

Abstract
Epithelioid sarcomas (ES) are mesenchymal neoplasms subclassified into distal and proximal subtypes based on their distinct clinical presentations and histologic features. Consistent loss of SMARCB1 nuclear expression has been considered as the hallmark abnormality for both subtypes, a feature shared with atypical teratoid/rhabdoid tumor of infancy (ATRT). While virtually all ATRTs harbor underlying SMARCB1 somatic or germline alterations, mechanisms of SMARCB1 inactivation in ES are less well defined. To further define mechanisms of SMARCB1 inactivation a detailed molecular analysis was performed on 40 ES (25 proximal and 15 distal ES, with classic morphology and negative SMARCB1 expression) for their genomic status of SMARCB1 and related genes encoding the SWI/SNF subunits (PBRM1, BRG1, BRM, SMARCC1/2 and ARID1A) by FISH using custom BAC probes. An additional control group was included spanning a variety of 41 soft tissue neoplasms with either rhabdoid/epithelioid features or selected histotypes previously shown to lack SMARCB1 by IHC. Furthermore, 12 ES were studied by array CGH (aCGH) and an independent TMA containing 50 additional ES cases was screened for Aurora Kinase A (AURKA) and cyclin D1 immunoexpression. Homozygous SMARCB1 deletions were found by FISH in 36/40 ES (21/25 proximal-type). One of the distal-type ES displayed homozygous SMARCB1 deletion in the tumor cells, along with a heterozygous deletion within normal tissue, finding confirmed by array CGH. None of the proximal ES lacking homozygous SMARCB1 deletions displayed alterations in other SWI/SNF subunits gene members. Among controls, only the SMARCB1-immunonegative myoepithelial carcinomas displayed SMARCB1 homozygous deletions in 3/5 cases, while no gene specific abnormalities were seen among all other histologic subtypes of sarcomas tested regardless of the SMARCB1 protein status. There was no consistent pattern of AURKA and Cyclin D1 expression. The array CGH was successful in 9/12 ES, confirming the SMARCB1 and other SWI/SNF genes copy numbers detected by FISH. Our study confirms the shared pathogenesis of proximal and distal ES, showing consistent SMARCB1 homozygous deletions. Additionally we report the first ES case associated with a SMARCB1 constitutional deletion, establishing a previously undocumented link with ATRT. Alternative mechanisms of SMARCB1 inactivation in SMARCB1-disomic ES remain to be identified, but appear unrelated to large genomic abnormalities in other SWI/SNF subunits.
Keywords: SMARCB1/SMARCB1, epithelioid sarcoma, rhabdoid tumor, SWI/SNF complex, rhabdoid
INTRODUCTION
Epithelioid sarcomas (ES) are rare but aggressive soft tissue neoplasms occurring in young individuals, being characterized by expression of epithelial markers along with loss of SMARCB1 (INI1, BAF47) nuclear expression (Fletcher et al., 2013). ES are sub-classified into distal (or conventional) type (Enzinger, 1970), typically occurring in the skin or superficial soft tissues of the distal extremities; and proximal type (Guillou et al., 1997), often more aggressive, located in the deep soft tissue of the perineum, and harboring distinctive rhabdoid cytology.
SMARCB1, which encodes the SMARCB1 protein, has been first identified as a tumor suppressor gene at the 22q11 locus, being inactivated in 95% of extracranial malignant rhabdoid tumors of the kidney (MRT) and atypical teratoid rhabdoid tumors (ATRT) of the central nervous system of infancy (Versteege et al., 1998; Jackson et al., 2009). Histologically, these lesions display similar rhabdoid features, express epithelial markers and show loss of SMARCB1 expression, features reminiscent of proximal-type ES. Accordingly, SMARCB1 deletions were later identified in proximal-type ES (Modena et al., 2005) and loss of SMARCB1 nuclear expression was subsequently detected by immunohistochemistry (IHC) in up to 93% of ES, both proximal and distal types (Chbani et al., 2009; Hornick et al., 2009). However, the prevalence of SMARCB1 gene alterations reported initially in distal ES by using standard molecular techniques, such as FISH or PCR, was significantly lower than in proximal type ES (Modena et al., 2005) or ATRT (Flucke et al., 2009; Kohashi et al., 2009; Gasparini et al., 2011).
Only more recently, a high frequency of SMARCB1 deletions was detected in a series of 12 ES of both subtypes by multiplex ligation-dependent probe amplification (Sullivan et al., 2013). Additionally, one family with rhabdoid tumor predisposition syndrome lacking SMACB1 gene abnormalities was linked to BRG1 inactivation, which encodes another subunit of the SWI/SNF complex tightly bound to SMARCB1 (Schneppenheim et al., 2010). This latter finding suggests that rhabdoid tumors may be more broadly related to SWI/SNF complex alterations. Therefore, we sought to reappraise the frequency of SMARCB1 deletions in a large series of ES and investigate alternative hits in the SWI/SNF complex-encoding genes. For this purpose, we screened a retrospective series of histologically and immunophenotypically typical 40 ES cases by fluorescence in situ hybridization (FISH) and array-comparative genomic hybridization (a-CGH). Additionally, as SMARCB1-loss of immunoexpression is not specific for ES and has been reported in other look-alike epithelioid malignancies (Hollmann and Hornick, 2011), we studied a control group of different histotypes, often entertained in the differential diagnosis of ES or previously reported to lose SMARCB1 expression, in order the investigate if their mechanism of SMARCB1 inactivation is also related to SMARCB1 homozygous deletions.
MATERIAL AND METHODS
Patient Selection and Tumor Characteristics
We retrieved 40 archival specimens of epithelioid sarcomas (ES), either distal or proximal types, from the Pathology Departments of Memorial Sloan Kettering Cancer Center (New York, USA) and Institut Bergonié (Bordeaux, France). Samples from Institut Bergonié were provided by the Biological Resources Center of Institut Bergonié (CRB-IB). In accordance with the French Public Health Code (articles L. 1243–4 and R. 1243–61), the CRB-IB received the agreement from the French authorities to deliver samples for scientific research (number AC-2008–812, on February 2011). Samples from MSKCC were collected and studied under 02–060 IRB protocol.
All cases were re-reviewed and loss of SMARCB1 expression by immunohistochemistry was required for inclusion in the study. Additionally, we included a control group composed of 41 tumors of either potential mimickers of ES or tumors previously reported to lose SMARCB1 expression. The following lesions were included: 12 soft tissue and visceral myoepithelial carcinomas (defined based on the increased nuclear pleomorphism and mitotic activity and selected based on either loss of SMARCB1-expression or distinctive rhabdoid phenotype); 1 chordoma periphericum (confirmed by expression of T-brachyury), 10 extraskeletal myxoid chondrosarcomas (selected based on either loss of SMARCB1-expression or distinctive rhabdoid phenotype), 4 ossifying fibromyxoid tumors (OFMT), 3 high grade sarcomas with rhabdoid features, 3 epithelioid malignant peripheral nerve sheath tumors (epMPNST), 7 pseudomyogenic (epithelioid sarcoma-like) hemangioendotheliomas (PHE), and one high grade epithelioid angiosarcoma with distinctive rhabdoid morphology.
Immunohistochemistry (IHC)
Immunohistochemistry for SMARCB1 were reviewed. Additional stains were performed with antibodies anti-BAF47 (mouse monoclonal, 1:30, clone 25, BD Bioscience), Aurora Kinase A (mouse monoclonal, 1:50, JLM28, Novocastra) and Cyclin D1 (rabbit monoclonal, 1:100, SP4, Lab Vision). Immunohistochemistry studies were performed on 4 μm-thick paraffin-embedded tissue sections with Benchmark-ultra Automated Ventana.
Fluorescence in situ hybridization (FISH)
FISH was performed on interphase nuclei using paraffin embedded 4 μm-sections. Slides pretreatment and FISH procedures were performed as previously described (Antonescu et al., 2010). Custom-made probes using bacterial artificial chromosomes (BAC) were designed covering and flanking genes of interest as well as control BAC probes targeting centromeric or telomeric part of the corresponding chromosome (Supplementary Table 1). BAC clones were obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA) (http://bacpac.chori.org). DNA extraction and labeling were performed according to manufacturer’s instructions. Slides were assessed using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis software (Metasystems, Watertown, MA, USA) and analyzed only when >80% of nuclei were hybridized. Copy number abnormalities were assessed on 100 nuclei by two independent observers. Normal copy number pattern was defined when two copies of the SMARCB1 gene or other members of the SWI/SNF complex were identified, with a 1:1 ratio to the control probe (i.e. telomeric-EWSR1 or 22q11). Heterozygous deletion was defined as only one copy of the gene of interest being present compared to the reference control probe on 22q (ratio 2:1). Homozygous deletion of SMARCB1 was interpreted when both copies of the gene were lost, compared to the control probes, either telomeric-EWSR1 or 22q11. A monosomy pattern (or large deletion) was defined if one allele copy of both the gene of interest and control were lost, with a ratio of 1:1. In cases where both SMARCB1 and EWSR1 signals were lost concurrently, two additional control probes were used as reference on 22q11 (RP11-960P21+RP11-81B3, see Supplementary Table 1).
Tissue micro-array (TMA)
The TMA included 50 ES samples (42 distal and 8 proximal types). The TMA also included 5 other neoplasms as controls. Each case was represented by three spots of 4 μm thick and 1 mm in diameter. IHC was performed with AURKA and cyclinD1 antibodies. CyclinD1 positivity was interpreted only if nuclear staining was detected, whereas both nuclear and cytoplasmic patterns were assessed for AURKA. We assessed the percentage of positive nuclear staining, as well as degree of staining intensity, as described previously (Ali et al., 2012).
Array comparative genomic hybridization (a-CGH)
aCGH was performed following manufacturer protocol modified by Hostetter et al. (Hostetter et al., 2010). One microgram of genomic DNA extracted from archival material and 400 ng of reference DNA were labeled with Cyanine 5 and 3, as described previously (Perot et al., 2012). Labeled DNA was hybridized to Agilent arrays (Agilent Technologies) with a 60k resolution across the genome. Slides were scanned on Agilent microarray scanner and analyzed using Feature extraction software, version 10.5.1.1 (Agilent Technologies) and Agilent genomic workbench lite 6.5.0.18. The ADM-2 algorithm was used to identify DNA copy number anomalies at the probe level. Homozygous deletion was considered when log2 ratios of targeting probes were below 1. Intermediate log2 ratios values between −1 and −0.25 do not allow to be conclusive as to whether the deletion is homo or heterozygous. A low-level copy number gain was defined as a log 2 ratio > 0.25. To further characterize the 22q11 somatic deletion identified in case ES#6, we used custom-designed aCGH 180k Agilent array with high density coverage of 22q11 locus (in which 200 oligonucleotide probes target SMARCB1).
RESULTS
Histological and Immunohistochemical Findings
Forty ES (25 proximal and 15 distal), showing a typical morphology and immunophenotype, including loss of SMARCB1 expression, were included in the study (Table 1). Patients mean age at diagnosis was 35 years old (range: 21–60 years) in the distal-type group and 36 years old (range: 14–71 years) in the proximal-type group. Among the control cases 5/12 myoepithelial carcinomas and 1/10 extraskeletal myxoid chondrosarcoma (EMC) showed diffuse loss of SMARCB1 expression. Six additional EMCs showed a mosaic pattern of loss of SMARCB1 expression. The remaining tumors with epithelioid or rhabdoid morphology showed retained SMARCB1 expression.
Table 1.
Clinicopathological data of the 40 distal and proximal epithelioid sarcoma patients.
| ES# | Age /Sex | Subtype | Site | SMARCB1 CN by FISH |
|---|---|---|---|---|
| 1 | 55/F | distal | forearm | 0 |
| 2 | 21/M | distal | hand | 0 |
| 3 | 49/M | distal | forearm | 0 |
| 4 | 27/F | distal | hand | 0 |
| 5 | 22/M | distal | hand | 0 |
| 6 | 25/F | distal | hand | 0 |
| 7 | 60/M | distal | thumb | 0 |
| 8 | 26/F | distal | hand | 0 |
| 9 | 44/M | distal | thumb | 0 |
| 10 | 36/F | distal | elbow | 0 |
| 11 | 24/M | distal | hand | 0 |
| 12 | 52/F | distal | wrist | 0 |
| 13 | 35/M | distal | finger | 0 |
| 14 | 38/F | distal | lower leg | 0 |
| 15 | 32/F | distal | lower leg | 0 |
| 16 | 23/M | proximal | groin | 0 |
| 17 | 37/M | proximal | perineum | 0 |
| 18 | 33/M | proximal | flank | 0 |
| 19 | 64/M | proximal | buttock | 0 |
| 20 | 49/F | proximal | suprapubic | 0 |
| 21 | 50/M | proximal | perineal | 0 |
| 22 | 52/M | proximal | axilla | 0 |
| 23 | 27/M | proximal | thigh | 0 |
| 24 | 30/M | proximal | inguinal | 0 |
| 25 | 17/M | proximal | arm | 0 |
| 26 | 38/M | proximal | perineum | 0 |
| 27 | 49/F | proximal | vulva | 0 |
| 28 | 31/M | proximal | perineum | 0 |
| 29 | 26/M | proximal | Perineum | 0 |
| 30 | 36/M | proximal | Perineum | 0 |
| 31 | 71/M | proximal | pubic area | 0 |
| 32 | 47/M | proximal | buttock | 0 |
| 33 | 17/M | proximal | cervical spine | 0 |
| 34 | 17/M | proximal | Pelvis | 0 |
| 35 | 9/M | proximal | arm/shoulder | 0 |
| 36 | 33/M | proximal | Buttock | 0 |
| 37 | 33/M | proximal | Groin | 1 |
| 38 | 40/M | proximal | Perineum | 2 |
| 39 | 38/M | proximal | cervical area | 2 |
| 40 | 14/M | proximal | axilla | 2 |
M, male; F, female; CN, copy number
SMARCB1 (SMARCB1) genomic status
Overall, SMARCB1 homozygous deletion was present in 36 of the 40 samples, including all 15 distal ES and 21/25 proximal ES (Table 1, Fig. 1A,B). The EWSR1 control probe was heterozygous co-deleted with SMARCB1 in 9 cases, accounting for larger deletions at the 22q locus. This finding was present in both distal (3) and proximal type (6) cases. Only one proximal ES showed a heterozygous deletion of SMARCB1. In this case the 22q11 deletion encompassed both SMARCB1 and EWSR1 loci, as illustrated by the additional reference 22q11 probes (Fig 1.C,D). Notably, one distal type ES displayed homozygous deletion in tumor cells along with heterozygous deletion in normal cells, a finding suggestive of constitutional deletion (Fig. 2).
Figure 1. Proximal-type ES with loss of SMARCB1 expression.

(A,B) Common homozygous deletions of SMARCB1 (no red signals) while preserving two copies of the control gene EWSR1 (green signals), except for the 2 normal cells in the center with 1:1 Ratio (ES21). (C,D) Three-color FISH showing loss of one SMARCB1 allele (red signal) as well as one EWSR1 copy (green signal) and retained the additional reference probe on 22q11 (yellow signal) in keeping with a heterozygous loss of SMARCB1 in ES37. (E,F) Rare cases of proximal ES showed no SMARCB1 gene abnormalities (ES40). (G) aCGH pan-genomic profile of tumor ES21 highlighting gains of chromosomes 7 and 8, on 16p and 16q and deletion on 22q. (H) Scatter Plot of the log2 ratio signals captured with the probes targeting chromosome 22. Probes targeting 22q11 locus encompassing SMARCB1 are highlighted in grey.
Figure 2. SMARCB1 homozygous deletion in a distal-type epithelioid sarcoma arising in a patient with constitutional SMARCB1 deletion (ES6).


(A,B). Loss of both SMARCB1 alleles (red signals), while retaining 2 copies of the control gene EWSR1 (green) in the tumor cells. (C) The adjacent normal tissue (keratinocytes, skin) shows loss of one SMARCB1 copy and retention of the two green control copies. (D) Scatter Plot of the log2 ratio signals captured with the probes targeting chromosome 22 (aCGH, Agilent) of tumor ES6. aCGH confirms the presence of deletions on 22q, one of which encompasses SMARCB1 (marked with a line) on 22q11. (E) Scatter Plot of the log2 ratio signals captured with the probes targeting 22q11 locus. Copy number altered regions are highlighted in brown; SMARCB1 location is indicated with *. (F) aCGH whole genome profile in normal fat of ES6 is unremarkable, except for a 3Mb somatic deletion in long arm of chromosome 22. (G) Scatter Plot of the log2 ratio signals captured with the probes targeting chromosome 22 in normal fat of ES6. Deletion is indicated in blue and SMARCB1 position with *. Data were achieved with a custom-designed180k Agilent array with high density coverage of 22q11 locus. (H) Scatter Plot of the log2 ratio signals captured with the probes targeting 22q11 locus (aCGH, Agilent) in normal muscle of ES6 patient. SMARCB1 position, indicated on the bottom part, is encompassed within the deleted area.
Three proximal ES cases showed a normal pattern, with two copies of the SMARCB1. The ES cases with either two copies or heterozygous deletion of SMARCB1 were also tested for abnormalities in other SWI/SNF gene subunits, however, no additional changes were noted. The three ES cases that retained two copies of SMARCB1 were all proximal ES located in the perineum of a 40 year-old male, axilla in a 14 year-old boy and cervical area in the 38 year-old man. The only tumor with heterozygous deletion of SMARCB1 occurred in the groin of a 33 year-old man. No difference in morphology or diagnostic immunophenotype was identified among the different SMARCB1 gene status cases.
From the control group all except one tumor with SMARCB1-retained protein expression by IHC showed 2 copies of SMARCB1. Three of the 5 SMARCB1-negative myoepithelial carcinomas displayed SMARCB1 homozygous deletions, all of which lacked EWSR1 gene rearrangements (Table 2). These 3 cases showed typical morphology with immunohistochemical support for myoepithelial differentiation (co-expression for S100 protein and epithelial markers), occurring in the foot, wrist and kidney (Fig. 3). All three tumors showed a distinctive rhabdoid histologic appearance, either as a sole pattern or admixed with a more nested, spindle or myxoid appearances. In contrast, EMC with either diffuse or mosaic pattern of SMARCB1 loss of immunoexpression showed no SMARCB1 gene abnormalities. Only one of the four ossifying fibromyxoid tumors showed one normal SMARCB1 copy and a large 22q11 deletion encompassing SMARCB1, EWSR1 and the additional reference probe, but showed retained SMARCB1 expression by immunohistochemistry.
Table 2.
Pathologic characteristic and FISH results in soft tissue neoplasms with rhabdoid/epithelioid features.
| CG# | Diagnosis | Age/Sex | Site | SMARCB1 IHC | SMARCB1 CN by FISH | Translocation status (FISH) |
|---|---|---|---|---|---|---|
| 1 | MEC | 25/M | foot | lost | 0 | EWSR1 neg |
| 2 | MEC | 54/M | kidney | lost | 0 | EWSR1 neg |
| 3 | MEC | 16/M | wrist | lost | 0 | EWSR1 neg |
| 4 | MEC | 16/F | groin node met | lost | 2 | EWSR1 pos |
| 5 | MEC | 1/M | H&N | lost | 2 | EWSR1 neg |
| 6 | MEC | 62/F | abd wall | retained | 2 | EWSR1 neg |
| 7 | MEC | 34/F | back | retained | 2 | EWSR1 neg |
| 8 | MEC | 45/F | pancreas | retained | 2 | EWSR1 neg |
| 9 | MEC | 48/F | breast | retained | 2 | EWSR1 neg |
| 10 | MEC | 47/F | submandibular gland | retained | 2 | EWSR1 neg |
| 11 | MEC | 88/M | parotid | retained | 2 | EWSR1 neg |
| 12 | MEC | 61/M | tongue | retained | 2 | EWSR1 neg |
| 13 | Chordoma periphericum | 20/F | tibia | retained | 2 | EWSR1 neg |
| 14 | EMC | 69/M | hip | Lost | 2 | EWSR1 & NR4A3 pos |
| 15 | EMC | 30/F | buttock | Lost | 2 | EWSR1 & NR4A3 pos |
| 16 | EMC | 69/M | chest wall/pleura | Lost | 2 | EWSR1 & NR4A3 pos |
| 17 | EMC | 55/F | buttock | Lost | 2 | EWSR1 & NR4A3 pos |
| 18 | EMC | 50/M | leg | Lost | 2 | EWSR1 & NR4A3 pos |
| 19 | EMC | 62/M | thigh | Lost | 2 | EWSR1 & NR4A3 pos |
| 20 | EMC | 80/M | thigh | Lost | 2 | EWSR1 & NR4A3 pos |
| 21 | EMC | 41/M | thigh | retained | 2 |
EWSR1 neg NR4A3 pos |
| 22 | EMC | 63/M | thigh | retained | 2 | TAF15 & NR4A3 pos |
| 23 | EMC | 12/F | buttock | retained | 2 | EWSR1 neg |
| 24 | OFMT | 71/M | hand | retained | 1* | PHF1 pos |
| 25 | OFMT | 56/F | shoulder | retained | 2 | EP400 & PHF1 pos |
| 26 | OFMT | 73/M | popliteal fossa | retained | 2 | MEAF6 & PHF1 pos |
| 27 | OFMT | 24/F | buttock | retained | 2 | EP400 & PHF1 pos |
| 28 | STS rhabdoid | 62/M | calf | retained | 2 | EWSR1 neg |
| 29 | STS rhabdoid | 30/F | chest wall | retained | 2 | EWSR1 neg |
| 30 | STS rhabdoid | 19/M | T8 | retained | 2 | EWSR1 neg |
| 31 | Epithelioid MPNST | 68/F | leg | retained | 2 | N/A |
| 32 | Epithelioid MPNST | 44/M | flank | retained | 2 | N/A |
| 33 | Epithelioid MPNST | 36/M | popliteal fossa | retained | 2 | N/A |
| 34 | PHE | 21/M | foot | retained | 2 | WWTR1& CAMTA1 neg |
| 35 | PHE | 24/M | abd wall | retained | 2 | WWTR1& CAMTA1 neg |
| 36 | PHE | 48/M | back | retained | 2 | WWTR1& CAMTA1 neg |
| 37 | PHE | 32/M | abd wall | retained | 2 | WWTR1& CAMTA1 neg |
| 38 | PHE | 19/M | foot | retained | 2 | WWTR1& CAMTA1 neg |
| 39 | PHE | 18/M | thigh | retained | 2 | WWTR1& CAMTA1 neg |
| 40 | PHE | 28/M | shoulder | retained | 2 | WWTR1& CAMTA1 neg |
| 41 | AS, rhabdoid morphology | 21/F | kidney | retained | 2 | WWTR1& CAMTA1 neg |
CG, control group#, M, male; F, female; MEC, myoepithelial carcinoma; OFMT, ossifying fibromyxoid tumor; EMC, extraskeletal myxoid chondrosarcoma; epMPNST, epithelioid malignant peripheral nerve sheath tumor; STS rhabdoid, soft tissue sarcoma with rhabdoid features; PHE, pseudomyogenic (epithelioid sarcoma-like) hemangioendothelioma; AS, angiosarcoma; H&N, head and neck; neg, negative; pos, positive; N/A, not applicable; 1*, FISH detected one copy of SMARCB1, EWSR1 and 22q11 reference probe (ratio 1:1) in keeping with a large deletion/ monosomy of 22q; custom BAC probes for WWTR1 CAMTA1, PHF1, EP400, and MEAF6, were previously published (Errani et al., 2011; Antonescu et al., 2013). Custom BAC probes spanning TAF15 and NR4A3 can be provided upon request.
Figure 3. SMARCB1 homozygous deletions in a subset of EWSR1 non-rearranged myoepithelial carcinomas (CG#1).
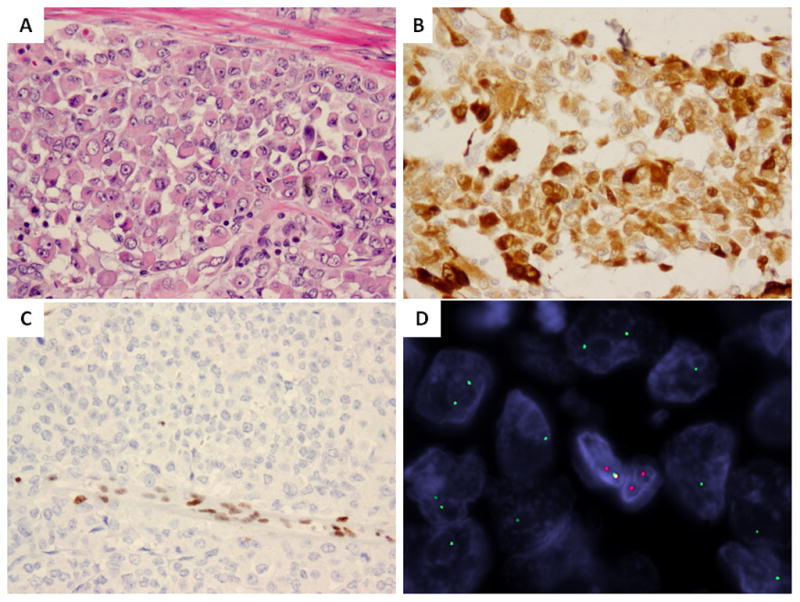
(A) Morphologic appearance of a foot myoepithelial carcinoma composed of a heterogenous patterns, including a predominant rhabdoid, nested, reticular, and focally spindle cell morphology. (B) The immunohistochemical profile was supportive of myoepithelial lineage with S100 protein staining. (C) The tumor showed complete loss of SMARCB1-expression (retained within the entrapped vessels). (D) FISH analysis showed loss of both SMARCB1 copies (red signals), while retaining the green EWSR1 control alleles.
Array-CGH results (Table 3)
Twelve samples were processed by aCGH. Deviation Log2 ratios (DLR) ranged from 0.19–0.82. Three cases were excluded due to a low tumor cellularity (50%) displaying flat profiles probably related to normal tissue contaminant. ES tumor displayed heterogeneous profiles, ranging from few copy number alterations (CNA) < 10 in 5 cases, to highly rearranged profiles in 6 cases. Genomic complexity did not correlate with ES subtype. The main recurrent event was a deletion of 22q11 encompassing SMARCB1 locus in 9 cases. In the patient with FISH evidence of SMARCB1 mono-allelic deletion in the normal tissue (ES6), aCGH was performed using genomic DNA extracted independently from normal and tumor tissue. Both profiles displayed a similar deletion of 3Mb at 22q11 locus covering SMARCB1 gene, thereby confirming the germline nature of the alteration (Fig. 2). The range of 22q11 deletions varied in size from 100 Kb to 18 Mb. aCGH data correlated with FISH results in all cases.
FISH screening for abnormalities in genes encoding SWI/SNF subunits
We hypothesized that alterations of other core members of the SWI/SNF complex might disrupt the complex with subsequent SMARCB1 protein loss or may be additionally involved in cases with one retained SMARCB1 allele or with SMARCB1-retained ES. Thus we screened for copy number of SWI/SNF-encoding genes, such as BRM, BRG1, PBRM1, SMARCC 1 and 2, ARID1A, in ES cases retaining 1 to 2 copies of SMARCB1 as well as 9 SMARCB1-null ES in comparison. In addition we tested all tumors included in the control group. In brief there were no homozygous or heterozygous deletions identified in any of genes and in any of the tumors tested. However, one distal ES (ES21) showed one copy of BRG1 (1:1 ratio of BRG1 to the control probe, in keeping with the presence of larger deletion/monosomy) in addition to a concurrent homozygous SMARCB1 deletion. By aCGH analysis there was no evidence of BRG1 deletion in this sample, possibly due to normal tissue contamination. Among the control cases, one case of myoepithelial carcinoma of the salivary gland (CG#12, Table 2) showed one copy of PBRM1 and BRG1 (both with 1:1 ratio to the control probe), while showing two normal signals for SMARCB1.
Tissue Micro-Array (TMA) Analysis (Supplementary Table 1)
As ATRT display hyperactivation and subsequent overexpression of Aurora kinase A (AURKA) and cyclin D1, we investigated whether SMARCB1-inactivated ES share a similar pattern of protein expression. However, AURKA and Cyclin D1 were not consistently overexpressed in ES, with only 13/50 ES showing positivity for AURKA, including 1/8 proximal and 12/42 distal types. Similarly, 22/50 ES showed Cyclin D1 (>5%, medium intensity), with 7/8 proximal and 15/42 of the distal cases. No clinicopathological features correlated with expression of either AURKA or Cyclin D1.
DISCUSSION
Underlying mechanisms of SMARCB1 protein loss in epithelioid sarcomas (ES) have long remained elusive. While previous series reported SMARCB1 loss of expression in roughly 90% of both proximal and distal types of ES (Chbani et al., 2009; Hornick et al., 2009), corresponding genetic alterations in SMARCB1 gene appeared rather infrequent (Jackson et al., 2009; Papp et al., 2013). This is in contrast with ATRT in which SMARCB1 loss is associated with SMARCB1 gene alterations in virtually all cases. Evidence for SMARCB1 abnormalities in ES was first demonstrated by Modena et al (Modena et al., 2005) exclusively in the proximal type. Subsequent conflicting data emerged from larger series highlighting that SMARCB1 deletions occur only a minority of proximal ES (Kohashi et al., 2009; Papp et al., 2013). Similarly, SMARCB1 alterations were occasionally reported in distal ES (Gasparini et al., 2011). In contrast, Sullivan et al. studying a group of 12 ES by FISH and multiplex ligation-dependent probe amplification showed deletions in all cases, equally represented in either proximal or distal types. Most cases showed homozygous deletions, in 10/12 (83%) cases, while the remaining two cases showing heterozygous deletions (Sullivan et al., 2013). In concordance with these results, the findings of our larger series using FISH, validated by aCGH, demonstrate that SMARCB1-inactivation occurs through homozygous deletions of SMARCB1 in the overwhelming majority of cases (36/40, 90% of cases). The combined results from these two series reconcile the previous conflicting data and demonstrate through different methodologies that proximal and distal types of ES show a similar mechanism of SMARCB1 inactivation, mainly through SMARCB1 homozygous deletions, which can readily be identified by FISH in clinical practice, in difficult to diagnose cases.
An additional novel finding is the identification of the first case of ES occurring in the setting of SMARCB1 constitutional deletion. So far, ES had never been reported in the setting of a rhabdoid predisposition syndrome or familial schwannomatosis. This particular case occurred in a 25 year-old woman without prior familial or personal history of cancer who developed a distal-type ES of the hand, treated with surgery and radiotherapy. The patient remains alive without recurrence at 5 years follow-up. aCGH highlighted a deletion of 3Mb on 22q11 encompassing SMARCB1 (Figs. 2C, D) present in both tumor and normal tissues. Additionally SMARCB1 heterozygous deletion was present in all normal cells examined by FISH (i.e. fibroblasts, lymphocytes, keratinocytes and endothelial cells). Histologically, the tumor showed classic clinical presentation and morphologic features, with mixed spindle and epithelioid appearance, distinct from a rhabdoid tumor of the soft tissue (Bourdeaut et al., 2007). In contrast, rhabdoid tumors typically occur in infancy and display at least focal rhabdoid features (Jackson et al., 2007; Bourdeaut et al., 2011). Most carriers of SMARCB1 constitutional alterations are prone to develop synchronous or metachronous rhabdoid tumors as well as multiple schwannomas and meningiomas (Hulsebos et al., 2007; Bacci et al., 2010). This finding expands the spectrum of tumors occurring in the setting of SMARCB1 germline alterations and strongly reaffirms the key role of SMARCB1 inactivation in ES pathogenesis. Smarcb1-deficient tumorigenesis has been extensively studied in mice models (Roberts et al., 2000; Roberts et al., 2002), which develop tumors recapitulating the features of rhabdoid tumors.
In rhabdoid tumors SMARCB1 inactivation drives tumorigenesis through deregulation of cell cycle G1-S transition (Imbalzano and Jones, 2005), subsequently inducing overactivation of Aurora kinase A (AURKA) and CyclinD1 (Tsikitis et al., 2005; Lee et al., 2011; Smith et al., 2011). By analogy, ES sharing SMARCB1 abnormalities could also show overexpression of CyclinD1 and AURKA. However, CyclinD1 and AURKA were not found to be consistently overexpressed in 50 additional ES cases tested, suggesting that their pathogenesis might rely on a different proliferation regulatory axis. Genomically, rhabdoid tumors display remarkably simple alterations, limited to SMARCB1 deletions, commonly without additional hits or other recurrent alteration (Lee et al., 2012; Hasselblatt et al., 2013). Likewise, except for consistent 22q11 deletions, no other recurrent alterations were detected in the 9 ES studied by aCGH.
Our results corroborate two previous studies (Gasparini et al., 2011; Sullivan et al., 2013) suggesting that the leading mechanism of SMARCB1 inactivation in both proximal and distal ES is bi-allelic deletions in 22q11 encompassing the SMARCB1 tumor suppressor gene locus, which in 90% of cases is within the resolution of FISH and can be reliably applied clinically in difficult diagnoses. Although our study design did not include sequencing analysis due to insufficient material, other studies have previously demonstrated either lack or very low incidence of SMARCB1 mutations/ intragenic deletions as detected through sequencing or MLPA (Kohashi et al., 2009; Papp et al., 2013; Sullivan et al., 2013), thus suggesting an infrequent event in ES pathogenesis. In a recent study Papp et al (Papp et al., 2013) investigated the possibility of SMARCB1-inactivation through epigenetic changes, such as promoter or histone methylation. However, neither SMARCB1 promoter methylation nor EZH2 overexpression (involved in gene silencing by histone methylation at H3K27me3) were identified in their study, suggesting that hyper-methylation is not a leading mechanism in ES. Furthermore, SMARCB1 mRNA loss was identified in all ES cases by Q-PCR regardless of the gene copy number status excluding the possibility of SMARCB1-inactivation through post-translational modifications. However, only 13% of their cases showed bi-allelic loss of SMARCB1 by FISH, a figure significantly lower than most other large studies using similar FISH methodology. The potential caveat was the use of a BCR-specific FISH probe, which could explain the significant difference in the incidence of SMARCB1 deletions.
An additional goal of this study was to investigate alternative alterations involved in SMARCB1-retained ES and SMARCB1-lost ES without homozygous SMARCB1 deletions. SMARCB1 is a subunit of the SWI/SNF complex, an ATP-dependent chromatin remodeling complex which regulates gene expression through modulation of chromatin structure (Roberts and Orkin, 2004). Chromatin remodeling has been the focus of intense investigations recently and many SWI/SNF-encoding genes have been linked to cancer, including BRG1, PBRM1 and ARID1A (Medina and Sanchez-Cespedes, 2008; Jones et al., 2012; Pena-Llopis et al., 2012). Of note, BRG1 has been shown to be inactivated both at somatic and germline levels in patients affected by SMARCB1-retained rhabdoid tumors (Schneppenheim et al., 2010). We speculated that deletions of SWI/SNF-encoding genes may be involved in the SMARCB1-retained ES or may represent an alternative mechanism for SMARCB1 loss by disruption of SWI/SNF complex. However, there was no evidence of recurrent deletions either by FISH or aCGH in genes encoding other SWI/SNF subunits.
Although SMARCB1 protein loss correlates with SMARCB1 gene alterations in most ES, SMARCB1 immunonegativity is not specific for ES diagnosis and has been reported in other sarcoma subtypes, displaying either epithelioid or rhabdoid morphology (Hollmann and Hornick, 2011). As no prior studies had investigated the mechanisms of SMARCB1 inactivation outside the ES spectrum of lesions, we sought to analyze a large number of sarcomas with either similar morphology or previously reported to show loss of SMARCB1 expression by immunohistochemistry. Except for a subset of myoepithelial carcinomas, the remaining tumors did not reveal SMARCB1 gene abnormalities by FISH regardless of SMARCB1 protein expression. These results confirm that loss of SMARCB1 expression is a non-specific finding and can be seen in other sarcomas with epithelioid morphology. Furthermore, SMARCB1 inactivation does not correlate with SMARCB1 gene abnormalities beyond ES diagnosis. The only notable exception was the presence of homozygous SMARCB1 deletions in a subset of EWSR1-fusion negative myoepithelial carcinomas with loss of SMARCB1 expression. This finding suggests that SMARCB1 abnormalities might be an important mechanism of tumorigenesis in myoepithelial carcinomas, alternative to EWSR1-associated fusions, and possibly related to ES pathogenesis. Intriguingly, the three SMARCB1-negative myoepithelial carcinomas with SMARCB1 biallelic deletions showed at least focally distinctive rhabdoid morphology. Furthermore, no abnormalities were identified in other SWI/SNF subunit members in any of these histologic mimics.
In summary, our results reveal that the dominant mechanism of SMARCB1 loss of expression in both distal and proximal ES is through homozygous deletions at the 22q11 locus, encompassing SMARCB1 in the overwhelming majority of cases (36/40, 90%). This shared genetic abnormality argues in favor of a single pathologic entity, for which the terminology of epithelioid sarcoma is preferred, regardless of its peripheral or proximal clinical presentation. As loss of SMARCB1 expression by immunohistochemistry is not specific and occurs in other look-alike tumors with epithelioid/rhabdoid phenotype, FISH for SMARCB1 gene abnormalities can be used in the clinical setting as a useful ancillary technique to document a homozygous deletion genomic pattern in challenging cases. In the small minority of ES with normal SMARCB1 copy number, we did not identify alternative recurrent alterations involving other genes in the SWI/SNF complex. Furthermore, our study reports the first patient with an ES occurring in the setting of SMARCB1 constitutional deletion. This finding further expands the spectrum of tumors occurring in the setting of SMARCB1 germline alterations. Lastly, similar homozygous SMARCB1 deletions were identified in EWSR1-fusion negative myoepithelial carcinomas with SMARCB1 loss of expression, suggesting a potential pathogenetic link with ES. No SMARCB1 or SWI/SNF complex gene abnormalities were identified in any other tumors with epithelioid histology studied, regardless of their SMARCB1 protein status.
Supplementary Material
Acknowledgments
Supported in part by: P01CA47179 (CRA, SS), P50 CA 140146-01 (CRA, SS).
The authors thank Milagros Soto for her excellent editorial assistance and Valerie Velasco (Institut Bergonié, Bordeaux) for her invaluable assistance in the Tissue Microarray study. We also thank Dr. Stephane Pinson (Hopital Edouard Herriot, Genetics Department, Lyon, France) for his fruitful suggestions.
Footnotes
Conflicts of interest: none
References
- Ali HR, Dawson SJ, Blows FM, Provenzano E, Pharoah PD, Caldas C. Aurora kinase A outperforms Ki67 as a prognostic marker in ER-positive breast cancer. Br J Cancer. 2012;106:1798–1806. doi: 10.1038/bjc.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonescu CR, Sung YS, Chen CL, Zhang L, Chen HW, Singer S, Agaram NP, Sboner A, Fletcher CD. Novel ZC3H7B-BCOR, MEAF6-PHF1, and EPC1-PHF1 fusions in ossifying fibromyxoid tumors-molecular characterization shows genetic overlap with endometrial stromal sarcoma. Genes Chromosomes Cancer. 2013 doi: 10.1002/gcc.22132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonescu CR, Zhang L, Chang NE, Pawel BR, Travis W, Katabi N, Edelman M, Rosenberg AE, Nielsen GP, Dal Cin P, Fletcher CD. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114–1124. doi: 10.1002/gcc.20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci C, Sestini R, Provenzano A, Paganini I, Mancini I, Porfirio B, Vivarelli R, Genuardi M, Papi L. Schwannomatosis associated with multiple meningiomas due to a familial SMARCB1 mutation. Neurogenetics. 2010;11:73–80. doi: 10.1007/s10048-009-0204-2. [DOI] [PubMed] [Google Scholar]
- Bourdeaut F, Freneaux P, Thuille B, Lellouch-Tubiana A, Nicolas A, Couturier J, Pierron G, Sainte-Rose C, Bergeron C, Bouvier R, Rialland X, Laurence V, Michon J, Sastre-Garau X, Delattre O. hSNF5/INI1-deficient tumours and rhabdoid tumours are convergent but not fully overlapping entities. J Pathol. 2007;211:323–330. doi: 10.1002/path.2103. [DOI] [PubMed] [Google Scholar]
- Bourdeaut F, Lequin D, Brugieres L, Reynaud S, Dufour C, Doz F, Andre N, Stephan JL, Perel Y, Oberlin O, Orbach D, Bergeron C, Rialland X, Freneaux P, Ranchere D, Figarella-Branger D, Audry G, Puget S, Evans DG, Pinas JC, Capra V, Mosseri V, Coupier I, Gauthier-Villars M, Pierron G, Delattre O. Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res. 2011;17:31–38. doi: 10.1158/1078-0432.CCR-10-1795. [DOI] [PubMed] [Google Scholar]
- Chbani L, Guillou L, Terrier P, Decouvelaere AV, Gregoire F, Terrier-Lacombe MJ, Ranchere D, Robin YM, Collin F, Freneaux P, Coindre JM. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol. 2009;131:222–227. doi: 10.1309/AJCPU98ABIPVJAIV. [DOI] [PubMed] [Google Scholar]
- Enzinger FM. Epitheloid sarcoma. A sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029–1041. doi: 10.1002/1097-0142(197011)26:5<1029::aid-cncr2820260510>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Errani C, Zhang L, Sung YS, Hajdu M, Singer S, Maki RG, Healey JH, Antonescu CR. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer. 2011;50:644–653. doi: 10.1002/gcc.20886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher C, Bridge JA, Hogendoorn PC, Mertens F. WHO Classification of Tumours of Soft Tissue and Bone. IARC; Lyon: 2013. [Google Scholar]
- Flucke U, Slootweg PJ, Mentzel T, Pauwels P, Hulsebos TJ. Re: Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: a useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor: Direct evidence of mutational inactivation of SMARCB1/INI1 in epithelioid sarcoma. Hum Pathol. 2009;40:1361–1362. doi: 10.1016/j.humpath.2009.04.018. author reply 1362–1364. [DOI] [PubMed] [Google Scholar]
- Gasparini P, Facchinetti F, Boeri M, Lorenzetto E, Livio A, Gronchi A, Ferrari A, Massimino M, Spreafico F, Giangaspero F, Forni M, Maestro R, Alaggio R, Pilotti S, Collini P, Modena P, Sozzi G. Prognostic determinants in epithelioid sarcoma. Eur J Cancer. 2011;47:287–295. doi: 10.1016/j.ejca.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Guillou L, Wadden C, Coindre JM, Krausz T, Fletcher CD. “Proximal-type” epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol. 1997;21:130–146. doi: 10.1097/00000478-199702000-00002. [DOI] [PubMed] [Google Scholar]
- Hasselblatt M, Isken S, Linge A, Eikmeier K, Jeibmann A, Oyen F, Nagel I, Richter J, Bartelheim K, Kordes U, Schneppenheim R, Fruhwald M, Siebert R, Paulus W. High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer. 2013;52:185–190. doi: 10.1002/gcc.22018. [DOI] [PubMed] [Google Scholar]
- Hollmann TJ, Hornick JL. INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol. 2011;35:e47–63. doi: 10.1097/PAS.0b013e31822b325b. [DOI] [PubMed] [Google Scholar]
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542–550. doi: 10.1097/PAS.0b013e3181882c54. [DOI] [PubMed] [Google Scholar]
- Hostetter G, Kim SY, Savage S, Gooden GC, Barrett M, Zhang J, Alla L, Watanabe A, Einspahr J, Prasad A, Nickoloff BJ, Carpten J, Trent J, Alberts D, Bittner M. Random DNA fragmentation allows detection of single-copy, single-exon alterations of copy number by oligonucleotide array CGH in clinical FFPE samples. Nucleic Acids Res. 2010;38:e9. doi: 10.1093/nar/gkp881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 2007;80:805–810. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbalzano AN, Jones SN. Snf5 tumor suppressor couples chromatin remodeling, checkpoint control, and chromosomal stability. Cancer Cell. 2005;7:294–295. doi: 10.1016/j.ccr.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Jackson EM, Shaikh TH, Gururangan S, Jones MC, Malkin D, Nikkel SM, Zuppan CW, Wainwright LM, Zhang F, Biegel JA. High-density single nucleotide polymorphism array analysis in patients with germline deletions of 22q11.2 and malignant rhabdoid tumor. Hum Genet. 2007;122:117–127. doi: 10.1007/s00439-007-0386-3. [DOI] [PubMed] [Google Scholar]
- Jackson EM, Sievert AJ, Gai X, Hakonarson H, Judkins AR, Tooke L, Perin JC, Xie H, Shaikh TH, Biegel JA. Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer Res. 2009;15:1923–1930. doi: 10.1158/1078-0432.CCR-08-2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Li M, Parsons DW, Zhang X, Wesseling J, Kristel P, Schmidt MK, Markowitz S, Yan H, Bigner D, Hruban RH, Eshleman JR, Iacobuzio-Donahue CA, Goggins M, Maitra A, Malek SN, Powell S, Vogelstein B, Kinzler KW, Velculescu VE, Papadopoulos N. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat. 2012;33:100–103. doi: 10.1002/humu.21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohashi K, Izumi T, Oda Y, Yamamoto H, Tamiya S, Taguchi T, Iwamoto Y, Hasegawa T, Tsuneyoshi M. Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: a useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol. 2009;40:349–355. doi: 10.1016/j.humpath.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR, Biegel JA, Getz G, Roberts CW. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest. 2012;122:2983–2988. doi: 10.1172/JCI64400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Cimica V, Ramachandra N, Zagzag D, Kalpana GV. Aurora A is a repressed effector target of the chromatin remodeling protein INI1/hSNF5 required for rhabdoid tumor cell survival. Cancer Res. 2011;71:3225–3235. doi: 10.1158/0008-5472.CAN-10-2167. [DOI] [PubMed] [Google Scholar]
- Medina PP, Sanchez-Cespedes M. Involvement of the chromatin-remodeling factor BRG1/SMARCA4 in human cancer. Epigenetics. 2008;3:64–68. doi: 10.4161/epi.3.2.6153. [DOI] [PubMed] [Google Scholar]
- Modena P, Lualdi E, Facchinetti F, Galli L, Teixeira MR, Pilotti S, Sozzi G. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012–4019. doi: 10.1158/0008-5472.CAN-04-3050. [DOI] [PubMed] [Google Scholar]
- Papp G, Changchien YC, Peterfia B, Pecsenka L, Krausz T, Stricker TP, Khoor A, Donner L, Sapi Z. SMARCB1 protein and mRNA loss is not caused by promoter and histone hypermethylation in epithelioid sarcoma. Mod Pathol. 2013;26:393–403. doi: 10.1038/modpathol.2012.190. [DOI] [PubMed] [Google Scholar]
- Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, Kinch L, Hambuch T, Jain S, Lotan Y, Margulis V, Sagalowsky AI, Summerour PB, Kabbani W, Wong SW, Grishin N, Laurent M, Xie XJ, Haudenschild CD, Ross MT, Bentley DR, Kapur P, Brugarolas J. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012;44:751–759. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perot G, Croce S, Ribeiro A, Lagarde P, Velasco V, Neuville A, Coindre JM, Stoeckle E, Floquet A, MacGrogan G, Chibon F. MED12 alterations in both human benign and malignant uterine soft tissue tumors. PLoS One. 2012;7:e40015. doi: 10.1371/journal.pone.0040015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci U S A. 2000;97:13796–13800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CW, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell. 2002;2:415–425. doi: 10.1016/s1535-6108(02)00185-x. [DOI] [PubMed] [Google Scholar]
- Roberts CW, Orkin SH. The SWI/SNF complex--chromatin and cancer. Nat Rev Cancer. 2004;4:133–142. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- Schneppenheim R, Fruhwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, Kreuz M, Leuschner I, Martin Subero JI, Obser T, Oyen F, Vater I, Siebert R. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010;86:279–284. doi: 10.1016/j.ajhg.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ME, Cimica V, Chinni S, Jana S, Koba W, Yang Z, Fine E, Zagzag D, Montagna C, Kalpana GV. Therapeutically targeting cyclin D1 in primary tumors arising from loss of Ini1. Proc Natl Acad Sci U S A. 2011;108:319–324. doi: 10.1073/pnas.0913297108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LM, Folpe AL, Pawel BR, Judkins AR, Biegel JA. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod Pathol. 2013;26:385–392. doi: 10.1038/modpathol.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsikitis M, Zhang Z, Edelman W, Zagzag D, Kalpana GV. Genetic ablation of Cyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proc Natl Acad Sci U S A. 2005;102:12129–12134. doi: 10.1073/pnas.0505300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.