results of pathological examination of liver sections (haematoxylin and eosin staining, ×400 magnification; Masson's trichrome staining, ×100 and ×200 magnification). Portal inflammation is shown by black arrowheads. Fibrosis is shown by yellow arrowheads.
Fig. 2.
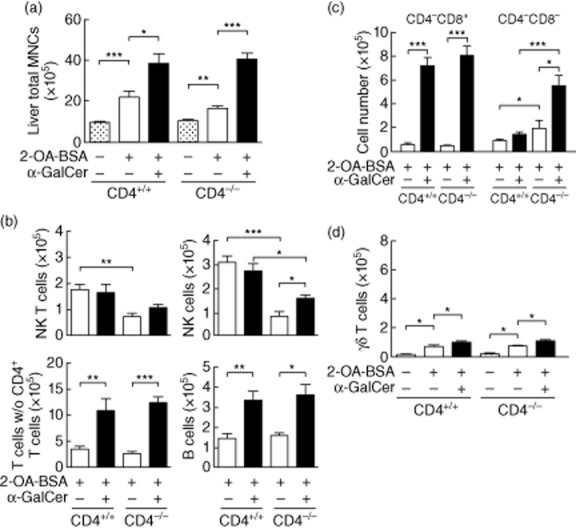
CD4 knock-out mice develop autoimmune cholangitis following immunization with 2-octynoic acid conjugated bovine serum albumin (2-OA–BSA). CD4−/− and CD4+/+ mice were immunized with either 2-OA–BSA/phosphate-buffered saline (PBS) or 2-OA–BSA/α-galactosylceramide (α-GalCer) and killed at week 12. (a) Liver total mononuclear cells (MNCs); (b) numbers of natural killer (NK) T (CD3+NK1.1+), NK (CD3−NK1.1+), T (CD3+ NK1.1−) excluding CD4+ T and B (CD19+) cells; (c) numbers of CD4−CD8+ and CD4−CD8− T cells; (d) number of γδ T cells were determined by flow cytometry. Results are expressed as mean ± standard error of the mean (s.e.m.); n = four to 16 per group. *P < 0·05; **P < 0·01; ***P < 0·005.
Administration of α-GalCer exacerbated autoimmune cholangitis in 2-OA–BSA immunized mice
Finally, in parallel and as a further positive control, we studied the effects of 2-OA–BSA immunization simultaneously with the injection of α-GalCer in wild-type C57BL/6 mice compared to mice immunized with 2-OA–BSA/PBS. Twelve weeks after such immunization, there were distinct differences in C57BL/6 mice immunized with 2-OA–BSA/α-GalCer compared to 2-OA–BSA/PBS. In the α-GalCer group, there was a significant increase in IgM and IgG autoantibodies to PDC-E2 (Tables 1 and 3) as well as a higher frequency of portal inflammation (Tables 2 and 4 and Fig. 3). In addition, there were fibrous septa extensions observed in the α-GalCer groups (Tables 2 and 4 and Fig. 3) and the total number of liver mononuclear cell infiltrates were elevated significantly (Figs 1a and 2a). There were also significant elevations of T and B cells in liver (Figs 1b and 2b), including the total numbers of CD4+ and CD8+ T cells (Figs 1c and 2c, P < 0·005).
Discussion
T cell infiltrates including CD4+ and CD8+ T cells are noted in the liver of both patients with PBC and murine models of autoimmune cholangitis [5–7,15,18,25–32]. In this study, 2-OA–BSA/α-GalCer-immunized mice had significant increases in CD4+ and CD8+ T cells. In an earlier study of this model, cytotoxic T lymphocyte antigen-4 (CTLA-4)-Ig treatment begun 1 day before 2-OA–BSA immunization inhibited the manifestations of cholangitis, including AMA production, intrahepatic T cell infiltrates and bile duct damage. CTLA-4-Ig binds to CD80/86, thus preventing CD80/86 from interacting with CD28 and thereby inhibiting the delivery of the second signal required for full T cell activation. This study indicates the importance of activating T cells in PBC [33]. Furthermore, adoptive transfer of CD8+ T cells but not CD4+ T cells from dnTGF-βRII PBC mice to Rag−/− mice leads to liver histopathology remarkably similar to PBC [8]. However, in this study our data reflect that the cholangitis was present in both CD4 and CD8 knock-out mice immunized with 2-OA–BSA/PBS or 2-OA–BSA/α-GalCer. These combined results suggest that conventional CD4+ and CD8+ T cells are important, but are not exclusively critical for development of autoimmune cholangitis.
Of note, there were significant increases in the number of γδ T cells in CD8−/− mice and CD4−CD8− double-negative αβ T cells in CD4−/− mice immunized with 2-OA–BSA/α-GalCer. This phenomenon has also been observed in other murine models of autoimmunity using CD4 and CD8 knock-out mice, i.e. collagen-induced arthritis [34]. A number of T cell subsets, including NK T cells, mucosal-associated invariant T cells, CD3+CD4−CD8− double-negative T cells and γδ T cells have both innate and adaptive characteristics and contribute significantly to the development and establishment of acute and chronic inflammatory diseases [35]. In fact, there is an expanded population of double-negative T cells and/or γδ T cells in patients with autoimmune diseases. These cells are associated with the pathogenesis of disease by producing cytokines such as IFN-γ and IL-17 and promoting inflammation [35–38]. Such expansion of double-negative T cells and/or γδ T cells are also reported in several autoimmune-prone and -induced mouse models, including those of autoimmune encephalitis, Murphy Roths large (MRL)-lpr/lpr, BXSB, non-obese diabetic (NOD) and New Zealand black/white (NZB/W) F1 mice [36,39]. We propose that 2-OA–BSA and/or α-GalCer administration activates double-negative T cells and/or γδ T cells in CD4−/− and CD8−/− mice and such activated double-negative T cells and/or γδ T cells mediate the effector functions that lead to liver damage.
CD4+ T cells are classified functionally into at least three distinct subsets based on the predominant cytokine being synthesized by each subset. This includes the T helper type 1 (Th1), Th2 and Th17 subsets. Th17, which secretes IL-17, IL-21 and IL-22, mediates host-defensive mechanisms to various infections, especially extracellular bacterial infections, and are involved in the pathogenesis of many autoimmune diseases [40]. The pathogenesis of organ-specific autoimmune diseases is thought to be orchestrated by Th1 and/or Th17, but not Th2 cells [41]. In the serum of patients with PBC the most significant increases were noted for IFN-γ and IL-17 compared to normal controls, but increased levels of IL-2, IL-4, IL-5 and IL-10 have also been reported [9–14,16,42]. In liver tissues from PBC patients, there is a significantly higher frequency of IFN-γ and IL-4 mRNA-positive cells compared with controls [9]. In addition, in PBC, there is an increase in the frequency of IL-17+ lymphocytic infiltration [11,13]. In our 2-OA–BSA/α-GalCer-immunized mice, IFN-γ production in liver tissue and liver mononuclear cells were all increased compared to controls (data not shown).
In humans, AMAs are the diagnostic hallmark of PBC. In our 2-OA–BSA/PBS- and 2-OA–BSA/α-GalCer-immunized mice, high levels of anti-PDC-E2 IgM and IgG autoantibodies were noted. However, 2-OA–BSA/α-GalCer-immunized CD4−/− mice manifest portal inflammation and fibrosis but without detectable anti-PDC-E2 IgG, as CD4+ T cells are essential for induction of a high-affinity antibody response and for efficient isotype switching from IgM to IgG production [43,44]. Treatment with CLTA-4-Ig, initiated after the development of autoimmune cholangitis in 2-OA–BSA immunization, reduces intrahepatic T cell infiltrates and bile duct damage, but levels of anti-PDC-E2 are not changed [33].
PBC exhibits several features characteristic of an autoimmune disease including the presence of the hallmark AMAs directed against the PDC-E2, autoreactive T cells in portal tracts and the specific immune targeting of cholangiocytes [45–48]. Thus PBC is an autoimmune disease and immunosuppressive agents, including corticosteroids, azathioprine, cyclosporin and methotrexate, should theoretically be successful to treat PBC; however, a clear benefit has not been demonstrated to date [4]. Our data highlight a major role for innate immune effector mechanisms and also provide a logical explanation for the recurrence of PBC following liver transplantation in the absence of MHC compatibility and explain the relative failure of immunosuppressive drugs to alter PBC, as such agents are relatively ineffective against innate immune effector mechanisms. Our results and the well-established role of adaptive immunity in PBC suggests that effective therapy of patients will be dependent upon agents that modulate both innate and adaptive responses.
Acknowledgments
This work was supported by grants from the National Health Research Institutes, Taiwan (NHRI-EX102-10027SC), National Taiwan University (NTU-CDP-102R7811) and National Institutes of Health grant DK067003.
Disclosure
No financial or commercial conflicts of interest exist.
Author contributions
All authors performed the experiments, designed the study and wrote the paper.
References
- 1.Silveira MG, Talwalkar JA, Lindor KD, Wiesner RH. Recurrent primary biliary cirrhosis after liver transplantation. Am J Transplant. 2010;10:720–726. doi: 10.1111/j.1600-6143.2010.03038.x. [DOI] [PubMed] [Google Scholar]
- 2.Manousou P, Arvaniti V, Tsochatzis E, et al. Primary biliary cirrhosis after liver transplantation: influence of immunosuppression and human leukocyte antigen locus disparity. Liver Transpl. 2010;16:64–73. doi: 10.1002/lt.21960. [DOI] [PubMed] [Google Scholar]
- 3.Gershwin ME, Mackay IR, Sturgess A, Coppel RL. Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol. 1987;138:3525–3531. [PubMed] [Google Scholar]
- 4.Silveira MG, Lindor KD. Treatment of primary biliary cirrhosis: therapy with choleretic and immunosuppressive agents. Clin Liver Dis. 2008;12:425–443. doi: 10.1016/j.cld.2008.02.008. x–xi. [DOI] [PubMed] [Google Scholar]
- 5.Shimoda S, Van de Water J, Ansari A, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest. 1998;102:1831–1840. doi: 10.1172/JCI4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kita H, Lian ZX, Van de Water J, et al. Identification of HLA-A2-restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J Exp Med. 2002;195:113–123. doi: 10.1084/jem.20010956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kita H, Matsumura S, He XS, et al. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002;109:1231–1240. doi: 10.1172/JCI14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang GX, Lian ZX, Chuang YH, et al. Adoptive transfer of CD8(+) T cells from transforming growth factor beta receptor type II (dominant negative form) induces autoimmune cholangitis in mice. Hepatology. 2008;47:1974–1982. doi: 10.1002/hep.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harada K, Van de Water J, Leung PS, et al. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology. 1997;25:791–796. doi: 10.1002/hep.510250402. [DOI] [PubMed] [Google Scholar]
- 10.Nagano T, Yamamoto K, Matsumoto S, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol. 1999;19:422–427. doi: 10.1023/a:1020511002025. [DOI] [PubMed] [Google Scholar]
- 11.Lan RY, Salunga TL, Tsuneyama K, et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun. 2009;32:43–51. doi: 10.1016/j.jaut.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rong G, Zhou Y, Xiong Y, et al. Imbalance between T helper type 17 and T regulatory cells in patients with primary biliary cirrhosis: the serum cytokine profile and peripheral cell population. Clin Exp Immunol. 2009;156:217–225. doi: 10.1111/j.1365-2249.2009.03898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harada K, Shimoda S, Sato Y, Isse K, Ikeda H, Nakanuma Y. Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin Exp Immunol. 2009;157:261–270. doi: 10.1111/j.1365-2249.2009.03947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krams SM, Cao S, Hayashi M, Villanueva JC, Martinez OM. Elevations in IFN-gamma, IL-5, and IL-10 in patients with the autoimmune disease primary biliary cirrhosis: association with autoantibodies and soluble CD30. Clin Immunol Immunopathol. 1996;80:311–320. doi: 10.1006/clin.1996.0129. [DOI] [PubMed] [Google Scholar]
- 15.Kawata K, Yang GX, Ando Y, et al. Clonality, activated antigen-specific CD8(+) T cells, and development of autoimmune cholangitis in dnTGFbetaRII mice. Hepatology. 2013;58:1094–1104. doi: 10.1002/hep.26418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rong GH, Yang GX, Ando Y, et al. Human intrahepatic biliary epithelial cells engulf blebs from their apoptotic peers. Clin Exp Immunol. 2013;172:95–103. doi: 10.1111/cei.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selmi C, Leung PS, Sherr DH, et al. Mechanisms of environmental influence on human autoimmunity: a National Institute of Environmental Health Sciences expert panel workshop. J Autoimmun. 2012;39:272–284. doi: 10.1016/j.jaut.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka H, Yang GX, Iwakoshi N, et al. Anti-CD40 ligand monoclonal antibody delays the progression of murine autoimmune cholangitis. Clin Exp Immunol. 2013;174:364–371. doi: 10.1111/cei.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian Z, Gershwin ME, Zhang C. Regulatory NK cells in autoimmune disease. J Autoimmun. 2012;39:206–215. doi: 10.1016/j.jaut.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 20.Kita H, Naidenko OV, Kronenberg M, et al. Quantitation and phenotypic analysis of natural killer T cells in primary biliary cirrhosis using a human CD1d tetramer. Gastroenterology. 2002;123:1031–1043. doi: 10.1053/gast.2002.36020. [DOI] [PubMed] [Google Scholar]
- 21.Tsuneyama K, Yasoshima M, Harada K, Hiramatsu K, Gershwin ME, Nakanuma Y. Increased CD1d expression on small bile duct epithelium and epithelioid granuloma in livers in primary biliary cirrhosis. Hepatology. 1998;28:620–623. doi: 10.1002/hep.510280303. [DOI] [PubMed] [Google Scholar]
- 22.Hudspeth K, Pontarini E, Tentorio P, et al. The role of natural killer cells in autoimmune liver disease: a comprehensive review. J Autoimmun. 2013;46:55–65. doi: 10.1016/j.jaut.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Chuang YH, Lian ZX, Yang GX, et al. Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor II dominant-negative mouse model of primary biliary cirrhosis. Hepatology. 2008;47:571–580. doi: 10.1002/hep.22052. [DOI] [PubMed] [Google Scholar]
- 24.Wu SJ, Yang YH, Tsuneyama K, et al. Innate immunity and primary biliary cirrhosis: activated invariant natural killer T cells exacerbate murine autoimmune cholangitis and fibrosis. Hepatology. 2011;53:915–925. doi: 10.1002/hep.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oertelt S, Lian ZX, Cheng CM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655–1660. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 26.Irie J, Wu Y, Wicker LS, et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med. 2006;203:1209–1219. doi: 10.1084/jem.20051911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wakabayashi K, Lian ZX, Moritoki Y, et al. IL-2 receptor alpha(−/−) mice and the development of primary biliary cirrhosis. Hepatology. 2006;44:1240–1249. doi: 10.1002/hep.21385. [DOI] [PubMed] [Google Scholar]
- 28.Wakabayashi K, Lian ZX, Leung PS, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48:531–540. doi: 10.1002/hep.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ando Y, Yang GX, Kenny TP, et al. Overexpression of microRNA-21 is associated with elevated pro-inflammatory cytokines in dominant-negative TGF-beta receptor type II mouse. J Autoimmun. 2013;41:111–119. doi: 10.1016/j.jaut.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayer CT, Tian L, Hesse C, et al. Anti-CD4 treatment inhibits autoimmunity in scurfy mice through the attenuation of co-stimulatory signals. J Autoimmun. 2014 doi: 10.1016/j.jaut.2013.08.010. epub. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Yang GX, Zhang W, et al. Escherichia coli infection induces autoimmune cholangitis and anti-mitochondrial antibodies in NOD.B6 (Idd10/Idd18) mice. Clin Exp Immunol. 2013;175:192–201. doi: 10.1111/cei.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang CY, Ma X, Tsuneyama K, et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology. 2014 doi: 10.1002/hep.26979. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhirapong A, Yang GX, Nadler S, et al. Therapeutic effect of cytotoxic T lymphocyte antigen 4/immunoglobulin on a murine model of primary biliary cirrhosis. Hepatology. 2013;57:708–715. doi: 10.1002/hep.26067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tada Y, Ho A, Koh DR, Mak TW. Collagen-induced arthritis in CD4- or CD8-deficient mice: CD8+ T cells play a role in initiation and regulate recovery phase of collagen-induced arthritis. J Immunol. 1996;156:4520–4526. [PubMed] [Google Scholar]
- 35.D'Acquisto F, Crompton T. CD3+CD4–CD8– (double negative) T cells: saviours or villains of the immune response? Biochem Pharmacol. 2011;82:333–340. doi: 10.1016/j.bcp.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 36.Blink SE, Miller SD. The contribution of gammadelta T cells to the pathogenesis of EAE and MS. Curr Mol Med. 2009;9:15–22. doi: 10.2174/156652409787314516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crispin JC, Oukka M, Bayliss G, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181:8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimonkevitz R, Colburn C, Burnham JA, Murray RS, Kotzin BL. Clonal expansions of activated gamma/delta T cells in recent-onset multiple sclerosis. Proc Natl Acad Sci USA. 1993;90:923–927. doi: 10.1073/pnas.90.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masuda T, Ohteki T, Abo T, et al. Expansion of the population of double negative CD4–8– T alpha beta-cells in the liver is a common feature of autoimmune mice. J Immunol. 1991;147:2907–2912. [PubMed] [Google Scholar]
- 40.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. 2008;31:252–256. doi: 10.1016/j.jaut.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowlus CL, Gershwin ME. The diagnosis of primary biliary cirrhosis. Autoimmun Rev. 2014;13:441–444. doi: 10.1016/j.autrev.2014.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–360. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- 44.Harris DP, Koch S, Mullen LM, Swain SL. B cell immunodeficiency fails to develop in CD4-deficient mice infected with BM5: murine AIDS as a multistep disease. J Immunol. 2001;166:6041–6049. doi: 10.4049/jimmunol.166.10.6041. [DOI] [PubMed] [Google Scholar]
- 45.Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: convenient and inconvenient truths. Hepatology. 2008;47:737–745. doi: 10.1002/hep.22042. [DOI] [PubMed] [Google Scholar]
- 46.He XS, Ansari AA, Ridgway WM, Coppel RL, Gershwin ME. New insights to the immunopathology and autoimmune responses in primary biliary cirrhosis. Cell Immunol. 2006;239:1–13. doi: 10.1016/j.cellimm.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 47.Chuang YH, Ridgway WM, Ueno Y, Gershwin ME. Animal models of primary biliary cirrhosis. Clin Liver Dis. 2008;12:333–347. doi: 10.1016/j.cld.2008.02.011. ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen RC, Naiyanetr P, Shu SA, et al. Antimitochondrial antibody heterogeneity and the xenobiotic etiology of primary biliary cirrhosis. Hepatology. 2013;57:1498–1508. doi: 10.1002/hep.26157. [DOI] [PMC free article] [PubMed] [Google Scholar]