

Abstract
Purpose
Preclinical and early clinical studies have demonstrated that initial therapy with combined BRAF and MEK inhibition is more effective in BRAFV600-mutant melanoma than single-agent BRAF inhibitors. This study assessed the safety and efficacy of dabrafenib and trametinib in patients who had received prior BRAF inhibitor treatment.
Patients and Methods
In this open-label phase I/II study, we evaluated the pharmacology, safety, and efficacy of dabrafenib and trametinib. Here, we report patients treated with combination therapy after disease progression with BRAF inhibitor treatment administered before study enrollment (part B; n = 26) or after cross-over at progression with dabrafenib monotherapy (part C; n = 45).
Results
In parts B and C, confirmed objective response rates (ORR) were 15% (95% CI, 4% to 35%) and 13% (95% CI, 5% to 27%), respectively; an additional 50% and 44% experienced stable disease ≥ 8 weeks, respectively. In part C, median progression-free survival (PFS) was 3.6 months (95% CI, 2 to 4), and median overall survival was 11.8 months (95% CI, 8 to 25) from cross-over. Patients who previously received dabrafenib ≥ 6 months had superior outcomes with the combination compared with those treated < 6 months; median PFS was 3.9 (95% CI, 3 to 7) versus 1.8 months (95% CI, 2 to 4; hazard ratio, 0.49; P = .02), and ORR was 26% (95% CI, 10% to 48%) versus 0% (95% CI, 0% to 15%).
Conclusion
Dabrafenib plus trametinib has modest clinical efficacy in patients with BRAF inhibitor–resistant melanoma. This regimen may be a therapeutic strategy for patients who previously benefited from BRAF inhibitor monotherapy ≥ 6 months but demonstrates minimal efficacy after rapid progression with BRAF inhibitor therapy.
INTRODUCTION
Oncogenic driver mutations at the V600 codon in the serine-threonine kinase BRAF induce constitutive activation of the mitogen-activated protein kinase (MAPK) signaling pathway and have been identified in 40% to 50% of cutaneous melanomas.1–3 Suppression of MAPK signaling by inhibiting BRAF or a downstream partner, MEK, has proven to be an effective therapeutic strategy in BRAFV600-mutant melanoma. Several selective BRAF inhibitors, including dabrafenib and vemurafenib, and the MEK inhibitor trametinib have each demonstrated improved progression-free survival (PFS) and, in some studies, overall survival (OS) as single agents compared with cytotoxic chemotherapy.4–7 Despite these advances, acquired resistance inevitably develops, with a median PFS of < 7 months in published trials.4–7 Furthermore, cutaneous squamous cell carcinomas (SCCs) and potentially other malignancies may be promoted and unmasked by BRAF inhibitor monotherapy through paradoxic MAPK pathway activation.8–10
Mechanisms of acquired resistance to BRAF inhibitor therapy include reactivation of MAPK signaling in the majority of cases.11–13 These include secondary NRAS or MEK mutations,14–16 amplification or alternate splicing of mutant BRAF,17,18 CRAF upregulation,19 or COT (MAP3K8) overexpression, among others.20 Additional adaptive mechanisms of resistance independent of MAPK reactivation have also been identified and include growth factor activation or receptor tyrosine kinase upregulation, metabolic reprogramming, and phosphoinositide 3-kinase (PI3K) –AKT pathway dysregulation.14,21–25 In preclinical models, combined BRAF and MEK inhibition achieves more thorough abrogation of MAPK signaling, thereby forestalling the development of acquired resistance and suppressing paradoxic activation of the MAPK pathway.18,26 We conducted a phase I/II study to evaluate the safety and clinical efficacy of combined inhibition with dabrafenib and trametinib in patients with BRAFV600-mutant metastatic melanoma. As previously reported, combination therapy demonstrated superior objective response rates (ORRs; 76% [95% CI, 40% to 67%] v 54% [95% CI, 62% to 87%]; P = .03) and median PFS (9.4 [95% CI, 9 to 17] v 5.8 months [95% CI, 5 to 7]; P < .001) compared with single-agent dabrafenib in BRAF inhibitor–naive patients in a randomized phase II study.27 Furthermore, cutaneous toxicities, including cutaneous SCCs, occurred less frequently with combination therapy (7% [95% CI, 2% to 18%] v 19% [95% CI, 9% to 32%]; P = .09) compared with dabrafenib alone.
Despite the compelling rationale for combined MAPK inhibition in BRAF inhibitor–naive melanoma, the clinical activity of dabrafenib in combination with trametinib in BRAF inhibitor–resistant patients has not been reported. In our phase I/II study, a subset of patients received dabrafenib and trametinib after tumor progression with dabrafenib or vemurafenib monotherapy. Here, we report the clinical efficacy and safety of combination therapy for this population of patients with BRAF inhibitor–resistant melanoma.
PATIENTS AND METHODS
Patient Selection
Inclusion criteria for this study included age ≥ 18 years, histologically confirmed BRAFV600E- or BRAFV600K-mutant melanoma, Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 to 1, and adequate organ function. BRAFV600 mutation status was determined at local laboratories. Patients with brain metastases that had been stable ≥ 3 months were permitted to enroll. Prior BRAF inhibitor therapy was permitted for a limited number of patients in part B (including vemurafenib, dabrafenib, or other experimental BRAF inhibitors), but patients in part C were BRAF- and MEK-inhibitor naive on initial enrollment (Fig 1). Up to one line of previous immune-based therapy was allowed. Patients with a history of central serous retinopathy (CSR), retinal vein occlusion, or serious cardiac comorbidities, including acute coronary syndrome in the preceding 6 months, congestive heart failure of New York Heart Association classes II to IV, and long QT interval were excluded.
Fig 1.

Study design. Gold areas show the study population reported in this article. BRAFi, BRAF inhibitor; HPMC, hydroxypropyl methylcellulose.
Study Design
This was an open-label study designed to assess the safety, clinical efficacy, and pharmacokinetic activity of combination therapy with dabrafenib and trametinib. The study was conducted in four parts; portions of parts B and C are reported here (Fig 1), and results for the other two parts were previously reported.27 Part B evaluated the safety and activity of escalating doses of dabrafenib (75 and 150 mg twice daily) and trametinib (1, 1.5, and 2 mg once daily); this included BRAF inhibitor–resistant and BRAF inhibitor–naive patients. Part C was a randomized, three-arm study in which patients were assigned at a ratio of 1:1:1 to receive dabrafenib 150 mg twice daily as monotherapy or in combination with either trametinib 1 mg once daily or 2 mg once daily. Patients assigned to dabrafenib monotherapy were eligible for cross-over to combination therapy (at time of tumor progression). Parts A and D are not described here. We report here the efficacy of combination therapy with dabrafenib and trametinib for patients previously treated with a BRAF inhibitor before enrollment (BRAF inhibitor–resistant portion of part B, referred to here as part B) and for those who received dabrafenib monotherapy in this study who then crossed over to combination therapy at disease progression (cross-over portion of part C, referred to here as part C). BRAF inhibitor resistance was defined as progression on prior single-agent BRAF inhibitor, either before study entry (part B) or with dabrafenib treatment during the study (part C). Patients in part C experienced disease progression by RECIST (version 1.1) criteria; patients in part B experienced progression as documented by history and imaging before study entry.
Disease assessment by cross-sectional imaging was performed at baseline and every 8 weeks (± 1 week) according to RECIST (version 1.1).28 Severity of toxicity was graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0). Tumor BRAFV600 mutations detected by Clinical Laboratory Improvement Amendment–approved tests at local laboratories were sufficient for enrollment.
Study Oversight
The protocol was approved by the institutional review board at each participating center and complied with country-specific regulatory requirements. The study was conducted in accordance with the provisions of the Declaration of Helsinki and Good Clinical Practice guidelines. The study was designed by the academic authors in conjunction with representatives of the sponsor, GlaxoSmithKline. Data were collected by the sponsor and analyzed in collaboration with the authors. The authors vouch for the accuracy and completeness of the data and the fidelity of the study to the protocol.
Statistical Analysis
Sample size calculations for part C were previously described.27 Primary efficacy end points for part C were ORR, PFS, and duration of response (DoR) to combination therapy for BRAF inhibitor–resistant patients as determined by the investigator (for parts B and C). OS from the initiation of combination therapy was a secondary end point. PFS, DoR, and OS were summarized with Kaplan-Meier methodology using medians and 95% CIs (estimated using Brookmeyer Crowley method). Follow-up time was calculated as the time from first dose of dabrafenib and trametinib during study (part B) and first dose of cross-over to dabrafenib and trametinib (part C) to the clinical cutoff date of January 15, 2014. Unplanned subgroup analyses were performed to identify factors predicting superior PFS (including duration of treatment with BRAF inhibitor monotherapy, BRAF mutation status [V600E v V600K], baseline lactate dehydrogenase [LDH], and ECOG PS), which were compared using the log-rank test. The magnitudes of individual responses by RECIST are displayed using waterfall plots. Duration of therapy for BRAF inhibitor monotherapy and subsequent combination therapy are displayed in a descriptive fashion.
RESULTS
Patient Characteristics
From March 26, 2010, through July 7, 2011, 443 patients at 16 centers were screened for eligibility, and of these, 103 and 162 patients were enrolled onto parts B and C, respectively. Of the 103 patients enrolled onto part B, 50 were treated with the recommended phase II dose (ie, dabrafenib 150 mg twice daily and trametinib 2 mg once daily); 26 of those treated at the recommended phase II dose had previously received a BRAF inhibitor and are described here. Of the 162 patients enrolled onto part C, 54 were assigned to receive dabrafenib monotherapy, and 45 (described here) crossed over to combination therapy. Baseline characteristics for BRAF inhibitor–resistant patients in parts B and C, at the time of initiation of combination therapy, are listed in Table 1. Patients in part B had more advanced disease compared with those in part C and had a higher incidence of elevated LDH (62% v 20%), American Joint Committee on Cancer stage M1c melanoma (92% v 67%), and history of brain metastases (23% v 9%). Other characteristics, including the percentage harboring a BRAFV600K mutation and response to prior BRAF therapy, were similar. In part C, cross-over patients received BRAF inhibitor monotherapy for a median of 6.1 months. In part B, the preceding BRAF inhibitor was dabrafenib in 46% and vemurafenib in 46% compared with dabrafenib in 100% based on the design of part C.
Table 1.
Patient Demographic and Clinical Characteristics
| Characteristic | Part B (n = 26) |
Part C (n = 45) |
||
|---|---|---|---|---|
| No. | % | No. | % | |
| Age, years | ||||
| Mean | 47.7 | 51 | ||
| Median | 48 | 49 | ||
| Range | 23-72 | 18-82 | ||
| Male sex | 12 | 46 | 23 | 51 |
| ECOG performance status* | ||||
| 0 | 10 | 38 | 26 | 58 |
| 1 | 16 | 62 | 19 | 42 |
| Stage | ||||
| IIIc | 0 | 0 | 1 | 2 |
| IVa | 0 | 0 | 9 | 20 |
| IVb | 2 | 8 | 5 | 11 |
| IVc | 24 | 92 | 30 | 67 |
| Prior brain metastases | 6 | 23 | 4 | 9 |
| Baseline LDH* | ||||
| < ULN | 10 | 38 | 36 | 80 |
| > ULN | 16 | 62 | 9 | 20 |
| BRAF mutation | ||||
| BRAFV600E | 23 | 88 | 38 | 84 |
| BRAFV600K | 3 | 12 | 7 | 16 |
| Prior systemic therapy | ||||
| Immunotherapy | 9 | 35 | 5 | 11 |
| Chemotherapy† | 14 | 54 | 4 | 9 |
| Duration of prior BRAF inhibitor, months‡ | ||||
| < 6 | 7 | 27 | 22 | 49 |
| ≥ 6 | 16 | 62 | 23 | 51 |
| Best response to prior BRAF inhibitor | ||||
| CR or PR | 10 | 38 | 26 | 58 |
| SD | 9 | 35 | 16 | 36 |
| PD | 7 | 27 | 3 | 7 |
| Time from treatment with prior BRAF inhibitor, months | ||||
| Median | 1.1 | § | ||
| Range | 0-12 | |||
| Prior BRAF inhibitor | ||||
| Dabrafenib | 12 | 46 | 45 | 100 |
| Vemurafenib | 12 | 46 | 0 | 0 |
| XL281 | 1 | 4 | 0 | 0 |
| Other | 1 | 4 | 0 | 0 |
Abbreviations: CR, complete response; ECOG, Eastern Cooperative Oncology Group; LDH, lactate dehydrogenase; PD, progressive disease; PR, partial response; SD, stable disease; ULN, upper limit of normal.
For part C, baseline is defined based on most recent assessment before cross-over treatment.
In advanced or metastatic setting.
Data for therapy duration were not available for three patients in part B.
For part C, patients received dabrafenib monotherapy until time of cross-over.
Efficacy
Median follow-ups for parts B and C (from initiation of cross-over treatment) were 35.3 and 27.4 months, respectively. In part B, the confirmed ORR was 15% (95% CI, 4% to 35%); all four responding patients had a partial response (Table 2). An additional 13 patients (50%) had stable disease ≥ 8 weeks. In part C, the confirmed ORR was 13% (one complete response and five partial responses; 95% CI, 5% to 27%), and an additional 20 patients (44%) had stable disease ≥ 8 weeks (Table 2). Among patients with evaluable tumor responses, 53% (34 of 64) had some degree of tumor shrinkage as best response to combination therapy after experiencing progression with single-agent BRAF inhibitor (Figs 2A and 2B). Several patients had unconfirmed responses (two each in parts B and C).
Table 2.
Clinical Efficacy
| Response | Part B (n = 26) |
Part C (n = 45) |
||
|---|---|---|---|---|
| No. | % | No. | % | |
| CR | 0 | 0 | 1 | 2 |
| PR | 4 | 15 | 5 | 11 |
| SD* | 13 | 50 | 20 | 44 |
| PD | 8 | 31 | 17 | 38 |
| Not evaluable | 1 | 4 | 2 | 4 |
| Response rate, % | 15 | 13 | ||
| 95% CI | 4 to 35 | 5 to 27 | ||
| Duration of response, months | ||||
| Median | 7.8 | |||
| Interquartile range | 4 to 12 | |||
Abbreviations: CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
For part C, this includes two patients with best response of non-CR/non-PD who had no baseline measurable disease at time of cross-over.
Fig 2.
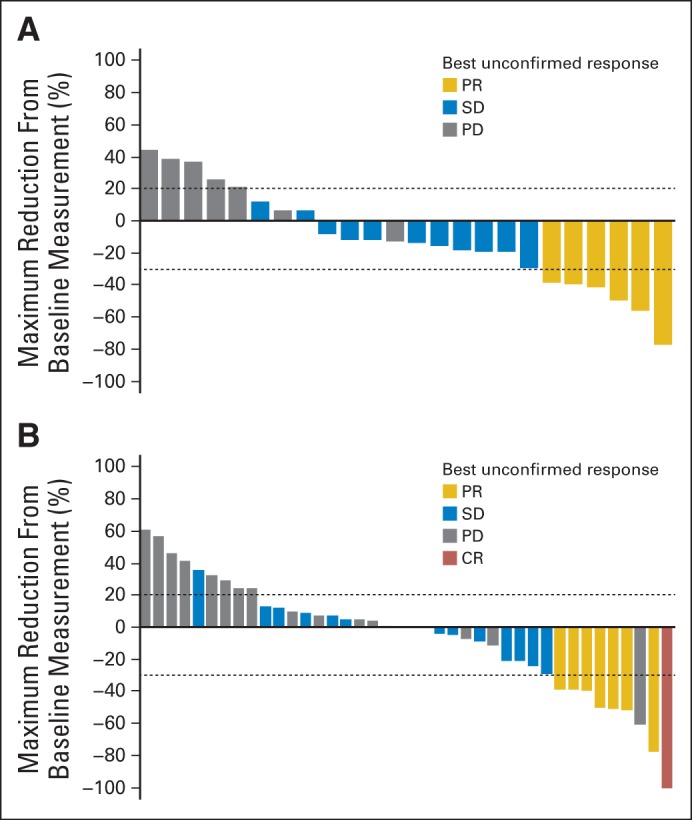
Maximum tumor reduction in BRAF inhibitor (BRAFi) –resistant patients in (A) parts B and (B) C. CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
From the first dose of combination therapy, median PFS was 3.6 months for both parts B and C (part B: 95% CI, 2 to 5 months; part C: 95% CI, 2 to 4 months; Fig 3A); median OS from start of combination therapy was 10 months (95% CI, 6 to 14 months) and 11.8 months (95% CI, 8 to 25 months) in parts B and C, respectively (Fig 3B). Median DoR for the 10 responding patients was 7.8 months (95% CI, 4 to 12 months), including one patient with an ongoing response ≥ 24 months. Duration of therapy with prior BRAF inhibitor monotherapy and with dabrafenib and trametinib for individual patients is displayed in Figure 3C.
Fig 3.

(A) Progression-free survival (PFS) for patients with BRAF inhibitor (BRAFi) –resistant melanoma (Part B 150/2 BRAFi failure: patients failed to respond to BRAFi off study, then received 150 mg dabrafenib twice per day and 2 mg of trametinib once per day); (B) overall survival (OS) from combination therapy for BRAFi-resistant patients (parts B and C); (C) duration of treatment with dabrafenib monotherapy followed by duration of treatment with combination therapy (part C); and (D) PFS with dabrafenib and trametinib for patients with rapid progression with BRAFi monotherapy versus patients with delayed resistance.
We then asked whether longer lasting benefit from BRAF inhibitor monotherapy would predict for extended PFS from subsequent combination therapy. Of the 45 patients in part C randomly assigned to dabrafenib alone who crossed over to combination therapy, 22 patients received dabrafenib monotherapy < 6 months (rapid resistance), and 23 received dabrafenib monotherapy ≥ 6 months (delayed resistance). In the rapid-resistance group, the ORR was 0% (95% CI, 0% to 15%), and 45% of patients had temporary stable disease lasting ≥ 8 weeks. In the delayed-resistance group, the ORR was 26% (including one complete response [95% CI, 10% to 48%]), and an additional 43% experienced stable disease. Median PFS was also superior in the delayed-resistance group (3.9 [95% CI, 3 to 7] v 1.8 months [95% CI, 2 to 4]; hazard ratio for progression, 0.49 [95% CI, 0.26 to 0.95]; log-rank P = .018; Fig 3D). Marginal or no improvements in PFS (all with nonsignificant P values) were observed for patients with normal compared with elevated LDH (3.7 [95% CI, 2 to 5] v 1.8 months [95% CI, 1 to 5]; P = .13), ECOG PS of 0 compared with ≥ 1 (3.7 [95% CI, 2 to 5] v 1.8 months [95% CI, 2 to 5]; P = .16), American Joint Committee on Cancer stage M1a/b compared with M1c (3.9 [95% CI, 2 to 7] v 2.8 months [95% CI, 2 to 4]; P = .45), and BRAFV600K versus BRAFV600E mutation (3.7 [95% CI, 2 to 7] v 3.0 months [95% CI, 2 to 4]; P = .88).
Safety
The most frequent adverse events (AEs) were pyrexia, nausea/vomiting, and fatigue (Table 3). Grade 4 AEs were relatively uncommon and included constipation, pulmonary embolism, back pain, tumor hemorrhage, and urosepsis. Two patients experienced grade 5 events (hyponatremia and neurologic decompensation, respectively). Pyrexia occurred in 44% and was managed by dose interruption, antipyretics, and, in some cases, corticosteroid administration; only one case was grade 3. No patients developed CSR or retinal vein occlusion. Six patients had a decreased ejection fraction (one grade 3; five were asymptomatic and reversible). Grade 3 hypotension occurred in three patients (4%) and was reversible with intravenous fluid administration and holding of the study drug; three patients developed grade 3 hypertension. Consistent with previous studies, cutaneous SCCs and keratoacanthomas were uncommon with combination therapy (five patients [7%]); other cutaneous manifestations also seemed to occur less frequently than previously reported with BRAF inhibitor monotherapy (hyperkeratosis in 4% and nonspecific rash in 17%).
Table 3.
AEs Occurring in > 15% of Patients in Part C and Events of Special Interest
| AE | Part B: Dabrafenib Plus Trametinib (n = 26) |
Part C: Cross-Over to Dabrafenib and Trametinib (n = 45) |
||||||
|---|---|---|---|---|---|---|---|---|
| All Grades |
Grade 3 or 4* |
All Grades |
Grade 3 or 4† |
|||||
| No. | % | No. | % | No. | % | No. | % | |
| Any event | 26 | 100 | 16 | 61 | 45 | 100 | 20 | 44 |
| Pyrexia | 15 | 58 | 0 | 0 | 16 | 36 | 1 | 2 |
| Nausea | 10 | 38 | 0 | 0 | 13 | 29 | 1 | 2 |
| Vomiting | 9 | 35 | 0 | 0 | 13 | 29 | 1 | 2 |
| Fatigue | 9 | 35 | 1 | 4 | 11 | 24 | 1 | 2 |
| Arthralgia | 2 | 8 | 0 | 0 | 11 | 24 | 0 | 0 |
| Diarrhea | 7 | 27 | 0 | 0 | 11 | 24 | 0 | 0 |
| Anemia | 3 | 12 | 1 | 4 | 9 | 20 | 1 | 2 |
| Chills | 6 | 23 | 0 | 0 | 9 | 20 | 0 | 0 |
| Back pain | 1 | 4 | 0 | 0 | 8 | 18 | 2 | 4 |
| Constipation | 8 | 31 | 0 | 0 | 8 | 18 | 1 | 2 |
| Headache | 6 | 23 | 0 | 0 | 8 | 18 | 0 | 0 |
| Rash | 4 | 15 | 0 | 0 | 8 | 18 | 0 | 0 |
| Peripheral edema | 3 | 12 | 0 | 0 | 8 | 18 | 0 | 0 |
| Urinary tract infection | 5 | 19 | 0 | 0 | 7 | 16 | 1 | 2 |
| Decreased appetite | 4 | 15 | 0 | 0 | 7 | 16 | 0 | 0 |
| Dizziness | 4 | 15 | 0 | 0 | 7 | 16 | 0 | 0 |
| Cutaneous SCC | 1 | 4 | 1 | 4 | 4 | 9 | 4 | 9 |
| Skin papilloma | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hyperkeratosis | 3 | 12 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hypertension | 1 | 4 | 0 | 0 | 4 | 9 | 3 | 7 |
| Hypotension | 7 | 27 | 3 | 12 | 0 | 0 | 0 | 0 |
| Decreased ejection fraction | 0 | 0 | 0 | 0 | 6 | 13 | 1 | 2 |
| Chorioretinopathy | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Blurred vision | 4 | 15 | 0 | 0 | 1 | 2 | 0 | 0 |
NOTE. Includes treatment- and non–treatment-related AEs.
Abbreviations: AE, adverse event; SCC, squamous cell carcinoma.
One patient in part B experienced fatal hyponatremia.
One patient in part C experienced fatal neurologic decompensation.
DISCUSSION
Pharmacologic inhibition of MAPK signaling has proven to be an effective therapeutic strategy in advanced BRAFV600-mutant melanoma. However, the development of acquired BRAF inhibitor resistance at a median of 4 to 7 months with BRAF or MEK inhibitors has limited the long-term benefit of single-agent targeted therapy.4–7 Because many acquired resistance mechanisms reactivate MAPK pathway activity, the combination of BRAF and MEK inhibitors was hypothesized to delay or even reverse acquired resistance to BRAF inhibition. As we reported in 2012, combination therapy with dabrafenib and trametinib seems to represent a therapeutic advance for patients with BRAFV600-mutant melanoma.27 In a BRAF inhibitor–naive cohort, ORR and PFS were significantly improved compared with patients treated with dabrafenib alone. In addition, incidence of hyperproliferative cutaneous lesions, including cutaneous SCCs, was decreased. Several other cohorts in this study allowed us to evaluate the role of combination therapy in patients with established acquired resistance to BRAF inhibitors.
In this population of BRAF inhibitor–refractory patients, we noted an ORR of 14% (95% CI, 7% to 24%), and an additional 46% of patients experienced stable disease ≥ 8 weeks; median PFS was 3.6 months (in parts B and C collectively). The activity of dabrafenib and trametinib in this BRAF inhibitor–resistant group was clearly inferior to that previously reported in the BRAF inhibitor–naive cohort (median PFS, 3.6 months [95% CI, 2 to 4] in this study v 9.4 months [95% CI, 9 to 17] in BRAF inhibitor–naive patients).27 Despite the comparative lack of efficacy, temporary disease regression or stabilization was observed in > 50% of this relatively small group of patients. Furthermore, patients occasionally experienced major clinical benefit, including one patient with an ongoing response at 24 months with therapy.
In view of the widely variable outcomes among patients, we investigated whether clinical factors would predict benefit from dabrafenib and trametinib in this population of BRAF inhibitor–refractory patients. Marginally better outcomes (without statistically significant P values) were noted for well-known melanoma prognostic factors, including normal LDH, stage M1a/b (v stage M1c), and ECOG PS of 0. However, delayed onset of resistance to prior single-agent BRAF inhibition seemed to be the best predictor of subsequent benefit from the combination. Patients in part C who received dabrafenib ≥ 6 months had more than double the PFS (3.9 v 1.8 months) and a superior ORR (26% v 0%) compared with those with prior rapid progression. The subgroup of patients with short duration of BRAF inhibitor monotherapy had no benefit, with almost universally rapid progression; no patients experienced objective responses.
This finding suggests that MAPK signaling addiction may be more dominant in melanomas with delayed onset of resistance. From these data, it could be hypothesized that mechanisms of resistance arising after prolonged BRAF inhibitor therapy more often primarily reactivate MAPK signaling, whereas earlier onset of resistance involves pathways more insensitive to combined BRAF/MEK inhibition. Several studies of resistance mechanisms provide preliminary support for this assertion, suggesting that most NRAS mutations arise after 6 months of therapy, whereas many non–MAPK-dependent mechanisms (eg, PI3K-AKT pathway or growth factors) occur early in the course of therapy.11–13 Early investigations of resistance to combined BRAF and MEK inhibition provide additional insight and complexity, demonstrating that multiple MAPK-reactivation mechanisms act in concert to drive tumor progression (BRAF hyperamplification, BRAF splice variant, and MEK1/2 mutations).29,30 Comprehensive genetic analysis at the time of resistance could define who may benefit from combination therapy.
The toxicity profile of dabrafenib plus trametinib in this BRAF inhibitor–resistant cohort was similar to that observed in previously untreated patients. Pyrexia was the most frequently observed AE; low-grade GI toxicities and blood pressure dysregulation (hyper- or hypotension) also occurred frequently. MEK inhibitor class toxicities were also observed, including temporary and reversible decreases in cardiac ejection fraction in six patients and peripheral edema in 15% of patients; no cases of CSR occurred. In addition, cutaneous SCCs were seen infrequently (five patients [7%]). As previously reported, the toxicity profile is distinct from that of BRAF inhibitor monotherapy, with decreased skin toxicity and a lower incidence of cutaneous SCCs.
This study has important clinical implications but also leaves several questions unanswered. For patients with BRAFV600-mutant melanoma who have had prolonged DoR or disease stability (≥ 6 months) with either vemurafenib or dabrafenib monotherapy, the combination of dabrafenib plus trametinib seems to be an appropriate and modestly active subsequent regimen. By contrast, our data suggest that the combination has little to no benefit for those experiencing rapid progression. How this combination regimen should be integrated with other pathway inhibitors (eg, PI3-AKT) and immune-based therapies and whether an intermittent dosing strategy should be tested remain important unanswered questions and are the subjects of active investigation. In addition, because nearly all patients will become resistant to BRAF and MEK inhibitors, the most appropriate therapy at the time of acquired resistance to BRAF and MEK inhibitor therapy remains unclear. This treatment hurdle warrants further investigation, and a similar question is now being addressed in a clinical trial incorporating comprehensive genetic analysis at the time of progression after a BRAF inhibitor (LGX818; LOGIC [LGX818 in Combination With Agents (MEK162; BKM120; LEE011; BGJ398; INC280) in Advanced BRAF Melanoma] trial; ClinicalTrials.gov No. NCT01820364). Even this strategy is likely unable to overcome the problems associated with the marked heterogeneity of resistant melanoma in the same patient or even at the same tumor site.11 Finally, our study should be viewed in context with the recently presented randomized phase III trial of dabrafenib and trametinib versus dabrafenib alone.31 Because the combination demonstrated improved PFS in that study, we would recommend dabrafenib plus trametinib as a first-line targeted therapy rather than a salvage strategy after a single-agent BRAF inhibitor.
Taken together, this phase I/II study suggests that combination therapy with dabrafenib and trametinib is a therapeutic option to delay the onset of acquired resistance when administered in the first-line setting but does not necessarily reverse established resistance to BRAF inhibitors. The data from this portion of the study do not support the universal use of dabrafenib and trametinib in BRAF inhibitor monotherapy–resistant patients. However, combination therapy for patients with prolonged disease control resulting from BRAF inhibitor monotherapy can be considered at the time of progression and, based on these results, can achieve a clinical benefit.
Supplementary Material
Acknowledgment
We thank the patients and their families for their participation. Authors from Westmead Hospital and Melanoma Institute Australia thank Arthur Clements and the clinical staff in oncology and dermatology at Westmead Hospital and Melanoma Institute Australia, and Vicky Wegener and the Clinical Trials Team, Crown Princess Mary Cancer Centre Westmead.
Glossary Terms
- BRAF:
an isoform of RAF. See Raf.
- BRAF V600E:
the most common oncogenic mutation of BRAF in cancer. The V600E amino acid change results in constitutive activation of the BRAF kinase and promotes cell transformation.
- MEK (MAPK-ERK kinase):
a protein kinase activated by c-Raf through phosphorylation of specific serine residues. Activation of ERK by activated MEK may lead to translocation of ERK to the nucleus, resulting in the activation of specific transcription factors.
Footnotes
Processed as a Rapid Communication manuscript.
Supported by GlaxoSmithKline, National Institutes of Health Grant No. K12 CA 0906525 (D.B.J.), and fellowships from the Cancer Institute New South Wales and the Royal Australasian College of Physicians (G.V.L.); editorial support provided by SciMentum, funded by GlaxoSmithKline.
Presented in part at the 49th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 31-June 4, 2013.
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT01072175.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Douglas B. Johnson, Keith T. Flaherty, Jeffrey R. Infante, Omid Hamid, Donald P. Lawrence, Georgina V. Long, Kiran Patel, Peng Sun, Shonda Little, Elizabeth Cunningham, Jeffrey A. Sosman
Administrative support: Howard A. Burris III
Provision of study materials or patients: Keith T. Flaherty, Richard F. Kefford, William H. Sharfman, Howard A. Burris III, Gerald S. Falchook, Alain Algazi, Georgina V. Long, Jeffrey A. Sosman, Adil Daud, Rene Gonzalez
Collection and assembly of data: Douglas B. Johnson, Keith T. Flaherty, Jeffrey S. Weber, Jeffrey R. Infante, Kevin B. Kim, Omid Hamid, Lynn Schuchter, Jonathan Cebon, William H. Sharfman, Mario Sznol, Donald P. Lawrence, Geoffrey T. Gibney, Howard A. Burris III, Gerald S. Falchook, Alain Algazi, Karl Lewis, Georgina V. Long, Kiran Patel, Peng Sun, Elizabeth Cunningham, Jeffrey A. Sosman, Adil Daud, Rene Gonzalez
Data analysis and interpretation: Douglas B. Johnson, Keith T. Flaherty, Jeffrey S. Weber, Jeffrey R. Infante, Richard F. Kefford, Omid Hamid, William H. Sharfman, Robert R. McWilliams, Howard A. Burris III, Gerald S. Falchook, Alain Algazi, Karl Lewis, Georgina V. Long, Kiran Patel, Nageatte Ibrahim, Peng Sun, Jeffrey A. Sosman, Adil Daud
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Douglas B. Johnson
No relationships to disclose
Keith T. Flaherty
Honoraria: GlaxoSmithKline
Consulting or Advisory Role: GlaxoSmithKline
Research Funding: GlaxoSmithKline
Jeffrey S. Weber
Stock or Other Ownership: Altor, Celldex
Honoraria: Bristol-Myers Squibb, Merck, Genentech, Abbvie, Astra Zeneca, Daiichi-Sankyo, GlaxoSmithKline, Eisai
Consulting or Advisory Role: Celldex, Ichor, cCAM, Lion Biotechnologies, Pieris, Altor
Research Funding: Bristol-Myers Squibb, Merck, GlaxoSmithKline, Genentech, Astellas Pharma
Travel, Accommodations, Expenses: Bristol-Myers Squibb, GlaxoSmithKline, Daiichi Sankyo, Pieris, cCAM
Jeffrey R. Infante
Consulting or Advisory Role: GlaxoSmithKline (Inst)
Research Funding: GlaxoSmithKline (Inst)
Kevin B. Kim
Honoraria: GlaxoSmithKline, Roche/Genentech, Bristol-Myers Squibb, Novartis
Consulting or Advisory Role: GlaxoSmithKline, Genentech/Roche, Bristol-Myers Squibb, Novartis
Research Funding: GlaxoSmithKline (Inst), Genentech/Roche (Inst), Bristol-Myers Squibb (Inst), Novartis (Inst), AstraZeneca (Inst), Eisai (Inst)
Richard F. Kefford
Honoraria: Roche, Merck, Novartis, Bristol-Myers Squibb, GlaxoSmithKline
Consulting or Advisory Role: Roche, GlaxoSmithKline, Bristol-Myers Squibb, Merck, Novartis
Speakers' Bureau: Bristol-Myers Squibb
Travel, Accommodations, Expenses: Roche, Bristol-Myers Squibb
Omid Hamid
Employment: Angeles Clinic and Research Institute
Leadership: Angeles Clinic and Research Institute
Stock or Other Ownership: Angeles Clinic and Research Institute
Honoraria: Bristol-Myers Squibb, Genentech
Consulting or Advisory Role: Merck, Genentech
Speakers' Bureau: Bristol-Myers Squibb, Genentech
Research Funding: Bristol-Myers Squibb, Merck, MedImmune, Genentech
Lynn Schuchter
Research Funding: GlaxoSmithKline (Inst), Merck (Inst), Bristol-Myers Squibb (Inst)
Jonathan Cebon
Honoraria: GlaxoSmithKline, Bristol-Myers Squibb, Novartis
Consulting or Advisory Role: Amgen (Inst), Bionomics (Inst), Bristol-Myers Squibb (Inst), Merck Sharp & Dohme (Inst), Bristol-Myers Squibb (Inst), Bristol-Myers Squibb (Inst), Merck Sharp & Dohme (Inst)
Research Funding: GlaxoSmithKline (Inst), CSL (Inst)
Patents, Royalties, Other Intellectual Property: GlaxoSmithKline
William H. Sharfman
Honoraria: Merck
Consulting or Advisory Role: Merck
Research Funding: Bristol-Myers Squibb, GlaxoSmithKline, Novartis
Robert R. McWilliams
No relationships to disclose
Mario Sznol
Stock or Other Ownership: Amphivena
Consulting or Advisory Role: Bristol-Myers Squibb, Genentech/Roche, Amgen, AstraZeneca/MedImmune, Symphogen, Merus, Immune Design, Anaeropharma, Kyowa-Hakko Kirin, Lion Biotechnologies, Nektar, Pfizer, Seattle Genetics
Other Relationship: Haymarket Media
Donald P. Lawrence
No relationships to disclose
Geoffrey T. Gibney
Consulting or Advisory Role: Roche/Genentech
Howard A. Burris III
No relationships to disclose
Gerald S. Falchook
Research Funding: GlaxoSmithKline (Inst), Merck Serono (Inst), Millennium/Tekada (Inst), Celgene (Inst), Circadian Technologies (Inst), AstraZeneca/Medimmune (Inst), Genmab (Inst)
Travel, Accommodations, Expenses: Sarah Cannon Research Institute
Alain Algazi
Research Funding: GlaxoSmithKline
Karl Lewis
Research Funding: GlaxoSmithKline (Inst)
Georgina V. Long
Honoraria: GlaxoSmithKline, Roche/Genentech, Bristol-Myers Squibb
Consulting or Advisory Role: GlaxoSmithKline, Bristol-Myers Squibb, Novartis, Roche/Genentech, Amgen, Merck Sharp & Dohme
Travel, Accommodations, Expenses: Roche/Genentech
Kiran Patel
Employment: GlaxoSmithKline
Stock or Other Ownership: GlaxoSmithKline
Nageatte Ibrahim
Employment: GlaxoSmithKline
Stock or Other Ownership: GlaxoSmithKline
Peng Sun
Employment: GlaxoSmithKline
Stock or Other Ownership: GlaxoSmithKline
Shonda Little
Employment: GlaxoSmithKline
Stock or Other Ownership: GlaxoSmithKline
Elizabeth Cunningham
Employment: GlaxoSmithKline
Jeffrey A. Sosman
Honoraria: GlaxoSmithKline
Consulting or Advisory Role: GlaxoSmithKline
Research Funding: Bristol-Myers Squibb, Novartis
Adil Daud
Stock or Other Ownership: Oncosec
Consulting or Advisory Role: Oncosec, Merck, GlaxoSmithKline
Research Funding: Merck/Schering Plough (Inst), GlaxoSmithKline (Inst), Pfizer (Inst), Genentech/Roche (Inst), Oncosec (Inst)
Rene Gonzalez
Honoraria: Roche/Genentech, GlaxoSmithKline
Consulting or Advisory Role: Roche/Genentech, Piramal Life Science
Research Funding: Roche/Genentech, GlaxoSmithKline, Novartis, Bristol-Myers Squibb, Millennium Pharmaceuticals
REFERENCES
- 1.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One. 2012;7:e35309. doi: 10.1371/journal.pone.0035309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 6.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 7.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Callahan MK, Rampal R, Harding JJ, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367:2316–2321. doi: 10.1056/NEJMoa1208958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrews MC, Behren A, Chionh F, et al. BRAF inhibitor–driven tumor proliferation in a KRAS-mutated colon carcinoma is not overcome by MEK1/2 inhibition. J Clin Oncol. 2013;31:e448–e451. doi: 10.1200/JCO.2013.50.4118. [DOI] [PubMed] [Google Scholar]
- 11.Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin Cancer Res. 2014;20:1965–1977. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- 14.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767–1774. doi: 10.1200/JCO.2012.44.7888. [DOI] [PubMed] [Google Scholar]
- 17.Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014;4:69–79. doi: 10.1158/2159-8290.CD-13-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haq R, Shoag J, Andreu-Perez P, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23:302–315. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paraiso KH, Fedorenko, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–1730. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 29.Wagle N, Van Allen EM, Treacy DJ, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61–68. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villanueva J, Infante JR, Krepler C, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4:1090–1099. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long GV, Stroyakovsky DL, Gogas H, et al. COMBI-d: A randomized, double-blinded, phase III study comparing the combination of dabrafenib and trametinib to dabrafenib and trametinib placebo as first-line therapy in patients (pts) with unresectable or metastatic BRAFV600E/K mutation-positive cutaneous melanoma. J Clin Oncol. 2014;32(suppl 15s):574s. abstr 9011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.