

Abstract
Peripheral myelin protein 22 (PMP22) resides in the plasma membrane and is required for myelin formation in the peripheral nervous system. Many PMP22 mutants accumulate in excess in the endoplasmic reticulum (ER) and lead to the inherited neuropathies of Charcot-Marie-Tooth (CMT) disease. However, the mechanism through which PMP22 mutants accumulate in the ER is unknown. Here, we studied the quality control mechanisms for the PMP22 mutants L16P and G150D, which were originally identified in mice and patients with CMT. We found that the ER-localised ubiquitin ligase Hrd1/SYVN1 mediates ER-associated degradation (ERAD) of PMP22(L16P) and PMP22(G150D), and another ubiquitin ligase, gp78/AMFR, mediates ERAD of PMP22(G150D) as well. We also found that PMP22(L16P), but not PMP22(G150D), is partly released from the ER by loss of Rer1, which is a Golgi-localised sorting receptor for ER retrieval. Rer1 interacts with the wild-type and mutant forms of PMP22. Interestingly, release of PMP22(L16P) from the ER was more prominent with simultaneous knockdown of Rer1 and the ER-localised chaperone calnexin than with the knockdown of each gene. These results suggest that CMT disease-related PMP22(L16P) is trapped in the ER by calnexin-dependent ER retention and Rer1-mediated early Golgi retrieval systems and partly degraded by the Hrd1-mediated ERAD system.
Charcot-Marie-Tooth disease (CMT) is the most commonly inherited neurological disorder of the peripheral nervous system and has an estimated frequency of 1/2,5001,2. CMT is classified into types 1 and 21,2. CMT type 1A (CMT1A) is an autosomal dominant demyelinating neuropathy that accounts for approximately 70% of CMT cases. Approximately 70% of patients with CMT1A harbour the duplication of a 1.4-Mb region of chromosome 17p11.2-12, which comprises the gene encoding peripheral myelin protein 22 (PMP22)3. PMP22 is a tetraspan membrane protein expressed at high levels by the myelinating Schwann cells of peripheral neurons, and it plays crucial roles in the development and maintenance of compact myelin sheaths4. Overproduction of wild-type (WT) PMP22 caused by gene duplication induces the apoptotic death of Schwann cells and demyelination of peripheral nerves5,6,7. Loss of PMP22 causes hereditary neuropathy with predisposition to pressure palsies, which are mild variants of peripheral neuropathy8. Approximately 20 missense mutations in PMP22 have also been identified in patients with CMT1A, and many of these mutants harbour an amino acid residue substitution in the transmembrane domain (TMD)2,9. These point mutations often cause more severe effects than those resulting from gene duplication or nonsense mutations of PMP2210. Interestingly, these mutations lead to high levels of mutant proteins that mainly localize to the endoplasmic reticulum (ER), indicating that misfolded PMP22 in the ER correlates with neuropathy11,12,13,14.
A substantial number of studies have focused on the missense mutations L16P and G150D9, which were originally identified in the spontaneously occurring mouse models of CMT1, Trembler J (TrJ) and Trembler (Tr), respectively, and subsequently in patients with CMT1A or the more severe Dejerine-Sottas syndrome, respectively1,15. These mutant proteins exert a dominant gain-of-function or dominant-negative effect on their cognate WT alleles and lead to severe neuropathies10,13,16. PMP22(L16P) is primarily retained in the ER in Schwann cells12,14, and when it is expressed in cultured cells, it localizes to the ER-Golgi intermediate compartment (ERGIC) as well as the ER13. PMP22(G150D) localizes mainly to the ER in Schwann cells and cultured cells11,12,14. The transcription of unfolding protein response-related genes is up-regulated in the sciatic nerves of TrJ mice, suggesting that the ER retention of mutant PMP22 induces ER stress17. Furthermore, curcumin, which mitigates ER retention of PMP22(L16P), significantly attenuates the apoptosis of Schwann cells and ameliorates the neuropathological phenotype of TrJ mice18. Thus, ER retention of mutant PMP22 may cause peripheral neuropathy by triggering the apoptosis of Schwann cells. However, the mechanism of ER retention of such PMP22 mutants is unknown.
Calnexin is one of the candidates involved in ER retention of mutant PMP22 because it associates with WT PMP22 and acts as an ER chaperone for the folding of PMP22 under physiological conditions19. Intracellular myelin-like structures containing PMP22(L16P) are observed in the Schwann cells of TrJ mice19. Interestingly, calnexin associates with PMP22(L16P) with a higher affinity compared with WT PMP22, and it colocalizes with PMP22(L16P) in the myelin-like structures of the Schwann cells of TrJ mice, implying that calnexin sequesters PMP22(L16P) in intracellular compartments.
Mammalian homolog of yeast Rer1p (Rer1) is another candidate involved in the retention of mutant PMP22 in the ER. In yeast, Rer1p localizes to the cis-Golgi at steady state and functions as a sorting receptor that recycles various membrane proteins to the ER via a coat protein I (COP I)-dependent pathway20,21,22,23. Rer1p is also required for the retention of an unassembled subunit of the iron transporter complex in the ER24. In mammalian cells, Rer1 is involved in the ER retention of an unassembled subunit of the γ-secretase complex and the muscle acetylcholine receptor complex and modulates their assembly and function25,26,27. Notably, yeast Rer1p is involved in the retention of mutant membrane proteins such as the sex pheromone receptor Ste2p and glycosylphosphatidylinositol-anchored Gas1p in the ER28. However, whether calnexin and Rer1 mediate the ER retention of disease-related mutant membrane proteins in mammalian cells remains elusive.
Misfolded membrane proteins that accumulate in the ER are generally recognized, ubiquitinated, and translocated to the cytosol for proteasome-mediated degradation by the ER-associated degradation (ERAD) system29,30. This process is consistent with findings that treatment with proteasome inhibitors induces the formation of ubiquitin-positive cytoplasmic aggregates, including WT or mutant PMP22, in cultured cells31. Furthermore, ubiquitin-positive cytoplasmic aggregates including PMP22 are present in the Schwann cells of TrJ mice and patients with CMT31,32, suggesting that a portion of PMP22 is degraded by the ubiquitin-proteasome pathway. By contrast, autophagy also mediates the removal of PMP22-positive aggregates from the ER or cytosol33. However, the precise mechanisms responsible for the ubiquitination of PMP22 and its removal from the ER are unknown.
In the present study, we show that the pools of WT and mutant PMP22 are degraded, in part, by the ERAD system. We also report that Rer1 is involved in the ER retention of the TrJ mutant L16P. Furthermore, we show that PMP22(L16P) is drastically released from the ER by the simultaneous depletion of calnexin and Rer1. These results provide new insights into the mechanisms of degradation and retention of PMP22 in the ER and suggest new therapeutic targets for CMT.
Results
PMP22(L16P) and PMP22(G150D) are mainly retained in the ER
To determine the molecular mechanisms through which PMP22 mutants are retained and degraded in the ER, we studied the PMP22 mutants L16P and G150D, which are retained in the ER12. The L16P (TrJ) mutation is present in the first TMD and causes CMT1A (Fig. 1A). The G150D (Tr) mutation is present in the fourth TMD and causes Dejerine-Sottas syndrome (Fig. 1A). These mutants accumulate mainly in the ER and form cytoplasmic aggregates when overexpressed in Schwann cells and other cell lines11,12,13. To determine the fate of each PMP22 mutant, we constructed genes encoding WT or individual PMP22 mutants with C-terminal green fluorescent protein (GFP) tags and used them to transfect HeLa cells. PMP22(WT)-GFP localized mainly to the plasma membrane and overlapped partly with the late endosome/lysosome marker Rab7 in perinuclear punctate structures (Fig. 1B, Supplemental Fig. S1). By contrast, PMP22(L16P)-GFP was detected as a reticular pattern that largely colocalized with the ER marker protein disulfide isomerase (PDI), indicating that it is largely retained in the ER (Fig. 1B). PMP22(L16P)-GFP was also observed on punctate structures partly colocalized with Rab7, suggesting that a fraction of the pool of mutant proteins is transported to the post-Golgi compartment (Fig. 1B, Supplemental Fig. S1). PMP22(G150D)-GFP mainly localized to the ER (Fig. 1B).
Figure 1. Disease-associated PMP22 mutants are retained in the ER and degraded in part by the proteasome-dependent degradation pathway.

(A) Structure of PMP22-GFP. The positions of each mutation in PMP22 are illustrated. (B) Localization of PMP22-GFP in HeLa cells. HeLa cells stably expressing wild-type (WT) or mutant PMP22-GFP (green) were immunostained using an anti-PDI antibody (red) and DAPI (blue) and observed using confocal laser scanning microscopy. Scale bar, 10 μm. (C) The effect of a proteasome inhibitor on the stability of PMP22. HeLa cells stably expressing WT or mutant PMP22-GFP were cultured for 8 h in the presence or absence of 10 μM MG132. The cell lysates were immunoblotted with the indicated antibodies. (D) Lysosomal degradation of WT PMP22. HeLa cells stably expressing WT or mutant PMP22-GFP were cultured for 16 h in the presence or absence of 100 nM bafilomycin A1 (Baf. A1). The immunoblots of cell lysates were probed with the indicated antibodies. Note that cropped western blots are shown, and full-length images are presented in the supplementary information.
We next characterized the PMP22-GFP proteins using immunoblot analysis with an anti-GFP antibody (Fig. 1C). PMP22(WT)-GFP migrated diffusely at approximately 47–75 kDa, indicating that it is highly glycosylated (Fig. 1C, post-ER form; see also Fig. 3B). PMP22(G150D)-GFP was detected as a sharp band at approximately 46 kDa, which is predicted to represent the ER form (Fig. 1C, ER form). PMP22(L16P)-GFP was largely detected as the ER form, but a fraction migrated diffusely at the position of the post-ER form.
Figure 2. E3 ubiquitin ligases Hrd1 and gp78 are involved in the degradation of PMP22 localized to the ER.

(A) Cells transfected with Hrd1-siRNA accumulated mutant PMP22. HeLa cells stably expressing WT or mutant PMP22-GFP were transfected with control or Hrd1 (#2) siRNAs for 3 days. Immunoblots of cell lysates were probed with the indicated antibodies. (B) The signal intensities of each PMP22-GFP derivative and α-actin were quantified using image J software, and the amount of each PMP22-GFP derivative was normalised to the amount of α-actin. To compare the amounts of PMP22-GFP derivatives in control cells with that in Hrd1-knockdown cells, we calculated fold changes by expressing each normalised value relative to the normalised value obtained with each PMP22-GFP derivative in control cells. Values indicate the mean ± standard error of the mean (SEM) of three independent experiments. *P < 0.05 (Student's t-test). (C) Cells transfected with gp78-siRNA accumulated PMP22(G150D) mutant. HeLa cells stably expressing WT or mutant PMP22-GFP were transfected with control or gp78 siRNAs for 3 days. Immunoblots of cell lysates were probed with the indicated antibodies. (D) Loss of gp78 increased the protein level of the G150D mutant. The signal intensities of each PMP22-GFP derivative and α-actin were quantified using image J software, and the amount of each PMP22-GFP derivative was normalised to the amount of α-actin. The fold changes (PMP22-GFP/actin) were analysed as described in panel B. (E) PMP22 mutants bind to Hrd1 and gp78. Extracts prepared from HeLa cells stably expressing WT or mutant PMP22-GFP were immunoprecipitated (IP) using an anti-GFP antibody. The resulting precipitates were examined with immunoblot analysis using the indicated antibodies. (F) Additive effect of Hrd1 and gp78 coinactivation on PMP22(G150D) degradation. HeLa cells stably expressing PMP22(G150D)-GFP were transfected with control or siRNAs against Hrd1 (#1 or #2), gp78, or both for 3 days. Lysates prepared from these cells were immunoblotted using the indicated antibodies. Note that a half amount of siRNA used in panels A and C was used for knockdown of Hrd1, gp78, or both. (G) Quantitative analysis of the levels of PMP22(G150D)-GFP. The signal intensities of each PMP22(G150D)-GFP and α-actin were quantified using image J software, and the amount of each PMP22(G150D)-GFP was normalised to the amount of α-actin. The fold changes (PMP22-GFP/actin) were analysed as described in panel B. Histograms show the average value ± SEM from triplicate tests. Different lettered superscripts (a, b, ab) indicate significant differences at P < 0.05 (one-way analysis of variance [ANOVA]). Note that cropped western blots are shown, and full-length images are presented in the supplementary information.
Figure 3. A portion of PMP22 (L16P) is retained in the ER by Rer1.

(A) Knockdown of Rer1 increases the post-ER form of the PMP22(L16P) mutant. HeLa cells stably expressing WT or mutant PMP22-GFP were transfected with control or Rer1 siRNAs for 3 days. Immunoblots of total cell lysates were probed with the indicated antibodies. (B) Glycosylation states of PMP22-GFP in Rer1-knockdown cells. HeLa cells stably expressing WT or mutant PMP22-GFP were treated with control or Rer1 siRNA for 3 days. Cell lysates were treated with or without endoglycosidase H (Endo H) or protein:N-glycanase F (PNGase F) and subjected to immunoblotting using the indicated antibodies. * and ** indicate the Endo H-resistant form of PMP22-GFP and the deglycosylated (Endo H-sensitive) form of PMP22-GFP, respectively. An arrow indicates the ER form of PMP22-GFP. (C) The signal intensities of Endo H-resistant (left graph) or sensitive (right graph) fractions of each PMP22-GFP derivative and α-actin were quantified using Image J software, and the amounts of the fractions of each PMP22-GFP were normalised to the amount of α-actin. To compare the transport efficiencies of WT and mutant PMP22-GFP, we calculated the fold changes by expressing each normalised value relative to the normalised value obtained with each PMP22-GFP derivative in control cells. Values indicate the mean ± SEM of four independent experiments. Two-way ANOVA was used to determine the significance of the differences. *P < 0.05 (ANOVA). (D) Knockdown of Rer1 releases a portion of the PMP22(L16P) mutant from the ER. HeLa cells stably expressing WT or mutant PMP22-GFP were treated with control or Rer1 siRNA for 3 days and then cells were immunostained using an anti-Lamp1 (late endosome/lysosome marker) antibody and observed using confocal laser scanning microscopy. Scale bars, 10 μm. (E) The graph shows the average number of PMP22-positive puncta per cell. More than 30 cells were counted in each experiment. Three independent experiments were conducted. Error bars represent the SEM; *P < 0.05 (Student's t-test). (F) The graph shows the percentages of cells containing more than 10 PMP22-positive puncta. More than 30 cells were counted in each experiment. Three independent experiments were conducted. Error bars represent the SEM; **P < 0.01 (Student's t-test). Note that cropped western blots are shown, and full-length images are presented in the supplementary information.
The population of PMP22 molecules localized to the ER is degraded partly by ER-localized ubiquitin ligase-mediated ERAD
Most of newly synthesized PMP22 is rapidly degraded34, and WT and mutant PMP22 accumulate in cells treated with proteasome inhibitors31, suggesting that part of the PMP22 pool is degraded through the ERAD pathway. By contrast, other evidence suggests that the lysosome-autophagy system mediates the degradation of mutant PMP2233,35. Therefore, we investigated whether PMP22 is degraded by these systems. Because substrates of the ERAD system are degraded in a proteasome-dependent manner, we treated cells with the proteasome inhibitor MG132 and detected the accumulation of the ER and post-ER forms of the WT and mutant PMP22-GFP proteins (Fig. 1C). Conversely, treating cells with bafilomycin A1 (BafA1), which inhibits the autophagy-lysosome pathway by inhibiting v-ATPase, increased the level of the post-ER form of PMP22(WT)-GFP, indicating that part of the PMP22(WT)-GFP pool is transported to the post-ER compartments and is degraded in lysosomes (Fig. 1D). BafA1 treatment did not significantly alter the levels of each mutant protein (Fig. 1D), suggesting that a portion of the PMP22 mutants that accumulate in the ER is degraded by the proteasome system.
ERAD of misfolded proteins is mediated by ER-localized E3 ubiquitin ligases such as Hrd1/SYVN1 and gp78/AMFR29,30. To determine whether PMP22 is degraded by these ubiquitin ligases in the ER, we treated HeLa cells expressing each form of PMP22-GFP with small interfering RNAs (siRNA) against Hrd1 (#2 siRNA for Fig. 2A and B, #1 siRNA for Supplemental Fig. S2) or gp78 (Fig. 2C and D). Knockdown of Hrd1 resulted in the accumulation of the ER forms of mutant PMP22-GFP (Fig. 2A, Supplemental Fig. S2) and an increase in the total level of mutant proteins (Fig. 2B), suggesting that Hrd1 is involved in the degradation of ER-localized PMP22. Interestingly, knockdown of gp78 increased only the level of the G150D mutant, indicating substrate specificity for gp78 (Fig. 2C and D). We further examined whether knockdown of Hrd1 or gp78 affected PMP22 stability with pulse-chase experiments using cycloheximide (CHX; Supplemental Fig. S3). We found that knockdown of either Hrd1 or gp78 delayed PMP22(G150D) degradation to the same extent (approximately 1.5 ~ 2-fold increase at steady state; see Fig. 2B and D, Supplemental Fig. S3), suggesting that Hrd1 and gp78 mediate the degradation of PMP22(G150D) similarly. Conversely, we found that gp78 knockdown significantly inhibited the degradation of the ER form of PMP22(G150D) but not PMP22(L16P) (Supplemental Fig. S3C–F).
We also analysed the interaction of these ubiquitin ligases with each form of PMP22-GFP through coimmunoprecipitation experiments (Fig. 2E). Hrd1 and gp78 bound strongly to the G150D mutant and less well to the L16P mutant. Hrd1 also bound to PMP22(WT)-GFP, albeit at a low level.
Because both Hrd1 and gp78 bound strongly to the G150D mutant, we next examined the effect of knockdown of both Hrd1 and gp78 on the accumulation of the G150D mutant. We found that the level of PMP22(G150D) additively increased when both Hrd1 and gp78 were knocked down, suggesting that Hrd1 and gp78 mediate the degradation of PMP22(G150D) individually. These results imply that when PMP22 is localized to the ER, it is degraded by Hrd1- or gp78-mediated ERAD.
The sorting receptor Rer1 is involved in the ER accumulation of PMP22(L16P)
Because yeast Rer1p is involved in the ER retention of various membrane proteins, including mutant proteins, we investigated whether Rer1 sequestered PMP22 mutants in the ER (Fig. 3). We first examined the effect of Rer1 knockdown on the retention of the L16P mutant in the ER with immunoblot analysis (Fig. 3A). In Rer1-knockdown cells, the levels of the post-ER forms of PMP22(L16P)-GFP were increased. By contrast, loss of Rer1 did not significantly affect the levels of the post-ER and ER forms of PMP22(G150D)-GFP.
To examine the trafficking of the WT and mutant forms of PMP22-GFP through the secretory pathway, we treated cell lysates with endoglycosidase H (Endo H) and protein:N-glycanase F (PNGase F). PNGase F cleaves all N-linked glycans, and Endo H digests high-mannose N-linked oligosaccharides linked to proteins that are not transported beyond the cis-Golgi compartment. In untreated lysates, PMP22(WT)-GFP migrated as a diffuse band corresponding to approximately 47–75 kDa (Fig. 3B). After Endo H treatment, the 46-kDa PMP22(WT)-GFP band was undetectable, and a new 42-kDa band appeared (Fig. 3B, double asterisk), indicating that the 46-kDa band represents an ER/cis-Golgi form (Fig. 3B, arrow). The 47- to 75-kDa PMP22(WT)-GFP band was Endo H resistant, indicating that this population was transported beyond the cis-Golgi compartment (asterisk). All species of the PMP22-GFP derivatives shifted to 42 kDa after PNGase F treatment, indicating that these forms were glycosylated. The level of Endo H-resistant PMP22(L16P)-GFP was significantly increased by Rer1 knockdown (Fig. 3B and C), suggesting that the ER retention of PMP22(L16P) depends in part on Rer1. By contrast, the glycosylation of PMP22(G150D)-GFP was not affected by Rer1 knockdown (Fig. 3A–C).
We also analysed the subcellular localization of the PMP22-GFP proteins when the expression of Rer1 was inhibited (Fig. 3D). PMP22(WT)-GFP localized to the plasma membrane and cytoplasmic punctate structures in both control and Rer1-knockdown cells. PMP22(L16P)-GFP displayed the ER pattern and, in part, formed punctate structures in control cells. By contrast, the signals specific for PMP22(L16P)-GFP significantly increased in punctate structures when Rer1 expression was inhibited (Fig. 3D–F). Because these punctate structures were labelled with late endosome/lysosome marker (Lamp1), they represent endosomal/lysosomal compartments (Fig. 3D). PMP22(G150D)-GFP exhibited the typical ER pattern in both control and Rer1-knockdown cells. Similar results were obtained when Rer1 was knocked down in other cultured cells such as COS1 (Supplemental Fig. S4). The redistribution of PMP22(L16P)-GFP to lysosomes in cells transfected with Rer1 siRNA was rescued by overexpression of an siRNA-resistant mouse Rer1, which was tagged with monomeric red fluorescent protein (mRFP) at its N-terminus, indicating that the phenotype induced by Rer1 siRNA is specifically caused by Rer1 knockdown (Supplemental Fig. S5).
Rer1 interacts preferentially with the ER/cis-Golgi form of PMP22
We performed coimmunoprecipitation experiments to determine whether Rer1 interacts with PMP22. HEK293T cells stably expressing WT and mutant forms of PMP22-GFP were transfected with 3xFLAG-Rer1. When 3xFLAG-Rer1 was immunoprecipitated with an anti-FLAG antibody, it preferentially bound to the ER/cis-Golgi form of the PMP22-GFP proteins (Fig. 4A). We next performed immunoprecipitation using cells overexpressing both PMP22-GFP and 3xFLAG-Rer1 (Fig. 4B). When 3xFLAG-Rer1 was immunoprecipitated with an anti-FLAG antibody, it bound to the ER/cis-Golgi form of the WT and mutant PMP22. The level of bound WT protein was comparable to that of the bound mutant PMP22. These results suggest that Rer1 directly interacts with PMP22, which is retained in the ER/cis-Golgi. Notably, when WT and PMP22(L16P)-GFP were overexpressed, they were mainly detected as ER forms, suggesting that the overexpression of PMP22 causes its accumulation in the ER (see Fig. 4A and 4B, ER form). In addition, co-overexpression of Rer1 further increased the levels of the ER forms of all PMP22-GFP derivatives, suggesting that Rer1 facilitates the ER accumulation of these proteins.
Figure 4. Rer1 interacts primarily with PMP22 mutants retained in the ER.
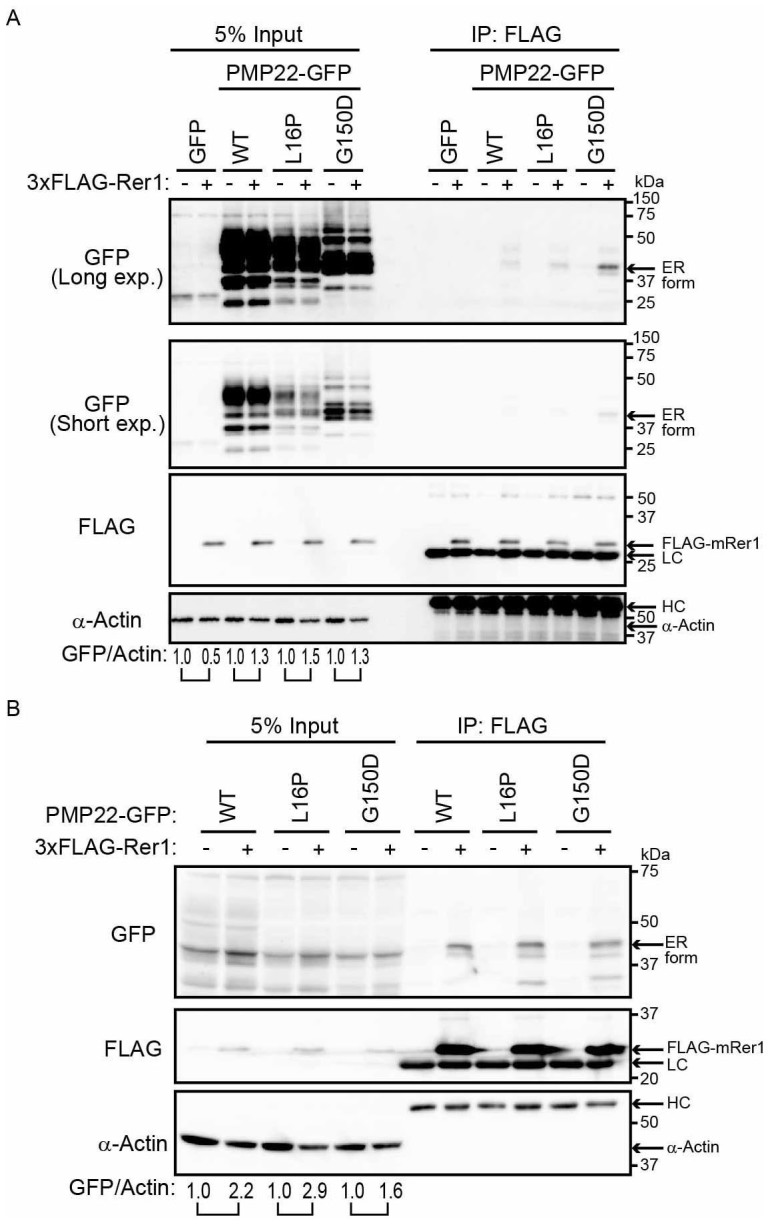
(A) Interaction of PMP22-GFP with FLAG-Rer1. HEK293T cells stably expressing WT or mutant PMP22-GFP were transiently transfected with empty vector (-) or 3xFLAG-mouse Rer1 (+). Cell lysates were immunoprecipitated with FLAG-M2 agarose beads. The immunoprecipitates (IP) and cell lysates were immunoblotted with the indicated antibodies. The signal intensities of each PMP22-GFP derivative and α-actin in each cell lysate were quantified using image J software, and the amount of each PMP22-GFP derivative was normalised to the amount of α-actin. To compare the amounts of PMP22-GFP derivatives in mock transfected cells (−) and FLAG-Rer1 transfected cells (+), we calculated the fold changes (GFP/actin) by expressing each normalised value relative to the normalised value obtained with each PMP22-GFP derivative in mock-transfected cells. (B) Interaction between PMP22-GFP and FLAG-Rer1 in cells overexpressing PMP22-GFPs. HEK293T cells were cotransfected with the indicated PMP22-GFP mutants, empty vector (−), or 3xFLAG-mouse Rer1 (+). Cell lysates were immunoprecipitated using FLAG-M2 agarose beads. The precipitates were analysed using western blotting with the indicated antibodies. The immunoglobulin G heavy (HC) and light (LC) chains are indicated. The fold changes (GFP/actin) were analysed as described in panel A. Note that cropped western blots are shown, and full-length images are presented in the supplementary information.
Calnexin and Rer1 cooperatively function in the ER retention of PMP22(L16P)
We further asked whether calnexin, which mediates the folding of PMP2236, was involved in the stability or ER retention of WT and mutant PMP22-GFPs (Fig. 5). We first examined whether calnexin was involved in the protein stability of mutant PMP22. The level of PMP22(WT)-GFP appeared to be increased by knockdown of calnexin (Fig. 5A and B). The ER forms of the L16P and G150D mutants were mainly detected, even in calnexin-knockdown cell lysates, although the total protein levels seemed to increase (see Fig. 5A and B). We also examined whether calnexin knockdown affected PMP22 stability with pulse-chase experiments using CHX (Supplemental Fig. S3). We found that calnexin knockdown delayed the decrease of the ER form of mutant PMP22 slightly, but the difference was not significant. We next examined subcellular localization of WT and mutant PMP22-GFPs in calnexin-knockdown cells. We found that the localization of the L16P and G150D mutants in the ER was not strongly affected by calnexin knockdown (Fig. 3C), suggesting that these mutants are retained largely in the ER even in the absence of calnexin.
Figure 5. Mutant PMP22 is primarily retained in the ER in calnexin-knockdown cells.

(A) The effect of calnexin knockdown on the stability of ER retention of WT and mutant PMP22-GFPs. HeLa cells stably expressing WT or mutant PMP22-GFP were transfected with control or siRNAs against calnexin for 3 days. Lysates prepared from these cells were immunoblotted using anti-GFP (top panel), anti-calnexin (CNX, middle panel), and anti-α-actin (bottom panel) antibodies. Note that cropped western blots are shown, and full-length images are presented in the supplementary information. (B) The signal intensities of each PMP22-GFP derivative and α-actin band were quantified using image J software, and the amount of each PMP22-GFP derivative was normalised to the amount of α-actin. To compare the amounts of PMP22-GFP derivatives in control cells with that in calnexin-knockdown cells, we calculated the fold changes by expressing each normalised value relative to the normalised value obtained with each PMP22-GFP derivative in control cells. Values indicate the mean ± SEM of three independent experiments. Student's t-test was used to determine the significance of the differences. (C) The effect of calnexin knockdown on the localization of PMP22-GFP derivatives. HeLa cells stably expressing WT or mutant PMP22-GFP were transfected with control or siRNAs against calnexin and cultured for 3 days. Then, cells were fixed and observed using confocal laser scanning microscopy. Scale bar, 10 μm.
We thus assumed that Rer1 might mask the leakage of mutant PMP22 from the ER in calnexin-deficient cells by returning it from the early Golgi to the ER. To test this possibility, we knocked down Rer1 and calnexin simultaneously and examined the glycosylation states and localization of WT and mutant PMP22 (Fig. 6 and Supplemental Fig. S6). We found that the level of the highly glycosylated form of PMP22(L16P) significantly increased when both calnexin and Rer1 were knocked down (Fig. 6A and B). In addition, the localization of PMP22(L16P) on lysosomes/endosomes and the plasma membrane became more prominent in Rer1/calnexin knockdown cells (Fig. 6C). These results suggest that PMP22(L16P) accumulates in the ER via the calnexin-dependent ER retention and Rer1-dependent Golgi retrieval systems. By contrast, the glycosylation of PMP22(G150D)-GFP and its localization to the ER was not affected even in the absence of calnexin and Rer1, suggesting a distinct mechanism for its ER retention (Fig. 6A–C, Supplemental Fig. S6).
Figure 6. Calnexin and Rer1 mediate the ER retention of PMP22(L16P).

(A) Simultaneous knockdown of Rer1 and calnexin drastically increases the post-ER form of the PMP22(L16P). HeLa cells stably expressing WT or mutant PMP22-GFP were transfected with control or siRNAs against Rer1, calnexin, or both for 3 days. Note that a half amount of siRNA used in Figs. 3 and 5 was used for knockdown of calnexin, Rer1, or both. Lysates prepared from these cells were immunoblotted using anti-GFP (top panel), anti-calnexin (CNX, second panel), anti-Rer1 (third panel), and anti-α-actin (bottom panel) antibodies. (B) The signal intensities of the post-ER form of WT or L16P mutant PMP22 and α-actin were quantified using image J software, and the amount of each post-ER form was normalised to the amount of α-actin. To compare the amounts of the post-ER form in control cells and each knockdown cell, we calculated the fold changes by expressing each normalised value relative to the normalised value obtained with the amount of the post-ER form in control cells. The histogram shows the average values ± SEM from triplicate tests. Two-way ANOVA was used to determine the significance of the differences. **P < 0.01, ***P < 0.001 (ANOVA). (C) Simultaneous knockdown of Rer1 and calnexin strongly releases ER-trapped PMP22(L16P) mutant. HeLa cells stably expressing WT or mutant PMP22-GFP were treated with control or siRNA against Rer1, calnexin, or both for 3 days. Then, the fixed cells were observed using confocal laser scanning microscopy. Scale bars, 10 μm. (D) Endogenous Rer1 and calnexin interact with PMP22-GFPs. HEK293T cells were transfected with an empty vector or PMP22-GFP constructs and treated with a chemical crosslinker, DSP. Cell lysates were immunoprecipitated (IP) using an anti-GFP antibody and the blots were probed with the indicated antibodies. Note that cropped western blots are shown, and full-length images are presented in the supplementary information.
We further tested whether endogenous calnexin and Rer1 interact with WT or mutant PMP22-GFPs. We treated the cell lysate with a chemical crosslinker, dithiobis succinimidyl propionate (DSP) and conducted coimmunoprecipitation experiments using anti-GFP antibody (Fig. 6D). We found that endogenous calnexin or Rer1 coimmunoprecipitated with WT and mutant PMP22-GFPs under this condition, suggesting that these proteins physically recognize PMP22 (Fig. 6D).
Discussion
In the present study, we showed that ERAD of PMP22(L16P) and PMP22(G150D) is mediated by ER-localized E3 ubiquitin ligases such as Hrd1/SYVN1 and gp78/AMFR. We also found that PMP22(L16P) was partly released from the ER by the loss of Rer1. Finally, we discovered that the depletion of both Rer1 and calnexin drastically released PMP22(L16P) from the ER to later compartments. These findings suggest that PMP22(L16P) is accumulated in the ER by the calnexin-mediated ER quality control system and Rer1-mediated early Golgi quality control system.
Newly synthesized membrane proteins translocate to the ER, where they undergo folding, posttranslational modification, or complex formation. Misfolded proteins are selectively recognized by the ER quality control system, and if they are not refolded correctly, they are degraded by proteasomes via the ERAD system29. We show herein that the ubiquitin ligases Hrd1 and gp78 mediate ERAD of mutant forms of PMP22 in a distinct manner. For example, knockdown of Hrd1 expression inhibited the degradation of both mutant forms of PMP22, suggesting that Hrd1 plays a central role in the degradation of misfolded PMP22. Conversely, loss of gp78 expression affected only the degradation of the G150D mutant, suggesting that the substrate specificity of gp78 differs from that of Hrd1. Because gp78 bound predominantly to the G150D mutant, it may selectively recognize the structural alteration induced by the G150D mutation. Alternatively, gp78 may function as a backup system when Hrd1 capacity is saturated by an excess of misfolded proteins. Interestingly, the G150D mutant bound strongly to Hrd1 and gp78 but was mildly affected by gp78 and Hrd1 knockdown, whereas the L16P mutant bound weakly to Hrd1 but was strongly stabilized when Hrd1 was silenced (Fig. 2). PMP22(G150D) also bound tightly to Rer1, although it localized to the ER in an Rer1-independent manner (Figs. 3 and 4). These observations imply that PMP22(G150D) has characteristics that enable it to bind to various membrane proteins nonspecifically, which may explain why this protein bound to both Hrd1 and gp78 more strongly than PMP22(L16P). The ERAD substrates should be degraded smoothly once they are recognized by ubiquitin ligases. Weak binding of Hrd1 to PMP22(L16P) at steady state may reflect the efficient removal of PMP22(L16P). Conversely, too much binding of PMP22(G150D) to Hrd1 or gp78 may interfere with these ubiquitin ligase activities to some extent. Such interactions may inhibit the disposal of other ERAD substrates as well as the mutant PMP22, leading to ER stress and cytotoxicity. Up-regulation of ERAD system activity may promote the degradation of ERAD substrates and mitigate cytotoxicity caused by the accumulation of mutant PMP22.
We also demonstrate that calnexin and Rer1 mediate the ER retention of PMP22(L16P) in the ER and early Golgi, respectively (Fig. 6). Previous studies have suggested that calnexin contributes to the retention of misfolded PMP22 in the ER because it binds to mutant PMP22 longer than it binds to WT PMP2219. However, whether mutant PMP22 is actually released by the depletion of calnexin remains to be determined. Unexpectedly, we found that the levels of mutant PMP22 proteins marginally increased, but their glycosylation and localization were not significantly affected by calnexin knockdown alone (see Fig. 5). Conversely, we found that loss of Rer1 released a portion of the L16P mutant pool from the ER to later compartments (Fig. 3). Strikingly, simultaneous depletion of calnexin and Rer1 led to a drastic increase of the post-ER fraction of the L16P mutant (Fig. 6), suggesting that the ER retention of this mutant is mediated by at least two mechanisms: calnexin-mediated static retention in the ER and Rer1-dependent retrieval in early Golgi. In this case, the L16P mutant could be returned to the ER by the Rer1-dependent retrieval system even if it escapes the calnexin-mediated ER quality control system. This may explain why the loss of calnexin has relatively mild effects on L16P mutant localization.
Calnexin binds the first TMD of the L16P mutant in a glycan-independent manner37. On the contrary, the recognition of target proteins by Rer1 depends on the position of polar residues within the TMD of target proteins38. The proline residue of the L16P mutant may break or kink α-helical structures within the TMD and expose polar residues to the hydrophobic milieu in the lipid bilayer to interact with Rer1. Interestingly, the L16P mutant was predominantly transported to endosomes/lysosomes in Rer1-knockdown cells, suggesting the existence of other quality control systems that target mutant proteins to lysosomes if they escape from the ER and early Golgi quality control systems39,40,41.
By contrast, although Rer1 bound tightly to the G150D mutant, it was not essential for retaining the G150D mutant in the ER, suggesting the existence of other ER retention systems. Interestingly, the G150D mutant bound calnexin, Hrd1, gp78, and Rer1 more strongly than WT and L16P PMP22 did (see Figs. 2 and 4). Because the G150D mutant is known to form aggregates—in contrast to the WT or L16P mutant proteins16,31—it may form complexes with various ER membrane proteins, preventing its entry into coat protein II (COP II) vesicles. Alternatively, the G150D mutant may be retained in the ER by unidentified static retention mechanisms implicated in the ER retention of Sec12p23 and cytochrome b542.
The TrJ mutation (L16P) is semi-dominant43, and heterozygous TrJ/+ mice exhibit peripheral myelin deficiency that is more severe than that in PMP22 null (−/−) and heterozygous (+/−) mice10,44, suggesting a dominant negative effect of the TrJ mutation on the WT allele. Furthermore, ER stress is induced in the sciatic nerves of TrJ mice, possibly by the accumulation of the L16P mutant in the ER. Down-regulation of Rer1 and calnexin may alleviate ER stress in the TrJ mutant through dominant-negative effects by releasing the L16P mutant from the ER.
Down-regulation of PMP22 expression is used as a mainstream therapeutic strategy for CMT1A in model systems45,46. Although these approaches are effective in model systems for treating CMT1 caused by PMP22 overexpression, additional strategies are required to cure other types of CMT1, such as those caused by the L16P and G150D mutations2. Our present findings provide insight into the mechanisms of CMT1 and reveal new targets for the treatment of CMT1 caused by the accumulation of mutant PMP22 in the ER.
Methods
Complementary DNA (cDNA) cloning and expression constructs
Human PMP22 cDNA was amplified from a human brain cDNA polymerase chain reaction (PCR)-ready cDNA library (3324G, Ambion, Austin, TX, USA) using PCR. DNA fragments encoding PMP22(L16P) and PMP22(G150D) were generated using PCR-based site-directed mutagenesis. Using Gateway recombination cloning technology (Life Technologies, Grand Island, NY, USA), cDNAs encoding the WT and mutant forms of PMP22 were cloned into the entry vector pDONR221 and then cloned into the destination vectors pMXs-IP-frameB-GFP or pcDNA3.1-frameB-GFP, which drive the expression of C-terminally GFP-tagged proteins under the control of long terminal repeat (LTR) or cytomegalovirus (CMV) promoters. To construct the destination vectors, we inserted a DNA fragment encoding GFP into the multiple cloning site (MCS) of the expression vector pMXs-IP (provided by T. Kitamura, University of Tokyo) or pcDNA3.1 (Life Technologies). Finally, the Gateway reading-frame B cassette (Life Technologies) was introduced between the MCS and the GFP coding region of these vectors. The cDNA-encoding mouse Rer1 was cloned into the entry vector pDONR221. The mouse Rer1 cDNA was then cloned into the destination vectors CMV10-rfB and pMXs-IP-mRFP-frameB, which express N-terminal FLAG-tagged and mRFP-tagged Rer1, respectively47. For construction of the pMXs-IP-mRFP-frameB vector, DNA fragments encoding mRFP and the Gateway reading-frame B cassette were inserted into the MCS of the expression vector pMXs-IP.
Cell culture and transfection
HeLa cells, HEK293T, and COS1 cells were cultured in Dulbecco's modified Eagle's medium (Wako Pure Chemical, Osaka, Japan) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin in an atmosphere containing 5% CO2. HeLa cells expressing PMP22-GFP were generated using retroviral transfection systems48. Human Plat-E cells were cotransfected with pMXs-IP-PMP22-GFP and pCG-VSV-G in the presence of Fugene HD (Promega, Madison, WI, USA) to generate retroviruses infectious to human cells. All cell lines were then infected with the recombinant retroviruses and selected in medium containing 1 μg/mL puromycin. Fugene HD (Promega) was used for transfection of HEK293T cells.
Antibodies and reagents
Antibodies used for western blotting were as follows: rabbit polyclonal anti-Rer1 (R4407, Sigma-Aldrich, St. Louis, MO, USA), anti-Hrd1 (H7915, Sigma), anti-gp78 (9590, Cell Signaling Technology, Danvers, MA, USA), a rabbit monoclonal anti-Calnexin (C5C7, Cell Signaling), goat polyclonal anti-GFP (RDI, Fitzgerald Industry, Concord, MA, USA), mouse monoclonal anti-α-actin (C4, Millipore, Billerica, MA, USA), anti-FLAG (M2, Sigma-Aldrich), and anti-GSK3β (610201, BD Transduction Laboratories, San Diego, CA, USA) antibodies. Antibodies used for immunoprecipitation were as follows: mouse monoclonal anti-GFP (3E6, Q-Biogene, Carlsbad, CA, USA), anti-α-tubulin (DM1A, Sigma-Aldrich), and anti-FLAG (M2, Sigma-Aldrich) antibodies. Antibodies used for immunocytochemistry were as follows: rabbit polyclonal anti-Rab7 (a gift from Y. Wada, Osaka University)49, anti-ERGIC-53 (E1031, Sigma-Aldrich), mouse monoclonal anti-PDI (1D3, Enzo Life Sciences, Plymouth Meeting, PA, USA), and anti-Lamp1 (H4A3, Santa Cruz Biotechnology, CA, USA) antibodies. Alexa Fluor® 555 or 594-conjugated anti-mouse or anti-rabbit immunoglobulin Gs (IgGs) (Life Technologies) were used as secondary antibodies.
MG132 was purchased from the Peptide Institute (Osaka, Japan). Bafilomycin A1 was purchased from Wako Pure Chemicals (Osaka, Japan). Cycloheximide was purchased from MP Biomedicals (Solon, OH, USA).
RNA interference (RNAi)
RNAi oligonucleotides (Qiagen, Valencia, CA, USA) were as follows: human Rer1 siRNA (#7) 5′-UGCGAGUUACAGAAUGUCUGA-3′, human Hrd1 (#1) siRNA 5′-AAUCAUCAAGGUUCUGCUGUA-3′, human Hrd1 (#2) siRNA 5′-AAGGTGATGGGCAAGGTGTTC-3′ human gp78 siRNA 5′-AAGACGGAUUCAAGUACCUUU-3′, human calnexin siRNA 5′-ACACUAGUCUGUGUAACUUUA-3′. AllStars Negative Control siRNA (Qiagen) was used as a control. The RNAi oligonucleotides were transfected into cells using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's protocols. Cells were used for subsequent experiments 72 h after transfection.
Immunoprecipitation (IP)
Cell lysates were prepared by incubating cells at 4°C for 30 min in a lysis buffer containing 40 mM HEPES (pH 7.4), 150 mM NaCl, 2 mM EDTA, 0.3% CHAPS, 10% Glycerol, 1 mM phenylmethanesulfonyl fluoride (PMSF), and complete protease inhibitors (Roche, Indianapolis, IN, USA), and the lysate was clarified by centrifugation. For IP of PMP22-GFP, the supernatant was incubated with an anti-GFP antibody and Dynabeads Protein G (Life Technologies). For IP of 3xFLAG-Rer1, the supernatant was incubated with M2 agarose beads (Sigma-Aldrich). Whole-cell lysates were subjected to immunoblotting.
Chemical cross-link experiments
HEK293T cells were transfected with an empty vector or PMP22-GFP constructs. After 30 h of transfection, the cells were washed with phosphate-buffered saline and incubated for 30 min with 0.5 mM cell-permeable crosslinker dithiobis succinimidyl propionate (DSP; Thermo Fisher Scientific, Madison, WI, USA). After three washes with phosphate-buffered saline, cells were immunoprecipitated (IP) using an anti-GFP antibody.
Endo H and PNGase F treatment
Cell lysates were prepared by incubating cells at 4°C for 30 min in a lysis buffer containing 10 mM Tris-HCl (pH 7.5), 1% Triton X-100, 0.1% sodium dodecyl sulphate (SDS), 0.1 M 2-mercaptoethanol, and complete protease inhibitors (Roche), and the lysate was clarified by centrifugation. The resulting supernatant was heated at 70°C for 10 min. For Endo H (Roche) treatment, sodium acetate (pH 5.5) was added to 20 μg of the cell lysate (final concentration 50 mM). After adding 1 μL of 1,000 U/mL Endo H, the reaction mixture (total volume 40 μL) was incubated for 3 h at 37°C. For PNGase F (Roche) treatment, Tris-HCl (pH 8.8) was added to 20 μg of the cell lysate (final concentration 50 mM). After adding 1 μL of 5 U/mL PNGase F, the reaction mixture (total volume 40 μL) was incubated for 1 h at 37°C. These samples were incubated in sample buffer at 37°C for 30 min and subjected to western blotting.
Fluorescence microscopy
HeLa cells expressing PMP22-GFP were cultured on coverslips and fixed with 4% paraformaldehyde, permeabilized with 0.05% saponin, and then treated with specific antibodies. Images were acquired with an FV1000 confocal microscope (Olympus Corporation, Tokyo, Japan) with a 100× PlanApo oil immersion lens (1.40 numerical aperture; Olympus).
Cycloheximide (CHX) chase experiment
HeLa cells expressing PMP22-GFP were treated with 10 μg/mL CHX, and the cell extracts were prepared at the time point indicated in the figures and subjected to immunoblot experiments with appropriate antibodies.
Statistical analysis
Data were analysed using GraphPad Prism 6 (GraphPad Software, Inc.), and were expressed as means ± standard error of the mean. Two-tailed Student's t-tests and one-way or two-way analysis of variance was used to evaluate significance and calculate P values. P values less than 0.001 (***), 0.01 (**), or 0.05 (*) were considered statistically significant.
Quantitation of PMP22 localization
To count PMP22(L16P)-GFP puncta, we analysed the images with MetaMorph software (Molecular Devices, Sunnyvale, CA, USA). The cell area, which was defined manually, was filtered using the TOP Hat algorithm, and the number of puncta was counted. Three independent experiments were conducted. In each experiment, more than 30 cells were analysed.
Author Contributions
T.H. and K.S. conceived and designed the experiments. T.H., Y.H., R.H., T.A. and H.K. performed the experiments. T.H. and K.S. analysed the data and wrote the manuscript.
Supplementary Material
Supplemental Info
Acknowledgments
We thank Drs. Toshio Kitamura (University of Tokyo) and Yoh Wada (Osaka University), Koichi Okamoto (Gunma University) for providing biological reagents and suggestions. We thank M. Sato, M. Tsunoda, and other members of the Sato laboratory for their technical assistance and discussions. Ken Sato was supported by the Funding Program for Next Generation World-Leading Researchers (NEXT program), JSPS KAKENHI Grant Number 26291036, the Naito Foundation, and the Mochida Memorial Foundation for Medical and Pharmaceutical Research (K.S.). Taichi Hara was supported by research grants from the Takeda Science Foundation, the Nakajima Foundation, the Astellas Foundation for Research on Metabolic Disorders, and MEXT JSPS KAKENHI grants 24116707 and 24590341.
References
- Houlden H. & Reilly M. M. Molecular genetics of autosomal-dominant demyelinating Charcot-Marie-Tooth disease. Neuromolecular Med 8, 43–62 (2006). [DOI] [PubMed] [Google Scholar]
- Li J., Parker B., Martyn C., Natarajan C. & Guo J. The PMP22 gene and its related diseases. Mol Neurobiol 47, 673–698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski J. R. et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66, 219–232 (1991). [DOI] [PubMed] [Google Scholar]
- Snipes G. J., Suter U., Welcher A. A. & Shooter E. M. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13). J Cell Biol 117, 225–238 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdem S., Mendell J. R. & Sahenk Z. Fate of Schwann cells in CMT1A and HNPP: evidence for apoptosis. J Neuropathol Exp Neurol 57, 635–642 (1998). [DOI] [PubMed] [Google Scholar]
- Brancolini C. et al. Rho-dependent regulation of cell spreading by the tetraspan membrane protein Gas3/PMP22. Mol Biol Cell 10, 2441–2459 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho S., Young P. & Suter U. Regulation of Schwann cell proliferation and apoptosis in PMP22-deficient mice and mouse models of Charcot-Marie-Tooth disease type 1A. Brain 124, 2177–2187 (2001). [DOI] [PubMed] [Google Scholar]
- Chance P. F. et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 72, 143–151 (1993). [DOI] [PubMed] [Google Scholar]
- Naef R. & Suter U. Impaired intracellular trafficking is a common disease mechanism of PMP22 point mutations in peripheral neuropathies. Neurobiol Dis 6, 1–14 (1999). [DOI] [PubMed] [Google Scholar]
- Adlkofer K., Naef R. & Suter U. Analysis of compound heterozygous mice reveals that the Trembler mutation can behave as a gain-of-function allele. J Neurosci Res 49, 671–680 (1997). [DOI] [PubMed] [Google Scholar]
- Naef R., Adlkofer K., Lescher B. & Suter U. Aberrant protein trafficking in Trembler suggests a disease mechanism for hereditary human peripheral neuropathies. Mol Cell Neurosci 9, 13–25 (1997). [DOI] [PubMed] [Google Scholar]
- D'Urso D., Prior R., Greiner-Petter R., Gabreels-Festen A. A. & Muller H. W. Overloaded endoplasmic reticulum-Golgi compartments, a possible pathomechanism of peripheral neuropathies caused by mutations of the peripheral myelin protein PMP22. J Neurosci 18, 731–740 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler A. R. et al. Transport of Trembler-J mutant peripheral myelin protein 22 is blocked in the intermediate compartment and affects the transport of the wild-type protein by direct interaction. J Neurosci 19, 2027–2036 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby J. et al. PMP22 carrying the trembler or trembler-J mutation is intracellularly retained in myelinating Schwann cells. Neurobiol Dis 7, 561–573 (2000). [DOI] [PubMed] [Google Scholar]
- Meekins G. D., Emery M. J. & Weiss M. D. Nerve conduction abnormalities in the trembler-j mouse: a model for Charcot-Marie-Tooth disease type 1A? J Peripher Nerv Syst 9, 177–182 (2004). [DOI] [PubMed] [Google Scholar]
- Tobler A. R., Liu N., Mueller L. & Shooter E. M. Differential aggregation of the Trembler and Trembler J mutants of peripheral myelin protein 22. Proc Natl Acad Sci U S A 99, 483–488 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y. et al. Curcumin facilitates a transitory cellular stress response in Trembler-J mice. Hum Mol Genet 22, 4698–4705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajavi M. et al. Oral curcumin mitigates the clinical and neuropathologic phenotype of the Trembler-J mouse: a potential therapy for inherited neuropathy. Am J Hum Genet 81, 438–453 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson K. M. et al. Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for “gain-of-function” ER diseases. Proc Natl Acad Sci U S A 99, 9852–9857 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K., Nishikawa S. & Nakano A. Membrane protein retrieval from the Golgi apparatus to the endoplasmic reticulum (ER): characterization of the RER1 gene product as a component involved in ER localization of Sec12p. Mol Biol Cell 6, 1459–1477 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K., Sato M. & Nakano A. Rer1p as common machinery for the endoplasmic reticulum localization of membrane proteins. Proc Natl Acad Sci U S A 94, 9693–9698 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K., Sato M. & Nakano A. Rer1p, a retrieval receptor for endoplasmic reticulum membrane proteins, is dynamically localized to the Golgi apparatus by coatomer. J Cell Biol 152, 935–944 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M., Sato K. & Nakano A. Endoplasmic reticulum localization of Sec12p is achieved by two mechanisms: Rer1p-dependent retrieval that requires the transmembrane domain and Rer1p-independent retention that involves the cytoplasmic domain. J Cell Biol 134, 279–293 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M., Sato K. & Nakano A. Endoplasmic reticulum quality control of unassembled iron transporter depends on Rer1p-mediated retrieval from the golgi. Mol Biol Cell 15, 1417–1424 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spasic D. et al. Rer1p competes with APH-1 for binding to nicastrin and regulates gamma-secretase complex assembly in the early secretory pathway. J Cell Biol 176, 629–640 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaether C. et al. Endoplasmic reticulum retention of the gamma-secretase complex component Pen2 by Rer1. EMBO reports 8, 743–748 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkova C. et al. Sorting receptor Rer1 controls surface expression of muscle acetylcholine receptors by ER retention of unassembled alpha-subunits. Proceedings of the National Academy of Sciences of the United States of America 108, 621–625 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur F. & Cosson P. Targeting to the endoplasmic reticulum in yeast cells by determinants present in transmembrane domains. J Biol Chem 273, 33273–33278 (1998). [DOI] [PubMed] [Google Scholar]
- Mayer R. J., Ciechanover A. J. & Rechsteiner M. Protein degradation. (Wiley-VCH, 2005). [Google Scholar]
- Nakatsukasa K. & Brodsky J. L. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 9, 861–870 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M. C., Shooter E. M. & Notterpek L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol Dis 10, 109–118 (2002). [DOI] [PubMed] [Google Scholar]
- Hanemann C. O., D'Urso D., Gabreels-Festen A. A. & Muller H. W. Mutation-dependent alteration in cellular distribution of peripheral myelin protein 22 in nerve biopsies from Charcot-Marie-Tooth type 1A. Brain 123 (Pt 5), 1001–1006 (2000). [DOI] [PubMed] [Google Scholar]
- Fortun J., Dunn W. A. Jr, Joy S., Li J. & Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci 23, 10672–10680 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek S. et al. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci 17, 7754–7762 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notterpek L., Shooter E. M. & Snipes G. J. Upregulation of the endosomal-lysosomal pathway in the trembler-J neuropathy. J Neurosci 17, 4190–4200 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J., Coe H. & Michalak M. Specialization of endoplasmic reticulum chaperones for the folding and function of myelin glycoproteins P0 and PMP22. FASEB J 25, 3929–3937 (2011). [DOI] [PubMed] [Google Scholar]
- Fontanini A. et al. Glycan-independent role of calnexin in the intracellular retention of Charcot-Marie-tooth 1A Gas3/PMP22 mutants. J Biol Chem 280, 2378–2387 (2005). [DOI] [PubMed] [Google Scholar]
- Sato K., Sato M. & Nakano A. Rer1p, a retrieval receptor for ER membrane proteins, recognizes transmembrane domains in multiple modes. Mol Biol Cell 14, 3605–3616 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzirusso M. & Chang A. Ubiquitin-mediated targeting of a mutant plasma membrane ATPase, Pma1-7, to the endosomal/vacuolar system in yeast. Mol Biol Cell 15, 2401–2409 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettema E. H., Valdez-Taubas J. & Pelham H. R. Bsd2 binds the ubiquitin ligase Rsp5 and mediates the ubiquitination of transmembrane proteins. EMBO J 23, 1279–1288 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T. et al. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329, 805–810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi P., Colombo S., Francolini M. & Borgese N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J Cell Biol 181, 105–118 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry E. W. & Sidman R. L. The murine mutation trembler-J: proof of semidominant expression by use of the linked vestigial tail marker. J Neurogenet 1, 39–52 (1983). [DOI] [PubMed] [Google Scholar]
- Waxman S. G. From neuroscience to neurology : neuroscience, molecular medicine, and the therapeutic transformation of neurology. (Elsevier Academic Press, 2005). [Google Scholar]
- Passage E. et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med 10, 396–401 (2004). [DOI] [PubMed] [Google Scholar]
- Sereda M. W., Meyer zu Horste G., Suter U., Uzma N. & Nave K. A. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med 9, 1533–1537 (2003). [DOI] [PubMed] [Google Scholar]
- Sato M. et al. Caenorhabditis elegans SNAP-29 is required for organellar integrity of the endomembrane system and general exocytosis in intestinal epithelial cells. Mol Biol Cell 22, 2579–2587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T. et al. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol 31, 1007–1014 (2003). [PubMed] [Google Scholar]
- Kawamura N. et al. Delivery of endosomes to lysosomes via microautophagy in the visceral endoderm of mouse embryos. Nat Commun 3, 1071 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Info