

Abstract
Use of biomarkers in the detection of early and preclinical Alzheimer’s disease (AD) has become of central importance following publication of the NIA-Alzheimer’s Association revised criteria for the diagnosis of AD, mild cognitive impairment (MCI) and preclinical AD. The use of in vivo amyloid imaging agents, such a Pittsburgh Compound-B and markers of neurodegeneration, such as fluoro-2-deoxy-D-glucose (FDG) are able to detect early AD pathological processes and subsequent neurodegeneration. Imaging with PiB and FDG thus has many potential clinical benefits: early or perhaps preclinical detection of disease and accurately distinguishing AD from dementias of other etiologies in patients presenting with mild or atypical symptoms or confounding comorbidities in which the diagnostic distinction is difficult to make clinically. From a research perspective, this allows us to study relationships between amyloid pathology and changes in cognition, brain structure, and function across the continuum from normal aging to MCI to AD. The present review focuses on use of PiB and FDG-PET and their relationship to one another.
Keywords: Amyloid, Glucose metabolism, Alzheimer’s disease, Pittsburgh Compound B, FDG, neuroimaging
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly with a worldwide prevalence estimated to quadruple over the next 50 years. AD is pathologically characterized by the presence of amyloid plaques, containing amyloid-beta (Aβ), and neurofibrillary tangles, containing hyperphosphorylated tau, as well as significant loss of neurons and deficits in neurotransmitter systems. A growing consensus points to deposition of Aβ plaques as a central event in the pathogenesis of AD. This “amyloid cascade hypothesis” (Hardy and Allsop, 1991; Hardy and Higgins, 1992) states that overproduction of Aβ, or failure to clear this peptide, leads to AD primarily through amyloid deposition, which triggers the production of NFT, cell death and, ultimately, the clinical symptoms such as memory loss and cognitive impairment (Hardy et al., 1998).
Definitive diagnosis of AD relies on the demonstration of sufficient amounts of Aβ plaques and NFT in autopsy brains (Mirra et al., 1991). Recently, the development of the PET tracer Pittsburgh Compound-B has made the in-vivo imaging of amyloid possible, with striking differences in PiB retention observed between control and AD subjects in brain areas known to contain significant amyloid deposits in AD (e.g., frontal cortex and parietal cortex) (Figure 2) (Klunk et al., 2004). Fluoro-2-deoxy-D-glucose (FDG) PET shows decreases in cerebral glucose metabolism, with a characteristic regional pattern of posterior temporoparietal > frontal hypometabolism in AD (Foster et al., 2007; Friedland et al., 1983; Herholz et al., 2007; Jagust et al., 2007) (Figure 2).
Figure 2.
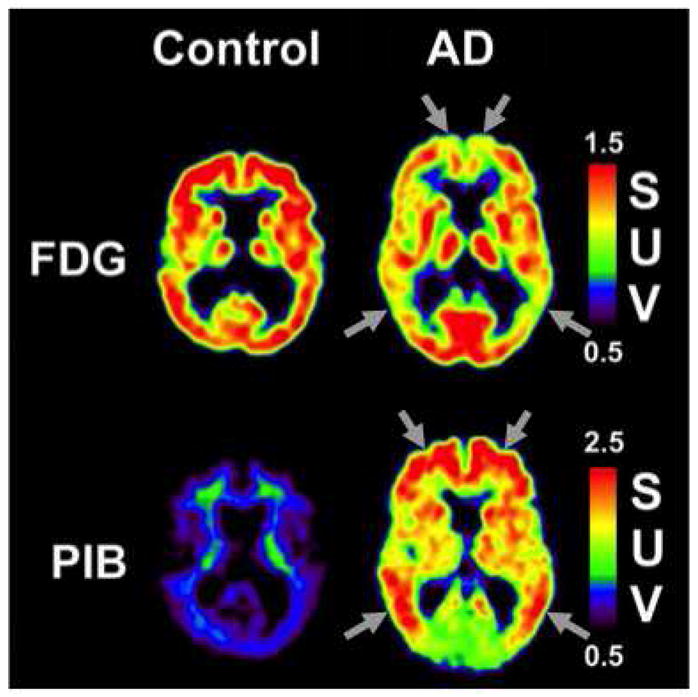
Representative PiB and FDG scans from control and AD participants. Arrows indicate areas of typical hypometabolism (FDG) or typical amyloid deposition (Head et al.).
Imaging AD pathology, using amyloid PET imaging agents such as PiB, and imaging AD neurodegenerative processes, using FDG PET (as well other markers such as structural MRI and CSF tau), have several potential clinical benefits: early or perhaps preclinical detection of disease and accurately distinguishing AD from non-AD dementia in patients with mild or atypical symptoms or confounding comorbidities (in which the distinction is difficult to make clinically). From a research perspective, these imaging techniques allow us to study relationships between amyloid, cognition and neurodegenerative processes across the continuum from normal aging to AD; and to monitor the biological effects of anti-Aβ drugs and relate them to effects on neurodegeneration and cognition. In particular, understanding biomarkers such as PiB and FDG in relation to normal aging has become critical given that we have entered the era of “prevention” trials in AD with two studies targeting autosomal dominant AD (DIAN and API), one study targeting homozygous APOE*4 carriers (API) and one study targeting typical late-onset disease (A4). All of these studies rely heavily on biomarkers in general and on Aβ biomarkers in particular. A key concept underlying these trials is the recently developed NIA-Alzheimer’s Association research criteria for preclinical AD, suggesting that Aβ deposition in cognitively normal individuals is in fact a preclinical stage of AD (Sperling et al., 2011). These criteria were recently operationalized by Jack et al. (Jack et al., 2012), and suggest amyloid biomarkers, including PiB-PET, become abnormal first and are followed by biomarkers of neuronal injury and degeneration, including FDG-PET, closer to the time when cognitive symptoms appear (Figure 1) (Jack et al., 2012). The present review focuses on use of PiB and FDG-PET and their relationship to one another.
Figure 1.

From Jack et al. (2012): Changes in AD biomarker data on the vertical axis vs. AD clinical stage, with preclinical staging highlighted in yellow Each biomarker is scaled from maximally normal (bottom) to maximally abnormal (top) with PET amyloid imaging (red line), biomarkers of neurodegeneration are FDG-PET or atrophy on MRI (blue line), and cognitive (purple line).
Amyloid imaging using PiB PET
The earliest studies with PiB in AD patients showed markedly increased PiB retention was observed in brain areas known to contain high levels of amyloid plaques when compared to HC subjects. In brain regions such as parietal and frontal cortices, the pattern of PiB retention was markedly different in AD patients compared to the HC subjects (Klunk et al., 2004). PiB retention in AD patients was generally most prominent in cortical areas and lower in white matter areas, in a manner most consistent with post-mortem studies of Aβ plaques in the AD brain (Thal et al., 2002). PiB retention was broadly observed in frontal cortex in AD, but also was observed in precuneus/posterior cingulate, temporal and parietal cortices. The occipital cortex and lateral temporal cortex were also significantly affected with a relative sparing of the mesial temporal areas. Significant striatal PiB retention also was observed, consistent with previous reports of extensive Aβ deposition in the striatum of AD patients (Braak and Braak, 1990; Brilliant et al., 1997; Suenaga et al., 1990; Wolf et al., 1999). These original studies provided a landmark description of the natural history of Aβ deposition in living subjects, and were later confirmed by additional studies using PiB in AD patients and cognitively normal subjects (Archer et al., 2006; Buckner et al., 2005; Edison et al., 2006; Fagan et al., 2006; Fagan et al., 2007; Kemppainen et al., 2006; Lopresti et al., 2005; Mintun et al., 2006; Nelissen et al., 2007; Pike et al., 2007; Price et al., 2005a; Rowe et al., 2007; Ziolko et al., 2006).
In early studies of mild cognitive impairment (MCI), PiB appeared to show a bimodal distribution, with 60%–75% of subjects showing a typical, AD-like pattern and burden of PiB retention, while the remaining subjects showed levels typical of PiB-negative [PiB(−)] controls (Jack et al., 2009; Lopresti et al., 2005; Price et al., 2005b; Rowe et al., 2007). Variations in PiB retention have also been explored when examining MCI subjects based on MCI subtype; subjects with non-amnestic MCI were much less likely to be PiB-positive [PiB(+)] than subjects with amnestic MCI (Jack et al., 2008; Kemppainen et al., 2006; Pike et al., 2007; Price et al., 2005a; Rowe et al., 2007), although other studies also found significant PiB retention in non-amnestic MCI (Wolk et al., 2008). These studies have suggested that the non-amnestic MCI subtype may include depression or incipient dementia where Aβ deposition is not a feature (e.g. frontotemporal or vascular dementia), or they may prove to be part of the 5–10% who have stable MCI, or the 20% who revert to apparent normality (Busse et al., 2006; Gauthier et al., 2006).
Longitudinal studies have suggested that MCI subjects with high PiB retention are much more likely to convert to AD than subjects with low PiB retention. In a study by Forsberg and colleagues (2007), all 7 MCI-to-AD converters were amyloid-positive at baseline and 9 of the 14 non-converters were amyloid-negative. In addition, none of the baseline PiB(−) MCI subjects converted to AD. This effect has also been observed is several subsequent studies, with MCI subjects with increased PiB retention showing much more frequent conversion to AD (Koivunen et al., 2011; Villemagne et al., 2011; Wolk et al., 2009). Therefore, amyloid PET is likely to have a prognostic role in the clinical evaluation of MCI, by identifying subjects who have underlying AD pathophysiology and are therefore at high risk for further clinical decline (Albert et al., 2011). Several studies have now demonstrated PiB retention in cognitively normal controls. Depending on the site, reports have ranged from a proportion of 10–30% of normal elderly subjects with significant PiB retention [i.e., PiB(+)] (Figure 3) (Aizenstein et al., 2008; Jack et al., 2008; Kantarci et al., 2012; Klunk et al., 2004; Mintun et al., 2006; Mormino et al., 2009; Mormino et al., 2011; Pike et al., 2007; Reiman et al., 2009; Rowe et al., 2010; Villemagne et al., 2008). Similarly, PiB-PET studies have found that ApoE4 genotype is associated with higher PiB retention in cognitively normal elderly in a dose-dependent manner (Morris et al., 2010; Reiman et al., 2009; Rowe et al., 2010), and ApoE4 carriers are more than twice as likely to convert from PiB(−) to PiB(+) over time (Reiman et al., 2009). Conversely, ApoE2 has been associated with lower PiB retention in normal elderly (Morris et al., 2010). This wide range likely depends on factors like the age of the cohort, proportion of subjects carrying the ApoE4 allele, definition of “cognitively normal” and the threshold for defining amyloid-positivity. The relationship between increased PiB retention and cognition in the normal elderly has been difficult to define. It is apparent that among cognitively normal subjects, significant plaque load is not related to broad differences in cognitive (Aizenstein et al., 2008; Jack et al., 2008; Mintun et al., 2006; Nebes et al., 2013; Rowe et al., 2010). In other studies, an increase in PiB retention has been associated with poorer performance on episodic memory tests (Kantarci et al., 2012; Mormino et al., 2009; Mormino et al., 2011; Pike et al., 2007; Villemagne et al., 2008). Most significantly, longitudinal studies have found that cognitively normal individuals with elevated PiB are at much higher risk for longitudinal cognitive decline and the emergence of clinically significant cognitive impairment than PiB(−) age and education matched subjects (Morris et al., 2010; Resnick and Sojkova, 2011; Storandt et al., 2009; Villemagne et al., 2011; Villemagne et al., 2008). Further, recent theoretical models suggest that the period of time from the first detection of Aβ deposition to levels typically seen in MCI is ~15 years, providing further evidence for an extended preclinical phase of AD (Villemagne et al., 2013).
Figure 3.
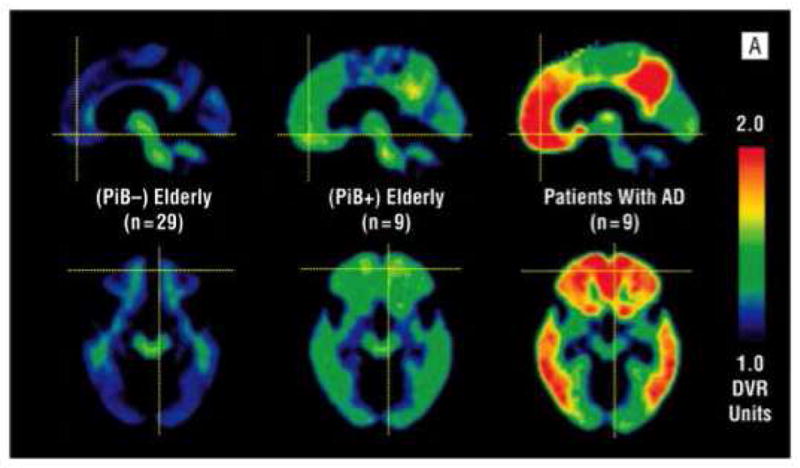
From Aizenstein et al. (2008): Mean distribution volume ratio images for PiB-negative clinically unimpaired participants (left), PiB-positive clinically unimpaired participants (center), and patients with Alzheimer disease (AD) (right).
Cerebral glucose metabolism imaging using FDG PET
Reductions of cerebral metabolism are well established in AD (Lopresti et al., 2005; Minoshima, 2003; Mosconi et al., 2007; Silverman and Alavi, 2005). Similar changes have been reported in cognitively normal individuals at high risk for AD due to expression of the ApoE4 alelle (Reiman et al., 1996; Small et al., 2000). Further, hypometabolism has been reported in cognitively normal individuals with a parent with AD (Mosconi et al., 2009; Mosconi et al., 2014; Mosconi et al., 2008a; Mosconi et al., 2013). Changes in cerebral metabolism also have been detected in MCI in many studies (Arnaiz et al., 2001; Chetelat and Baron, 2003; Chetelat et al., 2003; Del Sole et al., 2008; Garibotto et al., 2008; Li et al., 2008a; Li et al., 2008b; Mevel et al., 2007; Mosconi et al., 2006; Mosconi et al., 2008b; Perneczky et al., 2007). These early changes in suggest FDG could play a predictive role in detecting which normal controls or MCI patients are most likely to convert to AD (Yuan et al., 2008) Indeed, hypometabolism in AD-affected regions in normal, elderly controls has been related to global cognitive decline (de Leon et al., 2001; Jagust et al., 2006) and several studies have shown that abnormalities in FDG PET predict progression from MCI to AD (Anchisi et al., 2005; Drzezga et al., 2005; Mosconi et al., 2004).
Relationship between amyloid deposition and glucose metabolism
Comparisons of PiB and FDG PET data for detection of AD found PiB was more accurate than FDG both on visual reading (accuracy, 90% vs. 70%) and ROC analysis (95% vs. 83%). The authors concluded that the visual analysis of PiB images appears more accurate than visual reading of FDG for identification of AD (Ng et al., 2007). Similar results were found by Rabinovici et al.(Rabinovici et al., 2011) with inter-rater agreement significantly higher for PiB (kappa = 0.96) than FDG (kappa = 0.72), as was agreement between visual and quantitative classifications (average kappa = 0.90 for PiB, 0.66 for FDG). The authors concluded that PiB was the superior qualitative technique in that visual assessment was both more accurate and more precise. While PiB and FDG demonstrate high (94%) agreement in differentiating AD from normal controls, agreement is lower in classifying MCI subjects (54%), arguing for combining the two modalities (Li et al., 2008a). In addition, when exploring the use of PiB and FDG among both AD and MCI subtypes it was demonstrated that while PiB and FDG displayed similar diagnostic accuracy, PiB was significantly better at separating MCI subtypes (Lowe et al., 2009). These findings are not surprising since the two tracers provide complementary information, with PiB quantifying molecular pathology, and FDG demonstrating neuronal dysfunction. The complementary nature of the two techniques are reflected in the new diagnostic guidelines for MCI and AD dementia, which require biomarker evidence of both Aβ deposition (decreased CSF Aβ or elevated amyloid PET) and neurodegeneration (elevated CSF tau, hypometabolism on FDG-PET or atrophy on MRI) to diagnose AD pathophysiology with high-likelihood during life (McKhann AD criteria, Albert MCI criteria). Further, the lowered accuracy of FDG-PET in many of these studies may likely be due to the fact that FDG-PET may reflect hypometabolism and neurodegeneration resulting from non-AD syndromes, such as frontotemporal lobar degenerations (FTLD). In one of the largest comparison studies to date, Rabinovici et al. (2011) tested the diagnostic performance of PiB-PET in distinguishing clinically diagnosed AD (N=62) and FTLD (N=45) patients and compared it to the performance of FDG-PET. PET scans were rated visually (blinded to clinical diagnosis) as PiB-positive or PiB-negative and as consistent with the FDG patterns of AD (temporoparietal-predominant hypometabolism) or FTLD (frontal or anterior temporal-predominant hypometabolism). PiB visual reads were more sensitive for AD than FDG reads (89.5% vs. 77.5%) with similar specificity (83% vs. 84%). PiB outperformed FDG in a subset of 12 patients who underwent autopsy or carried a known pathogenic gene mutation, with an overall accuracy of 97% for PiB and 87% for FDG.
In the initial PiB-PET study, the largest and only significant difference in glucose metabolism (determined with FDG PET) between AD patients and control subjects was observed in parietal cortex. An inverse correlation between PiB retention and glucose metabolism was observed in most cortical areas, but this trend reached significance only in the parietal cortex. The lack of correlation between PiB and glucose metabolism in the frontal cortex suggests that Aβ deposition is not sufficient to locally reduce cerebral metabolism, suggesting that perhaps compensatory changes in neurotransmitter systems in the frontal cortex delay FDG hypometabolism in frontal brain regions (DeKosky et al., 2002; Ikonomovic et al., 2007). Edison et al. (2006) investigated the association between PiB and FDG PET in AD. AD subjects showed significant increases in PiB retention in cingulate, frontal, temporal, parietal, and occipital cortical areas and levels of temporal and parietal regional glucose metabolism were reduced by 20% in AD. Higher PiB retention correlated with lower regional glucose metabolism in temporal and parietal cortices, but not in frontal areas. While these typical negative correlations were observed in AD, subjects with MCI have been found to display positive correlations between PiB and FDG, reflecting increased brain reserve in those subjects who remain at the MCI level of cognitive impairment further into the process of Aβ deposition (Figure 4) (Cohen et al., 2009). Similarly, a positive relationship between PiB and FDG was also observed in cognitively normal adults (Oh et al., 2014). Recently, cortical hypermetabolism measured by FDG has been observed in cognitively normal controls with significant amyloid deposition in areas both likely (superior temporal gyrus) and unlikely (medial thalamus) to contain significant amyloid pathology (Figure 5) (Johnson et al., 2014) and independent of amyloid deposition in ApoE4 carriers (Yi et al., 2014). The phenomenon of hypermetabolism early in the course of Aβ deposition may be prove to be of great interest, particularly when considering the preclinical staging of AD, as those with significant amyloid and hypermetabolism will be less likely to be classified as neurodegeneration positive.
Figure 4.

From Cohen et al. (While we hypothesize that enrichment will enhance the function of the DMN et al.): Voxel-based correlations in AD and MCI. T values associated with negative correlations (blue) and positive correlations (red). Data are thresholded with FDR control at q = 0.1.
Figure 5.
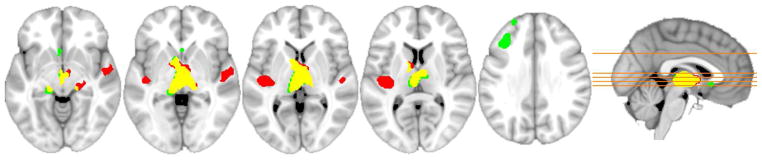
From Johnson et al. (2014): FDG group voxel wise differences, regions where the PiB-positive (red) and PiB-intermediate (green) groups, with yellow showing overlap, exhibited FDG hypermetabolism compared to the PiB-negative group.
It is interesting that there is considerable overlap between the regional distribution of amyloid deposition and the “default mode network” (Buckner et al., 2005), a group of brain areas that are more active during rest (McKiernan et al., 2003; Shulman et al., 2007). This spatial overlap has led to the hypothesis that there could be a relationship between brain activity patterns in early adulthood and later amyloid deposition (Buckner et al., 2005; Jagust and Mormino, 2011). This raises the possibility that when Aβ deposition starts, it becomes more pronounced in brain regions with high default mode activity. It has been shown in animals that Aβ levels are directly influenced by synaptic activity and are related to exocytosis of synaptic vesicles (Cirrito et al., 2005). Although much less direct, there is similar evidence in humans. Using intracerebral micro-dialysis in patients, positive correlations between Aβ levels and neurological status ( a proxy for neuronal activity) have been reported, suggesting that in patients with acute brain injury as neuronal activity increases, Aβ levels increase as well (Brody et al., 2008).
However, there are several studies that demonstrate the opposite effect in cognitively normal controls, with several groups demonstrating hypometabolism in cognitively normal individuals with significant amyloid deposition (Knopman et al., 2013; Lowe et al., 2009). Further, while Yi and colleagues demonstrated hypermetabolism in frontal and anterior temporal regions in cognitively normal ApoE4 carriers, they also demonstrated hypometabolism in temporoparietal regions. After adjusting for the effect of amyloid deposition most of the hypometabolic regions disappeared while the hypermetabolic regions persisted, suggesting that while hypometabolism may be amyloid dependent in this cohort, hypermetabolism was not (Yi et al., 2014). Additionally, longitudinal studies indicate that cognitively normal participants with significant amyloid deposition at baseline demonstrate significant increases in neurodegeneration markers at follow-up including, including FDG hypometabolism (Knopman et al., 2012a; Knopman et al., 2013). Further, individuals with both markers of amyloid deposition and neurodegeneration were more likely to develop cognitive impairments at follow-up (Knopman et al., 2012a; Landau et al., 2012).
Several groups also have demonstrated that there are a substantial number (~25%) of cognitively normal, elderly individuals without detectable amyloid deposition who display at least one significant marker of neurodegeneration, including FDG hypometabolism (Jack et al., 2012; Knopman et al., 2012b; Wirth et al., 2013). This group, defined as having abnormal neurodegeneration biomarkers, without significant amyloid deposition have been termed “Suspected Non-Alzheimer Pathology”, the SNAP category is suggested to be separate from preclinical AD and may represent other preclinical pathophysiologic processes, such as cerebrovascular disease, tauopathies or synucleinopathies (Jack et al., 2012). Interestingly, in subjects with SNAP, Wirth et al. (2013) observed hypometabolism in typical AD associated areas but also observed hypometabolism in regions less associated with AD, which they suggested was a more global pattern of hypometabolism. Wirth and colleagues (2013) also demonstrated a significant positive association between white matter lesions (a measure of brain cerebrovascular burden) and markers of neurodegeneration, consistent with the Jack et al (2012) definition of SNAP. Taken as a whole these data suggest that changes in glucose metabolism are clearly related to amyloid deposition but that early in the process of amyloid deposition this relationship is rather complex and that in some cases changes in amyloid and glucose metabolism may be independent of one another.
As has been reflected in the new diagnostic criteria for AD, MCI and “preclinical AD” the use of amyloid imaging, alone, or in conjunction with other biomarkers, will likely be critical to the identification of subjects at risk for AD and future decline (Sperling et al., 2011). One facet of Aβ deposition that has become clear from PiB-PET studies is how early in the spectrum of AD the full burden of amyloid plaques begins to develop. Therefore, major challenges for amyloid imaging will be; 1) how to determine the earliest signs of amyloid accumulation; 2) the associations of amyloid accumulation with cognitive impairments and, ultimately; 3) whether or not this early amyloid deposition will invariably lead to clinical dementia in a high percentage of individuals. This will likely require the field to focus on cognitively normal elderly and detection of the earliest signs of amyloid deposition along with markers of neurodegeneration such as FDG, in order to determine the clinical significance of pre-symptomatic pathology. Further, as anti-amyloid clinical trials begin in asymptomatic individuals, it will be critical to effectively identify the earliest changes in amyloid deposition and the significance of such changes on downstream neurodegenerative processes.
Highlights.
PiB and FDG are able to detect early AD pathological processes and subsequent neurodegeneration.
Imaging with PiB and FDG has potential clinical benefits, including preclinical detection of AD.
PiB and FDG allow the study of relationships of Aβ to changes in cognition and neurodegeneration.
The present review focuses on use of PiB and FDG-PET and their relationship to one another.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizenstein HJ, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Archives of Neurology. 2008;65:1509–17. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2011;7:270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anchisi D, et al. Heterogeneity of brain glucose metabolism in mild cognitive impairment and clinical progression to Alzheimer disease. Arch Neurol. 2005;62:1728–33. doi: 10.1001/archneur.62.11.1728. [DOI] [PubMed] [Google Scholar]
- Archer HA, et al. Amyloid load and cerebral atrophy in Alzheimer’s disease: an 11C-PIB positron emission tomography study. Ann Neurol. 2006;60:145–7. doi: 10.1002/ana.20889. [DOI] [PubMed] [Google Scholar]
- Arnaiz E, et al. Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment. Neuroreport. 2001;12:851–5. doi: 10.1097/00001756-200103260-00045. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Alzheimer’s disease: striatal amyloid deposits and neurofibrillary changes. Journal of Neuropathology & Experimental Neurology. 1990;49:215–224. [PubMed] [Google Scholar]
- Brilliant MJ, et al. The distribution of amyloid beta protein deposition in the corpus striatum of patients with Alzheimer’s disease. Neuropathology & Applied Neurobiology. 1997;23:322–325. [PubMed] [Google Scholar]
- Brody DL, et al. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–4. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–17. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse A, et al. Mild cognitive impairment: long-term course of four clinical subtypes. Neurology. 2006;67:2176–85. doi: 10.1212/01.wnl.0000249117.23318.e1. [DOI] [PubMed] [Google Scholar]
- Chetelat G, Baron JC. Early diagnosis of Alzheimer’s disease: contribution of structural neuroimaging. Neuroimage. 2003;18:525–541. doi: 10.1016/s1053-8119(02)00026-5. [DOI] [PubMed] [Google Scholar]
- Chetelat G, et al. Mild cognitive impairment: Can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology. 2003;60:1374–1377. doi: 10.1212/01.wnl.0000055847.17752.e6. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–22. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cohen AD, et al. Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:14770–8. doi: 10.1523/JNEUROSCI.3669-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon MJ, et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET) Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10966–71. doi: 10.1073/pnas.191044198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Annals of Neurology. 2002;51:145–55. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- Del Sole A, et al. Individual cerebral metabolic deficits in Alzheimer’s disease and amnestic mild cognitive impairment: an FDG PET study. Eur J Nucl Med Mol Imaging. 2008;35:1357–66. doi: 10.1007/s00259-008-0773-6. [DOI] [PubMed] [Google Scholar]
- Drzezga A, et al. Prediction of individual clinical outcome in MCI by means of genetic assessment and (18)F-FDG PET. J Nucl Med. 2005;46:1625–32. [PubMed] [Google Scholar]
- Edison P, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease. An [11C]PIB and [18F]FDG PET study. Neurology. 2006;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Fagan AM, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta(42) in humans. Ann Neurol. 2006;59:512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Fagan AM, et al. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–9. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- Forsberg A, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Foster NL, et al. FDG-PET improves accuracy in distinguishing frontotemporal dementia and Alzheimer’s disease. Brain. 2007;130:2616–35. doi: 10.1093/brain/awm177. [DOI] [PubMed] [Google Scholar]
- Friedland RP, et al. Regional cerebral metabolic alterations in dementia of the Alzheimer type: positron emission tomography with [18F]fluorodeoxyglucose. J Comput Assist Tomogr. 1983;7:590–598. doi: 10.1097/00004728-198308000-00003. [DOI] [PubMed] [Google Scholar]
- Garibotto V, et al. Education and occupation as proxies for reserve in aMCI converters and AD: FDG-PET evidence. Neurology. 2008;71:1342–9. doi: 10.1212/01.wnl.0000327670.62378.c0. [DOI] [PubMed] [Google Scholar]
- Gauthier S, et al. Mild cognitive impairment. Lancet. 2006;367:1262–70. doi: 10.1016/S0140-6736(06)68542-5. [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends in Pharmacological Sciences. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Hardy J, et al. Genetic dissection of Alzheimer’s disease and related dementias: amyloid and its relationship to tau. Nature Neuroscience. 1998;1:355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Herholz K, et al. Positron emission tomography imaging in dementia. Br J Radiol. 2007;80:S160–7. doi: 10.1259/bjr/97295129. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, et al. Superior frontal cortex cholinergic axon density in mild cognitive impairment and early Alzheimer disease. Archives of Neurology. 2007;64:1312–7. doi: 10.1001/archneur.64.9.1312. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, et al. An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Annals of Neurology. 2012;71:765–75. doi: 10.1002/ana.22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain : a journal of neurology. 2008;131:665–80. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W, et al. Brain imaging evidence of preclinical Alzheimer’s disease in normal aging. Annals of Neurology. 2006;59:673–81. doi: 10.1002/ana.20799. [DOI] [PubMed] [Google Scholar]
- Jagust W, et al. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology. 2007;69:871–7. doi: 10.1212/01.wnl.0000269790.05105.16. [DOI] [PubMed] [Google Scholar]
- Jagust WJ, Mormino EC. Lifespan brain activity, beta-amyloid, and Alzheimer’s disease. Trends in cognitive sciences. 2011;15:520–6. doi: 10.1016/j.tics.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, et al. Amyloid burden and neural function in people at risk for Alzheimer’s Disease. Neurobiology Of Aging. 2014;35:576–84. doi: 10.1016/j.neurobiolaging.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, et al. APOE modifies the association between Abeta load and cognition in cognitively normal older adults. Neurology. 2012;78:232–40. doi: 10.1212/WNL.0b013e31824365ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemppainen NM, et al. Voxel-based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease. Neurology. 2006;67:1575–80. doi: 10.1212/01.wnl.0000240117.55680.0a. [DOI] [PubMed] [Google Scholar]
- Klunk WE, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of Neurology. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Knopman DS, et al. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology. 2012a;78:1576–82. doi: 10.1212/WNL.0b013e3182563bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, et al. Brain injury biomarkers are not dependent on beta-amyloid in normal elderly. Annals of Neurology. 2012b doi: 10.1002/ana.23816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, et al. Selective worsening of brain injury biomarker abnormalities in cognitively normal elderly persons with beta-amyloidosis. JAMA neurology. 2013;70:1030–8. doi: 10.1001/jamaneurol.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen J, et al. Amyloid PET imaging in patients with mild cognitive impairment: a 2-year follow-up study. Neurology. 2011;76:1085–90. doi: 10.1212/WNL.0b013e318212015e. [DOI] [PubMed] [Google Scholar]
- Landau SM, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Annals of Neurology. 2012;72:578–86. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging. 2008;35:2169–2181. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopresti BJ, et al. Simplified Quantification of Pittsburgh Compound B Amyloid Imaging PET Studies: A Comparative Analysis. J Nucl Med. 2005;46:1959–72. [PubMed] [Google Scholar]
- Lowe VJ, et al. Comparison of 18F-FDG and PiB PET in cognitive impairment. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2009;50:878–86. doi: 10.2967/jnumed.108.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKiernan KA, et al. A Parametric Manipulation of Factors Affecting Task-induced Deactivation in Functional Neuroimaging. 2003;15:394–408. doi: 10.1162/089892903321593117. [DOI] [PubMed] [Google Scholar]
- Mevel K, et al. Detecting hippocampal hypometabolism in Mild Cognitive Impairment using automatic voxel-based approaches. Neuroimage. 2007;37:18–25. doi: 10.1016/j.neuroimage.2007.04.048. [DOI] [PubMed] [Google Scholar]
- Minoshima S. Imaging Alzheimer’s disease: clinical applications. Neuroimaging clinics of North America. 2003;13:769–80. doi: 10.1016/s1052-5149(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Mintun MA, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Mirra SS, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Mormino EC, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain : a journal of neurology. 2009;132:1310–23. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, et al. Relationships between Beta-Amyloid and Functional Connectivity in Different Components of the Default Mode Network in Aging. Cerebral cortex. 2011;21:2399–407. doi: 10.1093/cercor/bhr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Annals of Neurology. 2010;67:122–31. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, et al. Visual rating of medial temporal lobe metabolism in mild cognitive impairment and Alzheimer’s disease using FDG-PET. Eur J Nucl Med Mol Imaging. 2006;33:210–21. doi: 10.1007/s00259-005-1956-z. [DOI] [PubMed] [Google Scholar]
- Mosconi L, et al. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2009;72:513–20. doi: 10.1212/01.wnl.0000333247.51383.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, et al. Brain imaging of cognitively normal individuals with 2 parents affected by late-onset AD. Neurology. 2014 doi: 10.1212/WNL.0000000000000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, et al. MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET. Neurology. 2004;63:2332–40. doi: 10.1212/01.wnl.0000147469.18313.3b. [DOI] [PubMed] [Google Scholar]
- Mosconi L, et al. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Annals of the New York Academy of Sciences. 2008a;1147:180–95. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, et al. Amyloid and metabolic positron emission tomography imaging of cognitively normal adults with Alzheimer’s parents. Neurobiology Of Aging. 2013;34:22–34. doi: 10.1016/j.neurobiolaging.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, et al. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. J Nucl Med. 2008b;49:390–8. doi: 10.2967/jnumed.107.045385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, et al. (18)F-FDG PET database of longitudinally confirmed healthy elderly individuals improves detection of mild cognitive impairment and Alzheimer’s disease. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2007;48:1129–34. doi: 10.2967/jnumed.107.040675. [DOI] [PubMed] [Google Scholar]
- Nebes RD, et al. Cognitive aging in persons with minimal amyloid-beta and white matter hyperintensities. Neuropsychologia. 2013;51:2202–9. doi: 10.1016/j.neuropsychologia.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen N, et al. Abeta amyloid deposition in the language system and how the brain responds. Brain. 2007;130:2055–69. doi: 10.1093/brain/awm133. [DOI] [PubMed] [Google Scholar]
- Ng S, et al. Visual assessment versus quantitative assessment of 11C-PIB PET and 18F-FDG PET for detection of Alzheimer’s disease. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2007;48:547–52. doi: 10.2967/jnumed.106.037762. [DOI] [PubMed] [Google Scholar]
- Oh H, et al. Covarying alterations in Abeta deposition, glucose metabolism, and gray matter volume in cognitively normal elderly. Hum Brain Mapp. 2014;35:297–308. doi: 10.1002/hbm.22173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perneczky R, et al. Cerebral metabolic correlates of the clinical dementia rating scale in mild cognitive impairment. J Geriatr Psychiatry Neurol. 2007;20:84–8. doi: 10.1177/0891988706297093. [DOI] [PubMed] [Google Scholar]
- Pike KE, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain : a journal of neurology. 2007;130:2837–44. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- Price JC, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005a;25:1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- Price JC, et al. FDG and PIBPET imaging in Alzheimer’s disease and mild cognitive impairment. Neuropsychopharmacology. 2005b;30:S225–S225. [Google Scholar]
- Rabinovici GD, et al. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology. 2011;77:2034–42. doi: 10.1212/WNL.0b013e31823b9c5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. New England Journal of Medicine. 1996;96:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Reiman EM, et al. Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick SM, Sojkova J. Amyloid imaging and memory change for prediction of cognitive impairment. Alzheimer’s research & therapy. 2011;3:3. doi: 10.1186/alzrt62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe CC, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–83. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Rowe CC, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- Shulman GL, et al. Right TPJ deactivation during visual search: functional significance and support for a filter hypothesis. Cerebral cortex. 2007;17:2625–33. doi: 10.1093/cercor/bhl170. [DOI] [PubMed] [Google Scholar]
- Silverman DH, Alavi A. PET imaging in the assessment of normal and impaired cognitive function. Radiologic clinics of North America. 2005;43:67–77. x. doi: 10.1016/j.rcl.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Small GW, et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2000;97:6037–42. doi: 10.1073/pnas.090106797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storandt M, et al. Cognitive decline and brain volume loss as signatures of cerebral amyloid-beta peptide deposition identified with Pittsburgh compound B: cognitive decline associated with Abeta deposition. Archives of Neurology. 2009;66:1476–81. doi: 10.1001/archneurol.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga T, et al. Modified Bielschowsky stain and immunohistochemical studies on striatal plaques in Alzheimer’s disease. Acta Neuropathologica. 1990;80:280–286. doi: 10.1007/BF00294646. [DOI] [PubMed] [Google Scholar]
- Thal DR, et al. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12:357–67. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Annals of Neurology. 2011;69:181–92. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, et al. Abeta deposits in older non-demented individuals with cognitive decline are indicative of preclinical Alzheimer’s disease. Neuropsychologia. 2008 doi: 10.1016/j.neuropsychologia.2008.02.008. (in press) [DOI] [PubMed] [Google Scholar]
- Wirth M, et al. Associations between Alzheimer disease biomarkers, neurodegeneration, and cognition in cognitively normal older people. JAMA neurology. 2013;70:1512–9. doi: 10.1001/jamaneurol.2013.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DS, et al. Progression of regional neuropathology in Alzheimer disease and normal elderly: findings from the Nun study. Alzheimer Disease & Associated Disorders. 1999;13:226–231. doi: 10.1097/00002093-199910000-00009. [DOI] [PubMed] [Google Scholar]
- Wolk DA, et al. Amyloid imaging in mild cognitive impairment subtypes. Annals of Neurology. 2009;65:557–68. doi: 10.1002/ana.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi D, et al. Beta-Amyloid Associated Differential Effects of APOE epsilon4 on Brain Metabolism in Cognitively Normal Elderly. The American journal of geriatric psychiatry : official journal of the American Association for Geriatric Psychiatry. 2014 doi: 10.1016/j.jagp.2013.12.173. [DOI] [PubMed] [Google Scholar]
- Yuan Y, et al. Fluorodeoxyglucose-Positron-Emission Tomography, Single-Photon Emission Tomography, and Structural MR Imaging for Prediction of Rapid Conversion to Alzheimer Disease in Patients with Mild Cognitive Impairment: A Meta-Analysis. AJNR Am J Neuroradiol. 2008 doi: 10.3174/ajnr.A1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolko SK, et al. Evaluation of voxel-based methods for the statistical analysis of PIB PET amyloid imaging studies in Alzheimer’s disease. Neuroimage. 2006;33:94–102. doi: 10.1016/j.neuroimage.2006.05.063. [DOI] [PubMed] [Google Scholar]