

Abstract
Background
The homeodomain leucine zipper (HD-Zip) transcription factor family is one of the largest plant specific superfamilies, and includes genes with roles in modulation of plant growth and response to environmental stresses. Many HD-Zip genes are characterized in Arabidopsis (Arabidopsis thaliana), and members of the family are being investigated for abiotic stress responses in rice (Oryza sativa), maize (Zea mays), poplar (Populus trichocarpa) and cucumber (Cucmis sativus). Findings in these species suggest HD-Zip genes as high priority candidates for crop improvement.
Results
In this study we have identified members of the HD-Zip gene family in soybean cv. ‘Williams 82’, and characterized their expression under dehydration and salt stress. Homology searches with BLASTP and Hidden Markov Model guided sequence alignments identified 101 HD-Zip genes in the soybean genome. Phylogeny reconstruction coupled with domain and gene structure analyses using soybean, Arabidopsis, rice, grape (Vitis vinifera), and Medicago truncatula homologues enabled placement of these sequences into four previously described subfamilies. Of the 101 HD-Zip genes identified in soybean, 88 exist as whole-genome duplication-derived gene pairs, indicating high retention of these genes following polyploidy in Glycine ~13 Mya. The HD-Zip genes exhibit ubiquitous expression patterns across 24 conditions that include 17 tissues of soybean. An RNA-Seq experiment performed to study differential gene expression at 0, 1, 6 and 12 hr soybean roots under dehydration and salt stress identified 20 differentially expressed (DE) genes. Several of these DE genes are orthologs of genes previously reported to play a role under abiotic stress, implying conservation of HD-Zip gene functions across species. Screening of HD-Zip promoters identified transcription factor binding sites that are overrepresented in the DE genes under both dehydration and salt stress, providing further support for the role of HD-Zip genes in abiotic stress responses.
Conclusions
We provide a thorough description of soybean HD-Zip genes, and identify potential candidates with probable roles in dehydration and salt stress. Expression profiles generated for all soybean genes, under dehydration and salt stress, at four time points, will serve as an important resource for the soybean research community, and will aid in understanding plant responses to abiotic stress.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-950) contains supplementary material, which is available to authorized users.
Keywords: Soybean, HD-Zip, Transcription factor, Gene family, Whole-genome duplication, RNA-Seq, Dehydration stress, Salt stress, Abiotic stress
Background
Plants sense and respond to environmental variations in temperature, nutrient availability, water level, and light conditions. The homeodomain leucine zipper (HD-Zip) transcription factors play a significant role in regulating plant growth adaptation responses by integrating developmental and environmental signals. Homeodomain leucine zipper (HD-Zip) transcription factors have been found exclusively in the plant kingdom [1, 2], the only exception being the recent identification in the charophycean algae [3]. The characteristic feature of the HD-Zip gene family is the association of homeodomain (HD) and the leucine zipper (LZ) motif in a single protein. In other kingdoms, they are present as domains of distinct proteins. The homeodomain is a ~60 amino acid DNA binding domain composed of a helix-turn-helix structure that folds into three characteristic alpha-helices, capable of interacting specifically with DNA [2]. The LZ motif is a dimerization motif and is located immediately after the HD. The LZ motif allows the formation of homo- and hetero-dimers that are required for binding to DNA. The HD-Zip transcription factors can be subdivided into four subfamilies: HD-Zip I to IV, based on distinct sequence features (DNA-binding domains and additional conserved motifs that are specific to each of the subfamilies), and distinct functions of proteins from each of the subfamilies (for reviews, see [1, 4]).
The HD-Zip superfamily has been analyzed in several species including Arabidopsis (Arabidopsis thaliana) [1, 5, 6], rice (Oryza sativa) [4, 7], maize (Zea mays) [8], poplar (Populus trichocarpa) [9], and the HD-Zip I and IV genes in cucumber (Cucumis sativus) [10, 11]. However, functional characterization studies have been limited to the model plant Arabidopsis, while a few selected genes have been investigated in other species [1, 4]. A subset of the HD-Zip genes have recently been described in soybean (Glycine max) [12]. HD-Zip genes are involved in several abiotic stress responses, meristem regulation, photomorphogenesis, and root development [1, 4]. The HD-Zip I genes have been investigated for their roles in water deficit and salt stress responses. The HD-Zip I Arabidopsis genes ATHB7 and ATHB12, and their orthologs in other species, including HaHB4 from sunflower (Helianthus annuus), NaHD20 in Nicotiana attenuatta, and OsHOX6 in rice (Oryza sativa), have increased expression under water-stress conditions [7, 13–15]. ATHB7 and ATHB12 act as negative regulators of growth and development by reducing plant growth under water-deficit conditions [13, 16, 17]. HaHB4 delays the onset of senescence when expressed in Arabidopsis [18, 19]. The Arabidopsis HD-Zip I genes ATHB5 and ATHB6, and the homologs in Craterostigma plantagineum CpHB5, 6, 7 and CpHB8, are involved in water deficit stress [20]. ATHB5 acts as a positive regulator of ABA responsiveness at the seedling stage, with elevated levels of ATHB5 resulting in higher ABA responsiveness. On the contrary, ABA reduces the wild-type expression of ATHB5, indicating ATHB5 is part of a negative feedback loop regulating ABA sensitivity in the germinating seedlings [21]. This implies ATHB5 mediates an initial response of the seedling to an ABA signal imposed (for instance, seedling development under limited-term water-deficit conditions) - but reduces the response to extended water stress. The Arabidopsis gene ATHB6 has increased expression under water deficit stress [22]. Arabidopsis plants overexpressing ATHB6 display lowered stomatal closure and reduced inhibition of germination by ABA [23] - the characteristics of the ABA-insensitive mutant abi1 and abi2 [24]. Deng et al. [25] suggested that ATHB6 acts as a negative regulator of ABA response under water deficit stress.
A recent study in maize found all 17 HD-Zip I genes differentially expressed (DE) under drought stress [8]. The Arabidopsis genes ATHB21, ATHB40 and ATHB53 and the M. truncatula gene MTHB1 are induced by salt stress [4]. The over-expression of MTHB1 reduces lateral root emergence. Ariel et al. [26] proposed reduced lateral root growth as a mechanism to reduce the exposure of the roots to high saline soil. The Arabidopsis gene HAT22, the rice genes OsHOX11 and OsHOX27, and the C. plantagineum genes CpHB1 and CpHB2, all in HD-Zip II, are induced by water stress [7, 20, 27]. Thus, it is evident that members of the HD-Zip I and II are enriched for genes that are involved in water deficit and salt stress. The emphasis in the literature has focused on HD-Zip I proteins for their role in abiotic stress, while systematic characterization of genes from the other subfamilies has been lacking. A recent study in rice shows the importance of investigating genes from other subfamilies. Yu et al. [28] demonstrated the overexpression of a HD-Zip IV gene (HDG11) confers drought tolerance, and increases yield under both normal and drought conditions. With the utilization of high throughput sequencing techniques such as RNA-Seq, it is possible to investigate the expression of HD-Zip genes belonging to all subfamilies in the same experiment, and identify potential candidates for functional characterization studies.
The identification and classification of HD-Zip genes in prior studies has been based on homology searches, well-conserved domains and motifs in each of the subfamilies, and on conserved gene structures among subfamily members [5–11]. The availability of whole genome sequence information for increasing numbers of angiosperm species has enabled utilization of evolutionary relationships among the species to help characterize HD-Zip genes. Evolutionary relationships among species in a gene family analysis can be combined with whole genome duplication (WGD) histories. The eudicots Arabidopsis, grape, soybean and M. truncatula share a common “gamma” genome triplication event that occurred around 117 million years ago (Mya), early in the eudicot evolution [29, 30]. The Arabidopsis lineage shows a signal for two additional rounds of WGD events within the last 70 million years [30, 31]. Soybean and Medicago share a common legume-specific WGD event approximately 59 Mya [32, 33], and soybean has undergone an additional glycine-specific genome duplication event around 13 Mya [30, 32]. Rice shows evidence of two rounds of WGD events [30]. The grape genome has undergone a genome triplication event, but lacks a recent WGD event [34]. Conceptually, a single-copy gene in the ancestor of angiosperm plants and retained after every WGD event would give rise to the following numbers of homologous genes: 3 in grape, 6 in Medicago, 4 in rice, and 12 each in soybean and Arabidopsis. There is also evidence for two even older WGDs: one at around 320 Mya, prior to the separation of angiosperms and gymnosperms and referred to as the “ancestral seed plant WGD”; and another at around 190 Mya, predating the origin of angiosperms and termed the “ancestral angiosperm WGD” [31]. Per this model of WGDs, an angiosperm gene family is typically comprised of four old angiosperm clades, assuming a starting point of one gene copy in the ancestor of seed plants. We examine the HD-Zip family in the context of this hypothesized history of WGDs, and provide insights into evolutionary history of each of the subfamilies relative to these WGD events.
In this study, we have 1) identified all putative HD-Zip genes in the soybean genome and placed them into their respective subfamilies; 2) provided phylogenetic relationships among HD-Zip proteins from eight species that include six eudicots: poplar, cucumber, Arabidopsis, grape, soybean and M. truncatula, and two moncots: rice and maize; 3) characterized the structures of all HD-Zip genes; 4) described the genomic organization, tracing the expansion of the gene family through WGD events; 5) presented gene expression data for all HD-Zip genes in 24 conditions including at least 17 different tissues of soybean; 6) provided RNA-Seq based gene expression profiles of all soybean genes including HD-Zip genes, in the roots under normal conditions and dehydration and salt stress after 0, 1, 6 and 12 hr treatments; and 7) identified genes that may participate in HD-Zip gene pathways by screening HD-Zip promoters for conserved motif of transcription factor binding sites (TFBSs).
Methods
Homology searches, multiple sequence alignments, and phylogenetic analysis
The sequences of 47 HD-Zip (17 HD-Zip I, 9 HD-Zip II, 5 HD-Zip III and 16 HD-Zip IV) proteins of A. thaliana (TAIR8_genome_release, 11/30/09) described in Ariel et al. [1], were obtained from TAIR [35]. The proteomes of four other sequenced angiosperms G. max (assembly v1.01, JGI Glyma1.0 annotation), M. truncatula (v 3.5.1), O. sativa (MSU Release 6.0) and V. vinifera (12X March 2010 release) were obtained from the respective repositories for these genomes and BLAST databases were built for each of them on our local server. A BLASTP v2.2.22 (protein-protein BLAST) [36] search with a threshold of 1E-10 was used for initial identification of the homologous Arabidopsis HD-Zip genes in each of the genomes described above. The multiple sequence alignment of the homologous sequences from the five species was performed using MUSCLE v3.8.31 [37]. The alignment was manually inspected and trimmed using SeaView v4.2.5 [38, 39] and BBEdit v8.7.2 respectively. A preliminary phylogenetic tree (not shown) encompassing four HD-Zip subfamilies was built using CLUSTAL v2.0.12 [40] and the tree was visually examined using FigTree v1.3.1 [41].
The probable HD-Zip genes belonging to each of the four subfamilies were identified based on the clustering of sequences with known HD-Zip genes from Arabidopsis in the preliminary phylogenetic tree. The outlier sequences that did not cluster with any Arabidopsis genes were temporarily excluded. The probable HD-Zip genes were then aligned using MUSCLE to build a profile Hidden Markov Model (HMM) separately for each subfamily using the hmmbuild program, implemented in the package HMMER v3.0b2 [42]. The probable HD-Zip sequences were re-aligned to the profile HMM using hmmalign, available in the tool HMMER, and were viewed in SeaView. Sequence logos were generated for each subfamily using the web tool WebLogo [43] to identify conserved regions in the alignments (Additional file 1: Figure S1, Additional file 2: Figure S2, Additional file 3: Figure S3 and Additional file 4: Figure S4). The HMM alignments were trimmed to retain the conserved regions (HMM “match states”) using BBEdit. The trimmed alignments were used to build the phylogenetic trees for each subfamily using the maximum likelihood method implemented in PhyML v3.0 [44] available at iPlant Collaborative [45] using default settings. The approximate likelihood ratio test (aLRT) branch support values [46] are displayed on the branches in percentages. The phylogenetic tree for each subfamily was displayed using FigTree. The rooting was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies (data not shown).
The outlier sequences excluded based on the preliminary phylogenetic tree were used in a search against HMM of each subfamily using the hmmpfam available in the tool HMMER v2.3.2 and the membership of sequences in each subfamily was investigated. The process of generating a phylogenetic tree followed by excluding outlier sequences, re-alignments, building HMMs, re-aligning using HMM, and rebuilding the trees, was iterated several times for each subfamily. A phylogenetic tree with appropriate tree topology based on evolutionary relationship among the five species was then generated for each subfamily.
Lastly, we added HD-Zip I to IV sequences from maize and poplar, and HD-Zip I and IV sequences from cucumber that have recently been reported [8–11] to the final phylogenetic trees. This will allow the investigation of orthologous sequences from eight species that includes HD-Zip genes identified in all angiosperm species to date.
Validation, structural characterization, and duplication history of HD-Zip genes
The HD-Zip subfamilies have remarkably well-conserved domains, motifs, and gene structures [1, 2, 4] that can be utilized to validate genes identified using phylogenetic analysis. All sequences identified as HD-Zip genes as well as outlier sequences (excluded after preliminary phylogenetic tree construction) were used as queries in a batch search [47] against Pfam 27.0, with an E-value threshold of 1E-3 to identify the conserved domains. The conserved motifs were investigated by examining the sequence logos that were generated using HMM sequence alignments of each subfamily. The gene structure was studied using the exon-intron organization in the pre-mRNA. The gene structures were rendered using the G. max cv. Williams 82 gene models (assembly v1.01, JGI Glyma1.0 annotation) that were downloaded from Phytozome [48] and using a modified version of the Bio-Graphics 2.25 feature_draw.pl script [49]. The genomic locations were obtained from the GFF file of G. max assembly v1.01, JGI Glyma1.0 annotation, and were displayed using chromosome visualization tool (CViT) [50]. The homoeologous HD-Zip gene pairs that are a result of the early-legume WGD event (~59 Mya), and the Glycine-specific duplication event (~13 Mya), were inferred from the phylogeny, as well as from the syntenic paralog pair information available for all soybean genes from the Joint Genome Institute (JGI) at Phytozome [51]. Paralogous genes resulting from tandem duplication events were identified based on their proximity on the same chromosome [52] and pairing in the same clade in the phylogenetic tree.
Expression profiles of HD-Zip genes in 24 conditions (17 tissues) of soybean
An RNA-Seq atlas of G. max describing expression of genes in 24 conditions including at least 17 different tissues of soybean was reported by Severin et al. [53] and Libault et al. [54, 55]. The Reads/Kb/Million (RPKM) normalized data for 14 tissues investigated by Severin et al. are available for download and interactive analysis at SoyBase [56], and expression data for three additional tissues, and tissues infected with the bacterium Bradyrhizobium japonicum are available at SoyKB [57]. A gene was considered expressed if the RPKM value was greater than or equal to two in an expression atlas (modified criteria from [58]). The RPKM normalized read count data of expressed genes was log2-transformed and displayed in the form of heatmaps for each subfamily. The heatmap was generated in R [59] using the heatmap.2 function available in the gplots CRAN library. Genes in the heatmaps were ordered for consistency with the phylogeny.
Plant material and stress experiment
The seeds of G. max cv. Williams 82 were germinated on moist germination paper and were allowed to grow until the v1 stage (first trifoliolate stage) in a growth chamber maintained at 77 F and 60% humidity throughout the experiment. The temperature and humidity were continuously monitored and maintained in the growth chamber. The salt treatment was applied by transferring the seedlings into 100 mM NaCl solution. For the dehydration treatment, plants were removed from the germination paper and left in air under water-limiting conditions to impose dehydration stress. Root tissue was harvested after 0, 1, 6 and 12 hr of stress treatments. Five plants per time point were maintained for each of the stress treatments. In order to verify the gradual imposition of salt stress treatment, electrical conductivity was measured in two fragments of germination paper, after harvesting root tissue from plants exposed to salt stress at each of the time points (data not shown). Total RNA was isolated using Qiagen RNeasy® Plant mini kit from three biological replicates per time point per the manufacturer’s protocol. The RNA samples were treated with Ambion® TURBO DNA-free™ DNase to get rid of any DNA contamination in the RNA samples. The RNA samples were inspected for their quality and quantity using NanoDrop® spectrophotometer and Qubit® fluorometer.
Sequencing, data processing, gene expression analysis and annotation under stress conditions
Total RNA from 21 samples that includes three control samples (0 hr), and three biological replicates for each of the three time points 1, 6 and 12 hr under dehydration and salt stress was sent to the National Center for Genome Resources (Santa Fe, NM, USA) for sequencing on Illumina® HiSeq 2000. Seven randomly chosen samples were multiplexed in each lane and three lanes of HiSeq 2000 were utilized to generate single-end short-reads of 1×50 bp lengths. The reads were aligned with GSNAP [60] using default settings with a maximum of 4 mis-matches allowed against the Glycine max genome assembly and annotation v1.01 from Phytozome (JGI Glyma1.0 gene calls). The uniquely mapped reads that mapped to a single location in the genome were analyzed for differential gene expression between the control and treatment samples using the R package DESeq v1.7.10 [61]. A gene was considered to be DE if it satisfied the following three stringent filtering criteria: (1) P-value adjusted for multiple testing correction using Benjamini and Hochberg method [62] to be less than 0.05, (2) two fold or greater fold change, (3) residual variance quotients of both the control and treatment samples of less than 20. The residual variance criterion was used to filter genes that have significant variation between replicates, per recommendations in the DESeq manual (Released April 20, 2011). The raw and the normalized read counts, and the sequence data has been deposited in NCBI’s Gene Expression Omnibus [63, 64] and are accessible through the GEO series accession number GSE57252.
The DE genes were annotated using the top Arabidopsis hit, and the corresponding gene ontology (GO) biological process and molecular function terms were inferred [65]. The DE genes under dehydration and salt stress were then screened separately for overrepresented GO terms against all soybean genes using Fisher’s exact test [66] and Bonferroni [67] corrected significance value of less than 0.05. The overrepresented GO terms were enriched at the second level using BLAST2GO v.2.7.1 [68] and a reduced representation of enriched GO terms was obtained. The DE genes were also annotated using the SoyDB [69, 70] transcription factor (TF) database, and Fishers’s exact test followed by Bonferroni correction was utilized to determine the overrepresented TF classes under each of the stress conditions.
Screening of HD-Zip gene promoters for conserved motifs of transcription factor binding sites (TFBSs)
For the purpose of this study, the one kilobase (kb) region upstream of the annotated transcription start site for each gene was evaluated for promoter motifs. Promoter sequences were retrieved using custom Perl scripts for all gene models in the soybean genome. Promoter sequences that were either less than one kb or included two or more Ns were excluded from the analysis. The program Clover [71] was used to scan through a database of known motifs in TRANSFAC® v. 2010.4 [72]. Promoters of HD-Zip genes belonging to each subfamily were scanned separately for enriched motifs against a background of all soybean gene promoters, with a P-value threshold of 0.05 and an individual motif hit score of greater than or equal to 6. Similarly, promoters of genes that were DE in at least one of the three time points under dehydration and salt stress were screened to identify overrepresented motifs under each of the stress treatments. The overrepresented motifs were filtered to include only plant motifs. A comparison was made between motifs that were overrepresented in the promoters of HD-Zip genes belonging to each of the subfamilies and dehydration and salt stress treatments.
Results
Classification of HD-Zip genes using phylogenetic analysis
A BLASTP search with the Arabidopsis HD-Zip genes against soybean, M. truncatula, grape and rice, followed by reconstruction of the phylogeny, clustered the sequences into four previously defined HD-Zip subfamilies (I to IV). HMMs for each subfamily were used to determine subfamily membership and refine alignments. The outlier sequences excluded from the preliminary tree (see methods for details) included six sequences that belonged to the HD-Zip IV subfamily and these were included in the final phylogenetic trees of the four subfamilies (Figures 1, 2, 3 and 4).
Figure 1.
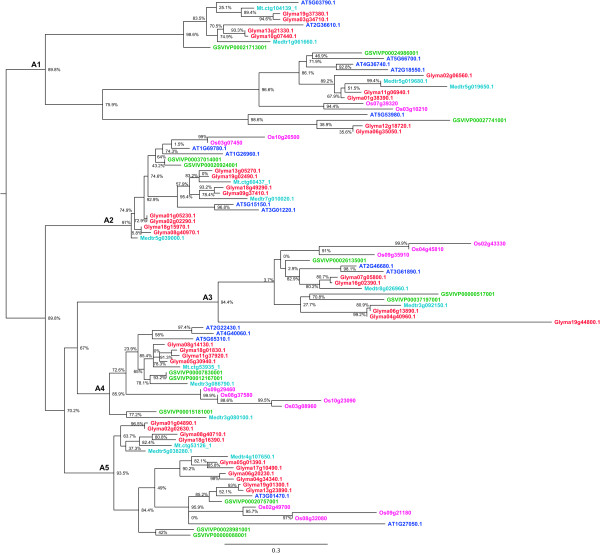
Phylogenetic relationships of HD-Zip I proteins from soybean, Medicago , Arabidopsis , grape and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A5 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. Genes from each of the species are highlighted in different colors, soybean (red), Medicago (light blue), Arabidopsis (dark blue), grape (green), and rice (Pink).
Figure 2.
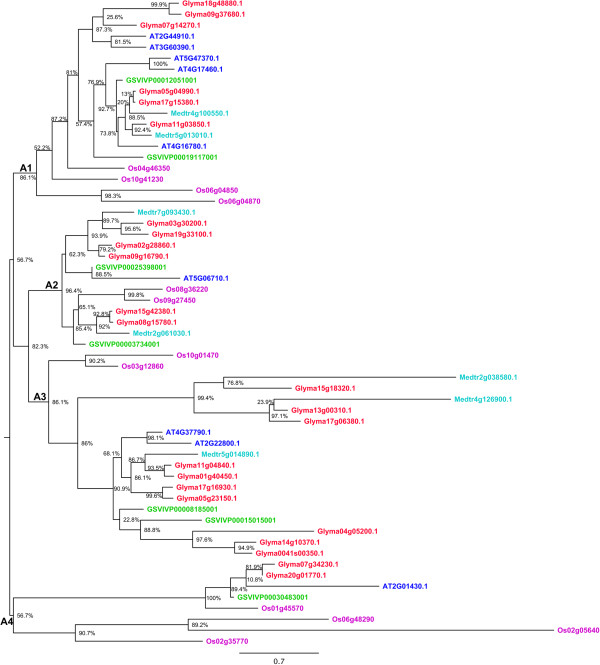
Phylogenetic relationships of HD-Zip II proteins from soybean, Medicago , Arabidopsis , grape and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A4 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. Genes from each of the species are highlighted in different colors, soybean (red), Medicago (light blue), Arabidopsis (dark blue), grape (green), and rice (Pink).
Figure 3.

Phylogenetic relationships of HD-Zip III proteins from soybean, Medicago , Arabidopsis , grape and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A4 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. Genes from each of the species are highlighted in different colors, soybean (red), Medicago (light blue), Arabidopsis (dark blue), grape (green), and rice (Pink).
Figure 4.
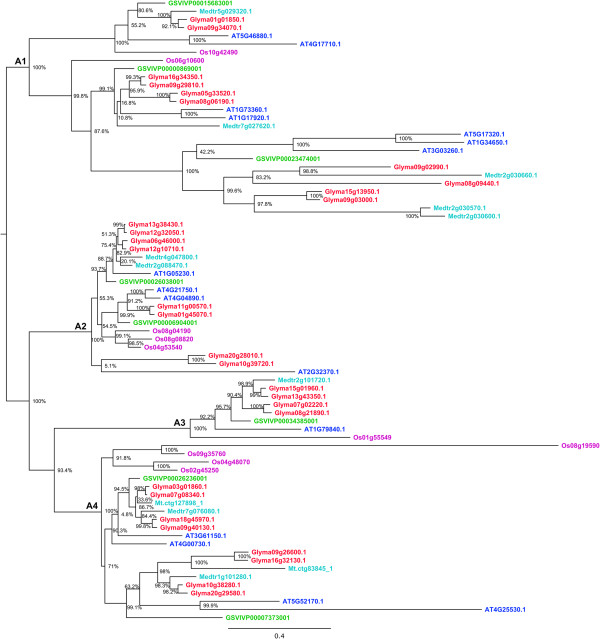
Phylogenetic relationships of HD-Zip IV proteins from soybean, Medicago , Arabidopsis , grape and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A4 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. Genes from each of the species are highlighted in different colors, soybean (red), Medicago (light blue), Arabidopsis (dark blue), grape (green), and rice (Pink). Genes Medtr5g005600.1 and Os01g57890 belong to the angiosperm clade “A2”. These two genes are not shown in the phylogeny because adding them significantly affects the topology.
Based on the species clustering patterns and the number of copies of genes belonging to each species, we identified four old angiosperm clades in HD-Zip II, III and IV, and five clades in HD-Zip I (Figures 1, 2, 3 and 4). The topology of most of the angiosperm clades is generally consistent with the species tree. Typically the two legume species (soybean and M. truncatula) form a clade, with Arabidopsis, grape, and rice each as increasingly distant outgroups from the legume sequences in the clade. The number of copies of genes of each species in each angiosperm clade reflects the number of WGD events the species has undergone. For instance, four of the five angiosperm clades in HD-Zip I phylogeny included exactly three grape sequences – likely the result of the “gamma” triplication event that occurred around 117 Mya [29, 30], and the angiosperm clade A1 in the HD-Zip I phylogeny contains nine of the 12 possible soybean sequences - possibly the result of “gamma” triplication event (~117 Mya), and the legume- (~59 Mya) and Glycine-specific (~13 Mya) WGD events [30, 32]. We identified 101 genes in soybean, 47 in Arabidopsis, 33 in grape, and 41 each in M. truncatula and rice (Table 1). The highest gene retention rate (52.7%) among the five species is in HD-Zip IV, whereas the HD-Zip III has the lowest (20.3%) retention rate (Table 1). Although soybean has the highest number of genes, grape and rice have relatively higher retention rate of 64.7% and 60.3% respectively (Table 1). Arabidopsis has the lowest retention rate of 23.0%, whereas soybean and M. truncatula have intermediate retention rates of 49.5% and 40.2% respectively (Table 1). The varying rate of retention across the five species reflects the changes in the genomes of each of the species after WGD events. Overall the phylogenetic analysis together with the WGD histories helps clarify our understanding of the evolution of each of the subfamilies.
Table 1.
Number of HD-Zip genes observed (O), expected (E) and retained (R) among five angiosperm species
| HD-Zip I (5)a | HD-Zip II (4)a | HD-Zip III (4)a | HD-Zip IV (4)a | Total - each species | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | O | E | R (%) | O | E | R (%) | O | E | R (%) | O | E | R (%) | O | E | R (%) |
| Arabidopsis thaliana (12)b | 17 | 60 | 28.3 | 9 | 48 | 18.8 | 5 | 48 | 10.4 | 16 | 48 | 33.3 | 47 | 204 | 23.0 |
| Vitis vinifera (3)b | 14 | 15 | 93.3 | 7 | 12 | 58.3 | 4 | 12 | 33.3 | 8 | 12 | 66.7 | 33 | 51 | 64.7 |
| Glycine max (12)b | 36 | 60 | 60.0 | 24 | 48 | 50.0 | 11 | 48 | 22.9 | 30 | 48 | 62.5 | 101 | 204 | 49.5 |
| Medicago truncatula (6)b | 15 | 30 | 50.0 | 7 | 24 | 29.2 | 6 | 24 | 25.0 | 13 | 24 | 54.2 | 41 | 102 | 40.2 |
| Oryza sativa (4)b | 14 | 20 | 70.0 | 12 | 16 | 75.0 | 4 | 16 | 25.0 | 11 | 16 | 68.8 | 41 | 68 | 60.3 |
| Total - Among five species | 96 | 185 | 51.9 | 59 | 148 | 39.9 | 30 | 148 | 20.3 | 78 | 148 | 52.7 | 263 | 629 | 41.8 |
aNumber of ancient angiosperm clades observed in each HD-Zip subfamily.
bNumber of genes expected in each ancient angiosperm clade based on the history of whole genome duplication events.
In the phylogeny generated with sequences from eight species, the eudictos (poplar, cucumber, Arabidopsis, grape, soybean and M. truncatula) usually clustered together, with the monocots (rice and maize) as an outgroup (Additional file 5: Figure S5, Additional file 6: Figure S6, Additional file 7: Figure S7 and Additional file 8: Figure S8).
Validation of HD-Zip genes using conserved domains, motifs and gene-structures
The HD-Zip I and II sequences contain the Homeobox (PF00046.24) domain and belong to the Homeobox associated leucine zipper family (HALZ; PF02183.13). In addition, the HD-Zip II sequences contain the conserved residues “CPSCE” at the carboxy terminal, and seven of the 24 HD-Zip II sequences contain a HD-ZIP_N (PF04618.7) domain at the N-terminal. The HD-Zip III sequences are highly conserved among all five species along the complete length of the coding sequence (Additional file 3: Figure S3). They contain the Homeobox (PF00046.24), START (PF01852.14) and MEKHLA (PF08670.6) domains. The HD-Zip IV sequences contain the Homeobox (PF00046.24) and the START (PF01852.14) domains. The presence of leucine zipper motif immediately following the homeodomain in HD-Zip III and IV sequences was confirmed using the sequence logos (Additional file 3: Figure S3, Additional file 4: Figure S4). Exon-intron structures are characteristic for each subfamily (Additional file 9: Figure S9, Additional file 10: Figure S10, Additional file 11: Figure S11 and Additional file 12: Figure S12). The HD-Zip III is particularly conserved, with each gene containing exactly 18 exons. The numbers of exons in genes in the HD-Zip I, II and IV subfamilies are in the ranges 1–5, 3–6, and 8–12. The HD-Zip I and II genes code for smaller proteins, with average peptide length of 265 and 275 amino acids, whereas HD-Zip III and IV genes code for average peptide lengths of 840 and 741 amino acids.
Genomic locations of HD-Zip genes in the soybean genome
The HD-Zip genes are distributed on all 20 chromosomes in the soybean genome, typically in the more gene-dense euchromatic regions near chromosome ends (Figure 5). One HD-Zip II gene (Glyma0041s00350) was found on an unanchored scaffold 41. The HD-Zip genes generally do not occur in clusters or arrays, with only three instances of tandemly duplicated genes.
Figure 5.
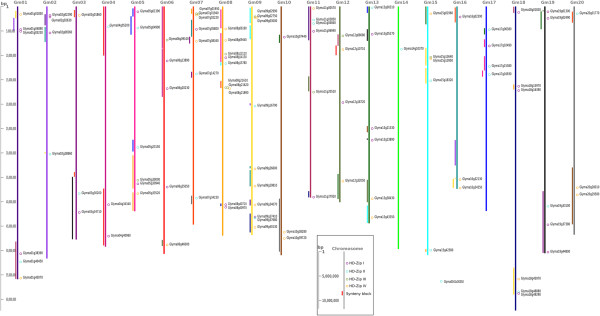
Chromosomal locations and synteny relationships of soybean HD-Zip genes. The chromosomal locations of the soybean HD-Zip genes were obtained from the GFF file of Glycine max assembly v1.01, annotation 1.0, and were displayed using chromosome visualization tool (CViT). All chromosomes and gene locations are shown to scale. Glyma0041s00350 located on scaffold 41 (149758–152298 bp) is included independently in the figure. The homoeologous gene pairs are identified with colored solid lines on the left side of the chromosomes. The chromosomes and the solid lines with identical colors are syntenic regions containing homoeologous genes. A detailed list of homoeologous HD-Zip genes is also provided in Additional file 13: Table S1.
Genome duplications and expansion of HD-Zip family in the soybean genome
Copy number expansion of the HD-Zip family in the soybean genome has primarily occurred through genome duplication events (Figure 5, Additional file 13: Table S1). Each angiosperm clade in each of the four subfamilies (Figures 1, 2, 3 and 4) contains two to four soybean gene copies that are a result of retention of genes after the legume WGD (~59 Mya) and/or the Glycine-specific WGD (~13 Mya). Retention of genes following these WGDs has been high, with retention of 32 of 36 HD-Zip I (88.9%), 20 of 24 HD-Zip II (83.3%), 10 of 11 HD-Zip III (90.9), and 26 of 30 (86.7%) HD-Zip IV genes (Additional file 13: Table S1). There are two tandemly duplicated HD-Zip pairs in subfamily III, and another pair in subfamily IV. Phylogenetic patterns indicate that the tandemly duplicated genes in subfamily III further duplicated during a WGD event, giving rise to Glyma07g01940 and Glyma07g01950 on chromosome 07 and Glyma08g21610 and Glyma08g21620 on homoeologous chromosome 8. Genes Glyma09g02990 and Glyma09g03000, in HD-Zip IV, are another pair of tandemly duplicated genes. The Glycine WGD event resulted in the homoeologous gene pair Glyma09g03000 and Glyma15gg13950, whereas the homoeologous gene for Glyma09g02990 has evidently either been lost following the WGD – or the Glyma09g02990 and Glyma09g03000 duplication occurred after the Glycine WGD. Overall, 88 of the 101 HD-Zip genes are members of homoeologous gene pairs in the soybean genome.
Expression of HD-Zip genes in 24 conditions including 17 tissues of soybean
The expression of HD-Zip genes was investigated using the G. max gene expression atlas reported by Severin et al. [53], and Libault et al. [54, 55]. Of the 44 homoeologous gene pairs, 41 show expression in identical tissues (Figures 6, 7, 8, and 9, Additional file 14: Figure S13, Additional file 15: Figure S14, Additional file 16: Figure S15 and Additional file 17: Figure S16). The remaining three show divergent patterns in different tissues between the WGD-derived paralogs (Figures 6 and 9). HD-Zip I gene Glyma06g20230 was expressed in each of the 14 tissues, whereas the homoeolog Glyma04g34340 was expressed in the roots, “pod.shell.10DAF” and “pod.shell.14DAF”. HD-Zip I gene Glyma19g01300 had expression in each of the 14 tissues, but the homoeolog Glyma13g23890 lacked expression in five “seed tissues” (10 DAF, 14 DAF, 21 DAF, 25 DAF and 28 DAF). HD-Zip IV gene Glyma11g00570 was expressed only in the flower, whereas the homoeolog Glyma01g45070 was expressed in young leaf, flower, “one.cm.pod” and “pod.shell.10DAF”. Similar divergent gene expression patterns between these homoeologous genes were also noticed in the gene expression atlas reported by Libault et al. [54] (Additional file 14: Figure S13, Additional file 17: Figure S16).
Figure 6.
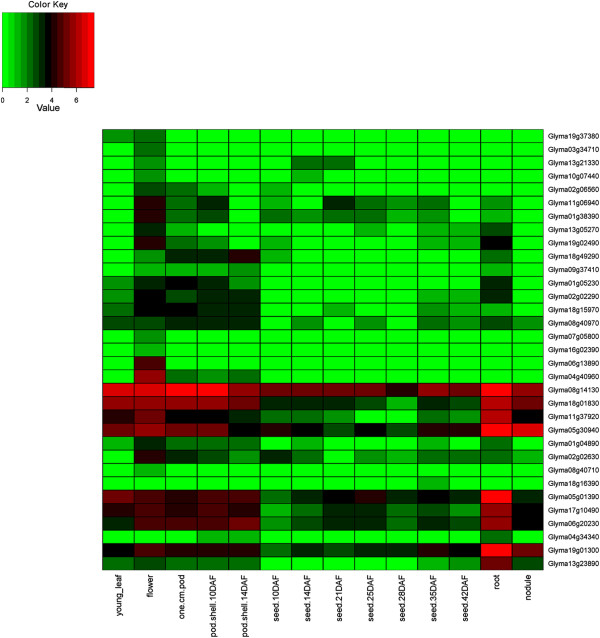
Expression profiles of HD-Zip I genes in 14 tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 1. The abbreviation “DAF” in the tissue label represents “Days after flowering”.
Figure 7.
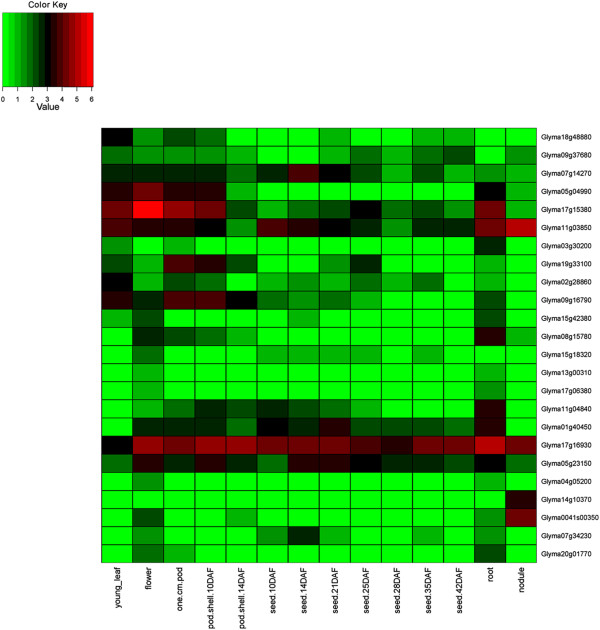
Expression profiles of HD-Zip II genes in 14 tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 2. The abbreviation “DAF” in the tissue label represents “Days after flowering”.
Figure 8.
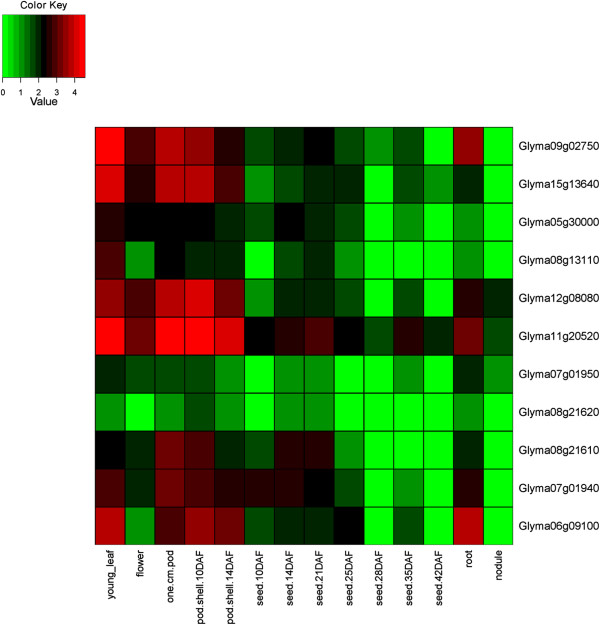
Expression profiles of HD-Zip III genes in 14 tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 3. The abbreviation “DAF” in the tissue label represents “Days after flowering”.
Figure 9.
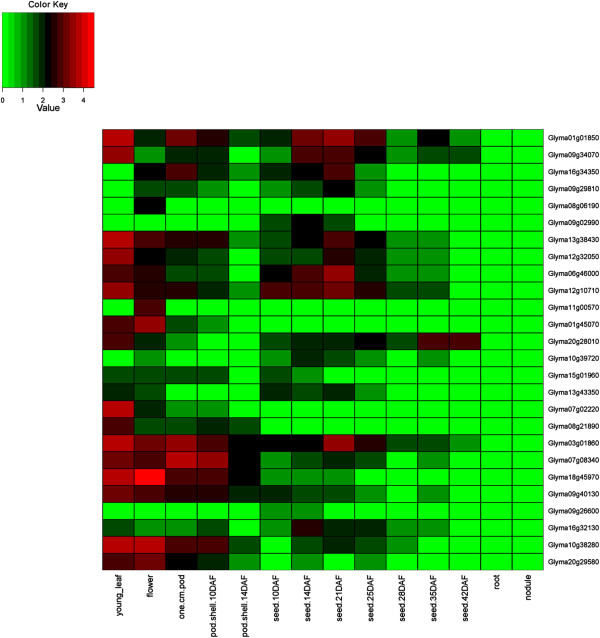
Expression profiles of HD-Zip IV genes in 14 tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 4. The abbreviation “DAF” in the tissue label represents “Days after flowering”.
Three HD-Zip I (Glyma12g18720, Glyma06g35050, and Glyma19g44800), and four HD-Zip IV (Glyma08g09440, Glyma15g13950, Glyma09g03000, and Glyma05g33520) genes showed no expression in any of the 14 tissues investigated by Severin et al. [53]. However, we found evidence of expression for Glyma12g18720 - HD-Zip I in the roots subjected to dehydration stress after 12 hr (data generated in this study). Glyma06g35050 - HD-Zip I showed expression in leaf, flower and root tip, whereas Glyma09g03000 and Glyma05g33520 - HD-Zip IV were expressed in green pods and shoot apical meristem respectively in Libault et al. [54]. The remaining three genes had no evidence for expression (Glyma19g44800 - HD-Zip I and Glyma08g09440, Glyma15g13950 - HD-Zip IV) in either of the two atlases. These three genes did not reveal any frame shift mutations when investigated at the sequence level. Hence, might be pseudogenes, or incorrectly predicted gene models – or they may only be expressed in certain tissues or under conditions that have not been sampled in this study.
Based on the mean expression of genes across 14 tissues investigated by Severin et al. [53], HD-Zip I and II genes had relatively higher expression in roots and flowers; HD-Zip III in young leaves, “one cm pod”, and “pod shell 10 days after flowering”; and HD-Zip IV in young leaves and flowers. Similar results were observed using the expression atlas generated by Libault et al. [54], with the exception of highest mean expression of genes belonging to each of the subfamilies was noticed in shoot apical meristem. Overall, HD-Zip genes had expression in each of the 17 tissues.
The screening of HD-Zip gene expression using mock-inoculated and B. japonicum-infected root hair cells at different time points highlighted HD-Zip genes with more than two fold expression differences between control and treatment samples (Additional file 18: Figure S17, Additional file 19: Figure S18, Additional file 20: Figure S19 and Additional file 21: Figure S20). More than 50% of the genes belonging to the HD-Zip III showed greater than two-fold difference between control and treatment samples at least at one time point (Additional file 20: Figure S19).
Expression of HD-Zip genes under dehydration and salt stress using RNA-Seq
To identify HD-Zip family members responsive to abiotic stress, we used an RNA-seq approach. Twenty-one samples were analyzed by RNA-seq including three control samples (0 hr), and three biological replicates for each of the three time points 1, 6 and 12 hr under dehydration and salt stress. The total number of reads generated in the RNA-Seq experiment from sequencing of 21 sample libraries was 238.8 million, of which 181.2 million (75.9%) uniquely mapped to a single location in the soybean genome (Table 2).
Table 2.
Experimental set-up and summary of read-count data from RNA-Seq analysis
| Treatment | Time point (hr) | Replicate (#) | Lane on HiSeq 2000 | Total reads | Uniquely mapped reads | Uniquely mapped reads (%) |
|---|---|---|---|---|---|---|
| Control | 0 | 1 | 4 | 10,150,369 | 8,047,650 | 79.3% |
| Control | 0 | 2 | 4 | 11,849,953 | 9,207,421 | 77.7% |
| Control | 0 | 3 | 3 | 11,272,789 | 9,164,808 | 81.3% |
| Dehydration | 1 | 1 | 3 | 6,875,669 | 5,571,406 | 81.0% |
| Dehydration | 1 | 2 | 3 | 6,744,882 | 4,948,665 | 73.4% |
| Dehydration | 1 | 3 | 3 | 8,650,675 | 6,612,402 | 76.4% |
| Dehydration | 6 | 1 | 3 | 11,828,271 | 9,624,573 | 81.4% |
| Dehydration | 6 | 2 | 3 | 11,355,361 | 8,599,941 | 75.7% |
| Dehydration | 6 | 3 | 4 | 10,038,099 | 8,009,975 | 79.8% |
| Dehydration | 12 | 1 | 4 | 9,270,260 | 7,194,931 | 77.6% |
| Dehydration | 12 | 2 | 4 | 5,555,797 | 4,050,429 | 72.9% |
| Dehydration | 12 | 3 | 4 | 5,105,827 | 4,087,108 | 80.0% |
| Salt | 1 | 1 | 2 | 38,214,261 | 30,683,738 | 80.3% |
| Salt | 1 | 2 | 2 | 9,046,880 | 7,428,387 | 82.1% |
| Salt | 1 | 3 | 2 | 9,423,474 | 7,202,416 | 76.4% |
| Salt | 6 | 1 | 2 | 7,445,356 | 5,580,211 | 74.9% |
| Salt | 6 | 2 | 2 | 5,890,968 | 4,422,058 | 75.1% |
| Salt | 6 | 3 | 2 | 25,296,306 | 14,687,424 | 58.1% |
| Salt | 12 | 1 | 2 | 7,481,184 | 5,253,438 | 70.2% |
| Salt | 12 | 2 | 3 | 14,201,579 | 11,170,436 | 78.7% |
| Salt | 12 | 3 | 4 | 13,124,265 | 9,645,045 | 73.5% |
| Total | 238,822,225 | 181,192,462 | 75.9% | |||
| Average | 11,372,487 | 8,628,212 | 76.5% |
We identified 4,389 and 8,077 genes to be DE in at least one of the three time points (1, 6 or 12 hr) under dehydration and salt stresses respectively (Additional file 22: Table S2, Additional file 23: Table S3, Additional file 24: Table S4, Additional file 25: Table S5, Additional file 26: Table S6 and Additional file 27: Table S7) (see Methods for the filtering criteria). Salt stress resulted in mostly upregulation of genes, whereas dehydration stress caused downregulation of genes (Additional file 28: Table S8). The number of genes discarded from the differential expression analysis due to significant amount of variation between the replicates under dehydration and salt stress at a given time point ranged from 119 to 220 (Additional file 28: Table S8). The raw and DESeq-normalized expression values for each gene model under both dehydration and salt stress at 1, 6 and 12 hr are provided in Additional file 29: Table S9 and Additional file 30: Table S10 respectively.
Six genes were DE at least at one of the three time points under dehydration stress (Figure 10): five in HD-Zip I, and one in HD-Zip II. Two genes were upregulated and the remaining four were downregulated under dehydration stress. Glyma01g04890 was significantly DE at two different time points. Three of the five DE HD-Zip I genes (Glyma17g10490, Glyma06g20230 and Glyma05g01390) belong to the angiosperm clade A5 (Figure 1), and were a result of the early-legume WGD and the recent Glycine WGD.
Figure 10.

RNA-Seq based expression profiles of HD-Zip genes that are differentially expressed in at least one time point under dehydration stress. The HD-Zip genes responsive to dehydration stress at the first trifoliolate stage in the roots of soybean cv. Williams 82 at least at one time point (1, 6 or 12 hr) are shown. The criteria for differential expression includes, (1) P-value corrected for multiple testing correction using Benjamini and Hochberg [62] to be less than 0.05, (2) two folder or greater fold change, (3) residual variance quotients of both control and treatment samples be less than 20. The criterion (3) filters genes that have significant variation between replicates.
We found sixteen genes DE at one of the three time points under salt stress (Figure 11): seven in HD-Zip I, four in HD-Zip II, one in HD-Zip III, and four in HD-Zip IV. Nine genes were upregulated and the remaining seven genes were downregulated under salt stress. Five of the 16 genes were significantly DE at two time points (HD-Zip I: Glyma01g04890, Glyma07g05800; HD-Zip II: Glyma15g18320, Glyma13g00310; HD-Zip IV: Glyma13g43350). Four of the seven DE HD-Zip I genes were two homoeologous gene pairs (Glyma07g05800/Glyma16g02390; Glyma01g38390/Glyma11g06940). One of the pairs is a member of angiosperm clade A3, and the other belongs to angiosperm clade A1 (Figure 1). One pair each from the HD-Zip II and HD-Zip IV DE genes (Glyma15g18320/Glyma13g00310 and Glyma13g43350/Glyma07g02220, respectively) resulted from the early-legume WGD. The HD-Zip IV gene Glyma13g38430 was not expressed under the control condition (0 hr time point), but was upregulated after 12 hr under salt stress.
Figure 11.
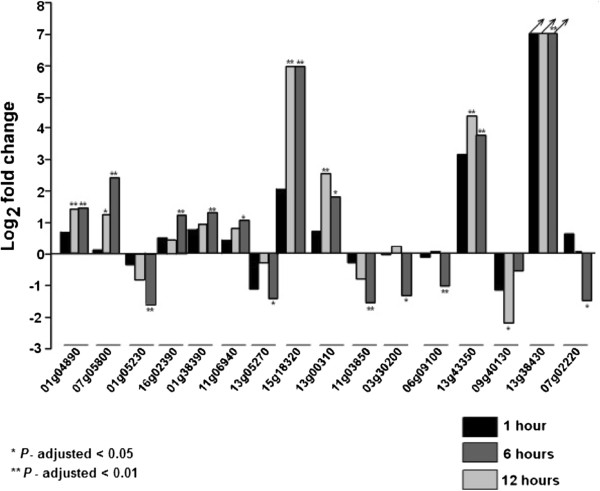
RNA-Seq based expression profiles of HD-Zip genes that are differentially expressed in at least one time point under salt stress. The HD-Zip genes responsive to salt stress at the first trifoliolate stage in the roots of soybean cv. Williams 82 at least at one time point (1, 6 or 12 hr) are shown. The criteria for differential expression includes, (1) P-value corrected for multiple testing correction using Benjamini and Hochberg [62] to be less than 0.05, (2) two folder or greater fold change, (3) residual variance quotients of both control and treatment samples be less than 20. The criterion (3) filters genes that have significant variation between replicates.
The two HD-Zip I genes, Glyma01g04890 and Glyma16g02390, were DE under both dehydration and salt stress. Glyma01g04890 was upregulated at the 6 hr and 12 hr time points under both stress treatments, whereas Glyma16g02390 was downregulated at the 6 hr time point under dehydration stress, and upregulated at the 12 hr time point under salt stress. In summary, 20 of the 101 HD-Zip genes in soybean were DE under either dehydration or salt stress, at least at one time point. Eleven of these 20 genes shared a common ancestor either before the early-legume or the Glycine WGDs, implying conservation of gene functions following these genome duplications.
Annotation of differentially expressed genes under dehydration and salt stress
In order to help evaluate and confirm results from the application of dehydration and salt stress treatments, GO and TF enrichment analysis were performed on the DE genes. Under dehydration stress, 28 “biological process” and 15 “molecular function” terms were significantly (corrected P <0.05) overrepresented, whereas 41 “biological process” and 27 “molecular function” terms were significantly (corrected P <0.05) overrepresented under salt stress (Additional file 31: Table S11). The enriched biological processes and molecular functions include terms such as - “GO:0009414 - response to water deprivation”, “GO:0015250 - water channel activity”, and “GO:0009651 - response to salt stress”, consistent with the experimental treatments (dehydration and salt stress). At the second level of GO analysis, the biological process category “response to stimulus” was the most prevalent one under both stress treatments, followed by “cellular process” and “metabolic process” (Figure 12A), while in the molecular function category, “catalytic activity” and “binding” were highly represented (Figure 12B).
Figure 12.
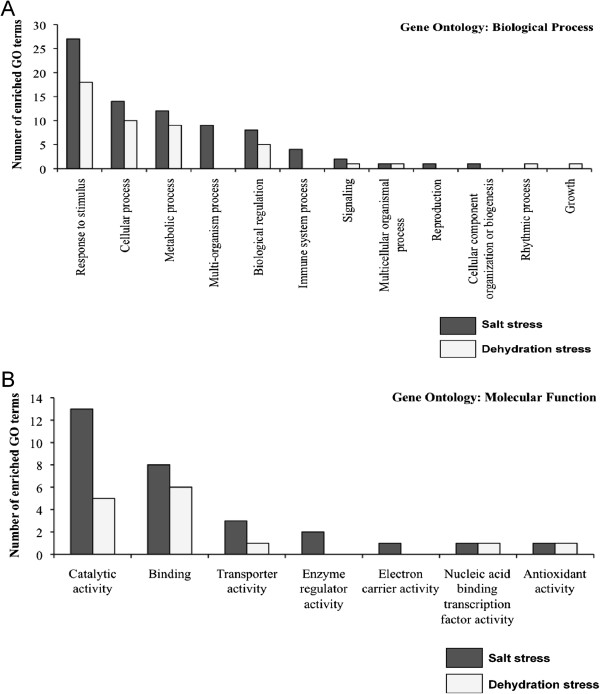
Gene ontology biological process (A) and molecular function (B) categories significantly (corrected P <0.05) overrepresented among differentially expressed genes under dehydration and salt stress. Differentially expressed genes under dehydration and salt stress were annotated using the top Arabidopsis hit, and then screened for overrepresented GO terms against all soybean genes using Fisher’s exact test [66] and Bonferroni [67] corrected significance value of less than 0.05 (Additional file 31: Table S11). The overrepresented GO terms were enriched at the second level using BLAST2GO v.2.7.1 [68] and are shown in the figure.
We identified 503 and 862 TFs among the DE genes under dehydration and salt stress treatments respectively (Additional file 32: Table S12). These TFs corresponded to 35 and 47 TF classes under dehydration and salt stress. Using the enrichment analysis, we identified four TF classes, “WRKY”, “AP2-EREBP”, “ZIM” and “C2C2 (Zn) CO-like” to be significantly (corrected P <0.05) overrepresented under both stress treatments, whereas the TF class “NAC” was overrepresented only under salt stress (Table 3).
Table 3.
Transcription factor class significantly (corrected P <0.05) overrepresented among the differentially expressed genes under dehydration and salt stress
| Transcription factor class | Genome count | Salt stress expression count | Corrected P-value | Dehydration stress expression count | Corrected P-value | Role in abiotic stress response |
|---|---|---|---|---|---|---|
| WRKY | 197 | 82 | 2.52E-21 | 34 | 3.45E-03 | [90–92] |
| AP2-EREBP | 381 | 111 | 1.67E-14 | 75 | 2.88E-10 | [93–95] |
| ZIM | 24 | 20 | 9.55E-13 | 16 | 1.28E-10 | [96–98] |
| C2C2 (Zn) CO-like | 72 | 33 | 9.94E-10 | 26 | 3.77E-09 | [99–101] |
| NAC | 208 | 49 | 3.48E-03 | NA | NA | [102–104] |
Promoter analysis
The enrichment analysis performed with the Clover program [71] and the TRANSFAC database [72] on the promoters of HD-Zip genes identified four different transcription factor binding sites (TFBSs) overrepresented in the promoters of HD-Zip I genes, and at least 9 different TFBSs in HD-Zip II to IV genes (Table 4). The genes belonging to the same subfamily had a diverse profile of TFBSs enriched in the promoters, suggesting the possible role of promoter sequences in functional diversification of the HD-Zip genes of the same subfamilies (Additional file 33: Table S13). The homoeologous genes in all subfamilies had reasonably different TFBSs enriched in their promoters, suggesting specific regulation of homoeologous genes under particular conditions (Additional file 33: Table S13).
Table 4.
Plant transcription factor binding sites significantly ( P <0.05) overrepresented in the promoters of HD-Zip genes belonging to each of the subfamilies
| Motif # | 1TFBS | 2Count | 3Proportion | 4TFBS_Dehydration | 5TFBS_Salt | 6TF_Class | 7DE_Dehydration | 8DE_Salt | |
|---|---|---|---|---|---|---|---|---|---|
| HD-Zip I | M00354 | Dof3 | 33 | 91.7 | - | - | Dof | + | + |
| M00700 | ROM | 31 | 86.1 | + | - | bZIP | + | + | |
| M01136 | Dof | 29 | 80.6 | - | - | Dof | + | + | |
| M00353 | Dof2 | 27 | 75.0 | - | - | Dof | + | + | |
| 9HD-Zip II | M00479 | Alfin1 | 21 | 91.3 | - | - | PHD | + | + |
| M01136 | Dof | 20 | 87.0 | - | - | Dof | + | + | |
| M00354 | Dof3 | 19 | 82.6 | - | - | Dof | + | + | |
| M00440 | CG1 | 18 | 78.3 | - | - | CAMTA | + | + | |
| M00506 | LIM1 | 18 | 78.3 | - | - | LIM | - | + | |
| M00502 | TEIL | 17 | 73.9 | - | + | 10AP2-EREBP | + | + | |
| M00653 | OCSBF-1 | 17 | 73.9 | + | + | bZIP | + | + | |
| M00788 | EmBP-1b | 17 | 73.9 | + | + | bZIP | + | + | |
| M01128 | SED | 17 | 73.9 | - | - | DOF | + | + | |
| M00942 | CPRF-1 | 16 | 69.6 | + | - | bZIP | + | + | |
| M00948 | PCF2 | 16 | 69.6 | + | - | TCP | + | + | |
| M00443 | Opaque-2 | 14 | 60.9 | + | + | bZIP | + | + | |
| M01133 | AG | 14 | 60.9 | - | - | MADS | + | + | |
| M00660 | RITA-1 | 13 | 56.5 | + | + | bZIP | + | + | |
| M01130 | PBF | 13 | 56.5 | - | - | Dof | + | + | |
| M01054 | bHLH66 | 12 | 52.2 | + | + | bHLH | + | + | |
| M00503 | ATHB-5 | 11 | 47.8 | + | - | HD-Zip I | + | + | |
| M00434 | PIF3 | 10 | 43.5 | + | + | bHLH | + | + | |
| HD-Zip III | M00479 | Alfin1 | 10 | 90.9 | - | - | PHD | + | + |
| M00438 | ARF | 9 | 81.8 | - | - | ARF | + | + | |
| M01021 | ID1 | 9 | 81.8 | + | - | C2H2 - zinc | + | + | |
| M01126 | BPC1 | 8 | 72.7 | - | - | BBR/BPC | - | - | |
| M00948 | PCF2 | 7 | 63.6 | + | - | TCP | + | + | |
| 11M00151 | AG | 7 | 63.6 | - | - | MADS | + | + | |
| M00820 | HAHB-4 | 6 | 54.5 | - | - | HD-Zip I | + | + | |
| 12M01061 | AGL2 | 6 | 54.5 | - | - | MADS | + | + | |
| M00392 | AGL3 | 5 | 45.5 | - | - | MADS | + | + | |
| M00949 | AGL15 | 5 | 45.5 | - | - | MADS | + | + | |
| HD-Zip IV | 13M00355 | PBF | 29 | 96.7 | + | - | Dof | + | + |
| M00438 | ARF | 25 | 83.3 | - | - | ARF | + | + | |
| M01126 | BPC1 | 25 | 83.3 | - | - | BBR/BPC | - | - | |
| M01136 | Dof | 25 | 83.3 | - | - | Dof | + | + | |
| M01128 | SED | 23 | 76.7 | - | - | DOF | + | + | |
| M01021 | ID1 | 22 | 73.3 | + | - | C2H2 - zinc | + | + | |
| M00702 | SPF1 | 20 | 66.7 | - | + | 10WRKY | + | + | |
| M00654 | OSBZ8 | 15 | 50.0 | + | + | bZIP | + | + | |
| M00089 | Athb-1 | 11 | 36.7 | + | - | HD-Zip I | + | + |
1TFBS: Transcription factor binding site (TFBS) significantly (P <0.05, motif score >5) overrepresented in the promoters of HD-Zip genes.
2Count: Number of HD-Zip genes within a subfamily that contain the TFBS significantly overrepresented in their promoters.
3Proportion: Percentage of HD-Zip genes within a subfamily that contain the TFBS overrepresented in their promoters.
4TFBS_Dehydration: " + " indicates that the respective TFBS is overrepresented in the promoters of genes that were differentially expressed under dehydration stress, and "-" represents not overrepresented.
5TFBS_Salt: " + " indicates that the respective TFBS is overrepresented in the promoters of genes that were differentially expressed under salt stress, and "-" represents not overrepresented.
6TF_Class: The membership of TFBS to a particular transcription factor (TF) class based on TRANSFAC [72] and UniprotKB [134].
7DE_Dehydration: " + " indicates members of the respective TF class are differentially expressed (DE) under dehydration stress, and “-” indicates otherwise.
8DE_Salt: " + " indicates members of the respective TF class are DE under salt stress, and “-” indicates otherwise.
9Although the HD-Zip II subfamily has 24 genes, the proportion is calculated using 23 genes. HD-Zip II gene Glyma05g23150 was excluded from the promoter analysis due to the selection criteria utilized (see methods for promoter selection criteria).
10TF class significantly (corrected P <0.05) overrepresented in the DE genes under dehydration and salt stress.
11AG TFBS has multiple motif identifiers - M00151, M01063, M01133, and M00950. Counts of AG TFBS’s irrespective of the identifier# were summed to estimate total count and proportion.
12AGL2 TFBS has two motif identifiers - M01061 and M01062. Counts of AGL2 TFBS’s irrespective of the identifier# were summed to estimate total count and proportion.
13PBF TFBS has two motif identifiers - M00355 and M01130. Counts of PBF TFBS’s irrespective of the identifier# were summed to estimate total count and proportion.
There are 14 TFBSs overrepresented in the promoters of HD-Zip genes as well as promoters of DE genes under dehydration stress. Similarly nine TFBSs are overrepresented in the promoters of HD-Zip genes and the promoters of DE genes under salt stress (Table 4, Additional file 34: Table S14). These TF classes are potential candidates that may influence both HD-Zip genes as well genes involved in dehydration and salt stress responses.
The TFBSs “Dof3” and “PBF” are overrepresented in more than 90% of the HD-Zip I and IV genes respectively, and “Alfin1” is overrepresented in more than 90% of HD-Zip II and III genes (Table 4). Hence, these transcription factors probably play an important role in regulating certain HD-Zip genes.
Finally, all but three TF classes corresponding to enriched TFBSs in the promoters of HD-Zip genes contain DE genes under dehydration and salt stress (Table 4). This observation is consistent with HD-Zip genes playing important roles under dehydration and salt stress-responses.
Discussion
Identification and phylogenetic analysis of HD-Zip genes
In this study we have identified and characterized 101 HD-Zip genes in the soybean genome. Recently, 88 HD-Zip genes have been described in soybean [12]. Chen et al. [12] used BLASTP to identify 100 putative HD-Zip transcription factors. SMART and PFAM analyses requiring both an HD and LZ domain were used to refine the number of HD-Zip genes to 88. Similarly, we initiated our study using BLASTP of Arabidopsis HD-Zip genes against the proteomes of soybean, M. truncatula, rice and grape. We then used phylogenetic analyses coupled with HMM searches, domain analyses, and known evolutionary relationships among the five species, to identify more diverse members of the HD-Zip family in each of these species. Using this approach, we were able to identify 13 additional novel HD-Zip genes in soybean and identify the HD-Zips in M. truncatula and grape, which had previously been unreported. Not surprisingly, our approach had the biggest impact on the largely uncharacterized HD-Zip IV genes. While Chen et al. [12] reported 19 genes in HD-Zip IV, we have found 30 genes. These genes may have novel biological functions.
By including multiple species in our search for HD-Zip genes, we also improved the classification of the different family members in soybean and other species. The clustering of Arabidopsis genes in the HD-Zip subfamilies was consistent with the results of Ariel et al. [1]. The HD-Zip I and II subfamilies can be classified into nine (α, β1, β2, γ, δ, ϵ, ϕ1, ϕ2 and ζ) and four (α, β, γ and δ) clades respectively that have been previously described in studies on Arabidopsis, rice and maize [4, 7, 8] (data not shown). Although the results in our study are consistent with the later classification, we suggest that the later strategy be used with discretion. One instance where it can lead to conflicting results is that the ζ clade has been described as monocot-specific clade in all previous studies [4, 7, 8], but this clade clearly contained dicot sequences as a part of an old angiosperm clade in our study. One potential reason for this conflict is that the previous studies included only Arabidopsis [4, 8] or Arabidopsis and C. plantagineum [7] as the dicot species. Sampling of additional dicot sequences of soybean, M. truncatula and grape in this study provided a clearer picture of the taxonomic contexts of the HD-Zip gene family.
We identified five ancient angiosperm clades in HD-Zip I, and four in the HD-Zip II, III and IV subfamilies. The presence of these multiple angiosperm clades in each subfamily is consistent with the recent discovery of two ancient WGD events, one occurring at the base of the angiosperm lineage (ancient angiosperm WGD) and the other before the angiosperm-gymnosperm split (ancestral seed plant WGD) [31]. Early diversification driven by multiple early-plant WGDs is also consistent with a previous study of the evolution of HD-Zip III subfamily in land plants [73]. The presence of five angiosperm clades in HD-Zip I (rather than the four that would be expected from two early WGDs) is intriguing and needs further investigation in the context of synteny analysis and inclusion of additional species in the phylogeny.
A phylogeny with eight species, including published HD-Zip sequences from maize, poplar and cucumber, was largely congruent with the phylogeny generated using five species. These phylogenetic relationships will help identify orthologous genes, and accelerate functional characterizations studies.
Conserved domains and gene structures for validation of HD-Zip genes
PFAM and sequence logos identify highly conserved domains and motifs in the HD-Zip gene family. These have been reported in previous studies [1, 2, 4, 12], but we note two exceptions: Arabidopsis HD-Zip I gene AT1G27050 had an additional “RRM_1” (RNA recognition motif), and Medicago HD-Zip IV gene Mt.ctg127898_1 had two START domains. Overall, the highly conserved domains and motifs are the signatures of the HD-Zip gene family and can be utilized to validate genes identified using several approaches.
Exon-intron structures are generally well conserved in each HD-Zip subfamily, particularly within each angiosperm clade. The HD-Zip III gene-structures were remarkably conserved, with each of the soybean genes having precisely 18 exons. Considering HD-Zip III gene structures reported in other species, all genes in poplar had 18 exons [9], and 4 of the 5 maize genes had 18 exons [8], but in rice only one of the four genes had 18 exons [7]. The generally well-conserved exon structure in HD-Zip III genes across different species highlights the possibility of conserved gene function and strict regulation of these genes. In a recent study involving identification of genes that are potential targets of miRNA in developing soybean seeds, all HD-Zip III genes were found to be targets of miRNA 166 [74]. Prigge and Clark [73], and Floyd and Bowman [75] have previously suggested that HD-Zip III sequences across all land plants produce transcripts that could be targeted by miRNA165 and miRNA166. DeRocher and Nguyen [76] overexpressed Arabidopsis HD-Zip III gene REVOLUTA in soybean embryo, leading to seed yield increase with no change in the seed composition. In short, the HD-Zip III genes appear to be both highly conserved and under intricate transcriptional regulation.
Expansion of HD-Zip gene family
The 101 HD-Zip genes in soybean is the highest number reported so far in any angiosperm species, comparing with 48 in Arabidopsis [2], 55 in maize [8], 47 in rice [2], and 63 in poplar [9]. The HD-Zip genes in soybean have expanded during the early-legume WGD event (~59 Mya), and the Glycine WGD event (~13 Mya), with high retention of paralogs. Expansion of the HD-Zip gene family due to WGDs has been previously reported in other species. The Arabidopsis, rice, maize and poplar have at least 75%, 50%, 62% and 81% homoeologous gene pairs respectively [5, 6, 8, 9, 77–79]. However, in cucumber, a species that lacks WGD events since eudicot radiation, there are no homoeologous gene pairs among HD-Zip I and IV (the two subfamilies described in cucumber) [10, 11]. These results imply that the HD-Zip gene family has expanded in a species-specific manner, with copy number generally depending on WGD events and high retention rates after duplications.
Gene families can be broadly categorized as having high rates of retention of segmental (WGD-derived) duplicates and low generation or retention of tandem duplicates – or vice versa (low segmental retention, high tandem generation and retention) [80]. The low-tandem/high-segmental duplication class of gene families has been reported to comprise highly conserved, housekeeping, and key regulatory gene families [80] – for example, transcription factor families such as heat shock and WRKY, housekeeping families such as mitochondrial carrier proteins [81, 82], and the proteasome 20S subunit family [83, 84]. Clearly, the HD-Zip superfamily falls in the “high segmental, low tandem” category, with only three tandem duplication events in the HD-Zip genes in soybean. The expansion and retention of the HD-Zip family during segmental duplication events will have consequences for functional characterization studies, due to the possibility of genetic redundancy in duplicated genes.
Gene expression patterns of HD-Zip genes in 24 conditions, including 17 tissues
The G. max expression atlas [53] was initially utilized for investigating gene expression patterns of HD-Zip genes in 14 tissues of soybean. The average expression values across 14 tissues for each subfamily was highly variable, and there were genes with extremely high expression relative to the average expression across tissues in each of the subfamily. Investigating gene expression patterns separately for each subfamily on a log2-transformed scale helped identify gene expression patterns that were unreported in Chen et al. [12]. Chen et al. [12] displayed expression of all four subfamilies on a single scale using average linkage clustering method. In addition we utilized two additional gene expression atlases developed by Libault et al. [54, 55], which allowed investigation of HD-Zip genes in three additional tissues, and seven different conditions.
All but three homoeologous gene pairs show consistent expression in the same tissues between the WGD-derived paralogs, suggesting retention of HD-Zip gene functions after genome duplications. The genome duplication events provide raw materials for new gene functions. The duplicated gene can evolve to have a new function (neofunctionalization) [85] or can acquire new deleterious mutations and become a pseudogene (pseudogneization); or both the ancestral and the newly formed gene can undergo reduction in their levels and patterns of activity, such that jointly their function matches with that of the ancestral gene (subfunctionalization) [86].
RNA-Seq based expression profiling of soybean genes during dehydration and salt stress
RNA-Seq analysis was utilized to investigate genes involved in dehydration and salt stress. The expression of all soybean genes including the 101 HD-Zip genes identified in this study was studied in the roots of soybean cv. Williams 82 at V1 stage, at four different time points, and under dehydration and salt stress. The evaluation of plants at the V1 stage may assist in identification of candidate genes involved in initiation of dehydration and salt stress. Recently, Chen et al. [12] reported the influence of drought and salinity stress on HD-Zip genes using publicly available microarray data sets available at National Center for Biotechnology Information under accession numbers GSE41125 and GSE40627. The microarray datasets facilitated investigation of the expression of 55 of the 88 HD-Zip genes identified in their study. The microarray dataset GSE40627 reports expression of genes in the leaves under drought stress imposed at late developmental stages (V6 and R2), whereas the dataset GSE41125 describes expression of genes in 14 d seedlings utilizing pooled RNA samples from 0, 3, 6, 12 and 24 hr of mock and salinity stressed plants. Thus, in the current study, the utilization of root tissue at the V1 stage, and investigation of gene expression separately at each of the four time points 0, 1, 6 and 12 hr provided clearly different and more precise insight into genes that are involved in dehydration and salt stress.
We identified 4,389 and 8,077 genes to be DE in the roots of soybean cv. Williams 82 at the V1 stage at least at one of the three time points (1, 6 or 12 hr) under dehydration and salt stress respectively. Partial validation of DE genes for their role in abiotic stress responses was obtained by performing GO and TF enrichment analysis. The highly represented biological process GO categories, “response to stimulus”, “cellular process”, and “metabolic process” as well as the molecular function categories, “catalytic activity” and “binding”, are generally found to be enriched during abiotic stress responses [87–89]. Similarly, the four TF classes WRKY [90–92], AP2-EREBP [93–95], ZIM [96–98] and C2C2 (Zn) CO-like [99–101] (all enriched under both dehydration and salt stresses), and NAC [102–104] (overrepresented under salt stress) are major TFs that have previously been shown to play critical role in stress responses, and are consistent with results reported in this study.
Expression profiling of HD-Zip genes under dehydration stress
RNA-Seq analysis identified 20 HD-Zip genes DE in the roots of soybean cv. “Williams 82”, under dehydration and salt stress. The role of HD-Zip genes in regulation of developmental adaptation under different environmental stress conditions has been previously established in Arabidopsis, Medicago, rice, sunflower, maize, cucumber, and poplar [4, 7–11, 19, 105–108].
All six genes identified as DE in the roots under dehydration stress in this study, were also, DE under drought stress in leaves [12]. Four of the five DE HD-Zip I genes belong to the angiosperm clade A5. This clade contains genes such as CPHB-5 from C. plantagineum, and Zmhdz1, −2, −3 from maize, that have previously been shown to have a role in water-stress response [4, 8, 20]. Chen et al. [109] showed Glyma06g20230 DE in this study was DE under dehydration stress, in the roots of drought-tolerant soybean genotype, “Jindou21”.
HD-Zip I gene Glyma16g02390 that is DE under dehydration stress belongs to the angiosperm clade A3. Genes in this clade have been extensively characterized for their role in water-stress responses in other species. For example, the Arabidopsis ATHB7 and ATHB12 genes have been shown to reduce plant growth under water-deficit condition [13, 16, 17]. The sunflower HaHB4 gene is strongly induced by water deficit stress [14], and when over-expressed in Arabidopsis the plants exhibit increased survival by a process that inhibits-drought related senescence [18, 19]. The N. attenuata NaHD20 gene is induced in roots under water-deficit conditions [15]. The rice Oshox6, 22 and 24 genes are involved in drought-responsiveness [7]. Hence, we hypothesize that the soybean gene Glyma16g02390 may have a role under water-deficit stress response and is a potential candidate for functional characterization.
HD-Zip II gene Glyma08g15780 that is DE under dehydration stress is an ortholog of rice genes Oshox11 and Oshox27, which have also been demonstrated to be involved in drought-response [7].
In summary, the HD-Zip I and II genes show differential expression patterns under dehydration stress that are consistent with the water-deficit stress response functions of orthologous genes previously identified in studies of water stress. These results support that HD-Zip I and II genes may generally have a role, conserved across many angiosperm species, in mediating water-stress responses; and that these genes may be viable targets for developing more drought-tolerant soybean cultivars.
Expression profiling of HD-Zip genes under salt stress
A subset of HD-Zip genes, from each of the four subfamilies, responded to salt (100 mM NaCl) stress in the roots, in at least one of the three time points. Six of the 16 genes (Glyma01g04890, Glyma07g05800, Glyma16g02390, Glyma13g05270, Glyma15g18320, Glyma03g30200) DE under salt stress have been recently shown to respond to salt stress, in 14 d old seedlings of soybean plant, in a microarray experiment [12].
The HD-Zip I gene Glyma13g05270 was downregulated under salt stress, which is similar to the expression of its Arabidopsis orthologs, ATHB3 and ATHB20, which are similarly downregulated under salt stress [5]. The homoeologous genes Glyma01g38390 and Glyma11g06940 were upregulated after 12 hr of salt stress, comparable to the Arabidopsis orthologs, ATHB20, ATHB50 and ATHB53, which are upregulated more than two-fold under salt stress [5].
Two of the four DE HD-Zip IV genes, Glyma13g43350 and Glyma13g38430, had nearly zero expression under control conditions, but were upregulated under salt stress, suggesting a possible role in root development under stress conditions. Glyma1343350 and Glyma07g02220 are orthologs of the Arabidopsis gene GLABRA2, which has been functionally characterized and shown to regulate root hair development, and cell specification of root epidermis in salt stressed plants [78, 110–112].
The two homoeologous HD-Zip I genes (Glyma07g05800 and Glyma16g02390) upregulated under salt stress belong to the angiosperm clade “A3”. This clade contains the functionally characterized Medicago gene MtHB1 (Medtr8g026960). MtHB1 is induced in the roots under ABA and salt stress, and regulates lateral root emergence in Medicago [26]. The reduction of lateral root emergence by MtHB1, under salt stress, is a mechanism to minimize the exposure of plant roots to excess salt in the soil.
The HD-Zip I gene Glyma01g04890 was upregulated at 6 and 12 hr time points under both salt and dehydration stress. This gene was also upregulated under both drought and salt stress in the leaves and seedlings, respectively, in two microarray experiments [12]. A BLASTP search with Glyma01g04890 protein sequence against the patent database [113, 114] found a match (E-value = 0; Similarity >99.4%; Coverage = 100%) with sequences in five “patent applications” (US_2012_0278947_A1; US_2012_0096584_A1; US_2007_0277269_A1; US_2012_0005773_A1; US_2009_0144847_A1) that described the role of this sequence in improving plant performance under abiotic stress.
Functional diversity and regulation of HD-Zip genes
The presence of highly diverse TFBSs enriched in the promoters of HD-Zip genes provides evidence for functional diversity. Previous studies have mainly focused on HD-Zip target-sequences, and regulatory regions adjacent to the DNA-binding domain of HD-Zip genes. All experimentally tested HD-Zip I genes have been shown to bind specifically, and with high affinity to target-sequences comprising of the same pseudopalindromic sequence CAAT(A/T)ATTG, under in vitro conditions [115–117]. Arce et al. [118] reported the presence of activation domain, sumoylation, and phosphorylation sites in the carboxy-terminal regions, and some putative regulatory regions in the amino-terminal regions, as being responsible for the functional diversity of HD-Zip I genes.
The “Dof3” and “PBF” TFBSs are enriched in more than 90% of HD-Zip I and IV gene promoters respectively. The “Dof” TFs like HD-Zip are plant-specific TFs and are involved in several process, for example stress-responses [119–121], phytochrome signaling [122], light-responses [123, 124], responses to plant hormones including auxin [125, 126] and gibberellin [127, 128], and seed germination [129, 130]. PBF also known as whirly family are known to regulate plant defense gene expression [131].
The TFBS “Alfin1” is overrepresented in more than 90% of HD-Zip II and III gene promoters. “Alfin1” TFs are shown to contribute toward salt tolerance in plants [132, 133].
Finally, the presence of highly diverse TFBSs enriched in the promoters of HD-Zip genes, both within and across subfamilies, suggests the complex integration of HD-Zip genes in various signal-transduction pathways, with a potential source for functional diversity of these highly conserved HD-Zip genes.
Conclusions
In this study we have described the soybean HD-Zip gene superfamily. Evolutionary histories, interpreted in the context of whole genome duplication events and analysis of gene structures, provide additional verification for the classification of the soybean HD-Zip genes. The HD-Zip genes in the soybean genome were preferentially retained after the legume-specific and/or Glycine-specific whole genome duplication events. The RNA-Seq experiment identified candidate genes that may be involved in dehydration and salt stress responses.
Electronic supplementary material
Additional file 1: Figure S1: Sequence logo of HD-Zip I displaying the conserved residues in HMM alignment. (TIFF 246 KB)
Additional file 2: Figure S2: Sequence logo of HD-Zip II displaying the conserved residues in HMM alignment. (TIFF 525 KB)
Additional file 3: Figure S3: Sequence logo of HD-Zip III displaying the conserved residues in HMM alignment. (TIFF 5 MB)
Additional file 4: Figure S4: Sequence logo of HD-Zip IV displaying the conserved residues in HMM alignment. (TIFF 3 MB)
Additional file 5: Figure S5: Phylogenetic relationships of HD-Zip I proteins from soybean, Medicago, Arabidopsis, grape, poplar, cucumber, maize and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A5 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The letters are ordered for consistency with the phylogeny in Figure 1. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. (TIFF 1 MB)
Additional file 6: Figure S6: Phylogenetic relationships of HD-Zip II proteins from soybean, Medicago, Arabidopsis, grape, poplar, maize and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A4 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The letters are ordered for consistency with the phylogeny in Figure 2. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. (TIFF 994 KB)
Additional file 7: Figure S7: Phylogenetic relationships of HD-Zip III proteins from soybean, Medicago, Arabidopsis, grape, poplar, maize and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A4 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The letters are ordered for consistency with the phylogeny in Figure 3. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. (TIFF 549 KB)
Additional file 8: Figure S8: Phylogenetic relationships of HD-Zip IV proteins from soybean, Medicago, Arabidopsis, grape, poplar, cucumber, maize and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A4 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The letters are ordered for consistency with the phylogeny in Figure 4. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. Genes Medtr5g005600.1 and Os01g57890 belong to the angiosperm clade “A2”. These two genes are not shown in the phylogeny because adding them significantly affects the topology. (TIFF 1 MB)
Additional file 9: Figure S9: Gene structure of HD-Zip I genes showing the exon-intron structure. (TIFF 676 KB)
Additional file 10: Figure S10: Gene structure of HD-Zip II genes showing the exon-intron structure. (TIFF 193 KB)
Additional file 11: Figure S11: Gene structure of HD-Zip III genes showing the exon-intron structure. (TIFF 114 KB)
Additional file 12: Figure S12: Gene structure of HD-Zip IV genes showing the exon-intron structure. (TIFF 633 KB)
Additional file 13: Table S1: List of homoeologous soybean HD-Zip genes. (XLSX 56 KB)
Additional file 14: Figure S13: Expression profiles of HD-Zip I genes in seven tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 1. The abbreviation “SAM” in the tissue label represents “shoot apical meristem”. (TIFF 328 KB)
Additional file 15: Figure S14: Expression profiles of HD-Zip II genes in seven tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 2. The abbreviation “SAM” in the tissue label represents “shoot apical meristem”. (TIFF 319 KB)
Additional file 16: Figure S15: Expression profiles of HD-Zip III genes in seven tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 3. The abbreviation “SAM” in the tissue label represents “shoot apical meristem”. (TIFF 233 KB)
Additional file 17: Figure S16: Expression profiles of HD-Zip IV genes in seven tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 4. The abbreviation “SAM” in the tissue label represents “shoot apical meristem”. (TIFF 326 KB)
Additional file 18: Figure S17: Expression profiles of HD-Zip I genes in mock-inoculated and Bradyrhizobium japonicum-infected root hair cells harvested at 12, 24, and 48 hr after inoculation (HAI), and stripped roots harvested at 48 HAI with B. japonicum. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 1. The abbreviation RH_UN and RH_IN in the tissue label represent mock-inoculated and B. japonicum infected root hair cells respectively. The sample RS_48HAI_IN represents stripped roots harvested at 48 HAI with B. japonicum. (TIFF 330 KB)
Additional file 19: Figure S18: Expression profiles of HD-Zip II genes in mock-inoculated and Bradyrhizobium japonicum-infected root hair cells harvested at 12, 24, and 48 hr after inoculation (HAI), and stripped roots harvested at 48 HAI with B. japonicum. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 2. The abbreviation RH_UN and RH_IN in the tissue label represent mock-inoculated and B. japonicum infected root hair cells respectively. The sample RS_48HAI_IN represents stripped roots harvested at 48 HAI with B. japonicum. (TIFF 319 KB)
Additional file 20: Figure S19: Expression profiles of HD-Zip III genes in mock-inoculated and Bradyrhizobium japonicum-infected root hair cells harvested at 12, 24, and 48 hr after inoculation (HAI), and stripped roots harvested at 48 HAI with B. japonicum. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 3. The abbreviation RH_UN and RH_IN in the tissue label represent mock-inoculated and B. japonicum infected root hair cells respectively. The sample RS_48HAI_IN represents stripped roots harvested at 48 HAI with B. japonicum. (TIFF 240 KB)
Additional file 21: Figure S20: Expression profiles of HD-Zip IV genes in mock-inoculated and Bradyrhizobium japonicum-infected root hair cells harvested at 12, 24, and 48 hr after inoculation (HAI), and stripped roots harvested at 48 HAI with B. japonicum. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 4. The abbreviation RH_UN and RH_IN in the tissue label represent mock-inoculated and B. japonicum infected root hair cells respectively. The sample RS_48HAI_IN represents stripped roots harvested at 48 HAI with B. japonicum. (TIFF 326 KB)
Additional file 22: Table S2: Soybean genes differentially expressed under dehydration stress at 1 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 396 KB)
Additional file 23: Table S3: Soybean genes differentially expressed under dehydration stress at 6 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 395 KB)
Additional file 24: Table S4: Soybean genes differentially expressed under dehydration stress at 12 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 326 KB)
Additional file 25: Table S5: Soybean genes differentially expressed under salt stress at 1 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 291 KB)
Additional file 26: Table S6: Soybean genes differentially expressed under salt stress at 6 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 741 KB)
Additional file 27: Table S7: Soybean genes differentially expressed under salt stress at 12 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 1 MB)
Additional file 28: Table S8: Summary statistics of RNA-Seq analysis under dehydration and salt stress. (XLSX 39 KB)
Additional file 29: Table S9: Raw read counts for each of the soybean gene under dehydration and salt stress at 0, 1, 6 and 12 hr generated in the RNA-Seq experiment. (TXT 4 MB)
Additional file 30: Table S10: DESeq normalized read counts for each of the soybean gene under dehydration and salt stress at 0, 1, 6 and 12 hr generated in the RNA-Seq experiment. (TXT 16 MB)
Additional file 31: Table S11: List of GO biological process and molecular function terms significantly (corrected P <0.05) overrepresented in differentially expressed genes under dehydration and salt stress. (XLSX 55 KB)
Additional file 32: Table S12: List of transcription factor classes significantly (corrected P <0.05) overrepresented in differentially expressed genes under dehydration and salt stress. (TXT 32 KB)
Additional file 33: Table S13: List of plant transcription factor binding sites (TFBSs) significantly (P <0.05, motif score >5) overrepresented in the promoters of HD-Zip genes, differentially expressed (DE) genes under dehydration and salt stress, and their respective counts. The TFBSs are provided separately for each of the HD-Zip gene and DE genes. (TXT 4 MB)
Additional file 34: Table S14: List of plant transcription factor binding sites (TFBSs) significantly (P <0.05, motif score >5) overrepresented in the promoters of differentially expressed genes under dehydration and salt stress, with relative proportion of each TFBS under each of the stress treatment. The list of genes that were excluded from the analysis because they did not meet the selection criteria (see Methods for selection criteria) are included. (XLSX 48 KB)
Acknowledgements
The authors are thankful to Dr. Randy C. Shoemaker for providing laboratory facilities, and to Rebecca Nolan for her invaluable support during laboratory experiments. This work was supported by USDA-ARS project funds to Steven B. Cannon.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VB and SBC conceived and planned the project. VB carried out the experiments. VB, SBC, NTW, AKB, ADF and MAG performed data analysis. VB and SBC wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Vikas Belamkar, Email: belamkar@iastate.edu.
Nathan T Weeks, Email: nathan.weeks@ars.usda.gov.
Arvind K Bharti, Email: arvindkbharti30@gmail.com.
Andrew D Farmer, Email: adf@ncgr.org.
Michelle A Graham, Email: michelle.graham@ars.usda.gov.
Steven B Cannon, Email: steven.cannon@ars.usda.gov.
References
- 1.Ariel FD, Manavella PA, Dezar CA, Chan RL. The true story of the HD-Zip family. Trends Plant Sci. 2007;12:419–426. doi: 10.1016/j.tplants.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Mukherjee K, Brocchieri L, Burglin TR. A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol Biol Evol. 2009;26:2775–2794. doi: 10.1093/molbev/msp201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zalewski CS, Floyd SK, Furumizu C, Sakakibara K, Stevenson DW, Bowman JL. Evolution of the class IV HD-zip gene family in streptophytes. Mol Biol Evol. 2013;30:2347–2365. doi: 10.1093/molbev/mst132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris JC, Hrmova M, Lopato S, Langridge P. Modulation of plant growth by HD-Zip class I and II transcription factors in response to environmental stimuli. New Phytol. 2011;190:823–837. doi: 10.1111/j.1469-8137.2011.03733.x. [DOI] [PubMed] [Google Scholar]
- 5.Henriksson E, Olsson AS, Johannesson H, Johansson H, Hanson J, Engstrom P, Soderman E. Homeodomain leucine zipper class I genes in Arabidopsis: expression patterns and phylogenetic relationships. Plant Physiol. 2005;139:509–518. doi: 10.1104/pp.105.063461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciarbelli AR, Ciolfi A, Salvucci S, Ruzza V, Possenti M, Carabelli M, Fruscalzo A, Sessa G, Morelli G, Ruberti I. The Arabidopsis homeodomain-leucine zipper II gene family: diversity and redundancy. Plant Mol Biol. 2008;68:465–478. doi: 10.1007/s11103-008-9383-8. [DOI] [PubMed] [Google Scholar]
- 7.Agalou A, Purwantomo S, Overnas E, Johannesson H, Zhu X, Estiati A, de Kam RJ, Engstrom P, Slamet-Loedin IH, Zhu Z, Wang M, Xiong L, Meijer AH, Ouwerkerk PB. A genome-wide survey of HD-Zip genes in rice and analysis of drought-responsive family members. Plant Mol Biol. 2008;66:87–103. doi: 10.1007/s11103-007-9255-7. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Zhou Y, Jiang H, Li X, Gan D, Peng X, Zhu S, Cheng B. Systematic analysis of sequences and expression patterns of drought-responsive members of the HD-Zip gene family in maize. PLoS One. 2011;6:e28488. doi: 10.1371/journal.pone.0028488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu R, Chi X, Chai G, Kong Y, He G, Wang X, Shi D, Zhang D, Zhou G. Genome-wide identification, evolutionary expansion, and expression profile of homeodomain-leucine zipper gene family in poplar (Populus trichocarpa) PLoS One. 2012;7:e31149. doi: 10.1371/journal.pone.0031149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu W, Fu R, Li Q, Li J, Wang L, Ren Z. Genome-wide identification and expression profile of homeodomain-leucine zipper class I gene family in cucumis sativus. Gene. 2013;531:279–287. doi: 10.1016/j.gene.2013.08.089. [DOI] [PubMed] [Google Scholar]
- 11.Fu R, Liu W, Li Q, Li J, Wang L, Ren Z. Comprehensive analysis of the homeodomain-leucine zipper IV transcription factor family in Cucumis sativus. Genome. 2013;56:395–405. doi: 10.1139/gen-2013-0143. [DOI] [PubMed] [Google Scholar]
- 12.Chen X, Chen Z, Zhao H, Zhao Y, Cheng B, Xiang Y. Genome-wide analysis of soybean HD-zip gene family and expression profiling under salinity and drought treatments. PLoS One. 2014;9:e87156. doi: 10.1371/journal.pone.0087156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsson A, Engstrom P, Soderman E. The homeobox genes ATHB12 and ATHB7 encode potential regulators of growth in response to water deficit in Arabidopsis. Plant Mol Biol. 2004;55:663–677. doi: 10.1007/s11103-004-1581-4. [DOI] [PubMed] [Google Scholar]
- 14.Gago GM, Almoguera C, Jordano J, Gonzalez DH, Chan RL. Hahb-4, a homeobox-leucine zipper gene potentially involved in abscisic acid-dependent responses to water stress in sunflower. Plant Cell Environ. 2002;25:633–640. doi: 10.1046/j.1365-3040.2002.00853.x. [DOI] [Google Scholar]
- 15.Re DA, Dezar CA, Chan RL, Baldwin IT, Bonaventure G. Nicotiana attenuata NaHD20 plays a role in leaf ABA accumulation during water stress, benzylacetone emission from flowers, and the timing of bolting and flower transitions. J Exp Bot. 2011;62:155–166. doi: 10.1093/jxb/erq252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hjellstrom M, Olsson ASB, Engstrom P, Soderman EM. Constitutive expression of the water deficit-inducible homeobox gene ATHB7 in transgenic Arabidopsis causes a suppression of stem elongation growth. Plant Cell Environ. 2003;26:1127–1136. doi: 10.1046/j.1365-3040.2003.01037.x. [DOI] [Google Scholar]
- 17.Son O, Hur YS, Kim YK, Lee HJ, Kim S, Kim MR, Nam KH, Lee MS, Kim BY, Park J, Lee SC, Hanada A, Yamaguchi S, Lee IJ, Kim SK, Yun DJ, Soderman E, Cheon CI. ATHB12, an ABA-inducible homeodomain-leucine zipper (HD-Zip) protein of Arabidopsis, negatively regulates the growth of the inflorescence stem by decreasing the expression of a gibberellin 20-oxidase gene. Plant Cell Physiol. 2010;51:1537–1547. doi: 10.1093/pcp/pcq108. [DOI] [PubMed] [Google Scholar]
- 18.Dezar CA, Gago GM, González DH, Chan RL. Hahb-4, a sunflower homeobox-leucine zipper gene, is a developmental regulator and confers drought tolerance to Arabidopsis thaliana plants. Transgenic Res. 2005;14:429–440. doi: 10.1007/s11248-005-5076-0. [DOI] [PubMed] [Google Scholar]
- 19.Manavella PA, Arce AL, Dezar CA, Bitton F, Renou JP, Crespi M, Chan RL. Cross-talk between ethylene and drought signalling pathways is mediated by the sunflower Hahb-4 transcription factor. Plant J. 2006;48:125–137. doi: 10.1111/j.1365-313X.2006.02865.x. [DOI] [PubMed] [Google Scholar]
- 20.Deng X, Phillips J, Meijer A, Salamini F, Bartels D. Characterization of five novel dehydration-responsive homeodomain leucine zipper genes from the resurrection plant Craterostigma plantagineum. Plant Mol Biol. 2002;49:601–610. doi: 10.1023/A:1015501205303. [DOI] [PubMed] [Google Scholar]
- 21.Johannesson H, Wang Y, Hanson J, Engstrom P. The Arabidopsis thaliana homeobox gene ATHB5 is a potential regulator of abscisic acid responsiveness in developing seedlings. Plant Mol Biol. 2003;51:719–729. doi: 10.1023/A:1022567625228. [DOI] [PubMed] [Google Scholar]
- 22.Soderman E, Hjellstrom M, Fahleson J, Engstrom P. The HD-Zip gene ATHB6 in Arabidopsis is expressed in developing leaves, roots and carpels and up-regulated by water deficit conditions. Plant Mol Biol. 1999;40:1073–1083. doi: 10.1023/A:1006267013170. [DOI] [PubMed] [Google Scholar]
- 23.Himmelbach A, Hoffmann T, Leube M, Hohener B, Grill E. Homeodomain protein ATHB6 is a target of the protein phosphatase ABI1 and regulates hormone responses in Arabidopsis. EMBO J. 2002;21:3029–3038. doi: 10.1093/emboj/cdf316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leung J, Merlot S, Giraudat J. The Arabidopsis ABSCISIC ACID-INSENSITIVE2 (ABI2) and ABI1 genes encode homologous protein phosphatases 2C involved in abscisic acid signal transduction. Plant Cell. 1997;9:759–771. doi: 10.1105/tpc.9.5.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng X, Phillips J, Brautigam A, Engstrom P, Johannesson H, Ouwerkerk PF, Ruberti I, Salinas J, Vera P, Iannacone R, Meijer A, Bartels D. A homeodomain leucine zipper gene from craterostigma plantagineum regulates abscisic acid responsive gene expression and physiological responses. Plant Mol Biol. 2006;61:469–489. doi: 10.1007/s11103-006-0023-x. [DOI] [PubMed] [Google Scholar]
- 26.Ariel F, Diet A, Verdenaud M, Gruber V, Frugier F, Chan R, Crespi M. Environmental regulation of lateral root emergence in Medicago truncatula requires the HD-Zip I transcription factor HB1. Plant Cell. 2010;22:2171–2183. doi: 10.1105/tpc.110.074823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang D, Wu W, Abrams SR, Cutler AJ. The relationship of drought-related gene expression in Arabidopsis thaliana to hormonal and environmental factors. J Exp Bot. 2008;59:2991–3007. doi: 10.1093/jxb/ern155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu L, Chen X, Wang Z, Wang S, Wang Y, Zhu Q, Li S, Xiang C. Arabidopsis enhanced drought tolerance1/HOMEODOMAIN GLABROUS11 confers drought tolerance in transgenic rice without yield penalty. Plant Physiol. 2013;162:1378–1391. doi: 10.1104/pp.113.217596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiao Y, Leebens-Mack J, Ayyampalayam S, Bowers JE, McKain MR, McNeal J, Rolf M, Ruzicka DR, Wafula E, Wickett NJ, Wu X, Zhang Y, Wang J, Zhang Y, Carpenter EJ, Deyholos MK, Kutchan TM, Chanderbali AS, Soltis PS, Stevenson DW, McCombie R, Pires JC, Wong GK, Soltis DE, Depamphilis CW. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012;13:R3. doi: 10.1186/gb-2012-13-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Proost S, Pattyn P, Gerats T, Van de Peer Y. Journey through the past: 150 million years of plant genome evolution. Plant J. 2011;66:58–65. doi: 10.1111/j.1365-313X.2011.04521.x. [DOI] [PubMed] [Google Scholar]
- 31.Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, Clifton SW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, DePamphilis CW. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473:97–100. doi: 10.1038/nature09916. [DOI] [PubMed] [Google Scholar]
- 32.Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- 33.Young ND, Debelle F, Oldroyd GED, Geurts R, Cannon SB, Udvardi MK, Benedito VA, Mayer KFX, Gouzy J, Schoof H, Van de Peer Y, Proost S, Cook DR, Meyers BC, Spannagl M, Cheung F, De Mita S, Krishnakumar V, Gundlach H, Zhou S, Mudge J, Bharti AK, Murray JD, Naoumkina MA, Rosen B, Silverstein KAT, Tang H, Rombauts S, Zhao PX, Zhou P, et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011;480:520–524. doi: 10.1038/480162a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 35.The Arabidopsis Information Resource [http://www.arabidopsis.org/]
- 36.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 37.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gouy M, Guindon S, Gascuel O. SeaView Version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 39.Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 40.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.FigTree [http://tree.bio.ed.ac.uk/software/figtree/]
- 42.HMMER [http://hmmer.janelia.org/]
- 43.WebLogo [http://weblogo.berkeley.edu/logo.cgi]
- 44.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 45.iPlant collaborative [http://www.iplantcollaborative.org/]
- 46.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 47.Pfam [http://pfam.sanger.ac.uk/search#tabview=tab1]
- 48.Phytozome [http://www.phytozome.net]
- 49.Bio-graphics [http://search.cpan.org/~lds/Bio-Graphics/]
- 50.Cannon EK, Cannon SB. Chromosome visualization tool: a whole genome viewer. Int J Plant Genomics. 2011;2011:373875. doi: 10.1155/2011/373875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phytozome v4.0 [ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v4.0/Gmax/misc_feature/Glyma1_domains/glyma1_syn_par.txt]
- 52.Wang L, Guo K, Li Y, Tu Y, Hu H, Wang B, Cui X, Peng L. Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 2010;10:282. doi: 10.1186/1471-2229-10-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, Graham MA, Cannon SB, May GD, Vance CP, Shoemaker RC. RNA-Seq Atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol. 2010;10:160. doi: 10.1186/1471-2229-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Libault M, Farmer A, Joshi T, Takahashi K, Langley RJ, Franklin LD, He J, Xu D, May G, Stacey G. An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant J. 2010;63:86–99. doi: 10.1111/j.1365-313X.2010.04222.x. [DOI] [PubMed] [Google Scholar]
- 55.Libault M, Farmer A, Brechenmacher L, Drnevich J, Langley RJ, Bilgin DD, Radwan O, Neece DJ, Clough SJ, May GD, Stacey G. Complete transcriptome of the soybean root hair cell, a single-cell model, and its alteration in response to Bradyrhizobium japonicum infection. Plant Physiol. 2010;152:541–552. doi: 10.1104/pp.109.148379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soybase [http://soybase.org/soyseq/]
- 57.soykb [http://soykb.org/]
- 58.Woody JL, Severin AJ, Bolon YT, Joseph B, Diers BW, Farmer AD, Weeks N, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, Graham MA, Cannon SB, May GD, Vance CP, Shoemaker RC. Gene expression patterns are correlated with genomic and genic structure in soybean. Genome. 2011;54:10–18. doi: 10.1139/G10-090. [DOI] [PubMed] [Google Scholar]
- 59.R: A language and environment for statistical computing [http://www.R-project.org/]
- 60.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- 63.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A. NCBI GEO: archive for functional genomics data sets - update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fisher RA. The design of experiments. 8. Edinburg: London Oliver and Boyd; 1966. [Google Scholar]
- 67.Bonferroni CE. Studi in Onore del Professore Salvatore Ortu Carboni. 1935. Il calcolo delle assicurazioni su gruppi di teste; pp. 13–60. [Google Scholar]
- 68.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 69.Wang Z, Libault M, Joshi T, Valliyodan B, Nguyen H, Xu D, Stacey G, Cheng J. SoyDB: a knowledge database of soybean transcription factors. BMC Plant Biol. 2010;10:1–12. doi: 10.1007/s10535-010-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.SoyDB [http://www.webcitation.org/6DoD8jYFU]
- 71.Frith MC, Fu Y, Yu L, Chen JÄ, Hansen U, Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 2004;32:1372–1381. doi: 10.1093/nar/gkh299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prigge MJ, Clark SE. Evolution of the class III HD-Zip gene family in land plants. Evol Dev. 2006;8:350–361. doi: 10.1111/j.1525-142X.2006.00107.x. [DOI] [PubMed] [Google Scholar]
- 74.Song QX, Liu YF, Hu XY, Zhang WK, Ma B, Chen SY, Zhang JS. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011;11:5. doi: 10.1186/1471-2229-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Floyd SK, Bowman JL. Gene regulation: Ancient microRNA target sequences in plants. Nature. 2004;428:485–486. doi: 10.1038/428485a. [DOI] [PubMed] [Google Scholar]
- 76.Google patents [http://www.google.com/patents/US8653325]
- 77.Prigge MJ, Otsuga D, Alonso JM, Ecker JR, Drews GN, Clark SE. Class III homeodomain-leucine zipper gene family members have overlapping, antagonistic, and distinct roles in Arabidopsis development. Plant Cell. 2005;17:61–76. doi: 10.1105/tpc.104.026161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakamura M, Katsumata H, Abe M, Yabe N, Komeda Y, Yamamoto KT, Takahashi T. Characterization of the class IV homeodomain-Leucine Zipper gene family in Arabidopsis. Plant Physiol. 2006;141:1363–1375. doi: 10.1104/pp.106.077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jain M, Tyagi AK, Khurana JP. Genome-wide identification, classification, evolutionary expansion and expression analyses of homeobox genes in rice. Febs J. 2008;275:2845–2861. doi: 10.1111/j.1742-4658.2008.06424.x. [DOI] [PubMed] [Google Scholar]
- 80.Cannon SB, Mitra A, Baumgarten A, Young ND, May G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004;4:10. doi: 10.1186/1471-2229-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuan J, Saier MH. The mitochondrial carrier family of transport proteins: structural, functional, and evolutionary relationships. Crit Rev Biochem Mol Biol. 1993;28:209–233. doi: 10.3109/10409239309086795. [DOI] [PubMed] [Google Scholar]
- 82.Borecky J, Maia IG, Arruda P. Mitochondrial uncoupling proteins in mammals and plants. Biosci Rep. 2001;21:201–212. doi: 10.1023/A:1013604526175. [DOI] [PubMed] [Google Scholar]
- 83.Parmentier Y, Bouchez D, Fleck J, Genschik P. The 20S proteasome gene family in Arabidopsis thaliana. FEBS Lett. 1997;416:281–285. doi: 10.1016/S0014-5793(97)01228-3. [DOI] [PubMed] [Google Scholar]
- 84.Vierstra RD. The ubiquitin/26S proteasome pathway, the complex last chapter in the life of many plant proteins. Trends Plant Sci. 2003;8:135–142. doi: 10.1016/S1360-1385(03)00014-1. [DOI] [PubMed] [Google Scholar]
- 85.Hughes AL. The evolution of functionally novel proteins after gene duplication. Proc Biol Sci. 1994;256:119–124. doi: 10.1098/rspb.1994.0058. [DOI] [PubMed] [Google Scholar]
- 86.Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yao L-M, Wang B, Cheng L-J, Wu T-L. Identification of key drought stress-related genes in the hyacinth bean. PLoS One. 2013;8:e58108. doi: 10.1371/journal.pone.0058108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu J, Yuan Y, Xu Y, Zhang G, Guo X, Wu F, Wang Q, Rong T, Pan G, Cao M, Tang Q, Gao S, Liu Y, Wang J, Lan H, Lu Y. Identification of candidate genes for drought tolerance by whole-genome resequencing in maize. BMC Plant Biol. 2014;14:83. doi: 10.1186/1471-2229-14-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Z-h D, Zheng LL, Wang J, Gao Z, Wu SB, Qi Z, Wang YC. Transcriptomic profiling of the salt-stress response in the wild recretohalophyte Reaumuria trigyna. BMC Genomics. 2013;14:29. doi: 10.1186/1471-2164-14-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tang J, Wang F, Wang Z, Huang Z, Xiong A, Hou X. Characterization and co-expression analysis of WRKY orthologs involved in responses to multiple abiotic stresses in Pak-choi (Brassica campestris ssp. chinensis) BMC Plant Biol. 2013;13:188. doi: 10.1186/1471-2229-13-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jing L. Role of WRKY Transcription Factors in Arabidopsis Development and Stress Responses. PhD thesis. 2014. [Google Scholar]
- 92.Chen L, Song Y, Li S, Zhang L, Zou C, Yu D. The role of WRKY transcription factors in plant abiotic stresses. Biochim Biophys Acta (BBA) Gene Regul Mech. 2012;1819:120–128. doi: 10.1016/j.bbagrm.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 93.Sharoni AM, Nuruzzaman M, Satoh K, Shimizu T, Kondoh H, Sasaya T, Choi I-R, Omura T, Kikuchi S. Gene structures, classification and expression models of the AP2/EREBP transcription factor family in rice. Plant Cell Physiol. 2011;52:344–360. doi: 10.1093/pcp/pcq196. [DOI] [PubMed] [Google Scholar]
- 94.Reddy DS, Mathur PB, Sharma KK. Regulatory role of transcription factors in abiotic stress responses in plants. In: Tuteja N, Gill SS, editors. Climate Change and Plant Abiotic Stress Tolerance. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2013. pp. 555–588. [Google Scholar]
- 95.Kizis D, Lumbreras V, Pagès M. Role of AP2/EREBP transcription factors in gene regulation during abiotic stress. FEBS Lett. 2001;498:187–189. doi: 10.1016/S0014-5793(01)02460-7. [DOI] [PubMed] [Google Scholar]
- 96.Ismail A, Riemann M, Nick P. The jasmonate pathway mediates salt tolerance in grapevines. J Exp Bot. 2012;63:2127–2139. doi: 10.1093/jxb/err426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vanholme B, Grunewald W, Bateman A, Kohchi T, Gheysen G. The tify family previously known as ZIM. Trends Plant Sci. 2007;12:239–244. doi: 10.1016/j.tplants.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 98.Jiang Y, Deyholos M. Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol. 2006;6:25. doi: 10.1186/1471-2229-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yao D, Zhang X, Zhao X, Liu C, Wang C, Zhang Z, Zhang C, Wei Q, Wang Q, Yan H, Li F, Su Z. Transcriptome analysis reveals salt-stress-regulated biological processes and key pathways in roots of cotton (Gossypium hirsutum L.) Genomics. 2011;98:47–55. doi: 10.1016/j.ygeno.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 100.Mishra S, Shukla A, Upadhyay S, Sanchita S, Sharma P, Singh S, Phukan UJ, Meena A, Khan F, Tripathi V, Shukla RK, Shrama A. Identification, occurrence, and validation of DRE and ABRE Cis-regulatory motifs in the promoter regions of genes of Arabidopsis thaliana. J Integr Plant Biol. 2014;56:388–399. doi: 10.1111/jipb.12149. [DOI] [PubMed] [Google Scholar]
- 101.Hiz MC, Canher B, Niron H, Turet M. Transcriptome analysis of salt tolerant common bean (Phaseolus vulgaris L.) under saline conditions. PLoS One. 2014;9:e92598. doi: 10.1371/journal.pone.0092598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nakashima K, Takasaki H, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. NAC transcription factors in plant abiotic stress responses. Biochim Biophys Acta (BBA) Gene Regul Mech. 2012;1819:97–103. doi: 10.1016/j.bbagrm.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 103.Mao X, Chen S, Li A, Zhai C, Jing R. Novel NAC Transcription Factor TaNAC67 confers enhanced multi-abiotic stress tolerances in Arabidopsis. PLoS One. 2014;9:e84359. doi: 10.1371/journal.pone.0084359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen X, Wang Y, Lv B, Li J, Luo L, Lu S, Zhang X, Ma H, Ming F. Plant Cell Physiol. 2014. The NAC family transcription factor OsNAP confers abiotic stress response through the ABA pathway. [DOI] [PubMed] [Google Scholar]
- 105.Meijer AH, Scarpella E, Van Dijk EL, Qin L, Taal AJC, Rueb S, Harrington SE, McCouch SR, Schilperoort RA, Hoge JHC. Transcriptional repression by Oshox1, a novel homeodomain leucine zipper protein from rice. Plant J. 1997;11:263–276. doi: 10.1046/j.1365-313X.1997.11020263.x. [DOI] [PubMed] [Google Scholar]
- 106.Lee Y-H, Chun J-Y. A new homeodomain-leucine zipper gene from Arabidopsis thaliana induced by water stress and abscisic acid treatment. Plant Mol Biol. 1998;37:377–384. doi: 10.1023/A:1006084305012. [DOI] [PubMed] [Google Scholar]
- 107.Scarpella E, Rueb S, Boot KJ, Hoge JH, Meijer AH. A role for the rice homeobox gene Oshox1 in provascular cell fate commitment. Development. 2000;127:3655–3669. doi: 10.1242/dev.127.17.3655. [DOI] [PubMed] [Google Scholar]
- 108.Scarpella E, Boot KJM, Rueb S, Meijer AH. The procambium specification gene Oshox1 promotes polar auxin transport capacity and reduces its sensitivity toward inhibition. Plant Physiol. 2002;130:1349–1360. doi: 10.1104/pp.009167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen L, Zhou X, Li W, Chang W, Zhou R, Wang C, Sha A, Shan Z, Zhang C, Qiu D, Yang Z, Chen S. Genome-wide transcriptional analysis of two soybean genotypes under dehydration and rehydration conditions. BMC Genomics. 2013;14:687. doi: 10.1186/1471-2164-14-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Di Cristina M, Sessa G, Dolan L, Linstead P, Baima S, Ruberti I, Morelli G. The Arabidopsis Athb-10 (GLABRA2) is an HD-Zip protein required for regulation of root hair development. Plant J. 1996;10:393–402. doi: 10.1046/j.1365-313X.1996.10030393.x. [DOI] [PubMed] [Google Scholar]
- 111.Wang Y, Zhang W, Li K, Sun F, Han C, Li X. Salt-induced plasticity of root hair development is caused by ion disequilibrium in Arabidopsis thaliana. J Plant Res. 2008;121:87–96. doi: 10.1007/s10265-007-0123-y. [DOI] [PubMed] [Google Scholar]
- 112.Wang Y, Li X. Salt stress-induced cell reprogramming, cell fate switch and adaptive plasticity during root hair development in Arabidopsis. Plant Signal Behav. 2008;3:436–438. doi: 10.4161/psb.3.7.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.The Lens [http://www.lens.org/lens/biological_search]
- 114.Jefferson OA, Kollhofer D, Ehrich TH, Jefferson RA. Transparency tools in gene patenting for informing policy and practice. Nat Biotech. 2013;31:1086–1093. doi: 10.1038/nbt.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Palena CM, Gonzalez DH, Chan RL. A monomer-dimer equilibrium modulates the interaction of the sunflower homeodomain leucine-zipper protein Hahb-4 with DNA. Biochem J. 1999;341:81–87. [PMC free article] [PubMed] [Google Scholar]
- 116.Palena CM, Tron AE, Bertoncini CW, Gonzalez DH, Chan RL. Positively charged residues at the N-terminal arm of the homeodomain are required for efficient DNA binding by homeodomain-leucine zipper proteins. J Mol Biol. 2001;308:39–47. doi: 10.1006/jmbi.2001.4563. [DOI] [PubMed] [Google Scholar]
- 117.Johannesson H, Wang Y, Engstrom P. DNA-binding and dimerization preferences of Arabidopsis homeodomain-leucine zipper transcription factors in¬†vitro. Plant Mol Biol. 2001;45:63–73. doi: 10.1023/A:1006423324025. [DOI] [PubMed] [Google Scholar]
- 118.Arce AL, Raineri J, Capella M, Cabello JV, Chan RL. Uncharacterized conserved motifs outside the HD-Zip domain in HD-Zip subfamily I transcription factors; a potential source of functional diversity. BMC Plant Biol. 2011;11:42. doi: 10.1186/1471-2229-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang B, Chen W, Foley RC, Buttner M, Singh KB. Interactions between distinct types of DNA binding proteins enhance binding to ocs element promoter sequences. Plant Cell Online. 1995;7:2241–2252. doi: 10.1105/tpc.7.12.2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen W, Chao G, Singh KB. The promoter of a H2O2-inducible, Arabidopsis glutathione S-transferase gene contains closely linked OBF- and OBP1-binding sites. Plant J. 1996;10:955–966. doi: 10.1046/j.1365-313X.1996.10060955.x. [DOI] [PubMed] [Google Scholar]
- 121.Kang H-G, Foley RC, Oñate-Sánchez L, Lin C, Singh KB. Target genes for OBP3, a Dof transcription factor, include novel basic helix-loop-helix domain proteins inducible by salicylic acid. Plant J. 2003;35:362–372. doi: 10.1046/j.1365-313X.2003.01812.x. [DOI] [PubMed] [Google Scholar]
- 122.Park DH, Lim PO, Kim JS, Cho DS, Hong SH, Nam HG. The Arabidopsis COG1 gene encodes a Dof domain transcription factor and negatively regulates phytochrome signaling. Plant J. 2003;34:161–171. doi: 10.1046/j.1365-313X.2003.01710.x. [DOI] [PubMed] [Google Scholar]
- 123.Yanagisawa S, Sheen J. Involvement of Maize Dof Zinc finger proteins in tissue-specific and light-regulated gene expression. Plant Cell Online. 1998;10:75–89. doi: 10.1105/tpc.10.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Papi M, Sabatini S, Altamura MM, Hennig L, Schafer E, Costantino P, Vittorioso P. Inactivation of the phloem-specific Dof Zinc finger GeneDAG1 affects response to light and integrity of the testa of Arabidopsis seeds. Plant Physiol. 2002;128:411–417. doi: 10.1104/pp.010488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.De Paolis A, Sabatini S, De Pascalis L, Costantino P, Capone I. A rolB regulatory factor belongs to a new class of single zinc finger plant proteins. Plant J. 1996;10:215–223. doi: 10.1046/j.1365-313X.1996.10020215.x. [DOI] [PubMed] [Google Scholar]
- 126.Kisu Y, Ono T, Shimofurutani N, Suzuki M, Esaka M. Characterization and expression of a new class of zinc finger protein that binds to silencer region of ascorbate oxidase gene. Plant Cell Physiol. 1998;39:1054–1064. doi: 10.1093/oxfordjournals.pcp.a029302. [DOI] [PubMed] [Google Scholar]
- 127.Washio K. Identification of Dof proteins with implication in the gibberellin-regulated expression of a peptidase gene following the germination of rice grains. Biochim Biophys Acta (BBA) Gene Struct Exp. 2001;1520:54–62. doi: 10.1016/S0167-4781(01)00251-2. [DOI] [PubMed] [Google Scholar]
- 128.Mena M, Cejudo FJ, Isabel-Lamoneda I, Carbonero P. A role for the DOF transcription factor BPBF in the regulation of gibberellin-responsive genes in barley aleurone. Plant Physiol. 2002;130:111–119. doi: 10.1104/pp.005561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Papi M, Sabatini S, Bouchez D, Camilleri C, Costantino P, Vittorioso P. Identification and disruption of an Arabidopsis zinc finger gene controlling seed germination. Genes Dev. 2000;14:28–33. [PMC free article] [PubMed] [Google Scholar]
- 130.Gualberti G, Papi M, Bellucci L, Ricci I, Bouchez D, Camilleri C, Costantino P, Vittorioso P. Mutations in the Dof Zinc finger Genes DAG2 and DAG1 influence with opposite effects the germination of Arabidopsis seeds. Plant Cell Online. 2002;14:1253–1263. doi: 10.1105/tpc.010491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Desveaux D, Marechal A, Brisson N. Whirly transcription factors: defense gene regulation and beyond. Trends Plant Sci. 2005;10:95–102. doi: 10.1016/j.tplants.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 132.Winicov I, Bastola DR. Transgenic overexpression of the transcription FactorAlfin1 enhances expression of the endogenous MsPRP2Gene in Alfalfa and improves salinity tolerance of the plants. Plant Physiol. 1999;120:473–480. doi: 10.1104/pp.120.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Winicov I. Alfin1 transcription factor overexpression enhances plant root growth under normal and saline conditions and improves salt tolerance in alfalfa. Planta. 2000;210:416–422. doi: 10.1007/PL00008150. [DOI] [PubMed] [Google Scholar]
- 134.UniProtKB [http://www.uniprot.org/uniprot/]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1: Sequence logo of HD-Zip I displaying the conserved residues in HMM alignment. (TIFF 246 KB)
Additional file 2: Figure S2: Sequence logo of HD-Zip II displaying the conserved residues in HMM alignment. (TIFF 525 KB)
Additional file 3: Figure S3: Sequence logo of HD-Zip III displaying the conserved residues in HMM alignment. (TIFF 5 MB)
Additional file 4: Figure S4: Sequence logo of HD-Zip IV displaying the conserved residues in HMM alignment. (TIFF 3 MB)
Additional file 5: Figure S5: Phylogenetic relationships of HD-Zip I proteins from soybean, Medicago, Arabidopsis, grape, poplar, cucumber, maize and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A5 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The letters are ordered for consistency with the phylogeny in Figure 1. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. (TIFF 1 MB)
Additional file 6: Figure S6: Phylogenetic relationships of HD-Zip II proteins from soybean, Medicago, Arabidopsis, grape, poplar, maize and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A4 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The letters are ordered for consistency with the phylogeny in Figure 2. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. (TIFF 994 KB)
Additional file 7: Figure S7: Phylogenetic relationships of HD-Zip III proteins from soybean, Medicago, Arabidopsis, grape, poplar, maize and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A4 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The letters are ordered for consistency with the phylogeny in Figure 3. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. (TIFF 549 KB)
Additional file 8: Figure S8: Phylogenetic relationships of HD-Zip IV proteins from soybean, Medicago, Arabidopsis, grape, poplar, cucumber, maize and rice. The phylogenetic tree was built using the maximum likelihood method implemented in PhyML. The letters A1- A4 represent ancient angiosperm clades, based on whole genome duplication events, and the copy number of genes from each of the species. The letters are ordered for consistency with the phylogeny in Figure 4. The branch support values estimated using approximate likelihood ratio test (aLRT) are displayed in percentages. Rooting of the tree was inferred from Ariel et al. [1], angiosperm clade composition, and outgroup sequences from other subfamilies. Genes Medtr5g005600.1 and Os01g57890 belong to the angiosperm clade “A2”. These two genes are not shown in the phylogeny because adding them significantly affects the topology. (TIFF 1 MB)
Additional file 9: Figure S9: Gene structure of HD-Zip I genes showing the exon-intron structure. (TIFF 676 KB)
Additional file 10: Figure S10: Gene structure of HD-Zip II genes showing the exon-intron structure. (TIFF 193 KB)
Additional file 11: Figure S11: Gene structure of HD-Zip III genes showing the exon-intron structure. (TIFF 114 KB)
Additional file 12: Figure S12: Gene structure of HD-Zip IV genes showing the exon-intron structure. (TIFF 633 KB)
Additional file 13: Table S1: List of homoeologous soybean HD-Zip genes. (XLSX 56 KB)
Additional file 14: Figure S13: Expression profiles of HD-Zip I genes in seven tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 1. The abbreviation “SAM” in the tissue label represents “shoot apical meristem”. (TIFF 328 KB)
Additional file 15: Figure S14: Expression profiles of HD-Zip II genes in seven tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 2. The abbreviation “SAM” in the tissue label represents “shoot apical meristem”. (TIFF 319 KB)
Additional file 16: Figure S15: Expression profiles of HD-Zip III genes in seven tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 3. The abbreviation “SAM” in the tissue label represents “shoot apical meristem”. (TIFF 233 KB)
Additional file 17: Figure S16: Expression profiles of HD-Zip IV genes in seven tissues of soybean. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 4. The abbreviation “SAM” in the tissue label represents “shoot apical meristem”. (TIFF 326 KB)
Additional file 18: Figure S17: Expression profiles of HD-Zip I genes in mock-inoculated and Bradyrhizobium japonicum-infected root hair cells harvested at 12, 24, and 48 hr after inoculation (HAI), and stripped roots harvested at 48 HAI with B. japonicum. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 1. The abbreviation RH_UN and RH_IN in the tissue label represent mock-inoculated and B. japonicum infected root hair cells respectively. The sample RS_48HAI_IN represents stripped roots harvested at 48 HAI with B. japonicum. (TIFF 330 KB)
Additional file 19: Figure S18: Expression profiles of HD-Zip II genes in mock-inoculated and Bradyrhizobium japonicum-infected root hair cells harvested at 12, 24, and 48 hr after inoculation (HAI), and stripped roots harvested at 48 HAI with B. japonicum. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 2. The abbreviation RH_UN and RH_IN in the tissue label represent mock-inoculated and B. japonicum infected root hair cells respectively. The sample RS_48HAI_IN represents stripped roots harvested at 48 HAI with B. japonicum. (TIFF 319 KB)
Additional file 20: Figure S19: Expression profiles of HD-Zip III genes in mock-inoculated and Bradyrhizobium japonicum-infected root hair cells harvested at 12, 24, and 48 hr after inoculation (HAI), and stripped roots harvested at 48 HAI with B. japonicum. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 3. The abbreviation RH_UN and RH_IN in the tissue label represent mock-inoculated and B. japonicum infected root hair cells respectively. The sample RS_48HAI_IN represents stripped roots harvested at 48 HAI with B. japonicum. (TIFF 240 KB)
Additional file 21: Figure S20: Expression profiles of HD-Zip IV genes in mock-inoculated and Bradyrhizobium japonicum-infected root hair cells harvested at 12, 24, and 48 hr after inoculation (HAI), and stripped roots harvested at 48 HAI with B. japonicum. The Reads/Kb/Million (RPKM) normalized values of expressed genes was log2-transformed and visualized as heatmaps. Genes in the heatmap are ordered for consistency with the phylogeny in Figure 4. The abbreviation RH_UN and RH_IN in the tissue label represent mock-inoculated and B. japonicum infected root hair cells respectively. The sample RS_48HAI_IN represents stripped roots harvested at 48 HAI with B. japonicum. (TIFF 326 KB)
Additional file 22: Table S2: Soybean genes differentially expressed under dehydration stress at 1 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 396 KB)
Additional file 23: Table S3: Soybean genes differentially expressed under dehydration stress at 6 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 395 KB)
Additional file 24: Table S4: Soybean genes differentially expressed under dehydration stress at 12 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 326 KB)
Additional file 25: Table S5: Soybean genes differentially expressed under salt stress at 1 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 291 KB)
Additional file 26: Table S6: Soybean genes differentially expressed under salt stress at 6 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 741 KB)
Additional file 27: Table S7: Soybean genes differentially expressed under salt stress at 12 hr. The table includes mean expression values under control and stress conditions; fold change and log2 fold change values, P-values and adjusted P-values, and residual variance quotients of control and treatment samples. See Methods for the criteria of differential expression. (TXT 1 MB)
Additional file 28: Table S8: Summary statistics of RNA-Seq analysis under dehydration and salt stress. (XLSX 39 KB)
Additional file 29: Table S9: Raw read counts for each of the soybean gene under dehydration and salt stress at 0, 1, 6 and 12 hr generated in the RNA-Seq experiment. (TXT 4 MB)
Additional file 30: Table S10: DESeq normalized read counts for each of the soybean gene under dehydration and salt stress at 0, 1, 6 and 12 hr generated in the RNA-Seq experiment. (TXT 16 MB)
Additional file 31: Table S11: List of GO biological process and molecular function terms significantly (corrected P <0.05) overrepresented in differentially expressed genes under dehydration and salt stress. (XLSX 55 KB)
Additional file 32: Table S12: List of transcription factor classes significantly (corrected P <0.05) overrepresented in differentially expressed genes under dehydration and salt stress. (TXT 32 KB)
Additional file 33: Table S13: List of plant transcription factor binding sites (TFBSs) significantly (P <0.05, motif score >5) overrepresented in the promoters of HD-Zip genes, differentially expressed (DE) genes under dehydration and salt stress, and their respective counts. The TFBSs are provided separately for each of the HD-Zip gene and DE genes. (TXT 4 MB)
Additional file 34: Table S14: List of plant transcription factor binding sites (TFBSs) significantly (P <0.05, motif score >5) overrepresented in the promoters of differentially expressed genes under dehydration and salt stress, with relative proportion of each TFBS under each of the stress treatment. The list of genes that were excluded from the analysis because they did not meet the selection criteria (see Methods for selection criteria) are included. (XLSX 48 KB)