

Abstract
A major impediment to the response of tumors to chemotherapy is that the large majority of cancer cells within a tumor are quiescent in G0/G1, where cancer cells are resistant to chemotherapy. To attempt to solve this problem of quiescent cells in a tumor, cancer cells were treated with recombinant methioninase (rMETase), which selectively traps cancer cells in S/G2. The cell cycle phase of the cancer cells was visualized with the fluorescence ubiquitination cell cycle indicator (FUCCI). At the time of rMETase-induced S/G2-phase blockage, identified by the cancer cells' green fluorescence by FUCCI imaging, the cancer cells were administered S/G2-dependent chemotherapy drugs, which interact with DNA or block DNA synthesis such as doxorubicin, cisplatin, or 5-fluorouracil. Treatment of cancer cells with drugs only, without rMETase-induced S/G2 phase blockage, led to the majority of the cancer-cell population being blocked in G0/G1 phase, identified by the cancer cells becoming red fluorescent in the FUCCI system. The G0/G1 blocked cells were resistant to the chemotherapy. In contrast, trapping of cancer cells in S/G2 phase by rMETase treatment followed by FUCCI-imaging-guided chemotherapy was highly effective in killing the cancer cells.
Keywords: cell cycle, FUCCI, imaging, S/G2 phase block, recombinant methioninase, rMETase, chemotherapy, HeLa cells, MCF-7 cells
INTRODUCTION
The problem of quiescent cancer cells within a tumor
The resistance of most solid tumors and metastases is a major problem in chemotherapy. The phase of the cell cycle determines to a great extent whether a cancer cell can respond to a given drug. We previously monitored real-time cell cycle dynamics of cancer cells throughout a live tumor intravitally using a fluorescence ubiquitination cell cycle indicator (FUCCI). Approximately 90% of cancer cells in the center and 80% of total cells of an established tumor are in G0/G1 phase. Similarly, approximately 75% of cancer cells far from (> 100 μm) tumor blood vessels of an established tumor are in G0/G1. FUCCI imaging demonstrated that cytotoxic agents killed only proliferating cancer cells at the surface, or near blood vessels, and had little effect on the majority of quiescent cancer cells within a tumor. Resistant quiescent cancer cells restarted cycling after the cessation of chemotherapy. Thus, the low chemo-sensitivity of most solid tumors is at least in part due to the large majority of cancer cells in solid tumors being quiescent [1].
FUCCI imaging was used for real-time visualization of the cell cycle kinetics of invading cancer cells in 3-dimensional (3D) Gelfoam® histoculture. Cancer cells in G0/G1 phase in Gelfoam® histoculture migrated more rapidly and further than the cancer cells in S/G2/M phase. After entry into S/G2/M phases, cancer cells ceased migrating and restarted migrating after division when the cells re-entered G0/G1. Migrating cancer cells were resistant to cytotoxic chemotherapy, since they were mostly in G0/G1 [2].
One solution to the problem of large numbers of cells in G0/G1 in a tumor is to block the cancer cells in S/G2 rather than G0/G1.
Methionine dependence
Methionine dependence, the elevated methionine requirement for cancer cell proliferation, is the property of the majority of cancer cell types [3]. There have been several therapeutic strategies to target the methionine dependence of cancer cells. Methionine-starvation therapy, such as with a methionine-free diet or methionine-depleted total parenteral nutrition, prolonged the survival time of tumor-bearing rodents [4, 5]. Methionine-free total parenteral nutrition in combination with chemotherapeutic drugs extended the survival of patients with high-stage gastric cancer [4]. Prostate-cancer patients have been treated with a methionine-depleted diet [5].
A reversible growth arrest of cancer cells has been produced by replacement of methionine in the growth medium by its immediate metabolic precursor, homocysteine, This growth arrest is accompanied by a reduction in the percentage of mitotic cells in the cell population. Furthermore, fluorescence-activated cell sorting demonstrated that the cells are arrested at the S/G2 phase of the cell cycle. This is in contrast to a G1-phase accumulation of cancer cells, which occurs only in methionine-supplemented medium at very high densities and which is similar to the G1 block seen in cultures of normal fibroblasts at high density [6]. The molecular mechanism of the S/G2 block has subsequently been investigated [7].
The S/G2 trap that that cancer cells enter upon methionine starvation was exploited for selective chemotherapy in vitro. In cultures that were initiated with equal amounts of cancer cells and human diploid fibroblasts, substitution of homocysteine and doxorubicin for methionine in the culture medium followed by methionine repletion with vincristine was totally effective at selectively eliminating a methionine-dependent human sarcoma and three methionine-dependent human carcinomas. This chemotherapeutic procedure used was not toxic to normal cells growing alongside the cancer cells and was ineffective when conducted totally in methionine-containing medium [8].
In the present report, we demonstrate that using recombinant methioninase (rMETase) to deplete methionine and thereby trap cancer cells in S/G2, and FUCCI imaging to detect the onset of the block, chemotherapy could become effective on the S/G2-trapped cancer cells.
RESULTS AND DISCUSSION
Recombinant methionine (rMETase) block of cancer cells in S/G2 visualized by FUCCI imaging
After seeding on 35 mm glass dishes and culture over night, HeLa cells were treated with rMETase at a dose of 1.0 unit/ml. rMETase blocked HeLa and MCF-7 cells in the S/G2 phase of the cell cycle as visualized by FUCCI imaging (Figure 1).
Figure 1. Time-lapse imaging of FUCCI-expressing HeLa cells treated with rMETase.

After seeding on 35 mm glass dishes and culture over night, HeLa cells were treated with rMETase at a dose of 1.0 unit/ml. All images were acquired with the FV1000 confocal microscope (Olympus, Tokyo, Japan). The cells in G0/G1, S, or G2/M phases appear red, yellow, or green, respectively. Scale bar: 50 μm.
After seeding on 35 mm glass dishes and culture over night, HeLa and MCF-7 cells were treated with rMETase either at 0.25 or 0.5 units/ml for 48 hours. FUCCI imaging showed that by 24 hours there was a large shift in the cancer-cell population from G0/G1 to S/G2/M (Figure 2). For HeLa cells, the percentage of cells in G0/G1 decreased from approximately 80% to approximately 20% by 48 hours in the presence of either 0.25 or 0.5 units/ml rMETase. Approximately 80% of the population became blocked in S/G2. For MCF-7 cells, approximately 40% of the untreated cells were in G0/G1. After 48 hours treatment with 0.25 units/ml rMETase, the percentage of cells in G0/G1 fell to 20% and with 0.5 units rMETase, the percentage decreased to approximately 15%. Approximately 85% of the cells became trapped in S/G2 (Figure 2).
Figure 2. rMETase traps cancer cells in S/G2 phase.

After seeding on 35 mm glass dishes and culture over night, HeLa cells and MCF-7 cells were treated with rMETase, at the indicated doses, for 48 hours. a. Representative images of control or rMETase-treated cells. b. Histogram shows the percentages of cells in G1 (red), early S (yellow), or late S/G2/M (green). Cells at each cell cycle phase were quantitatively assessed by counting the number of cells with each color. N=5 experiments were analyzed. Scale bars: 50 μm.
Cytotoxic chemotherapy drugs killed cancer cells trapped in S/G2 by rMETase
During rMETase-induced blockage in S/G2, doxorubicin (DOX) effectively killed the cancer cells. After overnight culture, HeLa cells and MCF-7 cells were treated with 0.25 unit/ml rMETase for 48 hours. The cancer cells were then treated with 0.5 μg/ml DOX (HeLa cells) or 2.5 μg/ml DOX (MCF-7) for 72 hours. HeLa cells were also treated with 0.5 μg/ml DOX, and MCF-7 cells were treated with 2.5 μg/ml DOX for 72 hours, both without rMETase. With HeLa cells, DOX treatment alone resulted in an increase of cells in G0/G1 from approximately 60% to 80%. With combination treatment of DOX and rMETase, the number of HeLa cells in G0/G1 was reduced to approximately 0 with almost all cells blocked in S/G2. For MCF-7 cells, approximately 40% of the untreated cells were in G0/G1 and increased to more than 80% after treatment with DOX alone for 72 hours. In the presence of rMETase and DOX for 72 hours, approximately 40% of the cells were in G0/G1 (Figure 3).
Figure 3. FUCCI cell cycle analysis during chemotherapy with and without rMETase.

After overnight culture, HeLa cells (a, b and c) and MCF-7 cells (d, e and f) were treated with 0.25 unit/ml rMETase for 48 hours and, then were treated with 0.5 μg/ml doxorubicin (HeLa cells) or 2.5 μg/ml doxorubicin (MCF-7) for 72 hours. For conventional chemotherapy, after culture for 48 hours, HeLa cells (a, b and c) and MCF-7 cells (d, e and f) were treated with 0.5 μg/ml DOX (HeLa cells) or 2.5 μg/ml DOX (MCF-7) for 72 hours. a, b, d, e Representative images acquired with the FV-1000 are shown. c, f, Histograms show the percentages of cells G1(red), early-S (yellow), or late-S/G2/M (green). Cells at each cell cycle phase were quantitatively assessed by counting the number of cells with each color. N=5 experiments were analyzed. Scale bars: 50 μm.
DOX alone, at 2.5 μg/ml, killed approximately 25% of the MCF-7 cells. The combination of DOX, at 2.5 μg/ml, and rMETase, at 0.25 units/ml, killed approximately 80% of the cells (P<0.01 compared to DOX alone). DOX, at 5 μ/ml, and rMETase, at 0.25 units/ml, killed approximately 90% of the cells (P<0.01 compared to DOX alone) (Figure 4).
Figure 4. Chemotherapy of S/G2-trapped cancer cells.
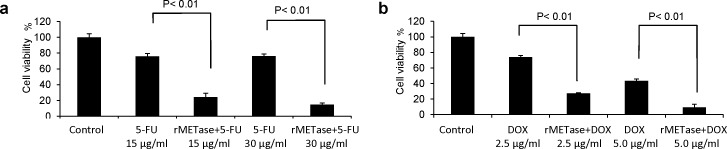
Efficacy of combination therapy of rMETase and 5-FU (a); rMETase and DOX (b); on FUCCI-expressing MCF-7 cells. Cell viability was assessed by counting living cells compared to control. Data bars means ± SD of triplicate samples.
For MCF-7 cells treated with 5-FU, at 15 μg/ml, approximately 40% of the cells were killed. With the combination of rMETase (0.25 units/ml) and 15 μg/ml 5-FU, approximately 80% of the cells were killed (P<0.01 compared to 5-FU alone). With 5-FU, at 30 μg/ml, approximately 40% of the cells were killed, and the combination of 5-FU, at 30 μg/ml, and 0.25 units/ml rMETase, approximately 90% of the cells were killed (P<0.01 compared to 5-FU alone) (Figure 4).
The strategy and technology described in this report, whereby cancer cells are selectively and synchronously trapped by rMETase in S/G2, the most drug-sensitive phase of the cell cycle, where they were identified by FUCCI imaging and then treated with S/G2-phase-specific-drugs was highly effective compared to standard chemotherapy.
Previously developed concepts and strategies of highly selective tumor-targeting [9-20] can take advantage of spatial–temporal cell-cycle imaging of cancer cells described in the present and previous [1, 2, 21] reports.
Previously, different methods of cancer-cell synchronization have been used in order to sensitize the cells to chemotherapy. Such methods include chronotherapy which attempts to target the time of day when most cancer cells in tumors are thought to be dividing [22]. Excess thymidine or its analogs have also been used to arrest cancer cells in S-phase, where they are sensitized to S-phase drugs such as 5-FU or methotrexate, and after release of the block, the cancer cells enter M-phase synchronously where they are sensitive to M-Phase drugs such as paclitaxel [23-25].
Cancer-cell synchronization with cell-cycle-phase-specific drugs, such as cytosine arabinoside, methotrexate and hydroxyorea have also been carried out [26-30], for example, to block cells in S-phase which can sensitize the cancer cells to an M-phase drug, such as paclitaxel, administered after the S-phase block is lifted [26-30].
The calcium channel blocker mibefradil has been used to synchronize glioblastoma cells at the G1/S checkpoint, thereby making the glioblastoma cells sensitive to first-line therapy temozolomide [31]. Statins, such as Lovastatin, can be used to synchronize cancer in G1 by preventing formation of an early intermediate in the cholesterol pathway essential for progression of cells through early G1 phase [32, 33]. After the block is lifted, the cancer cells can be effectively treated with an S-phase drug.
PDO332991, a pyridopyrimidine, has been shown to be a selective inhibitor of cyclin-dependent kinases 4 and 6 and induced early-G1 arrest in primary human myeloma cells and other cancer cell types, including breast cancer in vitro and in cancer xenograft models. As PDO0332991 acts reversibly, it can be used as a synchronizing agent and when used for sequence combination with cytotoxic agents is active against myeloma cells in vitro and in vivo [34]. A cyclin-dependent kinase inhibitor RO-3306 reversibly arrests 95% of treated cells in G2 phase. These cells rapidly enter mitosis after the block is lifted and become sensitive to M-phase drugs [35]. Growth factors such as EGF, G-CSF, and IL-6 can stimulate cancer cell out of G0, making them sensitive to chemotherapy agents such as docetaxel [36-38]. Reviews on cell synchronization are available [39-42].
The critical advantage of rMETase synchronization (blockage) is that, unlike the methods described above, it is cancer specific [3,6,8,43-51].
CONCLUSIONS
A major problem for successful chemotherapy is the very high percentage of quiescent G0/G1 cancer cells in a tumor. The present report has demonstrated a solution to the problem by selectively trapping cancer cells in S/G2, with recombinant methioninase (rMETase). The S/G2-trapped cancer cells became sensitive to chemotherapy which targets cells in this phase of the cell cycle, which are the majority of the most widely-used chemotherapy drugs. Alternatively, the rMETase-induced S/G2 block can be lifted and the cells can become sensitive to M-phase drugs. This approach has significant clinical potential since almost all cancer cell types tested are methionine dependent and arrest in S/G2 when deprived of methionine with an agent such as rMETase.
MATERIALS AND METHODS
Recombinant Methioninase (rMETase)
Recombinant L-methionine α-deamino-γ-mercaptomethane lyase (methioninase, METase) [EC 4.4.1.11] from Pseudomonas putida has been previously cloned and was produced in Escherichia coli (AntiCancer, Inc., San Diego, CA). rMETase is a homotetrameric PLP enzyme of 172-kDa molecular mass [52].
FUCCI (Fluorescence ubiquitination cell cycle indicator)
The FUCCI probe was generated by fusing mKO2 (monomeric Kusabira Orange2) and mAG (monomeric Azami Green) to the ubiquitination domains of human Cdt1 and geminin, respectively. These two chimeric proteins, mKO2-hCdt1(30/120) and mAG-hGem(1/110), accumulate reciprocally in the nuclei of transfected cells during the cell cycle, labeling the nuclei of G1 phase cells red and nuclei of cells in S/G2 phase green [53].
FUCCI-expressing HeLa cells and MCF-7 cells
Plasmids expressing mKO2-hCdt1 or mAG-hGem (MBL, Nagoya, Japan) were transfected into HeLa cells and MCF-7 cells. HeLa cells were grown in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin. MCF-7 were grown in MEM-supplemented with L-glutamine and 10% fetal bovine serum and penicillin/streptomycin [53].
Imaging of FUCCI-expressing cancer cells
Time-lapse images of HeLa and MCF-7 cells stably transfected with FUCCI vectors were acquired using a confocal laser scanning microscope (FV1000; Olympus, Tokyo, Japan) [1, 2, 21].
Cell viability
For cell viability determinations before and after chemotherapy, with and without rMETase, the cells were stained with crystal violet, and the relative number of cells was quantified using ImageJ (NIH, Bethesda, MD).
DEDICATION
This paper is dedicated to the memory of A. R. Moossa, MD.
Acknowledgments
This work was supported by National Cancer Institute grant CA132971.
Abbreviations
- rMETase
recombinant methioninase
- FUCCI
fluorescence ubiquitination cell cycle indicator
Footnotes
CONFLICTS OF INTEREST
S.L., Q.H. and Y.T. are employees of AntiCancer Inc. S.Y. and R.M.H. are unsalaried associates of AntiCancer Inc. There are no other competing financial interests.
REFERENCES
- 1.Yano S, Zhang Y, Miwa S, Tome Y, Hiroshima Y, Uehara F, Yamamoto M, Suetsugu A, Kishimoto H, Tazawa H, Zhao M, Bouvet M, Fujiwara T, Hoffman RM. Spatial-temporal FUCCI imaging of each cell in a tumor demonstrates locational dependence of cell cycle chemoresponsiveness. Cell Cycle. 2014;13:2110–2119. doi: 10.4161/cc.29156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yano S, Miwa S, Mii S, Hiroshima Y, Uehara F, Yamamoto M, Kishimoto H, Tazawa H, Bouvet M, Fujiwara T, Hoffman RM. Invading cancer cells are predominantly in G0/G1 resulting in chemoresistance demonstrated by real-time FUCCI imaging. Cell Cycle. 2014;13:953–960. doi: 10.4161/cc.27818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman RM. Altered methionine metabolism, DNA methylation and oncogene expression in carcinogenesis: a review and synthesis. Biochim Biophys Acta Reviews on Cancer. 1984;738:49–87. doi: 10.1016/0304-419x(84)90019-2. [DOI] [PubMed] [Google Scholar]
- 4.Goseki N, Yamazaki S, Shimojyu K, Kando F, Maruyama M, Endo M, Koike M, Takahashi H. Synergistic effect of methionine-depleting total parenteral nutrition with 5-fluorouracil on human gastric cancer: A randomized, prospective clinical trial. Jpn J Cancer Res. 1995;86:484–489. doi: 10.1111/j.1349-7006.1995.tb03082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epner DE, Morrow S, Wilcox M, Houghton JL. Nutrient intake and nutritional indexes in adults with metastatic cancer on a Phase I clinical trial of dietary methionine restriction. Nutrition and Cancer. 2002;42:158–166. doi: 10.1207/S15327914NC422_2. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman RM, Jacobsen SJ. Reversible growth arrest in simian virus 40-transformed human fibroblasts. Proc Natl Acad Sci USA. 1980;77:7306–7310. doi: 10.1073/pnas.77.12.7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu S, Epner DE. Molecular mechanisms of cell cycle block by methionine restriction in human prostate cancer cells. Nutrition and Cancer. 2000;38:123–130. doi: 10.1207/S15327914NC381_17. [DOI] [PubMed] [Google Scholar]
- 8.Stern PH, Hoffman RM. Enhanced in vitro selective toxicity of chemotherapeutic agents for human cancer cells based on a metabolic defect. J Natl Cancer Inst. 1986;76:629–639. doi: 10.1093/jnci/76.4.629. [DOI] [PubMed] [Google Scholar]
- 9.Blagosklonny MV. How cancer could be cured by 2015. Cell Cycle. 2005;4:269–78. [PubMed] [Google Scholar]
- 10.Blagosklonny MV. Tissue-selective therapy of cancer. Br J Cancer. 2003;89:1147–51. doi: 10.1038/sj.bjc.6601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blagosklonny MV. Matching targets for selective cancer therapy. Drug Discov Today. 2003;8:1104–7. doi: 10.1016/s1359-6446(03)02806-x. [DOI] [PubMed] [Google Scholar]
- 12.Blagosklonny MV. “Targeting the absence” and therapeutic engineering for cancer therapy. Cell Cycle. 2008;7:1307–12. doi: 10.4161/cc.7.10.6250. [DOI] [PubMed] [Google Scholar]
- 13.Blagosklonny MV. Teratogens as anti-cancer drugs. Cell Cycle. 2005;4:1518–21. doi: 10.4161/cc.4.11.2208. [DOI] [PubMed] [Google Scholar]
- 14.Blagosklonny MV. Treatment with inhibitors of caspases, that are substrates of drug transporters, selectively permits chemotherapy-induced apoptosis in multidrug-resistant cells but protects normal cells. Leukemia. 2001;15:936–41. doi: 10.1038/sj.leu.2402127. [DOI] [PubMed] [Google Scholar]
- 15.Blagosklonny MV. Target for cancer therapy: proliferating cells or stem cells. Leukemia. 2006;20:385–91. doi: 10.1038/sj.leu.2404075. [DOI] [PubMed] [Google Scholar]
- 16.Blagosklonny MV. Cancer stem cell and cancer stemloids: from biology to therapy. Cancer Biol Ther. 2007;6:1684–90. doi: 10.4161/cbt.6.11.5167. [DOI] [PubMed] [Google Scholar]
- 17.Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011;2:222–33. doi: 10.18632/oncotarget.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao B, van Leeuwen IM, Higgins M, Campbel J, Thompson AM, Lane DP, Lain S. Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget. 2010;1:639–50. doi: 10.18632/oncotarget.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blagosklonny MV. NCI's provocative questions on cancer: some answers to ignite discussion. Oncotarget. 2011;2:1352–67. doi: 10.18632/oncotarget.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blagosklonny MV. Antagonistic drug combinations that select against drug resistance: from bacteria to cancer. Cancer Biol Ther. 2007;6:1013–4. doi: 10.4161/cbt.6.7.4340. [DOI] [PubMed] [Google Scholar]
- 21.Yano S, Tazawa H, Hashimoto Y, Shirakawa Y, Kuroda S, Nishizaki M, Kishimoto H, Uno F, Nagasaka T, Urata Y, et al. A genetically engineered oncolytic adenovirus decoys and lethally traps quiescent cancer stem-like cells into S/G2/M phases. Clin Cancer Res. 2013;19:6495–505. doi: 10.1158/1078-0432.CCR-13-0742. [DOI] [PubMed] [Google Scholar]
- 22.Innominato PF, Roche VP, Palesh OG, Ulusakarya A, Spiegel D, Lévi FA. The circadian timing system in clinical oncology. Ann Med. 2014;46:191–207. doi: 10.3109/07853890.2014.916990. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Pan L, Mao N, Sun L, Qin X, Yin J. Cell-cycle synchronization reverses Taxol resistance of human ovarian cancer cell lines. Cancer Cell Int. 2013;13:77. doi: 10.1186/1475-2867-13-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandrasekaran B, Kute TE, Duch DS. Synchronization of cells in the S phase of the cell cycle by 3′-azido-3′-deoxythymidine: implications for cell cytotoxicity. Cancer Chemother Pharmacol. 1995;35:489–95. doi: 10.1007/BF00686833. [DOI] [PubMed] [Google Scholar]
- 25.Kufe DW, Egan EM, Rosowsky A, Ensminger W, Frei E., 3rd Thymidine arrest and synchrony of cellular growth in vivo. Cancer Treat Rep. 1980;64:1307–17. [PubMed] [Google Scholar]
- 26.Vogler WR, Kremer WB, Knospe WH, Omura GA, Tornyos K. Synchronization with phase-specific agents in leukemia and correlation with clinical response to chemotherapy. Cancer Treat Rep. 1976;60:1845–59. [PubMed] [Google Scholar]
- 27.Morris CM, Fitzgerald PH. An evaluation of high resolution chromosome banding of hematologic cells by methotrexate synchronization and thymidine release. Cancer Genet Cytogenet. 1985;14:275–84. doi: 10.1016/0165-4608(85)90193-1. [DOI] [PubMed] [Google Scholar]
- 28.Moran RE, Straus MJ. Synchronization of L1210 leukemia with hydroxyurea infusion and the effect of subsequent pulse dose chemotherapy. Cancer Treat Rep. 1980;64:81–6. [PubMed] [Google Scholar]
- 29.Dethlefsen LA, Sorensen SP, Riley RM. Effects of double and multiple doses of hydroxyurea on mouse duodenum and mammary tumors. Cancer Res. 1975;35:694–9. [PubMed] [Google Scholar]
- 30.Finzi L, Kraemer A, Capron C, Noullet S, Goere D, Penna C, Nordlinger B, Legagneux J, Emile JF, Malafosse R. Improved retroviral suicide gene transfer in colon cancer cell lines after cell synchronization with methotrexate. J Exp Clin Cancer Res. 2011;30:92. doi: 10.1186/1756-9966-30-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keir ST, Friedman HS, Reardon DA, Bigner DD, Gray LA. Mibefradil, a novel therapy for glioblastoma multiforme: cell cycle synchronization and interlaced therapy in a murine model. J Neurooncol. 2013;111:97–102. doi: 10.1007/s11060-012-0995-0. [DOI] [PubMed] [Google Scholar]
- 32.Keyomarsi K, Sandoval L, Band V, Pardee AB. Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res. 1991;51:3602–9. [PubMed] [Google Scholar]
- 33.Javanmoghadam-Kamrani S, Keyomarsi K. Synchronization of the cell cycle using lovastatin. Cell Cycle. 2008;7:2434–40. doi: 10.4161/cc.6364. [DOI] [PubMed] [Google Scholar]
- 34.Huang X, Di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, Shaffer AL, 3rd, Louie T, Chen I, Randolph S, Hahn WC, Staudt LM, Niesvizky R, Moore MA, Chen-Kiang S. Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood. 2012;120:1095–106. doi: 10.1182/blood-2012-03-415984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vassilev LT. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle. 2006;5:2555–6. doi: 10.4161/cc.5.22.3463. [DOI] [PubMed] [Google Scholar]
- 36.Dong XF, Berthois Y, Dussert C, Isnardon D, Palmari J, Martin PM. Mode of EGF action on cell cycle kinetics in human breast cancer cell line MCF-7: some evidence that EGF acts as a “progression factor”. Anticancer Res. 1992;12:2085–292. [PubMed] [Google Scholar]
- 37.Hambek M, Werner C, Baghi M, Gstöttner W, Knecht R. Enhancement of docetaxel efficacy in head and neck cancer treatment by G0 cell stimulation. Eur J Cancer. 2007;43:1502–7. doi: 10.1016/j.ejca.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 38.Hambek M, Werner C, Baghi M, Gstöttner W, Knecht R. Prestimulation of head and neck cancer cells with growth factors enhances treatment efficacy. Anticancer Res. 2006;26:1091–5. [PubMed] [Google Scholar]
- 39.Grdina DJ, Meistrich ML, Meyn RE. Cell synchrony techniques. A comparison of methods. In: Gray JW, Darzynkiewicz A, editors. Techniques in Cell Cycle Analysis. Humana Press Inc; Clifton, NJ: 1987. pp. 367–403. [Google Scholar]
- 40.Davis PK, Ho A, Dowdy SF. Biological methods for cell-cycle synchronization of mammalian cells. Biotechniques. 2001;30:1322–6. 1328, 1330–1. doi: 10.2144/01306rv01. [DOI] [PubMed] [Google Scholar]
- 41.Merril GF. Cell synchronization. Methods Cell Biol. 1998;57:229–249. [PubMed] [Google Scholar]
- 42.Amon A. Synchronization procedures. Methods Enzymol. 2002;351:457–67. doi: 10.1016/s0076-6879(02)51864-4. [DOI] [PubMed] [Google Scholar]
- 43.Hoffman R.M, Erbe R W. High in vivo rates of methionine biosynthesis in transformed human and malignant rat cells auxotrophic for methionine. Proc. Natl. Acad. Sci. USA. 1976;73:1523–7. doi: 10.1073/pnas.73.5.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoffman R.M, Jacobsen S J, Erbe R.W. Reversion to methionine independence by malignant rat and SV40-transformed human fibroblasts. Biochem. Biophys. Res. Commun. 1978;82:228–34. doi: 10.1016/0006-291x(78)90600-9. [DOI] [PubMed] [Google Scholar]
- 45.Hoffman R.M, Jacobsen S.J, Erbe R.W. Reversion to methionine independence in simian virus 40-transformed human and malignant rat fibroblasts is associated with altered ploidy and altered properties of transformation. Proc Nat!Acad Sci USA. 1979;76:I313–7. doi: 10.1073/pnas.76.3.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coalson D.W, Mecham J.O, Stem P.H, Hoffman R.M. Reduced availability of endogenously synthesized methionine for S-adenosylmethionine formation in methionine-dependent cancer cells. Proc Nat! Acad Sci USA. 1982;79:4248–51. doi: 10.1073/pnas.79.14.4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stem P.H, Mecham 1.0, Wallace C.D, Hoffman R.M. Reduced free-methionine in methionine-dependent SV40-transformed human fibroblasts synthesizing apparently normal amounts of methionine. J Cell Physiol. l983;117:9–14. doi: 10.1002/jcp.1041170103. [DOI] [PubMed] [Google Scholar]
- 48.Mecham 1.0, Rowitch D, Wallace C.D, Stem P.H, Hoffman R.M. The metabolic defect of methionine dependence occurs frequently in human tumor cell lines. Biochem, Biophys Res Commun. 1983;117:429–34. doi: 10.1016/0006-291x(83)91218-4. [DOI] [PubMed] [Google Scholar]
- 49.Stem P.H, Wallace C.D, Hoffman R.M. Altered methionine metabolism occurs in all members of a set of diverse human tumor cell lines. J Cell Physiol. 1984;I I 9:29–34. doi: 10.1002/jcp.1041190106. [DOI] [PubMed] [Google Scholar]
- 50.Stem P.H, Hoffman R.M. Elevated overall rates of transmethylation in cell lines from diverse human tumors. In Vitro. I984;20:663–70. doi: 10.1007/BF02619617. [DOI] [PubMed] [Google Scholar]
- 51.Tan Y, Xu M, Hoffman R.M. Broad selective efficacy of recombinant methioninase and polyethylene glycol-modified recombinant methioninase on cancer cells in vitro. Anticancer Res. 2010;30:1041–6. [PubMed] [Google Scholar]
- 52.Tan Y, Xu M, Tan X, Tan X, Wang X, Saikawa Y, Nagahama T, Sun X, Lenz M, Hoffman RM. Overexpression and large-scale production of recombinant methionine-α-deamino-γ-mercaptomethane-lyase for novel anticancer therapy. Protein Expression and Purification. 1997;9:233–245. doi: 10.1006/prep.1996.0700. [DOI] [PubMed] [Google Scholar]
- 53.Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell cycle progression. Cell. 2008;132:487–98. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]