

Abstract
Objective
After arterial injury, endothelial cell (EC) migration is essential for healing, but lipid oxidation products activate TRPC6 and TRPC5 ion channels leading to increased intracellular calcium and inhibition of EC migration in vitro. The objective of this study was to further evaluate the role of TRPC channels in EC migration in vitro and validate in vitro findings in an in vivo model.
Methods
Mouse aortic ECs were cultured and the effect of lysophosphatidylcholine (lysoPC), the major lysophospholipid in oxidized LDL, on migration was assessed in a razor scrape assay. EC healing after a carotid injury with electrocautery was evaluated in wild-type (WT), TRPC6-/-, and TRPC5-/- mice receiving either a chow or high cholesterol (HC) diet.
Results
LysoPC inhibited EC migration of WT ECs to 22% of baseline, TRPC5-/- EC migration to 53% of baseline but had minimal effect on TRPC6-/- EC migration. Hypercholesterolemia severely impaired EC healing in vivo, with 51.4±1.8% and 24.9±2.0% of the injury resurfaced with ECs at 5 days in chow-fed and HC-fed WT mice, respectively (P<.001). Hypercholesterolemia did not impair healing in TRPC6-/- mice, with coverage of 48.4±3.4% and 46.8±1.6% in chow-fed and HC-fed TRPC6-/- mice, respectively. Hypercholesterolemia had a reduced inhibitory effect in TRPC5-/- mice, with EC coverage of 51.7±3.0% and 37.7±1.4% in chow-fed and HC-fed TRPC5-/- mice, respectively.
Conclusions
Results suggest that activation of TRPC6 and TRPC5 channels are key contributors to impaired endothelial healing of arterial injuries in hypercholesterolemic mice.
Keywords: Endothelium, TRPC6, TRPC5, hypercholesterolemia, endothelial migration, arterial injury
Coronary and peripheral arterial diseases are prevalent disorders that have a major impact on quality of life. Angioplasty is one of the key interventions performed in the management of these disorders, but this causes endothelial cell (EC) denudation that contributes to thrombogenicity of the site and removes a regulator of smooth muscle cell proliferation. EC migration is critical for arterial healing after a denuding injury, but elevated cholesterol is associated with delayed healing.1 The mechanism is not clear, but several factors, including oxidatively modified lipids and lipoproteins and influx of calcium ions (Ca2+), have been identified as inhibiting EC migration in cell culture. A better understanding of factors inhibiting EC migration could suggest ways to improve healing of arterial injuries in vivo.
Oxidized low-density lipoprotein (LDL) and lysophosphatidylcholine (lysoPC), the major lysophospholipid in oxidized LDL, inhibit EC migration in vitro.2, 3 LysoPC is abundant in plasma and accumulates in atherosclerotic lesions, 4-6 and is associated with impaired EC healing after arterial injury in hypercholesterolemic mice.1 The mechanisms responsible for the antimigratory effect of oxidized LDL and lysoPC on ECs include increasing cellular production of reactive oxygen species,7 increasing cell membrane fluidity,8 altering membrane ion channels leading to an increase in the intracellular concentration of free Ca2+ ([Ca2+]i) in ECs,9 and disruption of the time- and site-specific regulation of adhesion complex assembly and disassembly required for normal cell migration.10
The impact of changes in [Ca2+]i on EC migration has been evaluated in multiple studies. EC monolayer disruption results in an influx of Ca2+ from the extracellular space, and a rise in [Ca2+]i is required for initiation of movement.11 A generalized, sustained increase in [Ca2+]i, such as that induced by lysoPC, activates calpain and disrupts the coordinated time- and site-specific changes in focal adhesions and cytoskeleton that are required for cell movement and monolayer repair.12 Thus, a brief, localized spike in [Ca2+]i is essential to initiate migration, but a prolonged elevation of [Ca2+]i inhibits EC migration.
Calcium can enter ECs by multiple pathways, including receptor-operated and store-operated calcium channels. Previous in vitro studies in our laboratory have shown that canonical transient receptor potential (TRPC) channels are important in lysoPC-induced Ca2+ entry.9 Specifically, exposure of ECs to lysoPC leads to TRPC6 activation with an increase in [Ca2+]i and externalization of TRPC5 with subsequent inhibition of EC migration. In TRPC6-/- ECs, lysoPC does not cause TRPC5 to translocate to the membrane and has little effect on migration.9 The use of knockout cells or mice to assess the role of TRPC channels in EC migration can be confounded by compensatory upregulation of other calcium channels. TRPC3, a member of the same subfamily of TRPC channels as TRPC6, is upregulated in TRPC6-/- mice,13 but no similar alteration has been identified in TRPC5-/- mice. The goals of the present study are to determine if lysoPC-induced TRPC6 and TRPC5 activation are co-dependent and to assess the relevance of in vitro findings to arterial healing in vivo following a denuding arterial injury in wild-type (WT), TRPC6-/-, and TRPC5-/- mice.
METHODS
Animal Use
WT, TRPC3-/-, TRPC6-/-, and TRPC5-/- mice on a 129Sv/C57BL/6J background, as previously described,9, 14, 15 were used for these studies. The Institutional Animal Care and Use Committee approved the animal use and study protocols. The animal protocols and care complied with the Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council. Washington: National Academy Press, 1996.
EC isolation and culture
Mouse aortic ECs (MAECs) were isolated from aortic rings as previously described 9. EC identity was verified by immunostaining with anti-human von Willebrand Factor polyclonal antibody (1:100, Dako, Carpinteria, CA). If other cells, including smooth muscle cells or fibroblasts, were mixed with ECs, pure ECs were cloned from the mixed cell population using a cloning disc (Millipore). MAECs in passage 3 or 4 were used for experiments.
MAEC migration
Prior to migration assays, ECs were made quiescent overnight in serum-free M199:F12 (4:1) medium with 0.1% gelatin. EC migration was studied using the razor-scrape method as previously described.12 The razor-scrape assay is a standard migration assay to evaluate early cell migration under conditions that impede cell proliferation.10 An observer blinded to the experimental condition quantified the migrating cells using NIH Image J software (http://rsb.info.nih.gov/ij/).
Biotinylation of cell surface proteins
Cell surface proteins were biotinylated as previously described.9 Briefly, ECs were grown to confluence, treated, then surface proteins biotinylated. Cells were lysed and incubated with streptavidin-agarose beads. Biotin-streptavidin complexes were collected and proteins of interest identified by immunoblot analysis.
TRPC channel detection by immunofluorescence
ECs were grown to approximately 60% confluence on 25-mm coverslips, then made quiescent for 12 hours. Cells were incubated with PBS or 10 μM lysoPC (1-palmitol-2-hydroxy-sn-glycerol-3- phosphocholine; Avanti Polar Lipids, Alabaster, AL), which produces consistent but nonlethal alterations in cells, then fixed and stained as previously described.9
Mouse carotid injury
Six-week old male mice were randomly assigned to receive either a chow diet or high cholesterol (HC) diet (Harlan Teklad, Madison, WI) containing 21% milk fat and 0.2% cholesterol by weight. The assigned diet was continued until the study concluded. When mice were 8 weeks of age, a perivascular electrical injury of the right carotid injury was created using bipolar electrocautery forceps with a 4 mm wide tip as previously described.1 Two watts of power were applied to the artery for 3 seconds.
Mouse carotid artery harvest and endothelial healing analysis
At 120 hours after electrical injury, when the injury is approximately 50% healed in chow-fed WT mice,1 animals were anesthetized, blood was collected from the inferior vena cava, and Evans Blue dye (5% in PBS, 100 μL) was injected into the inferior vena cava as previously described.1. After the mice were euthanized, arteries were perfusion-fixed with paraformaldehyde, excised, opened longitudinally, imaged, and healing measured as previously described.1
Plasma assays
The blood obtained at the time of carotid artery removal was used to measure total cholesterol, lysoPC, and thiobarbituric acid reactive substances (TBARS), which are markers of systemic oxidative stress, as previously described.1
Urine assays
Using a metabolic cage (Fisher Scientific, Pittsburgh, PA), urine was collected during the first day after injury and the day prior to carotid artery harvest for 8-isoprostane determination as an additional measure of systemic oxidative stress. Urine samples from two mice were pooled to provide an adequate volume for analysis. BHT added for a final concentration of 227 μmol/L. Samples were stored under N2 at -80°C until analysis. Prior to analysis, the samples were purified by affinity column (Cayman Chemical Co., Ann Arbor, MI). Urine creatinine and 8-isoprostane levels were determined by colorimetric assay and enzyme immunoassay, respectively, following the manufacturer’s protocol (Cayman Chemical Co.).
Immunostaining for macrophages
Immunohistochemistry was performed on paraffin-embedded specimens from the right and left carotid arteries of mice in each treatment group to determine the presence of macrophages using an antibody to Mac-3 (1:50, BD Pharmingen, Franklin Lakes, NJ) as previously described 1. Two observers blinded to the treatment group counted total cells and Mac-3 positive cells in the proximal uninjured portion and denuded area of the right common carotid artery (RCCA) and the uninjured left common carotid (LCCA). Data were reported as the percentage of macrophages in a 0.05 mm2 area of each region of the artery.
Statistical analysis
GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA) was utilized for statistical analysis. The results were expressed as the mean ± standard error (SE) of the mean, unless otherwise specified. Student t-test and analysis of variance (ANOVA) followed by Tukey’s posthoc multiple comparison test were used for the analysis. P < .05 was considered statistically significant.
RESULTS
LysoPC-induced TRPC6 externalization in the plasma membrane does not require TRPC5 activation
Our previous studies suggested that lysoPC inhibited EC migration in vitro by opening TRPC6 channels permitting the influx of Ca2+ and activation of TRPC5 channels.9 TRPC5 was not activated in TRPC6-/- ECs, but the sequential opening of TRPC6 and TRPC5 channels or co-dependence of their activation was not clarified. To further study the relationship, lysoPC-induced TRPC6 externalization in WT and TRPC5-/- ECs was determined by biotinylation assay. LysoPC increased TRPC6 externalization both in WT and TRPC5-/- ECs (n = 3, Fig 1A, top panel). Immunoblot analysis of the lysates from WT and TRPC5-/- cells showed no difference in total TRPC6, indicating that the increased in TRPC6 at the plasma membrane was not due to changes in TRPC6 levels (n = 3, Fig 1A, bottom panel).
Fig 1. Lysophosphatidylcholine (lysoPC) induces externalization of canonical transient receptor potential (TRPC) 6 protein in TRPC5-/- endothelial cells (ECs).
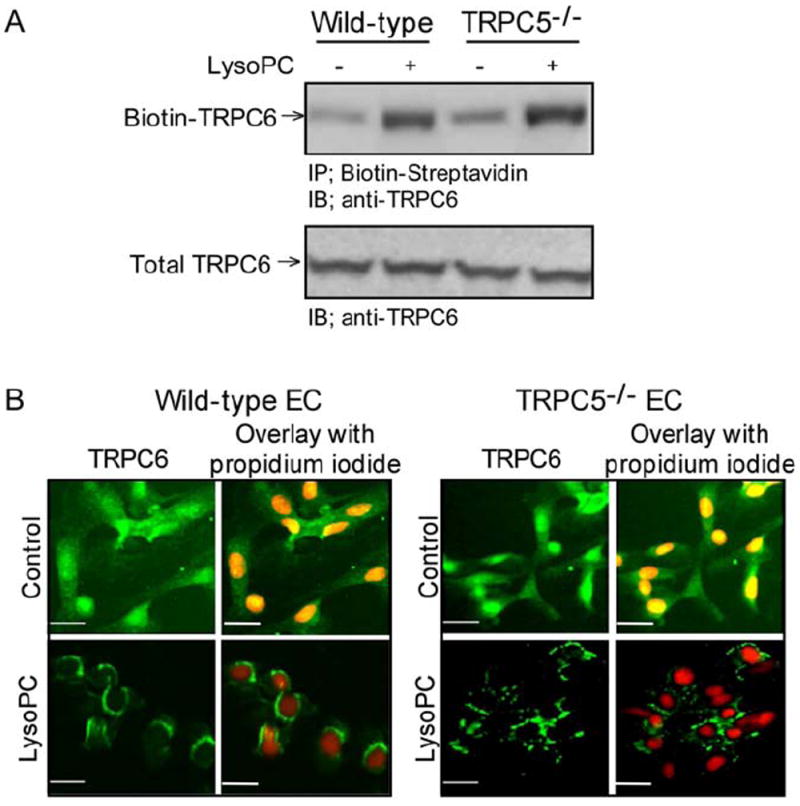
(A) Wild-type and TRPC5-/- ECs were incubated with or without lysoPC for 1 hour. Cell surface proteins were biotinylated and immunoblot analysis was performed for biotinylated TRPC6 (top panel). Prior to incubation with streptavidin-agarose beads, an aliquot of cell lysate was removed for immunoblot analysis to determine total TRPC6 protein level (bottom panel). (B) WT or TRPC5-/- ECs were incubated with or without lysoPC for 15 minutes, and then exposed to anti-TRPC6 antibody followed by Alexa 488 conjugated secondary antibody. TRPC6 location was assessed by fluorescence microscopy. Nuclei were detected by counterstaining with propidium iodide. Original magnification, x40. Bar, 40 μm.
LysoPC-induced translocation of TRPC6 to the cell surface was confirmed in WT and TRPC5-/- cells using fluorescence microscopy. Under control conditions, TRPC6 protein fluorescence pattern was diffuse throughout the cytoplasm, although it appeared to be more concentrated in the nuclear region (n = 3, Figure 1B, top panels). After incubation with lysoPC, TRPC6 fluorescence was associated with the plasma membrane in both WT and TRPC5-/- ECs (n = 3, Figure 1B, bottom panels). These studies showed that lysoPC-induced TRPC6 externalization was not dependent on TRPC5, and supported the hypothesis that TRPC6 activation precedes TRPC5 activation.
LysoPC-induced inhibition of EC migration is attenuated in TRPC5-/- MAECs
LysoPC’s inability to activate TRPC5 in TRPC6-/- ECs could account for the diminished inhibition of migration or this could be independent of TRPC5. To assess the role of TRPC5, migration of WT and TRPC5-/- MAECs was studied. Basal migration of WT and TRPC5-/- ECs was similar (Figure 2). After incubation with lysoPC (10 μM), WT EC migration was inhibited to 22% of baseline (n = 3, P < 0.001 compared with untreated), but TRPC5-/- EC migration was 53% of baseline (P < 0.001 compared with WT ECs with lysoPC). These results suggested that TRPC5 contributed but did not account for all the antimigratory effect of lysoPC.
Fig 2. The anti-migratory effect of lysophosphatidylcholine (lysoPC) is attenuated in canonical transient receptor potential (TRPC) 5-deficient (TRPC5-/-) ECs.
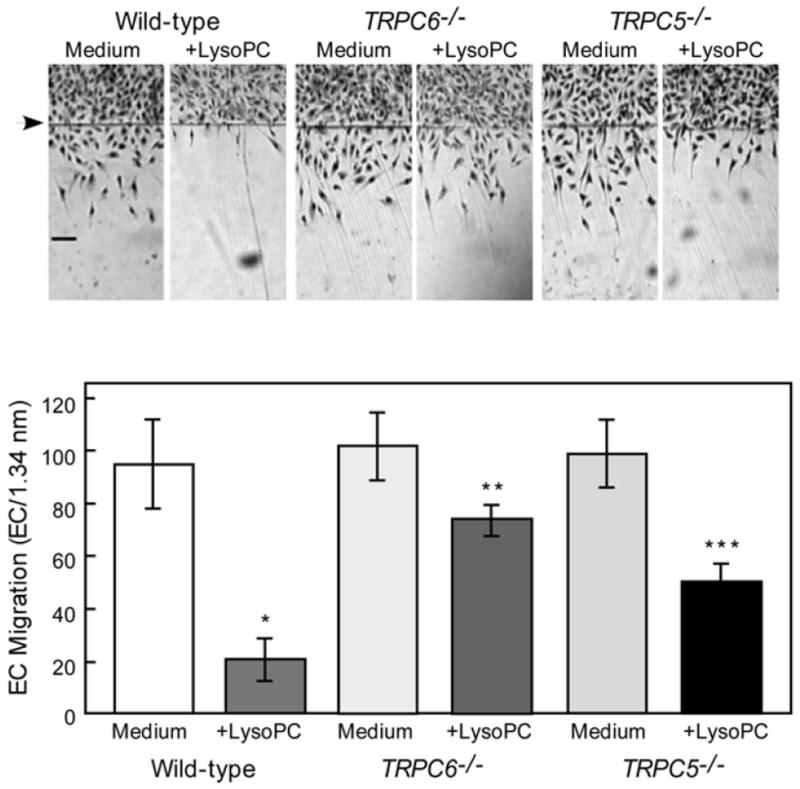
Migration assay was initiated in quiescent WT, TRPC6-/- (previously reported,9 but included for comparison), and TRPC5-/- ECs, lysoPC (10 μM) added, and migration quantitated at 24 hours. Arrow indicates starting line of migration. Original magnification, x40. Bar, 100 μm. The bottom panel represents migration results by mean ± SD (n = 3, * P < .001 compared with WT control ECs, ** P < .001 compared with TRPC6-/- control (previously reported,9 but included for comparison) and *** P < .001 compared with TRPC5-/- control).
LysoPC does not lead to activation or externalization of TRPC3 channels
TRPC3 channels are upregulated in mice that lack TRPC6 channels. To assess the potential impact of TRPC3 upregulation on EC migration, phosphorylation and externalization of TRPC3 in WT cells incubated with lysoPC was investigated. LysoPC did not cause TRPC3 phosphorylation in WT cells (n = 3, Supplemental Fig 1) or lead to TRPC3 externalization (n = 3, Supplemental Fig 1). These results suggest that TRPC3 channels did not contribute to the negative impact of lysoPC on EC migration.
Effect of TRPC channels on endothelial healing of arterial injuries
To study endothelial healing in vivo, 32 WT mice, 28 TRPC5-/- mice, 28 TRPC6-/- mice underwent a carotid injury at 8 weeks of age. Half of each group was on a chow diet and half on an HC diet. Demographics of mice are detailed in Table I. Mice on a HC diet tended to be heavier than the corresponding genotype on a chow diet, and TRPC5-/- mice were significantly heavier than the TRPC6-/- mice regardless of the dietary group. Mice tolerated the surgical procedure with no deaths or thrombosed arteries prior to the completion of the study.
Table I.
Mouse demographics and plasma cholesterol data, presented as mean ± standard error
| Group | N | Age (days) | Weight (g) | Cholesterol (mmol/L) |
|---|---|---|---|---|
| WT Chow | 16 | 57 ± 0.5 | 26.5 ± 0.5 | 2.42 ± 0.09a |
| WT High Cholesterol | 16 | 58 ± 0.5 | 28.3 ± 0.8 | 4.01 ± 0.09a |
| TRPC6-/- Chow | 14 | 56 ± 0.5 | 24.0 ± 0.4b | 2.29 ± 0.07a |
| TRPC6-/- High Cholesterol | 14 | 56 ± 0.5 | 26.3 ± 0.7c | 3.81 ± 0.07a |
| TRPC5-/- Chow | 14 | 57 ± 0.6 | 28.6 ± 0.3 | 2.40 ± 0.05a |
| TRPC5-/- High Cholesterol | 14 | 58 ± 0.4 | 29.6 ± 0.6 | 3.89 ± 0.12a |
Significant differences (P ≤ .05) are designated as:
Between any chow group and any high cholesterol group
Between TRPC6-/- Chow and TRPC5-/- Chow
Between TRPC6-/- High Cholesterol and TRPC5-/- High Cholesterol
Plasma cholesterol levels were significantly higher in mice fed the HC diet compared with those on the chow diet, regardless of genotype. Plasma cholesterol was 4.01 ± 0.09 mmol/L and 2.42 ± 0.09 mmol/L in WT mice on HC and chow diets, respectively (P < .001; Table I). In TRPC6-/- mice these values were 3.81 ± 0.07 mmol/L and 2.29 ± 0.07 mmol/L, and in TRPC5-/- mice 3.89 ± 0.12 mmol/L and 2.40 ± 0.05 mmol/L for mice on HC and chow diets, respectively (P < .001; Table I). Across all genotypes, plasma cholesterol levels were similar in mice receiving the chow diet and similar in mice receiving the HC diet.
At 120 hours after the electrocautery injury, carotid healing was quantitated.
Reendothelialization was significantly decreased in the WT mice fed a HC diet compared with a chow diet, endothelial coverage being 24.9 ± 2.0% and 51.4 ± 1.8% of the injured area, respectively (P < .0001, Fig 3). Similar to chow-fed WT mice, reendothelialization in the chow-fed TRPC6-/- mice was 48.4 ± 3.4%, but unlike the WT mice, HC feeding of TRPC6-/- mice did not inhibit reendothelialization, with coverage of 46.8 ± 1.6% (P < .0001 compared with WT HC, P = NS compared with chow-fed TRPC6-/- mice, Fig 3). Endothelial healing in chow-fed TRPC5-/- mice was 51.7 ± 3.0% (Fig 3), but reendothelialization in TRPC5-/- mice on a HC diet was significantly decreased with coverage of 37.7 ± 1.4% (P < .001, Fig 3). This was significantly improved, however, compared with WT mice on a HC diet (P = .0001). There was no significant difference in reendothelialization in the chow-fed animals of each genotype. In the hypercholesterolemic mice, endothelial healing was preserved in TRPC6-/- mice and partially preserved in TRPC5-/- mice compared with WT mice.
Fig 3. Endothelial healing after carotid artery injury.
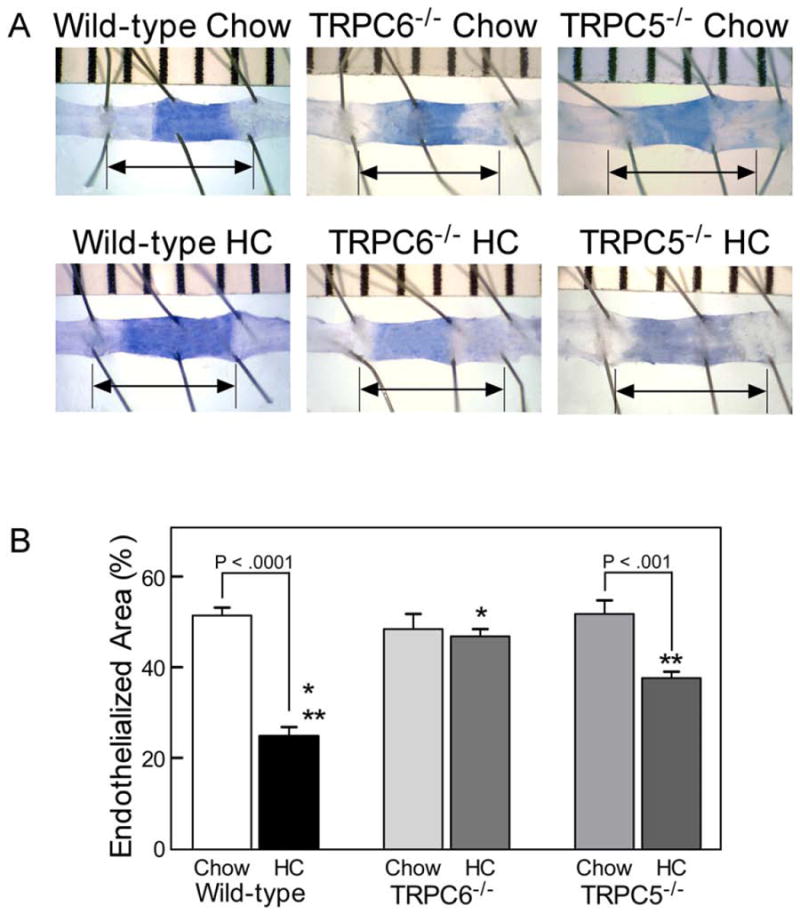
(A) Representative images 120 hours after carotid electrocautery. The area without an intact endothelial monolayer stained with Evans Blue. The arrow identifies the length of the original injury. (B) Reendothelialization results shown as the percent of reendothelialized area relative to the total injured area. Results are expressed as the mean ± standard error for each group: wild-type chow diet (n = 10), wild-type high cholesterol (HC) diet (n = 10), TRPC6-/- chow diet (n = 8), TRPC6-/- HC diet (n = 8, * P < .0001 compared with wild-type HC), TRPC5-/- chow diet (n = 8), and TRPC5-/- HC diet (n = 8, ** P = .0001 compared with wild-type HC).
Systemic oxidative stress markers
Markers of systemic oxidative stress, plasma lysoPC and TBARS levels, were measured in blood samples obtained 120 hours after injury. Plasma lysoPC levels were similar in normocholesterolemic mice of all genotypes. For each genotype, plasma lysoPC levels were significantly higher in hypercholesterolemic mice compared with normocholesterolemic mice (P = .002 for WT mice and P < .0001 for TRPC6-/- and TRPC5-/- mice, Table II). LysoPC levels were significantly higher in hypercholesterolemic TRPC6-/- and TRPC5-/- mice than in hypercholesterolemic WT mice (P < .0001 for TRPC6-/- mice and P < .001 for TRPC5-/- mice). Plasma TBARS levels were similar for all genotypes and significantly higher in hypercholesterolemic mice as compared with normocholesterolemic mice (P < .0001 for WT, TRPC6-/-, and TRPC5-/- mice, Table II).
Table II.
Mouse plasma and urine markers of oxidative stress, presented as mean ± standard error
| Group | N Plasma | Plasma LysoPC (μmol/L) | Plasma TBARS (μmol/L) | N Urine | Urine 8-Iso/Cr Day 1 (pg/mg) | Urine 8-Iso/Cr Day 5 (pg/mg) |
|---|---|---|---|---|---|---|
| WT Chow | 16 | 488.8 ± 18.2a | 0.432 ± 0.01a | 8 | 1258.9 ± 93.5a | 848.1 ± 70.5 |
| WT HC | 16 | 581.8 ± 20.7b, c | 0.750 ± 0.02 | 7 | 3896.8 ± 310.7b, c | 998.7 ± 83.6 |
| TRPC6-/- Chow | 14 | 483.6 ± 20.5d, e | 0.411 ± 0.01d | 6 | 1312.0 ± 134.6d | 776.6 ± 69.2d |
| TRPC6-/- HC | 14 | 721.3 ± 17.3 | 0.747 ± 0.01 | 5 | 2527.5 ± 256.7 | 1054.0 ± 63.7 |
| TRPC6-/- Chow | 14 | 444.6 ± 17e | 0.419 ± 0.01e | 5 | 1162.5 ± 64.1e | 764.3 ± 73.1 |
| TRPC6-/- HC | 14 | 691.1 ± 18.5 | 0.763 ± 0.01 | 5 | 2330.7 ± 219.5 | 1074.0 ± 132.5 |
HC = High cholesterol
LysoPC = Lysophosphatidylcholine
TBARS = Thiobarbituric acid reactive substances
8-iso/Cr = 8-isoprostane / Creatinine
Significant differences (P ≤ .05) are designated as:
Between WT Chow and WT HC
Between WT HC and TRPC6-/- HC
Between WT HC and TRPC5-/- HC
Between TRPC6-/- Chow and TRPC6-/- HC
Between TRPC5-/- Chow and TRPC5-/- HC
Urine 8-isoprostane levels reflect systemic oxidative stress. Levels were determined on day 1 and day 5 to assess the immediate and longer impact of the injury and diet on levels of oxidative stress. On day 1 after injury, the hypercholesterolemic mice of each genotype, had significantly higher 8-isoprostane levels than the corresponding chow-fed mice (P < .0001 for WT mice, P = .002 for TRPC6-/- mice, and P < .001 for TRPC5-/- mice, Table II). For chow-fed mice, 8-isoprostane levels on day 1 were not significantly different between genotypes. WT mice receiving the HC diet, however, had significantly higher 8-isoprostane levels on day 1 than did TRPC6-/- (P = .01) and TRPC5-/- (P = .003) mice (Table II). By day 5, 8-isoprostane levels were similar in all chow-fed groups and all HC-fed groups regardless of genotype (Table II). Comparing chow-fed and HC-fed mice, a difference was found only in the TRPC6-/- mice (Table II). Results suggested that surgical injury causes a significant oxidative stress.
Local inflammation markers
Macrophage infiltration and total cell number in the injured and uninjured arteries of WT, TRPC5-/-, and TRPC6-/- mice were assessed as markers of local inflammation. The total cell count for RCCA was lower than LCCA, perhaps reflecting cell death caused by dissection and injury (Table III). No significant differences were noted for the corresponding area within dietary groups across the genotypes. For all groups, the percentage of macrophages present was significantly higher in the injured RCCA and the non-injured RCCA compared with the corresponding LCCA (Table III). Macrophage infiltration into the injured right carotid artery was not significantly influenced by diet or genotype.
Table III.
Cellularity and macrophage infiltration of the injured right common carotid artery and non-injured left common carotid artery, expressed as mean ± standard error
| Group | RCCA Injured | RCCA Non-injured | LCCA | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| N | Total Cellsa | % Macrophageb | Total Cellsa | % Macrophageb | N | Total Cellsa | % Macrophageb | |
| WT Chow | 3 | 188 ± 25 | 51 ± 9.6c | 207 ± 18 | 54 ± 8.1c | 4 | 347 ± 34d | 1 ± 0.3 |
| WT HC | 6 | 247 ± 17 | 45 ± 5.7c | 245 ± 30 | 37 ± 6.0c | 6 | 331 ± 11d | 2 ± 0.5e |
| TRPC6-/- Chow | 2 | 151 ± 11 | 45 ± 17c | 246 ± 20 | 45 ± 7.7c | 6 | 344 ± 32 | 4 ± 1.1 |
| TRPC6-/- HC | 5 | 213 ± 28 | 52 ± 3.8c | 206 ± 10 | 58 ± 4.6c | 6 | 324 ± 22d | 2 ± 0.3f |
| TRPC5-/- Chow | 6 | 225 ± 17 | 40 ± 3.6c | 211 ± 14 | 37 ± 5.4c | 5 | 255 ± 21 | 3 ± 0.9g |
| TRPC5-/- HC | 5 | 249 ± 18 | 57 ± 3.8c | 200 ± 8 | 55 ± 4.2c | 5 | 250 ± 32 | 7 ± 0.9 |
RCCA = Right common carotid artery
LCCA = Left common carotid artery
HC = High cholesterol
Total cell count per 0.05 mm2
Percent of total number of cells identified as macrophages per 0.05 mm2
Between RCCA injured or RCCA non-injured and LCCA within same study group (P < .05)
Between LCCA and RCCA injured or RCCA within same study group (P < .05)
Between WT HC LCCA and TRPC5-/- HC LCCA (P < .05)
Between TRPC6-/- HC LCCA and TRPC5-/- HC LCCA (P < .05)
Between TRPC5-/- Chow LCCA and TRPC5-/- HC LCCA (P < .05)
TRPC3 influence on endothelial migration and healing
Because TRPC3 channels are upregulated in mice that lack TRPC6 channels, migration of TRPC3-/- ECs and carotid healing in TRPC3-/- mice were also assessed. Basal migration of TRPC3-/- ECs was similar to WT ECs. LysoPC inhibited migration to 38 % and 35 % of basal migration in WT and TRPC3-/- ECs, respectively (n = 3, Supplemental Fig 1). Ten TRPC3-/- mice underwent carotid injury, five on a chow diet and five on the HC diet. TRPC3-/- mice were significantly lighter than the TRPC5-/- mice regardless of the dietary group, chow-fed and HC-fed TRPC3-/- mice weighing 24.5 ± 0.4 g and 25.0 ± 0.9 g, respectively. Cholesterol levels were markedly higher in HC-fed TRPC3-/- mice, than in chow-fed mice, 3.84 ± 0.14 mmol/L and 2.69 ± 0.08 mmol/L, respectively (P < .001), similar to other genotypes. Reendothelialization in the TRPC3-/- was very similar to WT mice. In TRPC3-/- mice on a chow diet, coverage was 50.9 ± 1.7% (P = NS compared with chow-fed WT mice, Supplemental Fig 1). Like WT mice, TRPC3-/- mice on a HC diet had significantly less healing, 23.6 ± 1.2% (P = NS compared with HC-fed WT mice, Supplemental Fig 1). Measures of oxidative stress were similar those of other genotypes in the same dietary groups. In chow-fed and HC-fed TRPC3-/- mice, plasma lysoPC levels were 459.0 ± 30.6 and 671.4 ±17.6 μmol/L, respectively (P < .001), and plasma TBARS levels were 0.434 ± 0.01 and 0.770 ± 0.001 μmol/L, respectively (P < .001).
DISCUSSION
Our previous in vitro studies have shown that lysoPC activates TRPC6 and TRPC5 channels in mouse and bovine aortic ECs with externalization of the channels to the plasma membrane, subsequent increased [Ca2+]i, and inhibition of EC migration.9 LysoPC causes similar changes in TRPC externalization and cell migration in EA cells, a human EC line (Supplemental Fig 2). Downregulation of TRPC6 prevents TRPC5 externalization, increased [Ca2+]i, and inhibition of EC migration after exposure to lysoPC. These findings led to the hypothesis of a TRPC6-TRPC5 activation cascade.9 The current study shows that in TRPC5-/- ECs, exposure to lysoPC still results in TRCP6 externalization, supporting the premise that lysoPC-induced TRPC5 activation is downstream from TRPC6 activation. The inhibitory effect of lysoPC is diminished in TRPC5-/- ECs suggesting that both TRPC6 and TRPC5 participate in lysoPC’s inhibition of EC migration. Importantly, we show that EC migration in vitro and reendothelialization of carotid injuries follow the same pattern. Endothelial healing is a combination of migration and proliferation. In vitro studies reflect only migration, as shown by identical results when the proliferation is inhibited,10 but migration is essential in the early phase of healing. Oxidized LDL and lysoPC inhibit EC migration in vitro, and hypercholesterolemia inhibits EC healing of arterial injuries in WT mice. TRPC6-/- ECs are resistant to the inhibitory effects of lysoPC in vitro,9 and hypercholesterolemia does not inhibit endothelial healing in TRPC6-/- mice. The inhibitory effect of lysoPC on TRPC5-/- ECs is intermediate between the effect on WT and TRPC6-/- ECs.
Similarly, the inhibitory effect of hypercholesterolemia on endothelial healing of arterial injuries in TRPC5-/- mice is intermediate between the effect in WT and TRPC6-/- mice. We attribute this to sequential activation of TRPC6 and TRPC5. Early interruption of this signaling pathway by TRPC6 deficiency has a more pronounced effect than a later block caused by TRPC5 deficiency. We have previously reported the impaired EC migration in hypercholesterolemic WT mice.1 In hypercholesterolemic mice lacking TRPC6 or TRPC5 protein, cholesterol levels are similar to those in WT mice, but EC healing of arterial injuries in these mice is significantly improved. No changes in lipid handling or cellular uptake of cholesterol have been reported in the TRPC6-/- or TRPC5-/- mice. Hypercholesterolemia causes increased oxidative stress and oxidative stress inhibits EC migration,1, 7 but there are no significant differences in measures of oxidative stress between genotypes. LysoPC can activate TRPC6 and TRPC5 channels in vitro.9, 16 Furthermore, reactive oxygen species can activate TRPC5 in vitro.17 We have shown that TRPC6 and TRPC5 phosphorylation is increased in hypercholesterolemic animals compared with chow-fed animals, and the current study suggests that TRPC6 and TRPC5 channels play a significant role in the adverse effects of hypercholesterolemia on EC healing of arterial injuries. The improved EC healing in the hypercholesterolemic TRPC6-/- and TRPC5-/- mice compared with WT mice does not appear to be associated with an altered environment, but an altered cellular response to the environmental conditions.
Results using TRPC6-/- or TRPC5-/- ECs or mice to assess the role of TRPC6 and TRPC5, respectively, in EC migration can be confounded by upregulation of other calcium channels to compensate for the loss of TRPC6 or TRPC5 function. TRPC3 is upregulated in TRPC6-/- mice.13 Our in vitro studies show no change in TRPC3 phosphorylation or externalization in bovine aortic ECs or WT MAECs with exposure to lysoPC, suggesting that TRPC3 is not activated by lysoPC. LysoPC’s inhibitory effect on EC migration is similar in WT and TRPC3-/- MAECs. Furthermore, identical inhibition of healing of arterial injuries in HC-fed WT and TRPC3-/- mice suggests that TRPC3 is not affected by hypercholesterolemia. These data support the unique roles of TRPC6 and TRPC5 channels in impairment of EC healing in hypercholesterolemic animals.
The current study further explores the relationship between TRPC6 and TRPC5 activation and EC migration in vitro, and transitions the investigation to an in vivo model of reendothelialization of an arterial injury. The results support the relevance of our in vitro findings on EC migration to the healing of arterial injuries in vivo. Our results suggest that hypercholesterolemia activates TRPC6 and TRPC5, and this impairs endothelial healing. Reendothelialization after arterial injury is improved when TRPC6 or TRPC5 activation is blocked. Additional studies are required to more fully characterize the mechanism by which lysoPC activates TRPC6 and to better characterize the TRPC6-TRPC5 activation cascade with a goal of developing targeted inhibition to improve EC migration and healing after arterial injury.
Supplementary Material
CLINICAL RELEVANCE.
Arterial injury following coronary and peripheral arterial angioplasty and stenting is associated with oxidative stress. Oxidized lipids activate a cascade of TRPC channels leading an influx of calcium and impaired endothelial healing. Deficiency of TRPC6 or TRPC5 channels is associated with preservation of migration. Inhibition of TRPC channel activation following arterial intervention improves endothelial healing, which should decrease luminal thrombogenicity and intimal hyperplasia at the site of an intervention.
Acknowledgments
This project was supported by Grant Numbers HL64357 and F32HL090205 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
The authors acknowledge Lutz Birnbaumer, PhD, at the National Institute of Environmental Health Science, for generously providing the WT, TRPC5-/-, TRPC6-/-, and TRPC3-/- mice.
Footnotes
DISCLOSURE
The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosenbaum MA, Miyazaki K, Graham LM. Hypercholesterolemia and oxidative stress inhibit endothelial cell healing after arterial injury. J Vasc Surg. 2012 Oct 31;55(2):489–496. doi: 10.1016/j.jvs.2011.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murugesan G, Chisolm GM, Fox PL. Oxidized low density lipoprotein inhibits the migration of aortic endothelial cells in vitro. J Cell Biol. 1993;120:1011–1019. doi: 10.1083/jcb.120.4.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murugesan G, Fox PL. Role of lysophosphatidylcholine in the inhibition of endothelial cell motility by oxidized low density lipoprotein. J Clin Invest. 1996;97:2736–2744. doi: 10.1172/JCI118728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73:321–354. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 5.Portman OW, Alexander M. Lysophosphatidylcholine concentration and metabolism in aortic intima plus inner media: effect of nutritionally induced atherosclerosis. J Lipid Res. 1969;10:158–165. [PubMed] [Google Scholar]
- 6.Chen L, Liang BH, Froese DE, Liu SY, Wong JT, Tran K, Hatch GM, Mymin D, Kroeger EA, Man RYK, Choy PC. Oxidative modification of low density lipoprotein in normal and hyperlipidemic patients: Effect of lysophosphatidylcholine composition on vascular relaxation. J Lipid Res. 1997;38:546–553. [PubMed] [Google Scholar]
- 7.van Aalst JA, Zhang D-M, Miyazaki K, Colles SM, Fox PL, Graham LM. Role of reactive oxygen species in inhibition of endothelial cell migration by oxidized low-density lipoprotein. J Vasc Surg. 2004;40:1208–1215. doi: 10.1016/j.jvs.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh PK, Vasanji A, Murugesan G, Eppell SJ, Graham LM, Fox PL. Membrane microviscosity regulates endothelial cell motility. Nature Cell Biology. 2002;4:894–900. doi: 10.1038/ncb873. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhuri P, Colles SM, Bhat M, Van Wagoner DR, Birnbaumer L, Graham LM. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol Biol Cell. 2008;19:3203–3211. doi: 10.1091/mbc.E07-08-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaudhuri P, Colles SM, Fox PL, Graham LM. Protein kinase Cδ-dependent phosphorylation of syndecan-4 regulates cell migration. Circ Res. 2005;97:674–681. doi: 10.1161/01.RES.0000184667.82354.b1. [DOI] [PubMed] [Google Scholar]
- 11.Tran POT, Hinman LE, Unger GM, Sammak PJ. A wound-induced [Ca2+]i increase and its transcriptional activation of immediate early genes is important in the regulation of motility. Exp Cell Res. 1999;246:319–326. doi: 10.1006/excr.1998.4239. [DOI] [PubMed] [Google Scholar]
- 12.Chaudhuri P, Colles SM, Damron DS, Graham LM. Lysophosphatidylcholine inhibits endothelial cell migration by increasing intracellular calcium and activating calpain. Arterioscler Thromb Vasc Biol. 2003;23:218–223. doi: 10.1161/01.atv.0000052673.77316.01. [DOI] [PubMed] [Google Scholar]
- 13.Dietrich A, Mederos y, Schnitzler M, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008 Aug 14;59(3):392–398. doi: 10.1016/j.neuron.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dasari S, Abramowitz J, Birnbaumer L, Gulledge AT. Do canonical transient receptor potential channels mediate cholinergic excitation of cortical pyramidal neurons? Neuroreport. 2013 Jul 10;24(10):550–554. doi: 10.1097/WNR.0b013e3283621344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flemming PK, Dedman AM, Xu S-Z, Li J, Zeng F, Naylor J, Benham CD, Bateson AN, Muraki K, Beech DJ. Sensing of lysophospholipids by TRPC5 calcium channel. J Biol Chem. 2006;281:4977–4982. doi: 10.1074/jbc.M510301200. [DOI] [PubMed] [Google Scholar]
- 17.Naylor J, Al-Shawaf E, McKeown L, Manna PT, Porter KE, O’Regan D, Muraki K, Beech DJ. TRPC5 channel sensitivities to antioxidants and hydroxylated stilbenes. J Biol Chem. 2011 Feb 18;286(7):5078–5086. doi: 10.1074/jbc.M110.196956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.