

Abstract
Hereditary angioedema (HAE) is characterized by potentially life-threatening recurrent episodes of oedema. The open-label extension (OLE) phase of the For Angioedema Subcutaneous Treatment (FAST)-1 trial (NCT00097695) evaluated the efficacy and safety of repeated icatibant exposure in adults with multiple HAE attacks. Following completion of the randomized, controlled phase, patients could receive open-label icatibant (30 mg subcutaneously) for subsequent attacks. The primary end-point was time to onset of primary symptom relief, as assessed by visual analogue scale (VAS). Descriptive statistics were reported for cutaneous/abdominal attacks 1–10 treated in the OLE phase and individual laryngeal attacks. Post-hoc analyses were conducted in patients with ≥ 5 attacks across the controlled and OLE phases. Safety was evaluated throughout. During the OLE phase, 72 patients received icatibant for 340 attacks. For cutaneous/abdominal attacks 1–10, the median time to onset of primary symptom relief was 1·0–2·0 h. For laryngeal attacks 1–12, patient-assessed median time to initial symptom improvement was 0·3–1·2 h. Post-hoc analyses showed the time to onset of symptom relief based on composite VAS was consistent across repeated treatments with icatibant. One injection of icatibant was sufficient to treat 88·2% of attacks; rescue medication was required in 5·3% of attacks. No icatibant-related serious adverse events were reported. Icatibant provided consistent efficacy and was well tolerated for repeated treatment of HAE attacks.
Keywords: bradykinin B2 receptor antagonist, C1-inhibitor deficiency, FAST-1, hereditary angioedema, icatibant, OLE phase
Introduction
Hereditary angioedema (HAE) is a rare autosomal dominant disorder affecting approximately one in 50 000 people [1]. HAE attacks, characterized by recurrent episodes of oedema in the skin, abdomen, upper respiratory tract, and in rare cases other organs, are unpredictable in their onset, severity, frequency and duration [2]. Correct diagnosis and rapid treatment of HAE attacks can significantly reduce morbidity [3,4], and even mortality, due especially to untreated laryngeal attacks [4].
The majority of HAE patients experience numerous attacks over the course of their lifetime, ranging from fewer than one attack a year up to, and sometimes in excess of, 52 attacks per year [5–7]. The frequent need for medical intervention and disruption to normal daily life can significantly impact a patient's ability to work or attend school, having a serious detrimental effect on quality of life [8,9].
The underlying cause of HAE is a mutation in the C1-esterase inhibitor (C1-INH) gene, SERPING-1, resulting in reduced levels (HAE type I) or function (HAE type II) of the C1-INH protein [10]. C1-INH deficiency causes activation of several serum pathways (including the complement, contact, fibrinolytic and coagulation pathways), resulting in elevation of vascular mediators, such as bradykinin, the key mediator of oedema in HAE attacks. Once bound to bradykinin B2 receptors, bradykinin causes vasodilation, increased vascular permeability and fluid accumulation in interstitial tissues [11].
Icatibant (Firazyr®; Shire, Eysins, Switzerland), a bradykinin B2 receptor antagonist available for self-administration in adult patients with HAE types I and II, effectively blocks the binding of bradykinin to the bradykinin B2 receptor, thereby inhibiting oedema formation [12]. The efficacy of a single dose of icatibant (30 mg) administered subcutaneously in HAE patients was confirmed in three Phase III, randomized, controlled, multi-centre studies, the For Angioedema Subcutaneous Treatment (FAST)-1, -2 and -3 trials [13,14].
Following training, patients are able to self-administer icatibant without onsite health-care supervision; it is therefore important to demonstrate whether repeated icatibant self-treatment is associated with any loss of efficacy or unexpected adverse effects (AEs).
This paper describes the results of the open-label extension (OLE) phase and post-hoc analyses of the FAST-1 study, which investigated the efficacy and safety of repeated treatment with icatibant in HAE patients.
Methods
Study design
FAST-1 was a Phase III, randomized, double-blind, placebo-controlled study conducted at 32 centres between December 2004 and March 2008, in the United States, Canada, Australia, Argentina and Brazil.
FAST-1 comprised two phases: a controlled phase (previously reported by Cicardi et al. [13]) and an OLE phase. The controlled phase included patients treated for first attacks either as double-blind treatment of cutaneous and/or abdominal attacks with icatibant or placebo, or open-label treatment with icatibant of laryngeal attacks. All subsequent moderate-to-severe attacks that required treatment, independent of their location (cutaneous, abdominal or laryngeal), were treated in the OLE phase. Patients who were screened and found to be eligible, but who either did not experience an angioedema attack or did not experience an attack severe enough to require treatment during the controlled phase, were allowed to directly enter the modified OLE phase following the closure of the controlled phase.
The study was approved by the independent ethics committee or institutional review board at each site and conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonization Good Clinical Practice Guidelines and current local regulatory requirements. All patients provided written informed consent to be included in the study.
Patients
Inclusion and exclusion criteria for the OLE phase of FAST-1 were similar to the controlled phase of FAST-1 [13]. The main inclusion criteria were: age ≥18 years, documented diagnosis of HAE types I or II (confirmed by either functional or immunogenic C1-INH deficiency results from the central laboratory or by medical history), and an attack in the cutaneous, abdominal and/or laryngeal areas severe enough to warrant treatment.
The primary exclusion criteria included a diagnosis of other forms of angioedema, serious concomitant illness and pregnancy or lactation. Patients were also excluded if they received pain medication prior to treatment for the current attack, C1-INH products fewer than 3 days from the onset of the current angioedema attack or angiotensin-converting enzyme inhibitor treatment.
All patients who experienced an attack that was sufficiently severe to require treatment and did not receive treatment with replacement therapy, including C1-INH products within 3 days from the attack onset, were eligible for treatment with icatibant in the OLE phase of the study; a visual analogue scale (VAS) of ≥30 mm was not a prerequisite for treatment.
Treatment
During the OLE phase, initial treatment consisted of a single subcutaneous icatibant injection (30 mg) in the abdominal region, regardless of treatment received during the controlled phase. Further injections of icatibant (30 mg) were permitted if symptoms worsened within 48 h of the initial treatment (up to a maximum of three doses at least 6 h apart). Symptom worsening more than 48 h after initial treatment was considered a new attack. Rescue medication (e.g. C1-INH, anti-emetic agents or opiates) for relief of any symptom was permitted.
Symptom assessments
Prior to icatibant administration, the following assessments were made: patient symptom assessment using both a validated 100 mm VAS (with higher scores indicating increased severity; http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Pulmonary-AllergyDrugsAdvisoryCommittee/UCM260022.pdf), and separately using a five-point scale (where 0 = absence of symptoms and 4 = very severe symptoms), patient-reported time to initial symptom improvement, investigator global assessment (assessment of all cutaneous, abdominal and laryngeal symptoms combined using a five-point scale (as above), vital signs, physical examination and pregnancy status. Following icatibant treatment, patients were monitored in a hospital or outpatient clinical research centre for 4–5 h. Patients assessed their symptoms using a VAS every 30 min from 1–4 h post-treatment and then at 5, 6, 8, 10 and 12 h post-treatment. Patient symptom score assessment using the five-point scale was performed at 2, 4, 6 and 10 h post-treatment. The investigator global assessment was performed at 4 h post-treatment. Follow-up visits were scheduled 24–48 h post-icatibant injection and a final attack assessment was performed on day 14 (±2).
In addition to the pretreatment assessments, physicians recorded rescue medication use and AEs, and assessed local tolerability [injection site reaction (ISR)] during follow-up visits. Blood samples were collected pretreatment, during the attack at 0·5, 2·5 and 5·0 h and on day 14 to measure antibody activity, complement activation (c3a-des-Arg) and laboratory safety. For patients from whom at least one serum sample had been obtained after the start of the study, testing for anti-icatibant antibodies was performed with the use of enzyme-linked immunosorbent assays. Samples for complement activation were to be collected for only the first four attacks in the OLE phases.
Efficacy measures
Cutaneous and abdominal attacks
The attack was defined as either cutaneous or abdominal when patients experienced only cutaneous or only abdominal symptoms. If both types of symptoms were present, the attack was classified as abdominal if abdominal symptoms were considered moderate-to-very severe by the investigator, and cutaneous if abdominal symptoms were mild and at least one cutaneous symptom was moderate-to-very severe.
The primary end-point of FAST-1 was the time to onset of symptom relief based on a single primary symptom.
Other efficacy measures in patients with cutaneous and abdominal attacks in the OLE phase were time to almost complete symptom relief, investigators’ global assessment and patient-reported time to initial symptom improvement.
Time to onset of primary symptom relief was defined as the time from treatment to the first of three consecutive measurements in which there was a reduction below 0·86 × baseline VAS value – 16 for baseline VAS values ≥30 mm (corresponding to a 31 mm reduction for baseline VAS of 100 mm and 21 mm for baseline VAS of 30 mm). For abdominal attacks, the primary symptom was defined as abdominal pain; for cutaneous attacks, it was the more severe of either skin swelling or skin pain. Time to almost complete symptom relief was defined as the time from treatment to the first of three consecutive measurements in which all symptoms had VAS scores between 0 and 10 mm (of a possible 100 mm). Time to initial symptom improvement was defined as the time from treatment to when the patient first started to improve. Patients were asked to record the date and time when they felt their symptoms started to improve; this does not correspond to a symptom score.
Post-hoc analysis
The post-hoc analysis evaluated the time to onset of symptom relief based on the three-symptom composite patient-assessed symptom score (average VAS score for skin swelling, skin pain and abdominal pain). Time to onset of symptom relief was defined as the time from treatment to the first of three consecutive measurements with at least a 50% reduction from pretreatment in the three-symptom composite score.
Laryngeal attacks
The term ‘laryngeal attack’ was used for all cases with symptoms of upper airway obstruction. Patients presenting with laryngeal symptoms were always defined as such, irrespective of the severity of other symptoms of angioedema. The following outcome measures were used to assess laryngeal symptoms: patient-assessed time to initial symptom improvement, change in investigator-assessed symptom score severity and investigator global assessment of attack severity.
Safety
Safety was evaluated up to 24 weeks post-treatment by AE reporting, including the incidence and severity of AEs and ISRs.
An HAE attack was defined as any HAE symptoms occurring within 48 h after the onset of symptoms. Any clinically relevant worsening of the signs and symptoms of a treated attack, and any laryngeal attack which occurred during the course of the open-label phase, were considered to be an AE. Recurrence of HAE symptoms more than 48 h after an initial attack was to be considered a new attack, and was therefore not to be reported as an AE.
During the study, laboratory evaluations for safety were performed by local laboratories using standard methods. Haematology parameters included haemoglobin, haematocrit, red blood cell count, white blood cell count, platelet count, mean corpuscular haemoglobin and mean corpuscular volume. Coagulation parameters included prothrombin time and activated partial thromboplastin time. Clinical chemistry parameters included glucose, creatinine, uric acid, urea, total bilirubin, aspartate aminotransferase, alanine aminotransferase, gamma glutamyl transferase, creatinine phosphokinase and pregnancy testing. Urinalysis was performed using a dipstick test, and recorded as normal or abnormal.
Statistical analysis
Non-laryngeal attacks in the open-label extension phase
Efficacy and safety analyses included all patients treated in the OLE phase stratified by on-study attack number. Times to onset of primary symptom relief, almost complete symptom relief and initial symptom improvement were analysed using Kaplan–Meier methods where at least 10 patients were available for assessment. Patients not achieving the end-point were censored at the time of the last available assessment. Descriptive statistics, medians and 95% confidence intervals (CIs) are presented by on-study attack number for the first 10 attacks treated with icatibant in the OLE phase. The majority of patients experienced their first on-study attack (attack 1), treated with either icatibant or placebo during the controlled phase, and results from their first attack treated with icatibant in the OLE phase are categorized as attack 2 (second on-study attack). The results from patients who experienced their first on-study attack treated with icatibant in the OLE phase are categorized as attack 1.
Post-hoc analyses of non-laryngeal attacks
Post-hoc analyses were carried out to assess the efficacy of repeated treatment with icatibant. For the cohort of patients treated with icatibant for at least five attacks (including attacks treated in the controlled phase), descriptive statistics, medians and 95% CIs are presented stratified by icatibant-treated attack number. The first icatibant-treated attack could have occurred in either the controlled phase or the OLE phase. Time to onset of primary symptom relief, onset of symptom relief based on the composite symptom score, almost complete symptom relief and initial symptom improvement were analysed using Kaplan–Meier methods. Patients not achieving the end-point were censored at the time of the last available assessment. For the same cohort of patients, observation period AEs are also reported. These are defined as treatment-emergent AEs occurring prior to or on the date of the day 14 visit or study discontinuation (whichever occurs first) for each study drug-treated attack.
Analysis of laryngeal attacks
The efficacy of icatibant for the treatment of laryngeal attacks was evaluated using all icatibant-treated laryngeal attacks (including attacks treated in the controlled phase). Kaplan–Meier methods were employed for the analysis of the time to initial symptom improvement end-point stratified by on-study attack number. Patients not achieving the end-point were censored at the time of the last available assessment. Descriptive statistics, medians and 95% CIs are presented by on-study attack number. For the analysis of the investigator's global assessment and patient-assessed symptom scores, results are presented collapsed across all attacks.
Results
Patient characteristics
During the OLE phase, a total of 72 patients received icatibant for 340 attacks, including 20 patients who entered directly into the OLE phase and 52 of 64 patients from the controlled phase. Of these 72 patients, 42 were treated for 118 cutaneous attacks, 46 patients were treated for 184 abdominal attacks, 19 patients were treated for 37 laryngeal attacks and one patient had an unclassified attack. The date of the first treatment in the OLE phase was 2 March 2005 and the date of the last patient visit was 31 March 2008.
There were more female than male patients (68·1 versus 31·9%) and the majority of patients were Caucasian (63·9%). The mean age and weight of patients treated in the OLE phase (35·5 years, 76·6 kg) were similar to the icatibant-treated patients in the controlled phase (34·8 years, 80·3 kg) [13]. The highest number of attacks treated with icatibant in a single patient was 21 over a 2-year period.
Evaluations
Although 72 patients were treated in the OLE phase, the number of patients eligible for efficacy analyses based on VAS was lower due to a number of factors, including: patients with laryngeal attacks were not included in efficacy analyses based on VAS; and patients with missing baseline VAS or with a baseline VAS less than 30 mm were excluded from the analyses.
A total of 35 patients discontinued the study during the OLE phase: 28 patients in the safety population, two patients with laryngeal symptoms at baseline and five patients who did not receive treatment during the controlled phase.
The post-hoc analyses consisted of 26 patients with at least five icatibant-treated attacks. Of these 26 patients, 12 were treated for at least 10 attacks and five were treated for at least 15 attacks.
Clinical efficacy
Cutaneous and abdominal attacks
In the open-label extension phase, the median time to onset of primary symptom relief for cutaneous and/or abdominal attacks ranged between 1·0 and 2·0 h for attacks 1–10 (Table 1). The median time to onset of primary symptom relief for cutaneous attacks ranged between 2·0–4·8 h, and for abdominal attacks the range was 1·0–1·1 h (attacks 1–10).
Table 1.
Efficacy measures evaluated in patients treated in the open-label extension (OLE) phase of For Angioedema Subcutaneous Treatment (FAST)-1
| Time to onset of primary symptom relief†, h | Time to almost complete symptom relief§, h | Time to initial symptom improvement¶, h | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Icatibant-treated attack number | n‡ | Censored | Median (95% CI), h | n | Censored | Median (95% CI), h | n | Censored | Median (95% CI), h |
| 1 | 11 | 1 | 1·0 (1·0, 2·5) | 13 | 6 | 8·0 (4·0, 8·1) | 20 | 3 | 0·5 (0·2, 1·0) |
| 2 | 48 | 1 | 2·0 (1·5, 2·5) | 55 | 14 | 10·0 (3·6, 29·0) | 63 | 7 | 0·6 (0·4, 0·7) |
| 3 | 36 | 0 | 1·8 (1·0, 2·5) | 40 | 12 | 16·8 (6·0, 36·0) | 44 | 4 | 0·6 (0·4, 0·8) |
| 4 | 31 | 4 | 1·5 (1·0, 2·5) | 35 | 13 | 12·4 (5·0, 38·3) | 38 | 3 | 0·7 (0·4, 1·0) |
| 5 | 21 | 2 | 1·3 (1·0, 2·0) | 23 | 10 | 26·6 (2·5, 72·2) | 29 | 4 | 0·5 (0·4, 0·8) |
| 6 | 19 | 3 | 1·5 (1·0, 2·3) | 21 | 10 | 5·5 (3·2, n.e.) | 23 | 2 | 0·4 (0·3, 0·5) |
| 7 | 16 | 3 | 1·5 (1·0, 3·6) | 16 | 4 | 4·7 (1·6, 15·0) | 18 | 0 | 0·4 (0·3, 0·7) |
| 8 | 14 | 3 | 1·2 (1·0, 2·5) | 14 | 8 | 55·0 (3·0, 55·0) | 15 | 0 | 0·4 (0·3, 0·6) |
| 9 | 12 | 2 | 1·3 (1·0, 2·0) | 12 | 4 | 5·1 (2·5, 18·2) | 15 | 1 | 0·8 (0·4, 1·0) |
| 10 | 11 | 1 | 1·5 (1·0, 9·0) | 12 | 5 | 5·9 (3·0, n.e.) | 14 | 0 | 0·4 (0·3, 0·9) |
Onset of primary symptom relief was defined the first of three consecutive measures in which there was a reduction less than 0·86 × baseline VAS value – 16 for a single primary symptom VAS score where the baseline VAS value was ≥ 30 mm; n.e. = not estimable. For abdominal attacks, the primary VAS score was defined as abdominal pain, and for cutaneous attacks as the more severe of either skin swelling or skin pain. The median time to onset was calculated using Kaplan–Meier methodology.
Number of patients with pretreatment VAS score ≥ 30 mm.
Almost complete symptom relief was defined as the first of three consecutive measures in which all symptom scores were between 0 and 10 mm on the VAS scale. The median time to almost complete symptom relief was calculated using Kaplan–Meier methodology.
Time to initial symptom improvement was defined as the start of improvement of symptoms according to the patient. The median time to onset was calculated using Kaplan–Meier methodology. CI = confidence interval; VAS = visual analogue scale.
For cutaneous and/or abdominal attacks, the median time to almost complete symptom relief for attacks 1–10 ranged between 4·7 and 55·0 h (Table 1). The median time to almost complete symptom relief of 55·0 h was calculated for attack 8, where time to almost complete symptom relief was not documented for eight of 14 patients treated but censored.
The investigators’ global assessment demonstrated an improvement in symptom severity for both cutaneous and abdominal attacks within 4 h, regardless of pretreatment attack severity (Supporting information, Tables S1 and S2). A rapid reduction in symptom severity was reported similarly by the patient for most attacks following treatment with icatibant; the median time to initial symptom improvement ranged from 0·4 to 0·8 h (Table 1).
Post-hoc analyses of the cohort of 26 patients treated for at least five attacks in the controlled and OLE phases of FAST-1 demonstrated that the time to onset of symptom relief based on a three-symptom composite VAS score was similar (Fig. 1a), with no noticeable variation with repeated treatment with icatibant, ranging between 1·5 and 2·0 h (Table 2). Moreover, time to onset of primary symptom relief was comparable (Fig. 1b) with increasing attack number, ranging between 1·1 and 2·5 h (Table 2).
Fig. 1.
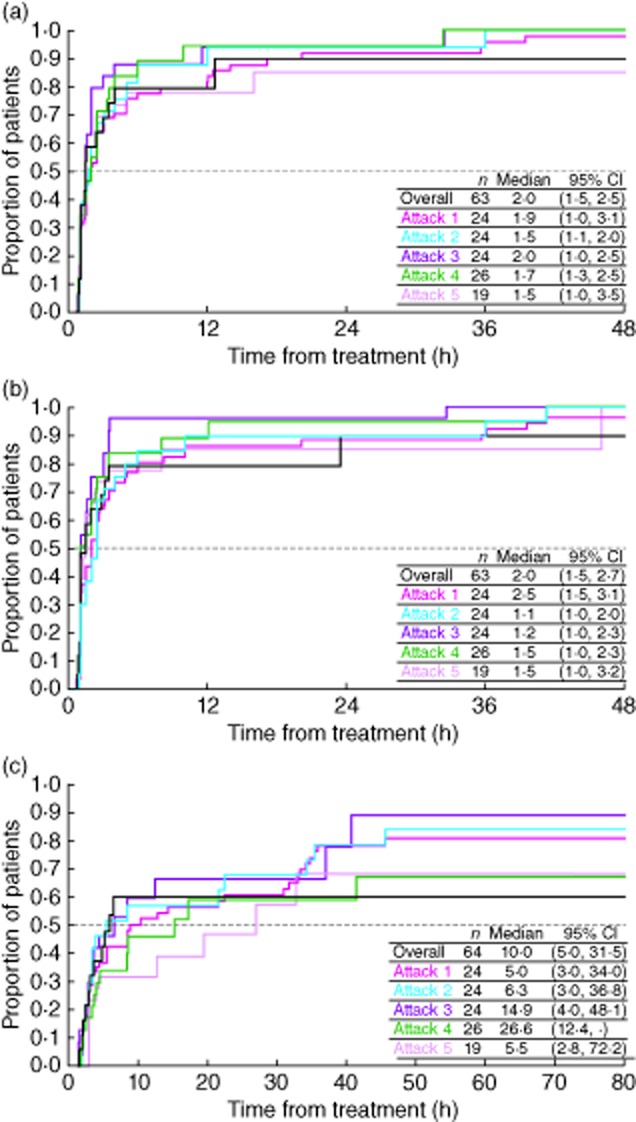
Post-hoc analyses of the proportion of patients achieving (a) onset of symptom relief† (b) onset of symptom relief for the primary symptomb and (c) almost complete symptom reliefc. The cohort of patients demonstrated here are from the post-hoc analyses of those treated for at least five attacks with icatibant during the controlled and open-label extension (OLE) phases of For Angioedema Subcutaneous Treatment (FAST-1). aOnset of symptom relief was defined as the first of three consecutive measures in which there was at least a 50% reduction from pretreatment in the three-symptom composite visual analogue scale (VAS) score. The median time to onset to onset is calculated using Kaplan–Meier methodology. bOnset of primary symptom relief was defined as the first of three consecutive measures in which there was a reduction less than 0·86 × baseline VAS value – 16 for a single primary VAS score, where the baseline VAS value was ≥30 mm. For abdominal attacks, the primary VAS score was defined as abdominal pain, and for cutaneous attacks as the more severe of either skin swelling or skin pain. The median time to onset is calculated using Kaplan–Meier methodology. cAlmost complete symptom relief was defined as the first of three consecutive measures in which all three symptoms scores were between 0 and 10 mm on the VAS scale. The median time to almost complete symptom relief was calculated using Kaplan–Meier methodology.
Table 2.
Post-hoc analyses of efficacy measures evaluated in patients treated for at least five attacks with icatibant in the controlled and open-label extension (OLE) phases of For Angioedema Subcutaneous Treatment (FAST)-1
| Time to onset of symptom relief†, h | Time to onset of primary symptom relief‡, h | Time to almost complete symptom relief§, h | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Icatibant-treated attack number | n | Censored | Median (95% CI), h | n | Censored | Median (95% CI), h | n | Censored | Median (95% CI), h |
| 1 | 24 | 2 | 1·9 (1·0, 3·1) | 24 | 1 | 2·5 (1·5, 3·1) | 24 | 5 | 5·0 (3·0, 34·0) |
| 2 | 24 | 1 | 1·5 (1·1, 2·0) | 24 | 0 | 1·1 (1·0, 2·0) | 24 | 7 | 6·3 (3·0, 36·8) |
| 3 | 24 | 1 | 2·0 (1·0, 2·5) | 24 | 1 | 1·2 (1·0, 2·3) | 24 | 7 | 14·9 (4·0, 48·1) |
| 4 | 26 | 4 | 1·7 (1·3, 2·5) | 26 | 4 | 1·5 (1·0, 2·3) | 26 | 14 | 26·6 (12·4, –) |
| 5 | 19 | 3 | 1·5 (1·0, 3·5) | 19 | 3 | 1·5 (1·0, 3·2) | 19 | 7 | 5·5 (2·5, 72·2) |
| Overall¶ | 63 | 5 | 2·0 (1·5, 2·5) | 63 | 5 | 2·0 (1·5, 2·7) | 64 | 16 | 10·0 (5·0, 31·5) |
The cohort of patients demonstrated here are from the post-hoc analyses of those treated for at least five attacks with icatibant during the controlled and OLE phases of FAST-1.
Onset of symptom relief was defined as the first of three consecutive measures in which there was at least a 50% reduction from pretreatment in the three-symptom composite VAS score. The median time to onset was calculated using Kaplan–Meier methodology.
Onset of primary symptom relief was defined as the first of three consecutive measures in which there was a reduction less than 0·86 × baseline VAS value – 16 for a single primary VAS score where the baseline VAS value was ≥30 mm. For abdominal attacks, the primary VAS score as defined as abdominal pain, and for cutaneous attacks as the more severe of either skin swelling or skin pain. The median time to onset was calculated using Kaplan–Meier methodology.
Almost complete symptom relief was defined as the first of three consecutive measures in which all symptom scores were between 0–10 mm on the VAS scale. The median time to almost complete symptom relief was calculated using Kaplan–Meier methodology.
The analyses of the ‘overall’ population are based on the cohort of patients with at least one icatibant-treated attack across the controlled and OLE phases. CI = confidence interval; FAST-1 = For Angioedema Subcutaneous Treatment 1 trial; OLE = open-label extension; VAS = visual analogue scale.
Time to almost complete symptom relief was also generally similar with increasing attack number (Fig. 1c), ranging between 5·0 and 26·6 h over the first five icatibant-treated attacks (Table 2). It should be noted that 14 of 26 patients (53·8%) in attack 4 were censored; censoring in the other attacks ranged from 20·8 to 36·8% of patients.
Laryngeal attacks
Icatibant also remained efficacious over repeated treatment of laryngeal attacks. Nineteen patients were treated for 37 laryngeal attacks in the OLE phase without the need for intubation; the patient-assessed time to initial symptom improvement ranged from 0·1 to 5·3 h across all attacks (Fig. 2). The investigator's global assessment showed a rapid improvement in symptom severity within 4 h post-dose and, by 24 h post-icatibant treatment, all laryngeal symptoms were reported as mild or absent (Supporting information, Fig. S1). This was supported by the patient-assessed symptom score, which showed a reduction of severity of the individual symptoms within 4 h, with the majority of symptoms reported as mild or absent by 4 h (Supporting information, Fig. S2).
Fig. 2.
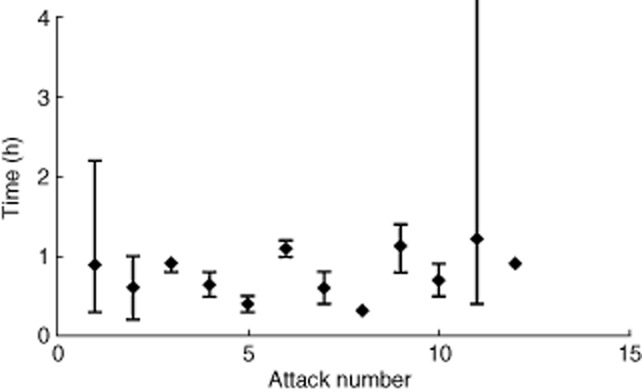
Time to initial symptom improvement for laryngeal attacks. Each dot represents a patient's report of the time of initial symptom improvement. There were three instances in which the time to initial symptom improvement was missing: one each at attacks 1, 8 and 152. Patient numbers for each attack were as follows: attack 1, n = 6; attack 2, n = 6; attack 3, n = 2; attack 4, n = 3; attack 5, n = 5; attack 6, n = 2; attack 7, n = 2; attack 8, n = 2; attack 9, n = 2; attack 10, n = 2; attack 11, n = 3; attack 12, n = 1); attack 15, n = 1.
Rescue medication and retreatment with icatibant
Open-label extension phase
A single icatibant injection was used in 300 of the 340 attacks (88·2%). Two injections were used in 36 attacks (10·6%) and three injections in four attacks (1·2%). Of the 40 attacks requiring additional icatibant injections, 23 were abdominal (12·5% of 184 abdominal attacks), 12 cutaneous (10·2% of 118 cutaneous attacks) and five laryngeal (13·5% of 37 laryngeal attacks); 29 attacks were associated with reported AEs of HAE. The second icatibant injection was administered on average 25·6 h (median = 24·0 h; IQR = 16·5–34·8 h) after the initial injection.
Rescue medication (including C1-INH in four patients and symptomatic therapy in 10 patients) was used in 18 of the 340 attacks (5·3% of attacks: nine abdominal, seven cutaneous and two laryngeal), two of which were associated with HAE AEs. In the majority of cases, rescue medication was administered 12–24 h after icatibant administration.
Post-hoc analyses
For patients treated with icatibant for at least five attacks, post-hoc analyses demonstrated that the proportion requiring more than one injection ranged between 0·0 and 23·1% for attacks 1–5 (Fig. 3).
Fig. 3.
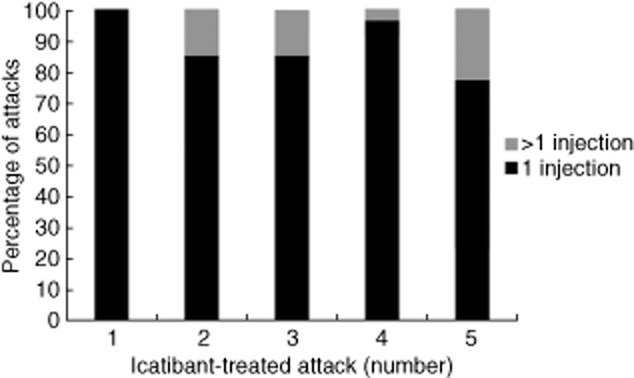
Post-hoc analyses of the overall number of attacks requiring more than one icatibant injection.
Safety
Open-label extension phase
During the OLE phase, 59 of the 72 patients (81·9%) reported 236 AEs, most of which were mild-to-moderate in severity. HAE attacks (either worsening of the current attack or a new attack) were the most commonly spontaneously reported AE (23 patients, 31·9%); 49% of these attacks were abdominal, 37% were cutaneous and 14% were laryngeal. Three patients had five events, one patient had four events, three patients had three events, four patients had two events and 12 patients had one event. Headache and upper respiratory tract infection were reported in 14 patients ([19·4%) and nine patients (12·5%), respectively. Nineteen patients (26·4%) reported 33 severe AEs: 11 patients (15·3%) reported severe HAE and two patients (2·8%) reported severe headache; all other severe AEs were each reported by just one patient. A total of four severe AEs were considered to be drug-related [headache (n = 3) in one patient, and HAE attack (n = 1) in another patient]. Overall, 31 drug-related AEs were observed in 16 patients (22·2%), with the most frequent being injection site pain and headache [five patients (6·9%) each]. Four serious AEs (SAEs) were reported in three patients (4·2%); two episodes of pancreatitis in one patient, a severe HAE attack (occurring 13 h post-icatibant injection) in one patient and severe chest pain in one patient. These were not considered related to icatibant, and all patients recovered.
ISRs were assessed separately from spontaneous reports of AEs. ISRs were reported in 70 (97·2%) patients, all of whom experienced erythema and the majority of whom also experienced swelling [66 patients (91·7%)]. Most ISRs were mild-to-moderate in severity and resolved spontaneously without further intervention. There were no withdrawals due to ISRs.
No antibody response towards icatibant was observed in patients with HAE, and there were no signs of complement activation as measured by C3a-des-Arg. There were also no clinically relevant changes in laboratory parameters during the OLE phase.
No patients discontinued due to AEs and no patient died during the study.
Post-hoc analyses
In the post-hoc analyses there was no observed trend towards increased number of AEs or treatment-related AEs with increasing number of icatibant administrations. No patients reported an SAE (Table 3).
Table 3.
Post-hoc analyses of the safety of repeated treatment with icatibant†
| Any AE | Any related AE | Any SAE | ||||
|---|---|---|---|---|---|---|
| Icatibant-treated attack number | n (%) | Events | n (%) | Events | n (%) | Events |
| 1 (n = 26) | 11 (42·3) | 16 | 2 (7·7) | 2 | 0 (0·0) | 0 |
| 2 (n = 26) | 7 (26·9) | 13 | 2 (7·7) | 3 | 0 (0·0) | 0 |
| 3 (n = 26) | 10 (38·5) | 12 | 2 (7·7) | 2 | 0 (0·0) | 0 |
| 4 (n = 26) | 5 (19·2) | 7 | 0 (0·0) | 0 | 0 (0·0) | 0 |
| 5 (n = 26) | 7 (26·9) | 9 | 1 (3·8) | 1 | 0 (0·0) | 0 |
Post-hoc analyses of safety included patients treated for at least five non-laryngeal attacks with icatibant during the controlled and OLE phases of FAST-1. Symptoms at injection site are not included in the AE categories. Analyses included treatment-emergent AEs occurring prior to or on the date of the day 14 visit or study discontinuation, whichever occurred first. AE = adverse event; FAST-1 = For Angioedema Subcutaneous Treatment 1 trial; OLE = open-label extension; SAE = serious AE.
Discussion
The results of the FAST-1 OLE phase and the post-hoc analyses presented here demonstrated that, over a mean period of 0·85 (0·03–2·32) years, icatibant provided consistent efficacy and was generally well tolerated for the repeat self-treatment of cutaneous, abdominal and laryngeal HAE attacks in adults. The efficacy observed in the controlled phase [13] was consistent throughout the OLE phase as evaluated up to attack 10, with no observed trend towards delayed onset of symptom relief associated with repeated icatibant treatment. Response to icatibant tended to be faster in patients with abdominal symptoms compared with those with cutaneous symptoms, as has been reported in previous studies with C-INH concentrate [15–17]. The overall median time to almost complete response (the closest measure to attack resolution in this study) was 10 h (95% CI = 5·0, 31·5) in the post-hoc analyses. A possible limitation of this study, however, was that almost half the patients were censored from the time to almost complete symptom relief analyses as they did not have three consecutive assessments with all VAS ≤10 mm. In patients with laryngeal attacks, treatment with icatibant was associated with a median time to initial symptom improvement in the range of 0·1–5·3 h across all attacks. Rapid symptom improvement is vital for patients who experience these types of HAE attacks due to the potential for airway obstruction [4].
Also supporting our results was the observation that the majority of patients (88·2%) were treated adequately with a single injection of icatibant, with only 40 of 340 (11·8%) attacks requiring additional injections and only 18 of 340 (5·3%) attacks requiring rescue medication (including C1-INH and symptomatic therapy). The reasons for administering rescue medication were not always indicated clearly by the investigator, but in the majority of cases rescue medication was given even when the severity of initial HAE symptoms had considerably subsided or the symptoms were recorded as absent. A limitation of this study was that the timing of the start of the AE was not recorded, and so we cannot draw any conclusions about the actual timing between icatibant dosing and worsening or recurrence of HAE symptoms. Reinjection and the use of rescue medication is an obvious concern to clinicians and patients, for reasons of both cost and patient convenience. Our results are similar to those of a recent Phase IIIb, open-label, multi-centre study of a single icatibant injection in adult patients with HAE types I and II, which reported that 91·8% of patients required only one icatibant injection and 6·6% of patients required rescue medication [18].
The relatively recent US approval of several therapies for the treatment of HAE attacks (pasteurized and nanofiltered C1-INH concentrate, ecallantide and icatibant) has resulted from controlled clinical trials demonstrating their efficacy in primarily single attacks [13,19–23]. As patients with HAE will continue to suffer attacks throughout their lives, sometimes on a weekly basis [5–7], they require treatments which demonstrate consistent efficacy with repeated use.
The findings reported here from the FAST-1 OLE phase and post-hoc analyses have demonstrated that icatibant maintained consistent efficacy in the treatment of cutaneous, abdominal and laryngeal HAE attacks, across a number of important clinical parameters including onset of symptom relief and almost complete symptom relief.
Acknowledgments
The FAST-1 trial was supported by Jerini AG/Shire and in part by NIH grant MO1 RR-00051. Medical writing support was provided by Kerren Davenport at Prime Medica Ltd, Knutsford, Cheshire, UK during the preparation of this paper, supported by Shire, Eysins, Switzerland. Data management and statistical analysis was performed by Hesperion, Allschwil, Switzerland. Statistical support in the preparation of the manuscript was provided by Jovanna Baptista, Shire Lexington, MA, USA.
Disclosure
A. M. has been an investigator in a company-sponsored scientific study for Jerini AG/Shire and CSL Behring. He has received a travel grant for presenting at a scientific congress from Jerini AG/Shire and CSL Behring and has lectured/spoken at a company-sponsored meeting for Jerini AG/Shire and CSL Behring. M. R. has performed research for CSL Behring, Dyax, Shire, Pharming and ViroPharma, participated on scientific advisory boards for BioCryst, CSL Behring, Dyax, Isis, Shire and ViroPharma, and been a speaker for CSL Behring, Dyax, Shire, and ViroPharma. B. R. has acted in a consultant/advisor capacity for Aventis, Bayer, Baxter, Bristol Myers Squibb, CSL Behring, Dyax, Novo Nordisk, OctaPharma, Pharming, Pfizer, Shire and Wyeth. He has received a research grant for participating in a company-sponsored scientific study from Baxter, CSL Behring, Novartis, Novo Nordisk, Pfizer and Wyeth. He has been an investigator in a company-sponsored scientific study for AstraZeneca, Aventis, Bayer, Baxter, Bristol Myers Squib, CSL Behring, Dyax, Novartis, Novo Nordisk, Pharming, Pfizer and Shire. He has received a travel grant for presenting at a scientific congress from Baxter, Bayer, Novo Nordisk, Pfizer, Dyax and Wyeth, and has lectured/spoken at a company-sponsored meeting for CSL Behring and Pfizer. W. B. S. has received a travel grant from Jerini AG/Shire to attend a conference, participated on scientific advisory boards for Shire, AstraZeneca and Novartis, and has acted in a consultant/advisory capacity for CSL Behring and ViroPharma. He has been an investigator in company-sponsored scientific studies for Jerini AG/Shire, CSL Behring, ALK-Abello and AstraZeneca. W. Y. has acted in a consultant/adviser capacity for ALK-Abello, Takeda (formerly Altana/Nycomed), Merck (Schering), GlaxoSmithKline, Novartis, CSL Behring, KOS Pharmaceuticals and Paladin Lab. He has also given lectures on behalf of Merck (Schering), GlaxoSmithKline, Novartis and Jerini AG/Shire, and has received unrestricted educational grants from Novartis, CSL Behring, Merck and Shire. In addition, he has received research grants from ALK-Abello, Pharming, Takeda (formerly Altana/Nycomed), AstraZeneca/Medimmune, Paladin Labs, Dyax Corp, GlaxoSmithKline, CSL Behring, Merck/Schering, Ajinomoto Inc, Novartis, Nutrasweet/Monsanto, Jerini AG/Shire, Pfizer, Sepracor, Ono Pharma, Mankind (Allucure), Apotex, Boehringer Ingelheim, Almirall, Allergy Therapeutics, Stallergenes, Forest Laboratories, Genzyme, DBV Technologies, Genentec, Amgen, Greer and Circassia. A. B. has participated on scientific advisory boards for Shire, CSL Behring and Dyax, and has been involved in research for Shire, CSL Behring, Viropharma and Dyax. J. H. has acted as consultant for CSL Behring, Shire, Novartis, Merck Canada and Stallergene/Paladin. He has also performed research for Merck, Novartis, GlaxoSmithKline, Circassia, Boehringer Ingelheim, Forest, CSL Behring, Dyax and Shire, and has lectured at company-sponsored meetings for Novartis and CSL Behring. G. G. is a consultant to GlaxoSmithKline and a board member of the American Partnership for Eosinophilic Diseases. He holds equity in Immune Design Corporation, is a founder of ImmViz, receives royalties from the Mayo Foundation and can receive royalties from Teva/Cephalon. His wife, Kristin M. Leiferman, receives honoraria from Beiersdorf and MBL International. D. H. is a primary investigator for Shire/Jerini, Dyax, ViroPharma and CSL Behring, and also sits on an advisory board for CSL Behring, Dyax, and ViroPharma. K. W. J. is an investigator for Shire, Dyax, Pharming, CSL Behring and ViroPharma. J. A. B. is an investigator for Shire, Dyax, Pharming, CSL Behring and ViroPharma. He is also a consultant for Shire, Dyax, CSL Behring, Santaurus and ViroPharma. He is a speaker for Dyax, CSL Behring and ViroPharma. D. A. K. has performed research for Shire and is a speaker for ViroPharma. C. H. K. has received research support from Jerini and Pharming for clinical trials and is a consultant for Dyax. D. R. has declared no conflicts of interest. H. L. was previously an employee of Shire, Lexington, MA, USA. W. L. has consultant arrangements with CSL Behring, Dyax, Merck, Novartis, Shire and ViroPharma Pharmaceuticals. He has received grants/research support from Biotest, Boehringer Ingelheim, CSL Behring, Dyax, Genentech, Green Cross, Meda, Merck, NIH, Novartis, Pharming, Shire, Teva Pharmaceuticals and ViroPharma. He is on the speaker's bureau for CSL Behring, Dyax, Meda, Merck, Novartis, Shire and ViroPharma.
Author contributions
Responsibility for opinions, conclusions and interpretation of data lies with the authors. A. M., M. R., B. R., W. B. S., W. Y., A. B., J. H., G. J. G., D. H., K. W. J., J. A. B., D. A. K., C. H. K., D. R., D. S. F. R. and W. L. were involved in the conception and design of the study as well as in its conduct and the generation of data. H. L. was the study statistician and was involved in the analysis and interpretation of the study data. All authors critically reviewed the manuscript and approved the final version for submission.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Investigator's global assessment of laryngeal patients treated in the controlled and open-label extension (OLE) phase of For Angioedema Subcutaneous Treatment (FAST-1).
Fig. S2. Patient-assessed symptom score in laryngeal patients during the open-label extension (OLE) phase of For Angioedema Subcutaneous Treatment (FAST-1).
Table S1. Investigator's global assessment of cutaneous patients treated in the open-label extension (OLE) phase of For Angioedema Subcutaneous Treatment (FAST-1).
Table S2. Investigator's global assessment of abdominal patients treated in the open-label extension (OLE) phase of For Angioedema Subcutaneous Treatment (FAST-1).
References
- 1.Zuraw BL. Hereditary angioedema. N Engl J Med. 2008;359:1027–1036. doi: 10.1056/NEJMcp0803977. [DOI] [PubMed] [Google Scholar]
- 2.Weis M. Clinical review of hereditary angioedema: diagnosis and management. Postgrad Med. 2009;121:113–120. doi: 10.3810/pgm.2009.11.2071. [DOI] [PubMed] [Google Scholar]
- 3.Bork K, Harct J, Schicketanz K-H, Ressel N. Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency. Arch Intern Med. 2003;163:1229–1235. doi: 10.1001/archinte.163.10.1229. [DOI] [PubMed] [Google Scholar]
- 4.Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012;130:692–697. doi: 10.1016/j.jaci.2012.05.055. [DOI] [PubMed] [Google Scholar]
- 5.Winnewisser J, Rossi M, Spath P, Bürgi H. Type I hereditary angio-oedema. Variability of clinical presentation and course within two large kindreds. J Intern Med. 1997;241:39–46. doi: 10.1046/j.1365-2796.1997.76893000.x. [DOI] [PubMed] [Google Scholar]
- 6.Agostoni A, Cicardi M. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Balt) 1992;71:206–215. doi: 10.1097/00005792-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Gompels A, Lock RJ, Abinun M, et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol. 2005;139:379–394. doi: 10.1111/j.1365-2249.2005.02726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lumry WR, Castaldo AJ, Vernon MK, Blaustein MB, Willson DA, Horn PT. The humanistic burden of hereditary angioedema: impact on health-related quality of life, productivity, and depression. Allergy Asthma Proc. 2010;31:407–414. doi: 10.2500/aap.2010.31.3394. [DOI] [PubMed] [Google Scholar]
- 9.Bygum A, Andersen KE, Mikkelsen CS. Self-administration of intravenous C1-inhibitor therapy for hereditary angioedema and associated quality of life benefits. Eur J Dermatol. 2009;19:147–151. doi: 10.1684/ejd.2008.0603. [DOI] [PubMed] [Google Scholar]
- 10.Tosi M. Molecular genetics of C1 inhibitor. Immunobiology. 1998;199:358–365. doi: 10.1016/S0171-2985(98)80040-5. [DOI] [PubMed] [Google Scholar]
- 11.Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio-oedema. Lancet. 1998;351:1693–1697. doi: 10.1016/S0140-6736(97)09137-X. [DOI] [PubMed] [Google Scholar]
- 12.Bork K, Frank J, Grundt B, Schlattmann P, Nussberger J, Kreuz W. Treatment of acute edema attacks in hereditary angioedema with a bradykinin receptor-2 antagonist (Icatibant) J Allergy Clin Immunol. 2007;119:1497–1503. doi: 10.1016/j.jaci.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 13.Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363:532–541. doi: 10.1056/NEJMoa0906393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lumry WR, Li HH, Levy RJ, et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol. 2011;107:529–537. doi: 10.1016/j.anai.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 15.Bork K, Barnstedt SE. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med. 2001;161:714–718. doi: 10.1001/archinte.161.5.714. [DOI] [PubMed] [Google Scholar]
- 16.Bork K, Meng G, Staubach P, Hardt J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion. 2005;45:1774–1784. doi: 10.1111/j.1537-2995.2005.00602.x. [DOI] [PubMed] [Google Scholar]
- 17.Bork K, Staubach P, Hardt J. Treatment of skin swellings with C1-inhibitor concentrate in patients with hereditary angio-oedema. Allergy. 2008;63:751–757. doi: 10.1111/j.1398-9995.2007.01577.x. [DOI] [PubMed] [Google Scholar]
- 18.Aberer W, Maurer M, Reshef A, et al. Open-label, multicenter study of self-administered icatibant for attacks of hereditary angioedema. Allergy. 2013;69:305–314. doi: 10.1111/all.12303. [DOI] [PubMed] [Google Scholar]
- 19.Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119:267–274. doi: 10.1016/j.amjmed.2005.09.064. [DOI] [PubMed] [Google Scholar]
- 20.Cicardi M, Levy RJ, McNeil DL, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523–531. doi: 10.1056/NEJMoa0905079. [DOI] [PubMed] [Google Scholar]
- 21.Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol. 2009;124:801–808. doi: 10.1016/j.jaci.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 22.Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104:523–529. doi: 10.1016/j.anai.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 23.Wasserman RL, Levy RJ, Bewtra AK, et al. Prospective study of C1 esterase inhibitor in the treatment of successive acute abdominal and facial hereditary angioedema attacks. Ann Allergy Asthma Immunol. 2011;106:62–68. doi: 10.1016/j.anai.2010.10.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Investigator's global assessment of laryngeal patients treated in the controlled and open-label extension (OLE) phase of For Angioedema Subcutaneous Treatment (FAST-1).
Fig. S2. Patient-assessed symptom score in laryngeal patients during the open-label extension (OLE) phase of For Angioedema Subcutaneous Treatment (FAST-1).
Table S1. Investigator's global assessment of cutaneous patients treated in the open-label extension (OLE) phase of For Angioedema Subcutaneous Treatment (FAST-1).
Table S2. Investigator's global assessment of abdominal patients treated in the open-label extension (OLE) phase of For Angioedema Subcutaneous Treatment (FAST-1).