

Abstract
Cancer stem cells (CSC) may take advantage of the Warburg effect-induced siphoning of metabolic intermediates into de novo fatty acid biosynthesis to increase self-renewal growth. We examined the anti-CSC effects of the antifungal polyketide soraphen A, a specific inhibitor of the first committed step of lipid biosynthesis catalyzed by acetyl-CoA carboxylase (ACACA). The mammosphere formation capability of MCF-7 cells was reduced following treatment with soraphen A in a dose-dependent manner. MCF-7 cells engineered to overexpress the oncogene HER2 (MCF-7/HER2 cells) were 5-fold more sensitive than MCF-7 parental cells to soraphen A-induced reductions in mammosphere-forming efficiency. Soraphen A treatment notably decreased aldehyde dehydrogenase (ALDH)-positive CSC-like cells and impeded the HER2's ability to increase the ALDH+-stem cell population. The following results confirmed that soraphen A-induced suppression of CSC populations occurred through ACACA-driven lipogenesis: a.) exogenous supplementation with supraphysiological concentrations of oleic acid fully rescued mammosphere formation in the presence of soraphen A and b.) mammosphere cultures of MCF-7 cells with stably silenced expression of the cytosolic isoform ACACA1, which specifically participates in de novo lipogenesis, were mostly refractory to soraphen A treatment. Our findings reveal for the first time that ACACA may constitute a previously unrecognized target for novel anti-breast CSC therapies.
Keywords: Acetyl-CoA Carboxylase, Cancer Stem Cells, Lipogenesis, Warburg effect, metabolism, breast cancer, Soraphen A
INTRODUCTION
Accumulating evidence indicates that the metabolic state of cancer stem cells (CSCs), a population of cancer cells capable of enhanced self-renewal, governs their tumor-initiating ability. Additionally, CSCs are resistant to conventional therapy and are significantly different from their differentiated cellular counterparts. Because metabolic reprogramming may control the ability of CSCs to avoid treatment and promote recurrence, successful metabolic therapy may eliminate these key drivers of tumor formation, progression, and recurrence [1-6].
Several attempts have been made to therapeutically modulate the metabolic state of cancer cells by treating with compounds that inhibit the most recognizable bioenergetic feature of tumor cells, i.e., aerobic glycolysis (the Warburg effect) [7-11]. However, efforts to inhibit glycolysis using the glucose analog 2-deoxyglucose (2-DG), which accumulates in cells and inhibits glycolytic hexokinase (KH), or the small molecule dichloroacetate (DCA), which inhibits mitochondrial pyruvate dehydrogenase kinase (PDK) and forces pyruvate into the mitochondria to increase mitochondrial metabolism, remain unsatisfactory. In addition, these approaches are not selective for either CSCs or more differentiated bulk tumor cells, and drugs that inhibit glycolysis do not necessarily result in increased mitochondrial metabolism and could result in the disruption of energy production and non-selective cell death. Thus, glycolysis inhibitors may be undesirably toxic to non-cancerous tissues that depend on glycolysis for energy production (i.e., skeletal muscle or brain tissues). CSCs are known to contain lower reactive oxygen species (ROS) levels than their cancerous epithelial-like progeny cells [12]. Therefore, one therapeutic alternative to consider is the re-activation of mitochondrial function and biogenesis, which in turn would impact the suppression of ROS-induced killing in CSCs, as opposed to acutely inducing energy starvation and cell death in all tissues utilizing glycolysis for energy production. In particular, the mitochondrial regulator metformin has been increasingly recognized as a strong therapeutic capable of targeting CSCs in pre-clinical models of human cancer [13-23].
Another possible treatment approach is related to the commonly observed upregulation of endogenous lipid biosynthetic pathways in cancer tissues. This so-called lipogenic phenotype fuels membrane biogenesis in rapidly proliferating cancer cells and renders cancer membrane lipids more saturated. The lipogenic phenotype also impacts fundamental cellular processes associated with cancer cell transformation, including signal transduction, gene expression, ciliogenesis, and response to therapy [24-30]. In the fatty acid synthesis pathway, acetyl-CoA is carboxylated to malonyl-CoA by acetyl-CoA carboxylase (ACACA). Both acetyl-CoA and malonyl-CoA are then used in a condensation reaction by the main lipogenic enzyme fatty acid synthase (FASN) to produce long-chain fatty acids. Of note, it is known that higher expression levels of lipogenic genes and proteins such as FASN are found in CSC subpopulations of breast cancer cell lines and that upregulation of de novo fatty acid biogenesis is a pre-requisite for the formation of pre-malignant lesions due to increased CSC survival [31-35]. Moreover, recent studies performed in induced pluripotent stem cells (iPSCs) have revealed that when activities of the ACACA and FASN lipogenic enzymes are inhibited, the efficiency of somatic reprogramming to stemness is decreased [30]. Coincidentally, ACACA and FASN are highly expressed in iPSCs. We recently hypothesized that the stemness features of cancer cells may take advantage of the Warburg effect-related ability of tricarboxylic acid (TCA) cycle intermediates to be siphoned into lipid biosynthesis metabolism for CSC self-renewal and survival.
To test the hypothesis that the therapeutic targeting of endogenous lipogenesis may impact the CSC cellular state in heterogeneous breast cancer cell populations, we examined the polyketide soraphen A, which was chosen for these studies because its mechanism of ACACA inhibition is well defined [36-43]. Unlike RNA interference-based approaches [44], the rapidity of soraphen A-induced inhibition of lipid metabolism minimizes non-specific or adaptive changes caused by changes in cell fatty acid composition and cell growth. Our current results are the first to show that soraphen A treatment can inhibit the formation of mammospheres in a fatty acid-dependent manner, highlighting the potential value of ACACA as a novel metabolic target in breast CSC.
RESULTS
Soraphen A decreases mammosphere formation in MCF-7 breast cancer cells
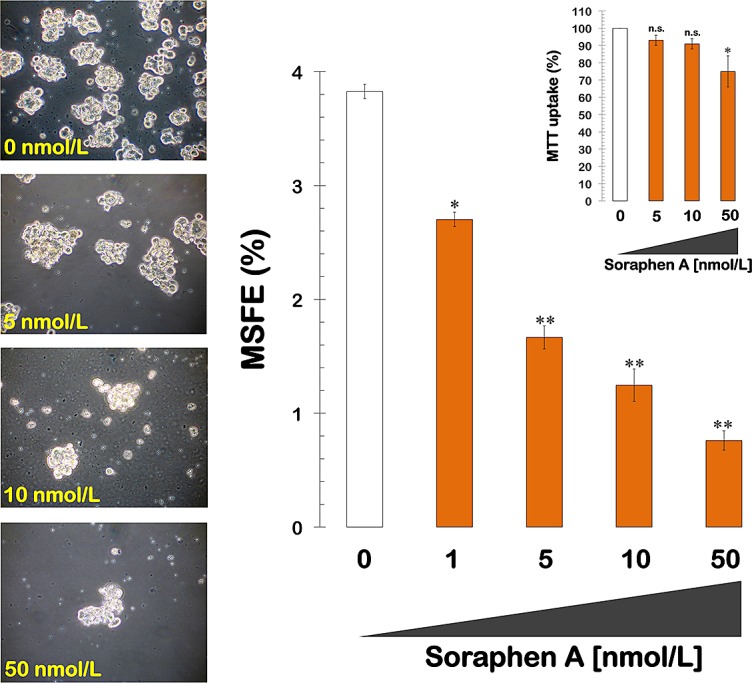
We first tested the ability of MCF-7 breast cancer cells to form tumor spheres when grown in suspension cultures in the presence of a range of concentrations of soraphen A (1, 5, 10, and 50 nmol/L). The MSFE was calculated as the number of sphere-like structures (diameter >50 μm) divided by the original number of cells seeded and expressed as the mean percentage (±SD). A subset (2.0 ± 0.01%) of untreated MCF-7 breast cancer cells formed bona fide mammospheres upon initial plating, and this result confirms previous reports that MCF-7 cell cultures intrinsically contain a SC-like population. Interestingly, the spheroid formation capability of MCF-7 cells was significantly reduced following treatment with increasing concentrations of soraphen A in a dose-dependent manner (50% reduction at 15 nmol/L soraphen A; Fig. 1, left and middle panels). Although the ability of some CSC-like cellular states to survive and proliferate as floating spherical colonies under anchorage-independent conditions at low frequencies (1-3%) is commonly regarded as an in vitro surrogate of the self-renewal and tumor-initiating capacity exclusively possessed by CSCs, it should be acknowledged that mammosphere formation assays can also be viewed as bona fide assays for evaluating the number of anoikis-resistant cells within heterogenous cancer populations. To unambiguously validate the anti-CSC effects of soraphen A, anoikis was induced by plating MCF-7 cells into culture dishes that had been coated with poly-2-hydroxyethyl methacrylate (Poly-HEME) in the absence or presence of soraphen A (50 nmol/L; 48 h) prior to replating a viable fraction of the suspended samples in serum- and soraphen A-free mammosphere medium. Remarkably, soraphen A-treated anoikis cultures reduced by 50% their mammosphere-formation efficiency after removal of the drug (Fig. 1, right panel). Importantly, the soraphen A-induced reduction in mammosphere formation was not due to non-specific toxicity, as MTT-based experiments in monolayer cultures of MCF-7 cells showed that cell viability remained as high as 90% in the presence of identical nanomolar concentrations of soraphen A (Fig. 1, middle panels).
Figure 1. Pharmacological blockade of ACACA activity inhibits mammosphere formation in MCF-7 cells.

Left. Figure shows representative light microscope representations of mammospheres formed by MCF-7 cells growing in sphere medium for 7 days in the absence or presence of graded concentrations of Soraphen A, as specified (20X magnifications). Middle. MSFE of MCF-7 cells was calculated as the number of mammospheres (diameter >50 μm) formed in 7 days divided by the original number of cells seeded and expressed as percentage means (columns) ± SD (bars). Re-feeding of mammospheres cultures with Soraphen A and/or sphere medium was performed on days 3 and 5. The metabolic status of monolayer cultures of MCF-7 cells treated with increasing concentrations of Soraphen A was measured using MTT uptake assays, and cell viability is expressed as % uptake relative to untreated control cells (= 100% cell viability). Right. MCF-7 cells were trypsinized into a single cell suspension, and 2 mL was cultured on poly-HEME-coated plates at a density of ~105 cells/mL (total of 2 x 105 cells/well) in the absence or presence of 50 nmol/L soraphen A and then incubated at 37 oC for an additional 48 h. Cells that were visibly anoikis-resistant with intact plasma membranes, i.e., excluding trypan blue stain, were then cultured in soraphen A-free mammosphere medium for 7 days following the same procedure as described above. The results are presented as the mean (columns) ± SD (bars) of 2 independent experiments performed in triplicate. *P < 0.01 and **P < 0.001, statistically significant differences from the control group. n. s. not statistically significant.
Soraphen A eliminates the HER2-enhanced formation of mammospheres
We next examined whether treatment with soraphen A was sufficient to prevent the well-recognized ability of the oncogene HER2 to expand the breast CSC population. MCF-7 cells engineered to overexpress HER2 (MCF-7/HER2) exhibited a significantly increased ability to form mammospheres (3.8 ± 0.1%) compared to parental MCF-7 cells. However, this enhanced spheroid formation capability of MCF-7/HER2 cells was drastically reduced following treatment with soraphen A, which decreased the MSFE by 50% at ~3 nmol/L (i.e., a Soraphen A concentration 5 times lower than that needed to decrease MSFE by 50% in MCF-7 parental cells). The highest dose of the soraphen A (50 nmol/L) elicited inhibitory effects of ~80% compared to the basal MSFE found in untreated MCF-7/HER2 tumor sphere cultures (Fig. 2). Mammosphere formation was also drastically decreased by soraphen A treatment in SKBR3 cells, which are a natural model of HER2 overexpression and HER2 dependency for cell proliferation and survival (data not shown).
Figure 2. Pharmacological blockade of ACACA activity inhibits mammosphere formation in MCF-7/HER2 cells.
Left. Figure shows representative light microscope representations of mammospheres formed by MCF-7/HER2 cells growing in sphere medium for 7 days in the absence or presence of graded concentrations of Soraphen A, as specified (20X magnifications). Right. MSFE of MCF-7/HER2 cells was calculated as the number of mammospheres (diameter >50 μm) formed in 7 days divided by the original number of cells seeded and expressed as percentage means (columns) ± SD (bars). Re-feeding of mammospheres cultures with Soraphen A and/or sphere medium was performed on days 3 and 5. The metabolic status of monolayer cultures of MCF-/HER2 cells treated with increasing concentrations of Soraphen A was measured using MTT uptake assays, and cell viability is expressed as % uptake relative to untreated control cells (= 100% cell viability). The results are presented as the mean (columns) ± SD (bars) of 2 independent experiments performed in triplicate. *P < 0.01 and **P < 0.001, statistically significant differences from the control group. n. s. not statistically significant.
Similar to the results obtained with MCF-7 cells, the reductions in mammosphere formation were not due to non-specific soraphen A toxicity, as identical concentrations of the drug had no drastic impact on MCF-7/HER2 cell viability under adherent culture conditions. These findings strongly suggest that the mechanism of action targeted by nanomolar concentrations of soraphen A is not essential for the bulk breast cancer cell population.
Soraphen A decreases the percentage of breast cancer cells expressing the CSC marker ALDEFLUOR
The drastic decrease in mammosphere formation rates observed following treatment of MCF-7 and MCF-7/HER2 cancer cell populations with Soraphen A certainly provided preliminary insight as to the Soraphen A's mechanism of action, in particular to its putative ability to specifically remove CSCs from the bulk population. We therefore envisioned that Soraphen A might specifically suppress aldehyde dehydrogenase (ALDH)-positive CSC-like cells. Using flow cytometry and the ALDEFLUOR® reagent, we first confirmed the presence of an enhanced aldefluor-positive sub-population in MCF-7 and MCF-7/HER2 cells. In Soraphen A-naïve MCF-7 cell populations, approximately 2.4% of the cells expressed high ALDH activity (Fig. 3A). Interestingly, in Soraphen A-treated MCF-7 cell populations, solely 1.5% of the cells remained ALDH-positive. We confirmed that overexpression of HER2 increased the aldefluor-positive population almost twofold compared to MCF-7 parental cells; remarkably, the aldefluor-positive cell content drastically decreased from 4% in Soraphen A-naïve MCF-7/HER2 cell populations to 1.8% in Soraphen A-treated MCF-7/HER2 cells (Fig. 3B).
Figure 3. Pharmacological blockade of ACACA activity impedes HER2-induced expansion of stem cell population.
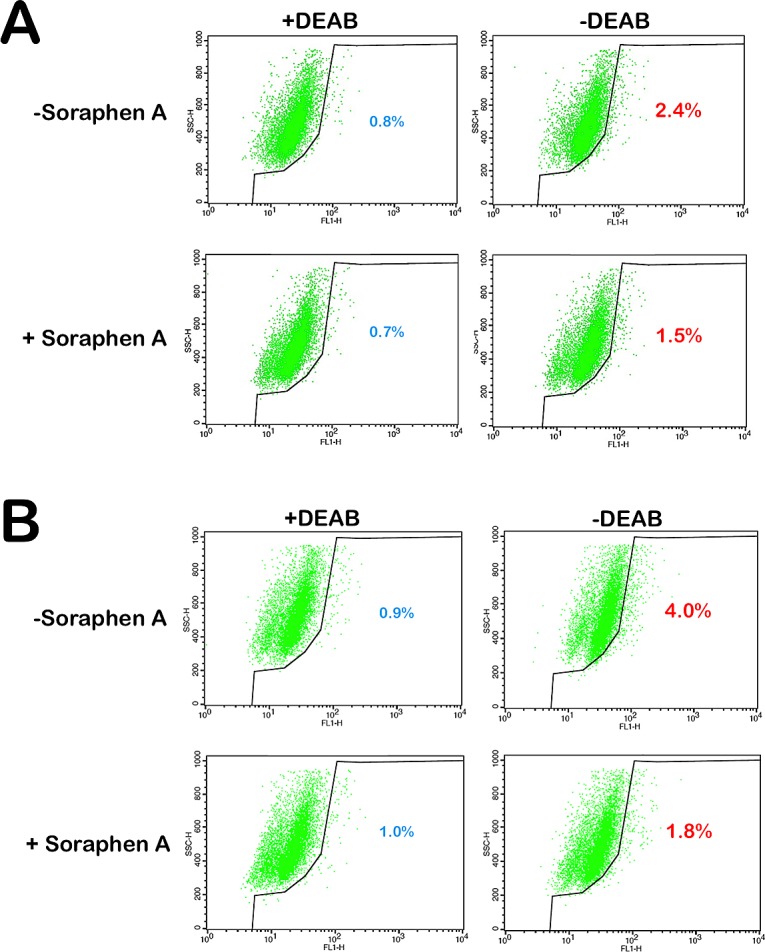
MCF-7 cells (A) and HER2-overexpressing MCF-7/HER2 cells (B) were subjected to the ALDEFLUOR® assay to identify cells with high ALDH activity in the absence or presence or Soraphen A (10 nmol/L, 3 days with daily re-feeding). The ALDH inhibitor DEAB was used as a negative control. The cells without inhibitor shifted to the right and were considered ALDH-positive cells.
Soraphen A suppresses HER2-enhanced mammosphere formation by blocking endogenous lipogenesis
We next sought to unambiguously establish that soraphen A suppresses mammosphere formation by inhibiting de novo fatty acid biogenesis (Fig. 4A, left panel). Therefore, we determined the rescue potential of oleic acid when the lipogenic pathway was specifically blocked through treatment with soraphen A. The strong inhibitory effects of soraphen A on the mammosphere formation capability of MCF-7/HER2 cells were fully counteracted by supplementation of the mammosphere medium with micromolar concentrations of oleic acid (100 μmol/L; Fig. 4A, middle panel).
Figure 4. Soraphen A suppresses self-renewal growth of CSC-like cells by blocking ACACA-catalyzed endogenous lipogenesis.

A. Right. MSFE of MCF-7/HER2 cells was calculated as the number of mammospheres (diameter >50 μm) formed in 7 days in the absence or presence of graded concentrations of Soraphen A and co-exposed to 100 μmol/L oleic acid, as specified, divided by the original number of cells seeded and expressed as percentage means (columns) ± SD (bars). Re-feeding of mammospheres cultures with sphere medium, Soraphen A, and/or oleic acid was performed on days 3 and 5. Left. MSFE of MCF-7 cells was calculated as the number B. MSFE of MCF-7 and MCF-7shACACA1 cells was calculated as the number of mammospheres (diameter >50 μm) formed in 7 days in the absence or presence of 100 nmol/L Soraphen A divided by the original number of cells seeded and expressed as percentage means (columns) ± SD (bars). Re-feeding of mammospheres cultures with Soraphen A and/or sphere medium was performed on days 3 and 5. The results in A and B are presented as the mean (columns) ± SD (bars) of 2 independent experiments performed in triplicate. *P < 0.01 and **P < 0.001, statistically significant differences from the control group. n. s. not statistically significant. C. MCF-7 and MCF-7/HER2 cells, untreated or treated for 3 days with 50 nmol/L soraphen A, were exposed for 60 min to H2DCF-DA and their fluorescence intensity was measured by flow cytometry. Figure shows representative scatter plot histograms of DCF fluorescence. The columns at the interior the histograms show relative measurements of ROS levels in soraphen A-treated MCF-7 and MCF-7/HER2 cells compared to untreated controls (=100%). *P < 0.01 and **P < 0.001, statistically significant differences from the control group.
Notably, in the presence of a gradient of non-cytotoxic micromolar concentrations of the FASN inhibitor C75, MCF-7 cells lost their ability to develop mammospheres in a dose-dependent manner with the highest dose of C75 (10 μmol/L) eliciting consistently greater inhibitory effects than the lowest dose (0.1 μmol/L). Indeed, 10 μmol/L C75 dramatically suppressed the MSFE by > 90% (Fig. 4A, right panel).
Soraphen A suppresses mammosphere formation via ACACA
We next evaluated whether soraphen A-induced inhibition of mammosphere formation occurred through the specific targeting of ACACA. To do so, we reevaluated the effects of soraphen A on the mammosphere capability of MCF-7 cells in which the ACACA gene was stably silenced via lentiviral-delivered small hairpin RNA (MCF-7shACACA1). First, we confirmed differences in mammosphere formation between conditions of ACACA chemical inhibition versus ACACA gene silencing. A previous study reported that knockdown of ACACA recapitulated AMPK activation and facilitated anchorage-independent growth [45]. Our data indicated that ACACA silencing elicited stronger mammosphere formation in MCF-7shACACA1 cells (Fig. 4B). In addition, we observed that the enhanced mammosphere formation ability of MCF-7shACACA1 cell was mostly refractory to soraphen A treatment even at concentrations of 100 nmol/L, which were able to fully suppress the mammosphere formation capability of MCF-7 parental cells.
DISCUSSION
Frustrated by the gene-centric limitations of conventional approaches used to identify molecular markers associated with rare tissue-specific sub-populations of CSCs, we recently postulated that CSCs may display unique metabolic features that distinguish them from the bulk of tumor cells. We hypothesized that these metabolic properties may constitute a basis for developing new therapeutic strategies to eliminate CSCs [1]. One of the properties of stem/progenitor cells is their ability to survive under anchorage-independent conditions and generate mammospheres. Our current data are the first to show a highly significant decrease in mammosphere formation in the presence of the polyketide fungicide soraphen A. These results confirm that the lipogenic enzyme ACACA might represent a novel target for anti-breast CSC therapies. These findings, together with the ability of chemical inhibitors of FASN activity such as C75 to recapitulate the anti-mammosphere activity of the ACACA inhibitor soraphen A, also confirm that de novo fatty acid biogenesis, a pathway that is frequently deregulated during breast carcinogenesis, plays a functional role in CSC self-renewal and survival.
Therapeutic exploitation of the lipogenic pathway in cancer should consider the effects of blocking one metabolic enzyme on the expression and activity of other metabolic genes. In particular, it is essential to consider that alternate mechanisms/pathways might compensate for the inhibition of any enzyme in the lipogenic pathway. For example, silencing of ATP citrate lyase (ACLY), a cytosolic enzyme that converts citrate into a shared precursor for fatty acid and mevalonate synthesis such as acetyl-CoA, is sufficient to counteract stem cell characteristics induced in diverse cancer cell systems. Moreover, the expression of FASN, which catalyzes the rate-limiting reaction for fatty acid synthesis, and HMGCR, which is the rate-limiting enzyme in the mevalonate pathway, were both shown to be significantly enhanced after ACLY knockdown. These results suggest that ACLY deficiency forces cancer cells to upregulate the expression of downstream genes in fatty acid and cholesterol synthesis pathways to compensate for the loss of ACLY. Of note, this type of compensation that occurs during the therapeutic targeting of lipogenesis-related enzymes such as ACLY might be unacceptable for cancer treatment [43, 46]. We acknowledge that our study failed to evaluate whether the chemical perturbation of ACACA promoted major changes in the expression profile of other metabolic genes. However, the rapidity of soraphen A-induced inhibition of de novo fatty acid biogenesis appeared to minimize the occurrence of non-specific or adaptive changes that could be switched on in response to changes in cell fatty acid composition and cell growth. In fact, the chemical inhibition of ACACA mediated by soraphen A promoted the opposite effect on mammosphere formation as stable genetic silencing of the cytosolic isoform ACACA1, which specifically participates in de novo lipogenesis. As reported in the study by Jeon et al. [45], knockdown of ACACA was found to recapitulate AMPK activation and facilitate anchorage-independent growth. Thus, MCF-7 cells with stably silenced expression of the cytosolic isoform ACACA1 demonstrate enhanced mammosphere formation capacity. Interestingly, MCF-7shACACA1 cells were largely refractory to concentrations of soraphen A that completely suppressed mammosphere formation in MCF-7 parental cells. This finding, as well as the data showing that exogenous supplementation with supraphysiological concentrations of the monounsaturated fatty acid oleic acid fully rescued the ability of MCF-7/HER2 cells to form mammospheres in the presence of soraphen A, mechanistically confirm that soraphen A-induced suppression of breast CSC populations specifically occurs via blockade of ACACA1-driven lipogenesis.
Soraphen A-induced inhibition of ACACA has been shown to augment the accumulation of unsaturated fatty acids derived from saturated and polyunsaturated fatty acid precursors by impacting both fatty acid elongation and by disrupting pathways where both elongases and desaturases are utilized. The inhibition of ACACA leads to a significant shift in fatty acid metabolism, whereby saturated fatty acids and essential fatty acids are desaturated but not elongated [42]. Thus, it is tempting to speculate that unlike RNA interference-based approaches [45], the rapid ability of soraphen A to acutely disrupt a strong functional link between ACACA activity and the synthesis of saturated-, monounsaturated-, and polyunsaturated fatty acids through the control of fatty acid elongase activity might underlie soraphen A-mediated elimination of breast CSCs. Other studies have found that reversing the lipogenic switch in cancer cells with soraphen A treatment leads to a marked decrease in saturated and mono-unsaturated phospholipid species and an increase in the relative degree of polyunsaturation. Because polyunsaturated acyl chains are more susceptible to peroxidation, soraphen A-induced acute inhibition of lipogenesis may increase the levels of peroxidation end products and render cells more susceptible to oxidative stress-induced cell death. One of the properties of anoikis-resistant stem/progenitor cells is their ability to survive in anchorage-independent conditions and generate mammospheres. Thus, the soraphen A-induced acute blockade of endogenous lipogenesis may impair the intrinsic ability of CSCs to escape anoikis due to increased oxidative damage and starvation and cell death in detached microenvironmental conditions. In this regard, exposure of breast cancer cells to soraphen A led to a marked augmentation of cells ability to oxidize H2DCF-DA (Fig. 4C); because H2DCF oxidation by ROS results in formation of strongly fluorescent DCF which is considered to be a marker of ROS abundance and low levels of ROS maintain characteristics of ROS, it might be tempting to suggest that increasing ROS might be part of the pharmacological mechanism through which soraphen A promotes the loss of breast CSCs. Additionally, soraphen A may alter the formation and functioning of membrane microdomains such as lipid rafts, a recently recognized mechanism involved in the escape from anoikis-induced cell death [47-52].
Chemoresistant cell subpopulations from breast cell lines have been shown to possess high levels of ALDH activity. By virtue of those high levels of ALDH activity, FACS can track chemoresistant CSC-like subpopulations as ALDH-positive cells. Indeed, in several types of tumors including breast cancer, cancer cell subpopulations that are enriched for cancer-initiating activity have been readily identified by flow cytometry analysis using the ALDEFLUOR® reagent to identify cells with high levels of ALDH activity [53-56]. We confirmed earlier studies showing that HER2-overexpressing breast cancer cells contain readily detectable higher amounts of ALDH-positive cells [57]. What we believe is remarkable here is that treatment with no cytotoxic concentrations of Soraphen A notably reduced the number of ALDH-positive cells to those levels commonly found in breast cancer cell lines expressing the lowest level of ALDH activity. Moreover, Soraphen A treatment was sufficient to impede the HER2's ability to increase the stem cell population. Therefore, in the presence of Soraphen, the decrease in ALDH activity may account, at least in part, for the loss of stemness of breast CSCs. This is very interesting since the ALDH-positive cells represent the chemosensitive fraction of biologically aggressive breast tumors and it indicates the potential of Soraphen A-like molecules for eliminating ALDH-positive chemoresistant cell subpopulations.
In summary, our findings confirm the importance of de novo fatty acid biogenesis in CSC self-renewal and survival and reveal for the first time that ACACA might constitute a previously unrecognized target for novel anti-breast CSC therapies. A large number of nanomolar small-molecule ACACA inhibitors have been developed, and several have been evaluated in clinical trials for metabolic diseases such as obesity and metabolic syndrome [58-60]. The potency and mechanism of ACACA inhibition by soraphen A strongly suggest that pharmacological targeting of the soraphen A binding subunit dimerization site (i.e., the biotin carboxylase (BC) domain of the enzyme) may be useful to identify potent, efficacious ACACA inhibitors for cancer intervention. Using state-of-the art structure-based drug design and crystal structures of human ACACA BC domain, Nimbus Discovery (Cambridge, MA) recently identified a unique series of allosteric inhibitors with low nanomolar potency (ND654, ND646) that bind to the soraphen A binding site. These inhibitors show anti-neoplastic properties against human non-small cell lung cancer (NSCLC) and hepatocarcinoma cells in vitro and in vivo [61, 62]. If further studies support the anti-CSC activity of these drug-like allosteric inhibitors to bind the BC domain of ACACA with high potency and selectivity, our current findings may open new avenues for exploring potential anti-lipogenesis treatments to successfully target breast CSCs.
MATERIALS and METHODS
Drugs and reagents
Soraphen A purified from the myxobacterium Sorangium cellulosum was kindly provided by Drs. Klaus Gerth and Rolf Jansen (Hemholtz Zentrum für Infektionsforschung GmbH, Braunschweig, Germany). Oleic acid was purchased from Sigma. C75 was purchased from Alexis Biochemicals (San Diego, CA). Soraphen A and C75 were dissolved in DMSO, and stored in the dark as stock solutions at -20 oC until utilization.
Culture conditions
MCF-7 human breast cancer cells were obtained from the American Type Culture Collection (ATCC). The cells were routinely grown in improved MEM (IMEM;BioSource International; Invitrogen S.A., Barcelona, Spain) supplemented with 5% fetal bovine serum (FBS) and 2 mmol/L L-Glutamine. The cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. The construction of pBABE/HER2 retroviruses and retroviral infection of MCF-7 cells to generate MCF-7/pBABE and MCF-7/HER2 cells have been described elsewhere [44]. The cells were screened periodically for Mycoplasma contamination.
Mammosphere culture
Mammospheres were generated from single cells of the MCF-7, MCF-7/HER2, and MCF-7shACACA1 cell lines seeded at 103 cells/cm2 in six-well ultralow attachment plates (Corning Inc.). The sphere medium consisted of F-12/DMEM containing 5 mg/mL insulin, 0.5 mg/mL hydrocortisone, 2% B27 (Invitrogen Ltd.), and 20 ng/mL epidermal growth factor. The medium was made semi-solid by the addition of 0.5% methylcellulose (R&D Systems, Minneapolis, MN) to prevent cell aggregation.
Mammosphere-forming efficiency
The mammosphere-forming efficiency (MSFE) was calculated as the number of sphere-like structures (large diameter >50 μm) formed in 7 days divided by the original number of cells seeded and expressed as a percentage (mean ± SD).
Anoikis Assay
6-well tissue plates were coated with 200 μl (12 mg/mL in 95% ethanol) of poly-(2-hydroxyethyl methacrylate) (poly-HEME; Sigma) by incubation overnight at 40 oC. To perform the anoikis assay, MCF-7 cells were trypsinized into a single cell suspension, and 2 mL was cultured in poly-HEME-coated plates at a density of ~105 cells/mL (total of 2 x 105 cells/well) in the absence or presence of 50 nmol/L soraphen A in Dulbecco's Modified Eagle's medium with 10% fetal bovine serum.
Metabolic status assessment (MTT-based cell viability assays)
Cell viability was determined using a standard colorimetric MTT (3-4,5-dimethylthiazol-2-yl-2, 5-diphenyl-tetrazolium bromide) reduction assay. Exponentially growing cells were harvested by trypsinization, seeded at a concentration of ~2.5 x 103 cells/200 μL/well in 96-well plates, and allowed to attach overnight. The medium was then removed, and fresh medium containing various concentrations of soraphen A was added to the cultures as specified. Control cells without soraphen A were cultured in parallel using the same conditions with comparable media changes. Following treatment (5 days), the medium was removed and replaced with fresh drug-free medium (100 μL/well), and MTT (5 mg/mL in PBS) was added to each well at a 1/10 volume. After incubation for 2–3 h at 37°C, the supernatants were carefully aspirated, and 100 μL of DMSO was added to each well. The plates were agitated to dissolve the crystal product. The optical density (OD) was measured at 570 nm in a multi-well plate reader. The cell viability effects resulting from exposure to soraphen A were analyzed as percentages of the control cell absorbances, which were obtained from control wells treated with appropriate concentrations of the soraphen A vehicle and processed simultaneously. For each treatment, cell viability was evaluated as a percentage using the following equation: (OD570 of treated sample/OD570 of untreated sample) x 100.
ALDEFLUOR® activity assay
The ALDEFLUOR® assay (Stem Cell Technologies) quantifies ALDH activity by measuring the conversion of the ALDH substrate BODIPY aminoacetaldehyde to the fluorescent product BODIPY aminoacetate. Briefly, cells were suspended in ALDEFLUOR assay buffer containing the fluorescent ALDH substrate BODIPY-aminoacetaldehyde (BAAA) and incubated for 45 min at 37 oC. The assay buffer also contained a transport inhibitor to prevent efflux of BAAA from the cells. BAAA passively diffuses into live cells and is then converted by intracellular ALDH into a negatively charged product (BODIPY-aminoacetate) that is retained inside cells, labeling the cells with a bright fluorescent signal. After a washing step, the brightly fluorescent ALDH-expressing cells (ALDHbright) were detected in the green fluorescence channel (FL1; 520–540 nm) on a FACSCalibur instrument (BD Biosciences). A sample of cells was further stained with a specific ALDH inhibitor, diethylaminobenzaldehyde (DEAB) (Sigma), to serve as a negative control for each experiment. Because only cells with an intact cellular membrane can retain the ALDH1 reaction product, only viable ALDHbright cells were identified. Cells incubated with BAAA and DEAB were used to establish the background signal and define the ALDHbright region. Incubation of cells with the substrate in the absence of DEAB induced a shift in the BAAA fluorescence and defined the ALDHbright population.
Lentiviral transduction
Pre-packaged lentiviral particles that either encoded a non-targeting shRNA (negative shRNA, sc-108080) or sequences specifically targeting the human ACCα(ACACA1) gene (sc-40312-V) were purchased from a commercial provider (Santa Cruz Biotechnology, Inc.). To infect MCF-7 cells with virus, the regular medium was replaced with culture medium containing 5 μg/mL polybrene (Santa Cruz Biotechnology, sc-124220). MCF-7 cells were then exposed to lentiviruses for 48 h. The lentiviral shRNA particles also encode a puromycin resistance gene for transduction selection. After infection, the cells were washed and grown in culture medium containing 10 μg/mL puromycin dihydrochloride (Sigma, P9620) for an additional 72 h. The MCF-7 cells were allowed to proliferate for at least 1 week before any experimental procedures. To monitor the lentiviral transduction efficiency and transgene expression during the experiment we incubated additional subsets of MCF-7 cells with lentiviral particles encoding a green fluorescence protein (GFP) reporter (sc-108084). The transduction efficiency (>90%) was calculated as the ratio of the number of GFP-positive cells to the total number of cells from five random visual fields in three independent culture experiments.
Reactive Oxygen Species (ROS) Detection
Untreated cells as well as cells treated with soraphen A were incubated 60 min with 10 μmol/L 2’,7’-dihydro-dichlorofluorescein-diacetate (H2DCF-DA) (Invitrogen/Molecular Probes) at 37°C. Cellular green fluorescence was then measured by flow cytometry. Following oxidation by ROS and peroxides within cells the non-fluorescent substrate H2DCF-DA is converted to the highly fluorescent derivative DCF [63-65]. The cell-permeant non-fluorescent H2DCF-DA upon cleavage of the acetate moiety by intercellular esterases and oxidation by ROS is converted to strongly fluorescent DCF and thus reports the ROS abundance.
Immunoblotting
Testing for the total expression level of ACACA1 was performed by immunoblotting procedures using the ACCα (H-76) sc-30212 rabbit polyclonal antibody (Santa Cruz Biotechnology, Inc.) according to the manufacturer's instructions.
Statistical analysis
The results are presented as the mean ± SD for at least three repeated individual experiments for each group. The analyses were performed using XLSTAT 2010 (AddinsoftTM). A P-value ≤ 0.01 was considered statistically significant.
Acknowledgments
This work was financially supported by the Ministerio de Ciencia e Innovación (SAF2012-38914), Plan Nacional de I+D+I, MICINN, Spain.
Footnotes
Conflict Of Interest Statement
The authors of this manuscript have no conflicts of interest to declare.
REFERENCES
- 1.Menendez JA, Joven J, Cufí S, Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, Martin-Castillo B, López-Bonet E, Alarcón T, Vazquez-Martin A. The Warburg effect version 2.0: metabolic reprogramming of cancer stem cells. Cell Cycle. 2013;12:1166–1179. doi: 10.4161/cc.24479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang G, Yang P, Guo P, Miele L, Sarkar FH, Wang Z, Zhou Q. Unraveling the mystery of cancer metabolism in the genesis of tumor-initiating cells and development of cancer. Biochim Biophys Acta. 2013;1836:49–59. doi: 10.1016/j.bbcan.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013;140:2535–2547. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao S, Liu H, Liu Y, Wu J, Wang C, Hou X, Chen X, Yang G, Zhao L, Che H, Bi Y, Wang H, Peng F, Ai J. miR-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013;333:253–260. doi: 10.1016/j.canlet.2013.01.039. [DOI] [PubMed] [Google Scholar]
- 5.Palorini R, Votta G, Balestrieri C, Monestiroli A, Olivieri S, Vento R, Chiaradonna F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J Cell Biochem. 2014;115:368–379. doi: 10.1002/jcb.24671. [DOI] [PubMed] [Google Scholar]
- 6.Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY, Cai S, Scheeren FA, Kuo AH, Diehn M. Targeting Unique Metabolic Properties of Breast Tumor Initiating Cells. Stem Cells. 2014 Feb 4; doi: 10.1002/stem.1662. doi: 10.1002/stem.1662. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuperlovic-Culf M, Culf AS, Touaibia M, Lefort N. Targeting the latest hallmark of cancer: another attempt at ‘magic bullet’ drugs targeting cancers’ metabolic phenotype. Future Oncol. 2012;8:1315–1330. doi: 10.2217/fon.12.121. [DOI] [PubMed] [Google Scholar]
- 8.Butler EB1, Zhao Y, Muñoz-Pinedo C, Lu J, Tan M. Stalling the engine of resistance: targeting cancer metabolism to overcome therapeutic resistance. Cancer Res. 2013;73:2709–2717. doi: 10.1158/0008-5472.CAN-12-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013 Mar 7;4:e532. doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- 12.Watson J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013;3:120144. doi: 10.1098/rsob.120144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat. 2011;126:355–364. doi: 10.1007/s10549-010-0924-x. [DOI] [PubMed] [Google Scholar]
- 14.Bao B, Wang Z, Ali S, Ahmad A, Azmi AS, Sarkar SH, Banerjee S, Kong D, Li Y, Thakur S, Sarkar FH. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila) 2012;5:355–364. doi: 10.1158/1940-6207.CAPR-11-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, Martin-Castillo B, Menendez JA. Metformin: multi-faceted protection against cancer. Oncotarget. 2011;2:896–917. doi: 10.18632/oncotarget.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cufi S, Corominas-Faja B, Vazquez-Martin A, Oliveras-Ferraros C, Dorca J, Bosch-Barrera J, Martin-Castillo B, Menendez JA. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget. 2012;3:395–398. doi: 10.18632/oncotarget.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato A, Sunayama J, Okada M, Watanabe E, Seino S, Shibuya K, Suzuki K, Narita Y, Shibui S, Kayama T, Kitanaka C. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl Med. 2012;1:811–824. doi: 10.5966/sctm.2012-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vazquez-Martin A, Cufi S, Lopez-Bonet E, Corominas-Faja B, Oliveras-Ferraros C, Martin-Castillo B, Menendez JA. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci Rep. 2012;2:964. doi: 10.1038/srep00964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Würth R, Pattarozzi A, Gatti M, Bajetto A, Corsaro A, Parodi A, Sirito R, Massollo M, Marini C, Zona G, Fenoglio D, Sambuceti G, Filaci G, Daga A, Barbieri F, Florio T. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle. 2013;12:145–156. doi: 10.4161/cc.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci U S A. 2013;110:972–977. doi: 10.1073/pnas.1221055110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lonardo E, Cioffi M, Sancho P, Sanchez-Ripoll Y, Trabulo SM, Dorado J, Balic A, Hidalgo M, Heeschen C. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS One. 2013;8:e76518. doi: 10.1371/journal.pone.0076518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu P, Davis M, Blackwelder AJ, Bachman N, Liu B, Edgerton S, Williams LL, Thor AD, Yang X. Metformin Selectively Targets Tumor-Initiating Cells in ErbB2-Overexpressing Breast Cancer Models. Cancer Prev Res (Phila) 2014;7:199–210. doi: 10.1158/1940-6207.CAPR-13-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohammed A, Janakiram NB, Brewer M, Ritchie RL, Marya A, Lightfoot S, Steele VE, Rao CV. Antidiabetic Drug Metformin Prevents Progression of Pancreatic Cancer by Targeting in Part Cancer Stem Cells and mTOR Signaling. Transl Oncol. 2013;6:649–659. doi: 10.1593/tlo.13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 25.Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer. 2009;100:1369–72. doi: 10.1038/sj.bjc.6605007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menendez JA. Fine-tuning the lipogenic/lipolytic balance to optimize the metabolic requirements of cancer cell growth: molecular mechanisms and therapeutic perspectives. Biochim Biophys Acta. 2010;1801:381–91. doi: 10.1016/j.bbalip.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Rajput S, Watabe K, Liao DF, Cao D. Acetyl-CoA carboxylase-a as a novel target for cancer therapy. Front Biosci (Schol Ed) 2010;2:515–526. doi: 10.2741/s82. [DOI] [PubMed] [Google Scholar]
- 28.Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010;6:551–62. doi: 10.2217/fon.10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zadra G, Priolo C, Patnaik A, Loda M. New strategies in prostate cancer: targeting lipogenic pathways and the energy sensor AMPK. Clin Cancer Res. 2010;16:3322–3328. doi: 10.1158/1078-0432.CCR-09-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vazquez-Martin A, Corominas-Faja B, Cufi S, Vellon L, Oliveras-Ferraros C, Menendez OJ, Joven J, Lupu R, Menendez JA. The mitochondrial H(+)-ATP synthase and the lipogenic switch: new core components of metabolic reprogramming in induced pluripotent stem (iPS) cells. Cell Cycle. 2013;12:207–218. doi: 10.4161/cc.23352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A, Xing F, Fukuda K, Hirota S, Sugai T, Wakabayashi G, Koeda K, Kashiwaba M, et al. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat. 2011;130:387–398. doi: 10.1007/s10549-010-1300-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li G, Zhao F, Cui Y. Proteomics using mammospheres as a model system to identify proteins deregulated in breast cancer stem cells. Curr Mol Med. 2013;13:459–463. [PubMed] [Google Scholar]
- 33.Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Araúzo-Bravo MJ, Kovacs WJ, Karalay O, Suter U, Machado RA, Roccio M, Lutolf MP, Semenkovich CF, et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. 2013;493:226–230. doi: 10.1038/nature11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Folmes CD, Park S, Terzic A. Lipid metabolism greases the stem cell engine. Cell Metab. 2013;17:153–155. doi: 10.1016/j.cmet.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Pandey PR, Xing F, Sharma S, Watabe M, Pai SK, Iiizumi-Gairani M, Fukuda K, Hirota S, Mo YY, Watabe K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene. 2013;32:5111–5122. doi: 10.1038/onc.2012.519. [DOI] [PubMed] [Google Scholar]
- 36.Vahlensieck HF, Pridzun L, Reichenbach H, Hinnen A. Identification of the yeast ACC1 gene product (acetyl-CoA carboxylase) as the target of the polyketide fungicide soraphen A. Curr Genet. 1994;25:95–100. doi: 10.1007/BF00309532. [DOI] [PubMed] [Google Scholar]
- 37.Weatherly SC, Volrath SL, Elich TD. Expression and characterization of recombinant fungal acetyl-CoA carboxylase and isolation of a soraphen-binding domain. Biochem J. 2004;380:105–110. doi: 10.1042/BJ20031960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen Y, Volrath SL, Weatherly SC, Elich TD, Tong L. A mechanism for the potent inhibition of eukaryotic acetyl-coenzyme A carboxylase by soraphen A, a macrocyclic polyketide natural product. Mol Cell. 2004;16:881–891. doi: 10.1016/j.molcel.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 39.Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007;67:8180–8187. doi: 10.1158/0008-5472.CAN-07-0389. [DOI] [PubMed] [Google Scholar]
- 40.Raymer B, Kavana M, Price A, Wang B, Corcoran L, Kulathila R, Groarke J, Mann T. Synthesis and characterization of a BODIPY-labeled derivative of Soraphen A that binds to acetyl-CoA carboxylase. Bioorg Med Chem Lett. 2009;19:2804–2807. doi: 10.1016/j.bmcl.2009.03.107. [DOI] [PubMed] [Google Scholar]
- 41.Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniëls VW, Machiels J, Vanderhoydonc F, Smans K, Waelkens E, Verhoeven G, Swinnen JV. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70:8117–8126. doi: 10.1158/0008-5472.CAN-09-3871. [DOI] [PubMed] [Google Scholar]
- 42.Jump DB, Torres-Gonzalez M, Olson LK. Soraphen A, an inhibitor of acetyl CoA carboxylase activity, interferes with fatty acid elongation. Biochem Pharmacol. 2011;81:649–660. doi: 10.1016/j.bcp.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zaidi N, Royaux I, Swinnen JV, Smans K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol Cancer Ther. 2012;11:1925–1935. doi: 10.1158/1535-7163.MCT-12-0095. [DOI] [PubMed] [Google Scholar]
- 44.Vazquez-Martin A1, Oliveras-Ferraros C, Colomer R, Brunet J, Menendez JA. Low-scale phosphoproteome analyses identify the mTOR effector p70 S6 kinase 1 as a specific biomarker of the dual-HER1/HER2 tyrosine kinase inhibitor lapatinib (Tykerb) in human breast carcinoma cells. Ann Oncol. 2008;19:1097–1109. doi: 10.1093/annonc/mdm589. [DOI] [PubMed] [Google Scholar]
- 45.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanai JI, Doro N, Seth P, Sukhatme VP. ATP citrate lyase knockdown impacts cancer stem cells in vitro. Cell Death Dis. 2013;4:e696. doi: 10.1038/cddis.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cai J, Niu X, Chen Y, Hu Q, Shi G, Wu H, Wang J, Yi J. Emodin-induced generation of reactive oxygen species inhibits RhoA activation to sensitize gastric carcinoma cells to anoikis. Neoplasia. 2008;10:41–51. doi: 10.1593/neo.07754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park EK, Park MJ, Lee SH, Li YC, Kim J, Lee JS, Lee JW, Ye SK, Park JW, Kim CW, Park BK, Kim YN. Cholesterol depletion induces anoikis-like apoptosis via FAK down-regulation and caveolae internalization. J Pathol. 2009;218:337–349. doi: 10.1002/path.2531. [DOI] [PubMed] [Google Scholar]
- 49.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guadamillas MC, Cerezo A, Del Pozo MA. Overcoming anoikis--pathways to anchorage-independent growth in cancer. J Cell Sci. 2011;124:3189–3197. doi: 10.1242/jcs.072165. [DOI] [PubMed] [Google Scholar]
- 51.Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, Aguirre-Ghiso JA. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. 2011;31:3616–3629. doi: 10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim YN, Koo KH, Sung JY, Yun UJ, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. Int J Cell Biol. 2012;2012:306879. doi: 10.1155/2012/306879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma I, Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2011;7:292–306. doi: 10.1007/s12015-010-9208-4. [DOI] [PubMed] [Google Scholar]
- 55.Greve B, Kelsch R, Spaniol K, Eich HT, Götte M. Flow cytometry in cancer stem cell analysis and separation. Cytometry A. 2012;81:284–293. doi: 10.1002/cyto.a.22022. [DOI] [PubMed] [Google Scholar]
- 56.Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res Treat. 2012;133:75–87. doi: 10.1007/s10549-011-1692-y. [DOI] [PubMed] [Google Scholar]
- 57.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27:6120–6130. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tong L, Harwood HJ., Jr Acetyl-coenzyme A carboxylases: versatile targets for drug discovery. J Cell Biochem. 2006;99:1476–88. doi: 10.1002/jcb.21077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang C, Rajput S, Watabe K, Liao DF, Cao D. Acetyl-CoA carboxylase-a as a novel target for cancer therapy. Front Biosci (Schol Ed) 2010;2:515–526. doi: 10.2741/s82. [DOI] [PubMed] [Google Scholar]
- 60.Luo DX, Tong DJ, Rajput S, Wang C, Liao DF, Cao D, Maser E. Targeting acetyl-CoA carboxylases: small molecular inhibitors and their therapeutic potential. Recent Pat Anticancer Drug Discov. 2012;7:168–184. doi: 10.2174/157489212799972918. [DOI] [PubMed] [Google Scholar]
- 61.DePeralta DK, Wei L, Harriman G, Greenwood J, Bhat S, Westlin W, Harwood HJ, Jr., Kapeller R, Tanabe KK, Fuchs BC. Liver selective Acetyl-CoA Carboxylase inhibition by ND-654 decreases hepatocellular carcinoma development in cirrhotic rats. AACR Meeting Abstracts 2014. Abstract#1427. [Google Scholar]
- 62.Svensson R, Harriman G, Greenwood J, Bhat S, Harwood HJ, Kapeller R, Shaw R. Acetyl-CoA carboxylase inhibition by ND646 reduces fatty acid synthesis and inhibits cell proliferation in human non-small cell lung cancer cells. AACR Meeting Abstracts 2014. Abstract#2679. [Google Scholar]
- 63.Bass TM, Weinkove G, Hourthoofd K, Gems D, Partridge L. Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech Ageing Dev. 2007;128:546–552. doi: 10.1016/j.mad.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 64.Halicka HD, Zhao H, Li J, Traganos F, Studzinski GP, Darzynkiewicz Z. Attenuation of constitutive DNA damage signaling by 1,25-dihydroxyvitamin D3. Aging (Albany NY) 2012;4:270–8. doi: 10.18632/aging.100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, Darzynkiewicz Z. Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage-signaling. Aging (Albany NY) 2012;4:952–965. doi: 10.18632/aging.100521. [DOI] [PMC free article] [PubMed] [Google Scholar]