

Abstract
Background
The Aquificales are a diverse group of thermophilic bacteria that thrive in terrestrial and marine hydrothermal environments. They can be divided into the families Aquificaceae, Desulfurobacteriaceae and Hydrogenothermaceae. Although eleven fully sequenced and assembled genomes are available, only little is known about this taxonomic order in terms of RNA metabolism.
Results
In this work, we compare the available genomes, extend their protein annotation, identify regulatory sequences, annotate non-coding RNAs (ncRNAs) of known function, predict novel ncRNA candidates, show idiosyncrasies of the genetic decoding machinery, present two different types of transfer-messenger RNAs and variations of the CRISPR systems. Furthermore, we performed a phylogenetic analysis of the Aquificales based on entire genome sequences, and extended this by a classification among all bacteria using 16S rRNA sequences and a set of orthologous proteins.
Combining several in silico features (e.g. conserved and stable secondary structures, GC-content, comparison based on multiple genome alignments) with an in vivo dRNA-seq transcriptome analysis of Aquifex aeolicus, we predict roughly 100 novel ncRNA candidates in this bacterium.
Conclusions
We have here re-analyzed the Aquificales, a group of bacteria thriving in extreme environments, sharing the feature of a small, compact genome with a reduced number of protein and ncRNA genes. We present several classical ncRNAs and riboswitch candidates. By combining in silico analysis with dRNA-seq data of A. aeolicus we predict nearly 100 novel ncRNA candidates.
Keywords: Aquificales, Thermophiles, ncRNA, Aquificaceae, Desulfurobacteriaceae, Hydrogenothermaceae
Background
Aquificales are gram-negative, non-sporulating bacteria that are thermophilic to hyperthermophilic [1,2], living in terrestrial and marine hot springs. They are autotrophs that primarily fix carbon by the tricarboxylic acid (TCA) cycle [3-5]. The hyperthermophile A. aeolicus, living under extreme temperatures of up to 95°C, has been proposed to have adopted 10% of its protein-coding genes by horizontal gene transfer [6,7] from Archaea. Accumulation of all the special properties of thermophiles (also referred to as accumulation profiles [8]) are rarely understood. Special protein-protective mechanisms have been analyzed [9,10], but we are far away from a comprehensive understanding of the molecular biology of extremophilic bacteria. Beyond idiosyncratic features of Aquificales genomes, our interest focussed on their transcriptomes. Experimentally, we performed a deep sequencing analysis on the model hyperthermophile A. aeolicus with the primary goal of identifying novel ncRNAs candidates. NcRNAs are known to have various functions in all domains of life. Apart from their general importance as gene expression regulators [11-13], ncRNAs are involved in processing [14] and translation [15] of other genes, in defending genomes from viral invasion [16], in shaping and maintenance of bacterial chromosome architecture [17], and they can even be multifunctional [18,19]. According to 16S rRNA analysis, the Aquificales constitute the most deeply rooted bacterial group [20]. However, protein-based phylogenetic reconstructions are not in line with this model [21-26].
We compared the genomes of the three Aquificales families, i.e. Aquificaceae, Hydrogenothermaceae and Desulfurobacteriaceae. We have extended the protein annotation of the mentioned Aquificales and reconstructed the phylogenetic position of these species based on 16S rRNAs as well as on a set of orthologous proteins. Moreover, we have identified ncRNAs based on known homologs and present a complete set of novel ncRNA candidates based on sequence analyses and deep sequencing data obtained for A. aeolicus. For selected ncRNA loci, we provide independent experimental evidence for their expression.
Methods
Genomes
We analyzed the genomes of the following species split into their respective families:
– Aquificaceae: Aquifex aeolicus VF5 (AAE), Hydrogenivirga sp. 128-5-R1-1 (HVI), Hydrogenobacter thermophilus TK-6 (HTH), Thermocrinis ruber (TRU), Thermocrinis albus DSM 14484 (TAL), Hydrogenobaculum sp. Y04AAS1 (HBA),
– Hydrogenothermaceae: Sulfurihydrogenibium sp. YO3AOP1 (SSP), Sulfurihydrogenibium azorense Az-Fu1 (SAZ), Persephonella marina EX-H1 (PMA), and
– Desulfurobacteriaceae: Desulfobacterium thermolithotrophum DSM 11699 (DTH), and Thermovibrio ammonificans HB-1 (TAM).
Accession numbers and sources of genomes are listed in the electronic Supplemental Material http://www.rna.uni-jena.de/supplements/aquificales/index.html. Whole-genome alignments were constructed using Pomago (v.1.0) [27] and TBA (v.11.2) (threaded blockset aligner) [28] with default parameters. Pomago alignments were computed separately for each species as reference. The TBA alignment was projected to each of the reference genomes. Coverage, alignment quality (Weighted sum-of-pairs score – WSoP [29]) and gap ratio are given in Figure 1.
Figure 1.
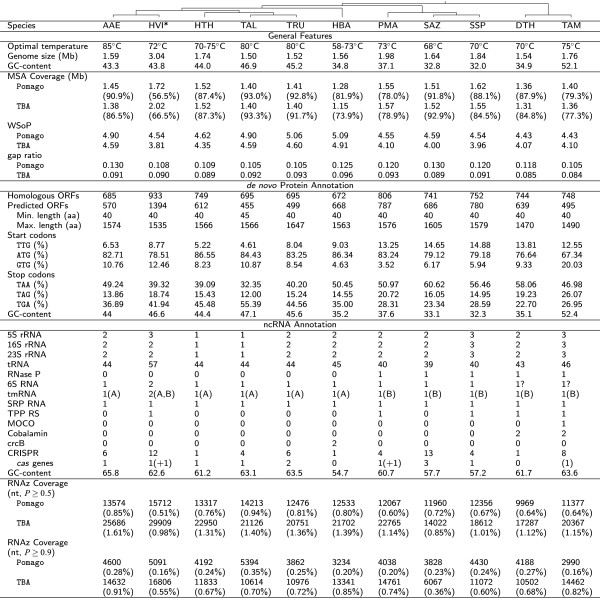
General genome features of the Aquificales. The genome size is given as the total number of nucleotides in the assembly. Multiple sequence alignments (MSA) were performed by Pomago and TBA. RNAz was applied to the Pomago- and TBA-derived MSAs. De novo protein annotation is based on statistics from BacProt, neglecting previously reported proteins for Aquificales. Annotation of ncRNAs shows the statistics for identified ncRNAs of known function. Details of CRISPR cassettes, number of repeats and associated proteins can be found in Figure 9 and in the Supplemental Material. TmRNAs are classified into two types (Figure 6). The phylogenetic tree shown at the top of the table is based on the whole genome as well as 16S rRNA analysis of the 11 Aquificales species. It reproduces the results presented in [30-32]. For further information, see Supplemental Material. AAE – A. aeolicus, HVI – Hydrogenivirga sp., HTH – H. thermophilus, HBA – Hydrogenobaculum sp., TAL – T. albus, TRU – T. ruber, PMA – P. marina, SAZ – S. azorense, SSP – Sulfurihydrogenibium sp., DTH – D. thermolithotrophum, TAM – T. ammonificans, RS – Riboswitch, WSoP – Weighted sum-of-pairs score [29], * denotes the Hydrogenivirga sp. genome of unfinished assembly.
Extension of protein annotation
We used BacProt (publication in progress, see [33] for details) to complement the present annotation of protein-coding genes for each Aquificales genome. It uses a database of groups of orthologous protein-coding genes present in most bacteria [34]. Matches in the genome of interest are annotated, and species-specific features like codon usage, Shine-Dalgarno sequences, Pribnow box motifs and Rho-independent terminators are used to predict additional protein-coding genes. To actually achieve a de novo annotation, we excluded all Aquificales genes from the reference database. Alternative start codons like ATT and CTG were considered as well [35-37]. Re-annotated and previously annotated proteins (genomic positions and sequences) and statistics (mono-/di-nucleotide distribution, position and occurrence of Shine-Dalgarno sequence motifs and Pribnow boxes) for each species are provided in the Supplemental Material.
Annotation of ncRNAs by homology
We used GORAP (v.1.0, publication in progress) to annotate ncRNAs in the following manner: transfer-RNAs (tRNAs) were detected by tRNAscan-SE (v.1.3.1) [38] with the option -B for bacteria. Split tRNAs were searched using SPLITS (v.1.1) [39]. By applying ARAGORN (v.1.2), we searched for tRNAs containing introns [40]. Searches for RNase P RNA were conducted with Bcheck (v.1.0) [41]. For the detection of putative CRISPR loci, crt (v1.2) [42] and CRISPRFinder[43] were used. We searched for cas protein genes by blast (v.2.2.26, E-value ≤10-4) [44] based on known cas genes (downloaded from UniProt (downloaded Jan. 2013) [45]).
To find further ncRNAs, we used blast and Infernal (v.1.1rc2) [46]. Seed sequences from the Rfam (v.11.0) database [47] and European Ribosomal RNA Database[48] were used as query with an E-value ≤0.001 for blast and the Rfam-provided family specific noise cutoffa for Infernal.
NcRNAs expected to escape from detection (e.g. 6S RNA) were searched in a second step with rnabob[49] for short motif search in combination with RNAsubopt, RNAduplex, RNAcofold, RNAalifold and RNAup from the RNA Vienna Package (v.2.0) [50-53]. For verification, we aligned candidates with ClustalW (v.2.0.10) [54] or Locarnate (v.1.7.7.1) [55]. Stockholm alignments were adjusted by hand in the Emacs Ralee mode[56].
Resulting Stockholm alignments are supplied in the Supplemental Material in the General Feature Format (gff) as well as in Fasta (fa) and Stockholm (stk) formats.
Phylogenetic reconstruction
Protein-based phylogeny was performed based on the official NCBI[57] annotations for 42 bacteria shown in the Supplemental Material. In addition to eleven Aquificales species, we included two Archaea as outgroup and a wide phylogenetic range of 29 bacterial species representing all bacterial clades.
Protein sequences were clustered using Proteinortho[34] in the blastp+-mode, thus performing a pairwise all-against-all comparison of sequences from different species to derive orthologous relationships. Whenever an orthologous group did not have a member in a certain species, we applied tblastn to the respective genome to complement for potentially incomplete annotations. The highest scoring alignment to an ORF above a fairly high E-value ≤ 10-20 was added to the initial protein annotation. Finally, Proteinortho was applied again using the expanded annotation.
For a high resolution phylogeny within the Aquificales, we created a whole genome alignment using Pomago. The alignment was analyzed using RAxML (v.7.4.2) [58] with a GAMMA model of rate heterogeneity with an estimate on the proportion of invariable sites and 100 rapid bootstraps.
In an additional phylogenetic analysis we used single-copy orthologous proteins present in at least 50% of all species in the set (189 groups in 42 species). Each protein group was aligned separately using dialign-tx[59]. Both ends of the group’s alignments were cropped to remove leading and tailing gaps. The remaining sequences were concatenated resulting in a 57,260 aa long alignment and applied to RAxML using the LG substitution model [60] as well as the GAMMA model of rate heterogeneity with 100 rapid bootstraps.
The 16S rRNA-based phylogeny was computed with Mafft (v.7.017) [61] using the L-INS-i method with 1000 iterations. We used different approaches: (1) Neighbor Joining with the Kimura correction model [62] (1000 bootstraps), (2) Bayesian inference with MrBayes (v.3.1.2) [63] with default parameters, (3) Maximum likelihood with RAxML (v.7.2.8) [64] (200 bootstraps) with the base substitution models (3a) GTRGAMMA (most accurate, 1000 steps) and (3b) GTRCAT for the bootstrapping phase. For all previously mentioned methods the Archaea Methanobacterium sp. AL-21 and Archaeoglobus fulgidus were used as outgroup. As state of the art, we have estimated a tree with (4) Sate (v.2.2.5) [65] (200 iterations). Related sequences were aligned with Mafft and subsequently merged by Muscle (v.3.7) [66]. The tree was computed using RAxML.
dRNA-seq of A. aeolicus total cellular RNA
Transcriptome analysis of A. aeolicus was based on cDNA libraries from a differential deep sequencing approach (dRNA-seq) [67,68]. A. aeolicus cells, provided by M. Thomm and R. Huber (Regensburg, Germany), were grown for 1 day (late exponential phase) and harvested as described [69]. For preparation of total cellular RNA, we used the hot phenol method [70]: cell pellets were resuspended in extraction buffer (10 mM sodium acetate pH 4.8, 150 mM sucrose) and incubated for 10 min at room temperature with 0.1 volumes of lysozyme (20 mg/ml, Roth, Karlsruhe, Germany). SDS was added to a final concentration of 1% followed by vigorous vortexing. After addition of 1 volume phenol (preheated to 65°C) and vortexing, the mixture was incubated for 5 min at 65°C, then cooled on ice for 5 min, and centrifuged for 30 min at 4°C and 8200 g. Phenol extraction was repeated, followed by chloroform (1+1) extraction and ethanol precipitation. Finally, the DNA was digested with 10 U Turbo DNase (Ambion, Austin, USA) for 30 min at 37°C, followed by addition of another 10 U DNase and incubation for another 30 min at 37°C. Subsequently, the RNA was subjected to phenol/chloroform extraction and ethanol precipitation. After redissolving the RNA in double-distilled water, its concentration was determined by UV spectroscopy. Before cDNA library construction, the RNA was split into two fractions; one fraction was treated with Terminator 5’ P-dependent exonuclease (Epicentre, Madison, USA) for depletion of transcripts carrying a 5’-monophosphate. Both fractions were treated with Tobacco Acid Phosphatase (TAP) before 5’-linker ligation, poly(A) tailing and conversion into cDNA (vertis Biotechnologie AG, Freising, Germany). The cDNA libraries were then sequenced on a Roche FLX sequencer and resulted in the (-)-library with 25,816 reads and the (+)-library (33,697 reads) containing the enriched primary transcripts.
Detection of novel ncRNAs
We used the IGB (Integrated Genome Browser) [71] to visualize the following features of A. aeolicus: (1) nucleotide sequence; (2) local GC-content (for each nucleotide 15 nt on both sides were included for the calculation of GC-content); (3) protein genes annotated by NCBI[72] and BacProt; (4) locally stable secondary structures: calculation was performed with RNALfold with options -d2 and -L120 for both strands with a maximum base-pair span of 120 nucleotides. Sequences with local structures of fewer than 50 nt were discarded. For the prediction of thermodynamically stable RNA structures, each sequence was shuffled 1000 times while preserving the dinucleotide frequencies; to classify extraordinarily stable RNA secondary structures, we chose to use a Z-score cutoff of -3.0 (∼ top 5% of stable structures); (5) conserved regions among the Aquificales: with default parameters of TBA and Pomago we aligned 11 genomes; the TBA alignment was projected to each of the reference genomes; coverage, WSoP and gap ratio are given in Figure 1; (6) novel ncRNAs: novel ncRNA candidates were predicted using RNAz. We used rnazWindow.pl –min-seqs=4 and RNAz -n -b -p 0.5 on the alignments of Pomago and TBA. As rnazWindow.pl assumes lower case nucleotides to be masked, the alignments were converted to upper case letters beforehand; (7) dRNA-seq: cDNA libraries were mapped with segemehl (v.0.0.9.3) [73] applying the parameters -m 12 -D 1 -e 2 -p 4 -X 8 -A 90 -E 5.0.
Northern blot experiments
Total RNA preparation
Total RNA was prepared from cell pellets using the hot phenol method as described [74].
Positive and negative controls
The positive and the negative controls for the Northern blot experiments were synthesized by in vitro transcription using the "TranscriptAid T7 High Yield Transcription Kit" (Thermo Scientific, Germany), according to the protocol supplied by the manufacturer. PCR products generated with the "Long PCR Enzyme Mix" (Thermo Scientific) served as templates for in vitro transcription. As positive controls for the antisense tRNA blots, chemically synthesized RNA oligonucleotides from "Integrated DNA Technologies" (IDT, Belgium) were used (for sequences, see Supplemental Material). RNA oligonucleotides were 5’-phosphorylated before gel electrophoresis. The in vitro transcribed full-length sense tRNAs (generated from PCR products) were used as negative controls for the Northern blots of antisense tRNAs.
Digoxigenin and LNA probes
For the Northern blot detection internally digoxigenin-labeled probes were transcribed using the DIG RNA Labeling Mix (Roche Diagnostics, Germany) as described [74]. The antisense tRNA transcripts were detected with chemically synthesized 5’-digoxigenin-labeled DNA/LNA mixmer probes (Exiqon, Denmark; for sequences, see Supplemental Material).
5’-Phosphorylation of RNA oligonucleotides
67 ng/ μl RNA oligonucleotide, 2.5 mM DTT, 2 mM ATP and 10 U T4 polynucleotide kinase (T4 PNK; Thermo Scientific) were incubated in 1 × T4 PNK buffer (Thermo Scientific) in a volume of 15 μl for 1 h at 37°C, followed by transfer to and storage at -20°C.
Electrophoresis
RNAs were separated on 8% or 10% denaturing (8 M urea) PAA gel with 1 × TBE as electrophoresis buffer [74].
Blotting, crosslinking, hybridization and detection
RNA blotting, hybridization (EDC crosslinking or baking at 80°C for 40 min) and immunological detection were performed as described [74], except that RNA blotting was carried out at 0.36 mA/cm 2 overnight. Prehybridization and hybridization were performed at 68°C (except for 50°C in the case of antisense tRNA 44) using 12 ml hybridization solution. 3.5 μl of in vitro transcribed, internally digoxigenin-labeled probe were added for overnight hybridization. 300 pmol of chemically synthesized, 5’-digoxigenin-labeled DNA/LNA mixmer probe were used for Northern detection of antisense tRNAs. Blotted membranes were stored at room temperature.
In vitro transcripts, probes and primers
Further details on in vitro transcripts, probes and primers are listed in the Supplemental Material.
Results and discussion
Genome analysis – general observations
The genomes of the Aquificales range from 1.50 Mb (T. albus) to 1.98 Mb (P. marina), thus being at the lower limit of bacterial genomes ranging in size from 0.14 to 14.38 Mb with a mean of ∼ 4 Mb [75]. The current annotation file of Hydrogenivirga sp. contains 3.04 Mb, which is considerably larger than the genome size of the other Aquificales, which might be an assembly artefact as discussed later.
Aquificales are known to be AT-rich with a GC-content of about 43% [72,76]. In Hydrogenobaculum sp., Sulfurihydrogenibium sp. and S. azorense even only one-third of the nucleotides are guanine or cytosine. For T. ammonificans an atypically high GC-content of more than 50% was observed.
Between 6.5% (S. azorense) and 28.5% (Hydrogenobaculum sp.) of the genomes were found to be unique to each member bacterium (Figure 1). The comparatively low coverage of Hydrogenivirga sp. is due to the currently assembled genome being almost twice as long as those of other Aquificales. 10.5% to 13.0% of the Pomago alignment, resp. 8.4% to 9.6% of the TBA alignment, consist of gaps. According to the WSoP each nucleotide from the alignment is conserved on average in slightly less than half of the other 10 species (4.43 to 5.09 out of 11 and 3.81 to 4.91 out of 11, for Pomago and TBA, respectively) indicating that the genomes diverged relatively fast. Genomic rearrangements among the Aquificales, underlining the diversity, can be seen in an overview of the Pomago alignment in the Supplemental Material.
Extended annotation of proteins
We extended the original NCBI annotation of proteins of the Aquificalesde novo using BacProt, revealing a number of additional proteins (Table 1). Since a large fraction of proteins are hypothetical or of unknown function, we added for each species a second row which exclusively depicts those with an associated function. The annotations of NCBI and BacProt were merged to generate an extended annotation of protein genes in the Aquificales.
Table 1.
Protein annotations
| NCBI | BacProt | Equal | Start shifted | End shifted | NCBI only | BacProt only | Extended | |
|---|---|---|---|---|---|---|---|---|
| AAE |
1560 |
1255 |
954 |
116 |
124 |
366 |
61 |
1621 |
| |
897 |
685 |
475 |
51 |
54 |
317 |
105 |
1002 |
| DTH |
1513 |
1383 |
1092 |
86 |
105 |
230 |
100 |
1613 |
| |
1115 |
744 |
561 |
58 |
74 |
422 |
51 |
1166 |
| HBA |
1629 |
1340 |
1040 |
119 |
126 |
344 |
55 |
1684 |
| |
1063 |
672 |
500 |
62 |
68 |
433 |
42 |
1105 |
| HTH |
1893 |
1361 |
1069 |
111 |
129 |
584 |
52 |
1945 |
| |
1343 |
749 |
594 |
62 |
84 |
603 |
9 |
1352 |
| PMA |
2051 |
1593 |
1286 |
129 |
122 |
514 |
56 |
2107 |
| |
1494 |
806 |
629 |
84 |
76 |
705 |
17 |
1511 |
| SAZ |
1723 |
1427 |
1190 |
90 |
99 |
344 |
48 |
1771 |
| |
1321 |
741 |
601 |
50 |
73 |
597 |
17 |
1338 |
| SSP |
1722 |
1532 |
1225 |
76 |
108 |
313 |
123 |
1845 |
| |
1145 |
752 |
573 |
38 |
70 |
464 |
71 |
1216 |
| TAL |
1593 |
1145 |
903 |
93 |
127 |
470 |
22 |
1615 |
| |
1144 |
691 |
514 |
59 |
85 |
486 |
33 |
1177 |
| TAM |
1814 |
1243 |
1014 |
90 |
99 |
611 |
40 |
1854 |
| |
1176 |
748 |
575 |
60 |
63 |
478 |
50 |
1226 |
| HVI |
3808 |
2327 |
1537 |
302 |
306 |
1663 |
182 |
3990 |
| 1960 | 933 | 595 | 102 | 92 | 1171 | 144 | 2104 |
Annotations obtained with NCBI (first column, bold font) and those identified with BacProt (second column) lead to an extended current annotation of Aquificales (last column, bold font). In the second lines, hypothetical proteins were removed. Equal – proteins equally identified by BacProt and NCBI; Start/End shifted – proteins identified by BacProt and NCBI vary in length (only 5’ or 3’); NCBI/BacProt only – proteins identified only by NCBI/BacProt. All gff files are available in the Supplemental Material. Species abbreviations as in Figure 1.
We added between 0.7% of H. thermophilus (1352/1343) and 10.6% of A. aeolicus (1002/897) protein-coding genes to the NCBI annotation.
For all proteins annotated by BacProt, we extracted the Shine-Dalgarno and Pribnow box (-10 box) motifs (see Figure 2) in order to facilitate the assignment of novel Aquificales-specific proteins. The Shine-Dalgarno sequence is rather conserved (GGAGG, but always NGAGN). In contrast, the Pribnow box is recognizable but less conserved, indicating more sequence variations among promoters. With the appropriate covariance models we searched for species-specific novel proteins and listed them as predicted proteins in the Supplemental Material.
Figure 2.
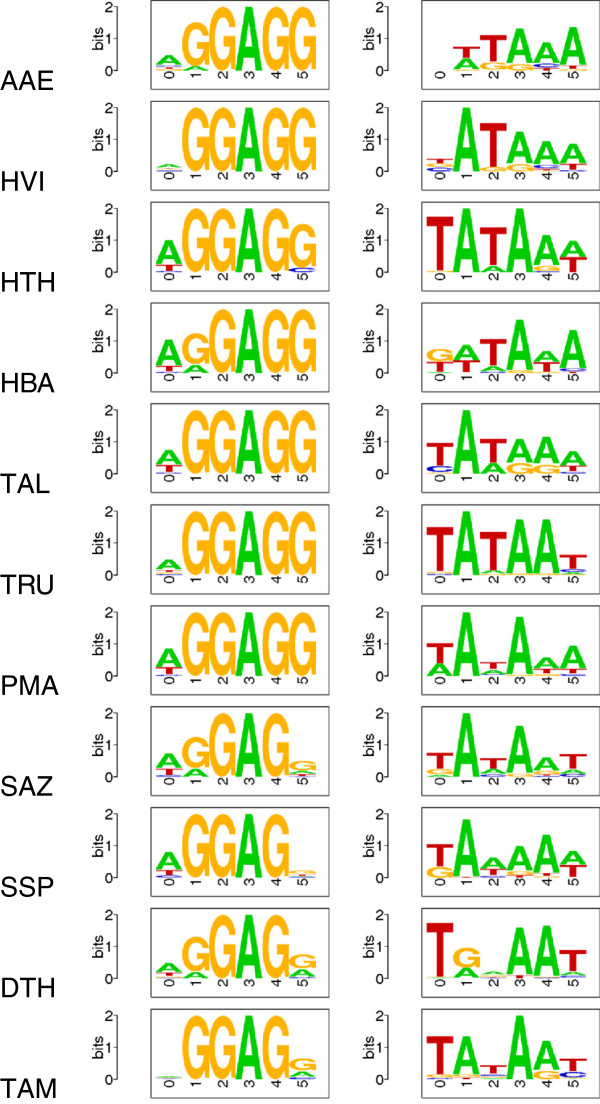
Shine-Dalgarno sequence motifs (left) and Pribnow box (-10 box) motifs (right) in the Aquificales. Details can be found in the Supplemental Material. For species abbreviations see Figure 1.
An overview of the codon usage of A. aeolicus is shown in Table 2. Complete data on all codon usage tables and mono/dinucleotide distributions are provided in the Supplemental Material. We observe a disproportionate usage of certain triplets: isoleucine is mostly (63%) encoded by AUA, tyrosine by UAC (82%) and histidine by CAC (84%). The four arginine codons with a cytosine at the first position of the triplet are rarely used, compared to the two adenine-containing triplets (9%/91%).
Table 2.
Codon usage of A. aeolicus
| Codon | aa | % | Fraction | Codon | aa | % | Fraction | Codon | aa | % | Fraction | Codon | aa | % | Fraction | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| U |
UUU |
Phe (F) |
2.9 |
0.56 |
UCU |
Ser (S) |
0.9 |
0.18 |
UAU |
Tyr (Y) |
0.8 |
0.18 |
UGU |
Cys (C) |
0.4 |
0.49 |
U |
| UUC |
Phe (F) |
2.3 |
0.44 |
UCC |
Ser (S) |
1.3 |
0.27 |
UAC |
Tyr (Y) |
3.4 |
0.82 |
UGC |
Cys (C) |
0.4 |
0.51 |
C |
|
| UUA |
Leu (L) |
1.7 |
0.16 |
UCA |
Ser (S) |
0.7 |
0.15 |
UAA |
stop |
0.1 |
0.49 |
UGA |
stop |
0.1 |
0.37 |
A |
|
| UUG |
Leu (L) |
0.8 |
0.08 |
UCG |
Ser (S) |
0.4 |
0.07 |
UAG |
stop |
0 |
0.14 |
UGG |
Trp (W) |
0.9 |
1 |
G |
|
| C |
CUU |
Leu (L) |
2.7 |
0.25 |
CCU |
Pro (P) |
1.1 |
0.26 |
CAU |
His (H) |
0.3 |
0.16 |
CGU |
Arg (R) |
0.2 |
0.03 |
U |
| CUC |
Leu (L) |
3.1 |
0.3 |
CCC |
Pro (P) |
1.8 |
0.42 |
CAC |
His (H) |
1.3 |
0.84 |
CGC |
Arg (R) |
0.1 |
0.03 |
C |
|
| CUA |
Leu (L) |
0.8 |
0.07 |
CCA |
Pro (P) |
0.6 |
0.14 |
CAA |
Gln (Q) |
0.7 |
0.35 |
CGA |
Arg (R) |
0.1 |
0.01 |
A |
|
| CUG |
Leu (L) |
1.4 |
0.14 |
CCG |
Pro (P) |
0.7 |
0.18 |
CAG |
Gln (Q) |
1.3 |
0.65 |
CGG |
Arg (R) |
0.1 |
0.02 |
G |
|
| A |
AUU |
Ile (I) |
1.7 |
0.23 |
ACU |
Thr (T) |
1 |
0.23 |
AAU |
Asn (N) |
1.1 |
0.3 |
AGU |
Ser (S) |
0.8 |
0.16 |
U |
| AUC |
Ile (I) |
1 |
0.13 |
ACC |
Thr (T) |
1.2 |
0.27 |
AAC |
Asn (N) |
2.5 |
0.7 |
AGC |
Ser (S) |
0.8 |
0.17 |
C |
|
| AUA |
Ile (I) |
4.6 |
0.63 |
ACA |
Thr (T) |
0.9 |
0.21 |
AAA |
Lys (K) |
4.4 |
0.48 |
AGA |
Arg (R) |
1.9 |
0.38 |
A |
|
| AUG |
Met (M) |
1.8 |
1 |
ACG |
Thr (T) |
1.2 |
0.29 |
AAG |
Lys (K) |
4.8 |
0.52 |
AGG |
Arg (R) |
2.6 |
0.53 |
G |
|
| G |
GUU |
Val (V) |
3 |
0.38 |
GCU |
Ala (A) |
1.6 |
0.26 |
GAU |
Asp (D) |
1.6 |
0.37 |
GGU |
Gly (G) |
1.6 |
0.23 |
U |
| GUC |
Val (V) |
0.9 |
0.11 |
GCC |
Ala (A) |
1.3 |
0.21 |
GAC |
Asp (D) |
2.7 |
0.63 |
GGC |
Gly (G) |
0.9 |
0.12 |
C |
|
| GUA |
Val (V) |
2.5 |
0.32 |
GCA |
Ala (A) |
1.7 |
0.29 |
GAA |
Glu (E) |
6.2 |
0.65 |
GGA |
Gly (G) |
3.4 |
0.5 |
A |
|
| GUG |
Val (V) |
1.5 |
0.19 |
GCG |
Ala (A) |
1.4 |
0.24 |
GAG |
Glu (E) |
3.3 |
0.35 |
GGG |
Gly (G) |
1 |
0.15 |
G |
|
| U | C | A | G | ||||||||||||||
Codon usage is based on 1,255 protein-coding genes comprising 431,072 codons. Codon usage of other Aquificales can be viewed in the Supplemental Material. aa – amino acid; the fraction of a particular amino acid encoded by the respective codon is given (1 for Trp encoded by a single codon).
Homology search and annotation of known ncRNAs
A search for ncRNA candidates with RNAz[77] predicted a relatively constant fraction of the genome to code for ncRNAs (between 0.36% for S. azorense and 0.91% for A. aeolicus). Besides the well-known and described rRNAs and tRNAs, only a handful of other wide-spread ncRNAs were detected (Figure 1).
rRNA operons
Most of the Aquificales genomes have two rRNA operons (Figure 1). H. thermophilus and T. albus appear to harbor only one operon. The genomes of T. ammonificans and Sulfurihydrogenibium sp. contain three operons, whereas Hydrogenivirga sp. appears to have two 16S, two 23S and three 5S rRNA genes.
tRNAs
With the exception of Hydrogenivirga sp. (see below), tRNAscan identified between 39 (S. azorense) and 46 tRNAs (T. ammonificans) per Aquificales species. With SPLITS and ARAGORN no split tRNAs were found.
All possible codons are utilized in the Aquificales (see Table 2 for A. aeolicus, and Supplemental Material for other Aquificales), but the number of tRNA genes is reduced to a minimum in contrast to reference bacteria such as E. coli which encodes multiple copies of many tRNA isoacceptors.
Figure 3 shows nearly no tRNA with 5’-A in the anticodon and only half of the Aquificales have some anticodons with 5’-C, where the non-Aquificaceae apparently favored the reduction of such tRNA genes (Figure 4). Important tRNA modification enzymes (TadA – tRNA adenosine deaminase and TilS – tRNA-Ile lysidine synthetase) are encoded in Aquificales and X-ray structures of TadA and TilS from A. aeolicus have been reported [78,79]. TadA converts A residues in the 5’-position of certain tRNA anticodons to inosine to expand wobble decoding, and TilS converts the 5’-C residue in the CAU anticodon of specific tRNA-Ile molecules to lysidine (2-lysyl cytidine; abbreviated as L or k2C) to decode 5’-AUA (Ile) codons instead of 5’-AUG (Met) codons [80]. Without this posttranscriptional modification, decoding of isoleucine AUA codons would be impossible [81-83]. Selenocysteine-specific tRNAs decoding 5’-UGA are present in the Aquificaceae (except for Hydrogenobaculum sp.) and in the Desulfurobacteriaceae (T. ammonificans and D. thermolithotrophum), but are absent from the Hydrogenothermaceae (P. marina, S. azorense, Sulfurihydrogenibium sp.; see Figure 3). The Aquificaceae (except Hydrogenivirga sp.), in contrast to the other Aquificales or mesophiles such as E. coli or B. subtilis, encode the lysine isoacceptor with the anticodon 5’-CUU to decode the AAG codon.
Figure 3.
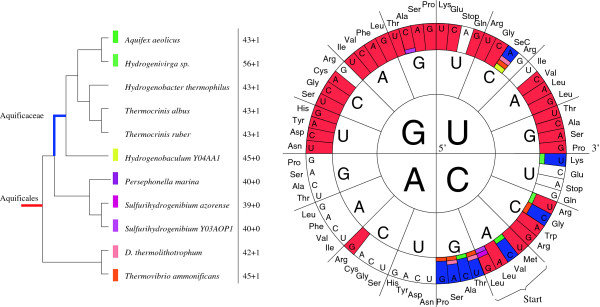
Distribution of tRNAs in the Aquificales. Left: The total numbers of encoded tRNAs including the absence (+0) or presence (+1) of selenocysteine tRNA (tRNA-SeC) are given. Phylogenetic tree as in Figure 1; Right: anticodons specified by the following colors: red – tRNA with this anticodon encoded in all Aquificales; blue – tRNA encoded in the Aquificaceae only. Other colors represent the absence or presence of a tRNA with this anticodon, as defined in the phylogenetic tree on the left. For example, tRNA-SeC is present in all Aquificaceae except for Hydrogenobaculum sp., and is additionally found in the non-Aquificaceae species D. thermolithotrophum and T. ammonificans.
Figure 4.
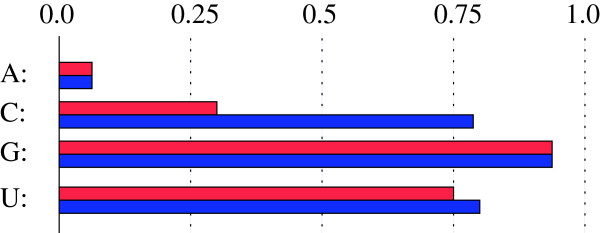
The distribution of the 5’ (wobble) position of a tRNA anticodon in all Aquificales (red) versus Aquificaceae only (blue) is U: 0.75/0.80%, G: 0.93/0.94%, C:0.30/0.78%, A:0.06/0.06%. Non-Aquificaceae show a low percentage of wobble C.
RNase P
The catalytic RNA subunit of the tRNA processing endoribonuclease RNase P was previously identified in P. marina and S. azorense[84]. Additionally, RNase P RNAs were easily identified here with Bcheck in Sulfurihydrogenibium sp., T. ammonificans and D. thermolithotrophum. In the Aquificaceae, RNase P RNA candidates were neither detected with Bcheck, rnabob nor by manual in silico search methods using cDNA libraries of A. aeolicus. This is consistent with the negative results of previous searches for RNase P RNA in A. aeolicus[85,86].
All identified RNase P RNAs lack the P18 element, which appears to be a general feature of type A RNase P RNAs in the Hydrogenothermaceae and Desulfurobacteriaceae. The Sulfurihydrogenibium sp., T. ammonificans and D. thermolithotrophum RNAs differ from their P. marina and S. azorense counterparts by a weaker L9-P1 tertiary contact (L9 5’-GYAA tetraloop docking on an A-U/G-C tandem bp instead of a G-C/G-C tandem which is a hallmark of RNase P RNAs from thermophiles [84,87]). Other differences are: (1) very short P12 stems in T. ammonificans and D. thermolithotrophum, (2) particularly weak P17 stems in Sulfurihydrogenibium sp. and D. thermolithotrophum, (3) a destabilized L8-P4 interaction, a destabilized P14 helix, but a stabilized L14-P8 interaction in T. ammonificans. For details, see RNase P RNA 2D structures in the Supplemental Material.
6S RNA
Bacterial 6S RNAs, about 200 nt in length, form a rod-shaped secondary structure with a central bulge region flanked by largely helical arms on both sides. Their structure is thought to mimic the structure of an open DNA promoter [88,89]. 6S RNAs bind to the housekeeping RNA polymerase holoenzyme to block transcription at DNA promoters, primarily upon entry of cells into stationary growth phase. When nutrients are resupplied (including NTPs), RNA polymerase massively synthesizes transcripts (so-called product RNAs – pRNAs) on 6S RNA as template, which lead to a structural rearrangement of 6S RNA and release of RNA polymerase. Thus, 6S RNA is a fast riboregulator that makes RNA polymerase instantly available for a new exponential growth when nutrients are resupplied [68,90-93].
In A. aeolicus the 6S RNA was clearly identified via an experimental RNomics approach [85]. 6S RNA candidates in the other Aquificales were predicted computationally using the Rfam covariance model and, as expected, vary substantially in primary, but less in secondary structure. For Hydrogenivirgia we found two copies. Predicted 6S RNAs for T. ammonificans and D. thermolithotrophum remain candidates since they differ substantially from those of other Aquificales.
The RNAalifold consensus structure for the 6S RNA candidates from all other Aquificales analyzed here is shown in the Supplement. Individual RNAfold predictions (see Supplemental Material for details) support the notion that they are bona fide 6S RNAs.
In the case of A. aeolicus 6S RNA, we proposed that formation of a "central bulge collapse" helix (Figure 5-Top, [85]) is the major component of the pRNA-induced rearrangement of this 6S RNA structure [90]. If at all, or to which extent, the adjacent hairpin structure forms in the pRNA-rearranged structure remains to be investigated. For the eight other 6S RNA candidates (Figure 5), we predicted rod-shaped structures with a destabilized central region that is not necessarily purely single-stranded (see Supplemental Material for further details). According to our proposals, pRNAs would start with a G residue in the Aquificaceae, whereas those of the Hydrogenothermaceae (P. marina, S. azorense and Sulfurihydrogenibium sp.) would initiate with an A residue.
Figure 5.
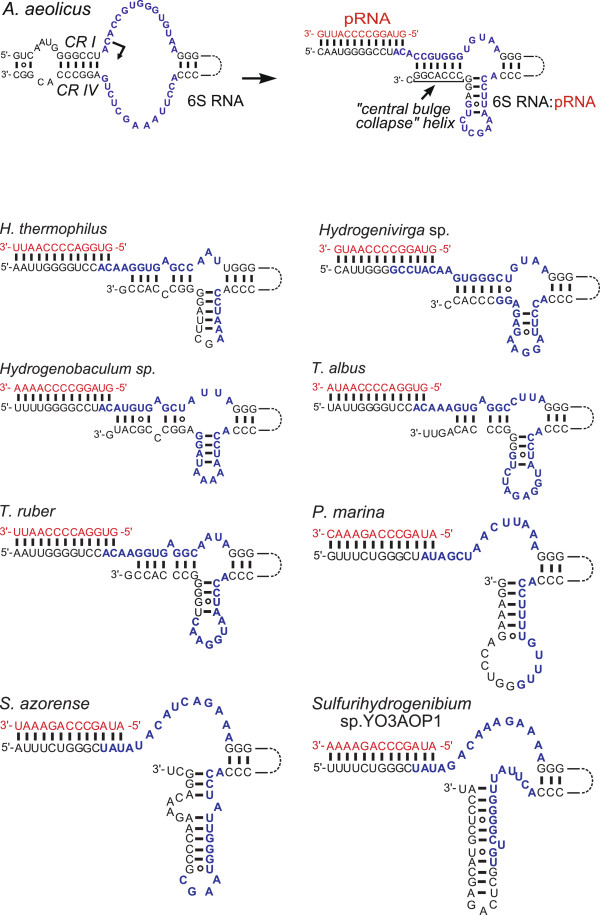
Aquificales 6S RNAs: predicted pRNA transcription initiation sites, and pRNA-induced structural rearrangements of 6S RNAs. Top: A. aeolicus 6S RNA; this 6S RNA was experimentally verified [85] and the pRNA transcription start site identified by deep sequencing (unpublished results); nucleotides of the central bulge region are marked in blue; during pRNA transcription on 6S RNA as template, the endogenous helix is disrupted, leading to the formation of new base-pairing interactions. Here, a 6S RNA hybrid with a pRNA 13-mer (red) is shown on the right; the proposed rearranged structure of the central 6S RNA region [90] has not yet been proven experimentally. Proposed structures of the central bulge regions and their pRNA-induced rearrangements of the other eight Aquificales 6S RNA candidates: rearranged structures upon duplex formation with putative pRNA 13-mers; the pRNA initiation sites are proposed on the basis of resemblance to A. aeolicus 6S RNA. For more details, see Supplemental Material.
tmRNA
In bacteria, stalling of translating ribosomes on truncated mRNAs is rescued through action of the dual-function transfer-messenger RNAs (tmRNAs) [94,95]. The tRNA-like domain is present and highly conserved in all Aquificales. An architectural feature of tmRNAs is their intricate structure consisting of four pseudoknots. Interestingly, we found two different types of tmRNAs, introduced here as type A (present in the Aquificaceae) and B (specific to Hydrogenothermaceae and Desulfurobacteriaceae). This classification is based on the observation that the lower stem of pseudoknot 1 (pk1) involves 4-5 bp in type A tmRNAs, but only 2–3 bp in type B variants (Figure 6, Supplemental Material). Pk1 is critical for tmRNA function and binds near the ribosomal decoding site [95]. Mutational analysis of E. coli tmRNA revealed that mutations disrupting the upper stem of pk1 are not tolerated, whereas the outer two base pairs of the lower stem (Figure 6) can be disrupted (resulting in a 3-bp stem) without loss of function [95]. On the other hand, the tmRNA of another thermophile, Thermotoga maritima, has a lower pk1 stem expanded to 7 bp [96]. This raises the question if the Aquificales type B tmRNAs, for which only a 2-bp lower pk1 stem is predicted (Sulfurihydrogenibium sp., P. marina and S. azorense), are still able to form this pseudoknot, or if the weakness or absence of this stem is compensated for by e.g. tmRNA ligand interactions that are idiosyncratic to the Aquificales encoding a type B tmRNA.
Figure 6.
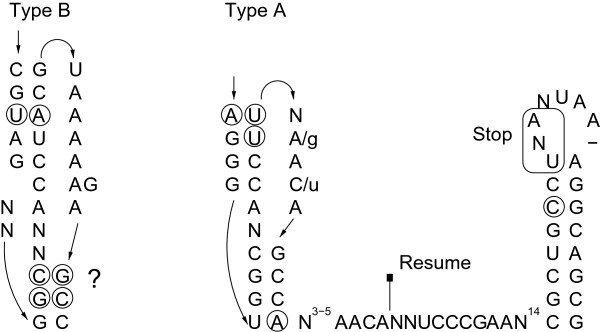
Pseudoknot 1 (pk1) of tmRNAs type A and B. Formation of the lower stem of pk1 is questionable. Circled nucleotides represent positions of compensatory mutations. The question mark shows an unclear interaction of two (in one case three) base pairs.
The messenger RNA-like regions (MLR), which are in close vicinity of pk1, encode tag preptides of 10 amino acids, with subphyla-specific signatures (Figure 7). For example, all Aquificaceae and Hydrogenothermaceae tmRNAs code for a proline at the second position, which is alanine in the Desulfurobacteriaceae. The genome of Hydrogenivirga sp. appears to encode both types of tmRNAs (type A and B). Whether this reflects a genuine tmRNA gene duplication rather than a genome contamination or assembly artefact remains to be clarified (see below).
Figure 7.
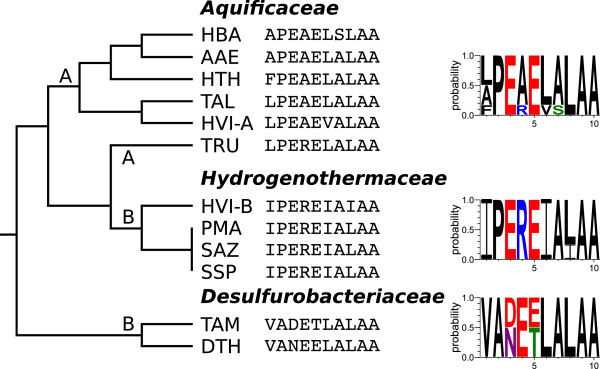
Proteolysis tags of tmRNA types (A/B). The encoded proteolysis tag as well as a probability logo for each family are shown. Two tmRNAs were identified in the genome assembly of Hydrogenivirga sp.
Furthermore, Hydrogenobaculum sp. carries a 78-nt hairpin-like insertion in the pseudoknot 4 (pk4) region, which however is compatible with formation of pk4 (Figure 8). Such a long extension within tmRNAs has been not reported yet.
Figure 8.
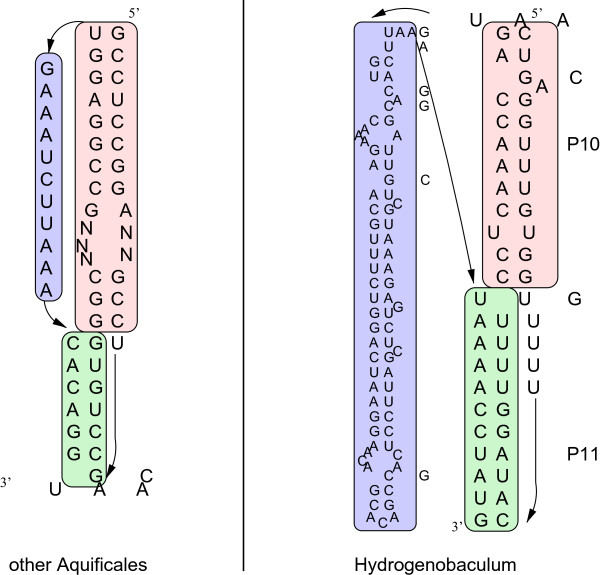
Hydrogenobaculum sp. has a 78-nt insertion downstream of P10, but pseudoknot 4 (pk4) is still predicted to form. The region is shown in comparison to T. albus as a representative of the other Aquificales. Detailed figures of all tmRNAs can be viewed in the Supplemental Material.
CRISPR system
For each member of the Aquificales we could identify at least one locus of clustered interspaced short palindromic repeat sequences (CRISPRs), which are involved in an immunity against viruses and plasmids [97]. Although the Aquificales have very compact genomes, the number of identified CRISPR clusters varied from one to thirteen (Figure 1), indicating the presence of thermostable viruses in extreme environments as reported for Archaea [98]. The number of CRISPR clusters does not seem to be clade-specific. Also, the number of repeats in a cluster varies strongly. For example, in T. albus we found in total four CRISPR systems containing 36, 41, 57 and 63 repeats, whereas in A. aeolicus the five CRISPR loci only had four to five repeats. For some, but not all of the CRISPR clusters, we could detect associated cas genes (Figure 9). The exact numbers of detected CRISPR clusters and Cas protein cassettes can be seen in Figure 1. In this table we included only CRISPR clusters that were found by both approaches (crt and CRISPRfinder). It has to be kept in mind that the genome of Hydrogenivirga sp. is in an unfinished state, so it is possible that some CRISPR loci and especially associated cas genes escaped detection.
Figure 9.

CRISPR clusters and associated cas protein genes in the Aquificales. Shown cas genes and CRISPR clusters (black filled boxes) are proportional in size. As only CRISPR clusters with associated cas genes are displayed in this figure, Hydrogenobaculum sp. and D. thermolithotrophum are not displayed here.
Other ncRNA
SRP RNA was found once per genome being highly conserved in sequence and structure (see Supplemental Material). Additionally, we show some riboswitch candidates: TPP, MOCO, Cobalamin and crcB (see Figure 1). The MOCO riboswitch found in T. ammonificans and the two crcB riboswitches identified in Hydrogenobaculum sp. conform well to the Rfam conservation model (see Supplemental Material). Riboswitches were only found sporadically among the Aquificales.
Novel ncRNAs in A. aeolicus
Besides the annotation of ncRNAs with known functions, we additionally aimed to detect novel ncRNAs, as they often regulate transcription or play an important role as posttranscriptional regulators. Here we combined in silico analysis of the A. aeolicus genome and dRNA-seq data from the same organism to identify novel ncRNA candidates, some of which were subsequently analyzed by Northern blot analysis.
In the in silico search, small ncRNAs (sRNAs) were distinguished from proteins by the following analysis steps: (1) The GC-content of the A. aeolicus genome is 43%. However, the ncRNAs described above show an average GC-content of 66%. We associated each nucleotide with a local GC-value. (2) The function of small ncRNAs, e.g. 6S RNA, is often determined by their stable secondary structure. To each position in the genome, we assigned the minimum free energy of the most stable local secondary structure including this nucleotide, using RNALfold. (3) Most ncRNAs are conserved among closely related organisms. We calculated genomewide multiple sequence alignments (MSA) with TBA and Pomago of all Aquificales genomes, which can be viewed in the Supplemental Material. (4) Based on the MSAs we performed a novel ncRNA prediction with RNAz and displayed their probability.
All ncRNA candidates with a minimum length of 25 nt and not overlapping protein-coding sequences, rRNA operons or tRNAs, were summarized in a full candidate table, containing all properties mentioned above (see Supplemental Material). A subset of these genes can be seen in Table 3. We identified 99 putative loci for ncRNAs, abbreviated n1 to n99. All above annotated ncRNAs, such as tmRNA (n74) or SRP RNA (n85) were mutually confirmed by our dRNA-seq and in silico approaches. Interestingly, known ncRNAs as well as novel ncRNA candidates show a significant level of antisense transcripts (see examples in Figures 10 and 11). For unknown ncRNAs the sense direction is not assignable. Putative ncRNAs, referring to one genomic location and having comparable numbers of cDNA read counts on both strands, are described with the same ID.
Table 3.
Selection of highly potential novel ncRNA candidates of A. aeolicus
|
ID |
Location |
GC |
cDNA |
Annotation |
Structure and Sequence |
Remarks |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5’ boundary | 3’ boundary | Strand | (+/-) | (NCBI/BacProt) | RNALfold | Cons_p | Cons_t | RNAz_p | RNAz_t | |||
| Known ncRNAs | ||||||||||||
| 45 |
567675 |
567915 |
- |
0.53 |
2237/899 |
murF/UDP |
-3.89 |
11 |
7 |
No |
0.9990 |
Downstream of 5S RNA |
| 74 |
1153499 |
1153856 |
- |
0.65 |
83/31 |
tmRNA/no |
-5.04 |
6 |
11 |
No |
0.9996 |
tmRNA |
| 78 |
1219679 |
1219903 |
+ |
0.55 |
382/1384 |
pheT/pheT |
-6.59 |
11 |
11 |
No |
No |
6S RNA |
| 85 |
1303758 |
1303875 |
+ |
0.57 |
5239/456 |
No/no |
-4.15 |
11 |
11 |
No |
0.7085 |
SRP RNA |
| Putative Novel ncRNAs | ||||||||||||
| 2 |
15301 |
15474 |
+ |
– |
0/0 |
No/no |
-3.66 |
No |
5 |
No |
No |
Plasmid region |
| 6 |
69101 |
69198 |
- |
0.37 |
809/545 |
No/no |
-4.71 |
11 |
11 |
No |
No |
|
| 25 |
328934 |
328995 |
+ |
0.37 |
582/250 |
No/no |
No |
9 |
9 |
No |
No |
|
| 48 |
620054 |
620211 |
- |
0.44 |
41/71 |
No/no |
-3.13 |
11 |
9 |
No |
No |
|
| 58 |
739705 |
739811 |
+ |
0.44 |
41/144 |
No/no |
-3.92 |
11 |
11 |
No |
No |
|
| 68 |
989704 |
989840 |
+ |
0.50 |
476/756 |
aq_1392/permease |
-4.53 |
4 |
2 |
No |
No |
Aae-65
[85] |
| 74 |
1153547 |
1153769 |
+ |
0.65 |
326/51 |
No/no |
-5.04 |
6 |
11 |
No |
0.9996 |
|
| 75 |
1168974 |
1169071 |
- |
0.55 |
158/79 |
aq_1666/no |
-3.84 |
3 |
3 |
No |
No |
|
| 80 |
1231909 |
1232006 |
+ |
0.38 |
860/2339 |
No/no |
-3.74 |
11 |
2 |
No |
No |
|
| 97 |
1491199 |
1491559 |
- |
0.40 |
10/297 |
rfaG/glycosyltransferase |
-4.60 |
11 |
11 |
No |
No |
|
| Tail to tail Transcripts (T2T) | ||||||||||||
| t2t10 |
608075 |
608182 |
+ |
0.52 |
60/20 |
aq_880/no |
-3.70 |
11 |
11 |
No |
No |
|
| |
608075 |
608308 |
- |
0.48 |
22/12 |
aq_881/DOXP synthase |
-3.70 |
11 |
11 |
No |
No |
|
| t2t17 |
1336433 |
1336708 |
+ |
0.46 |
380/87 |
aq_1896/predicted |
No |
11 |
11 |
No |
No |
|
| |
1336544 |
1336642 |
- |
0.51 |
100/55 |
folD/folD |
No |
11 |
11 |
No |
No |
|
| t2t20 |
1479248 |
1479345 |
+ |
0.44 |
180/117 |
prmA/prmA |
No |
11 |
8 |
No |
No |
|
| |
1479168 |
1479508 |
- |
0.43 |
12/62 |
acs’/predicted |
-3.19 |
11 |
8 |
No |
No |
|
| tRNAs with sense transcripts only | ||||||||||||
| t06;43 |
383154 |
383390 |
- |
0.52 |
9/2 |
recN; tRNA/predicted |
-3.53 |
11 |
10 |
No |
0.9943 |
|
| tRNAs with sense and various antisense transcripts | ||||||||||||
| t34;15 |
1356464 |
1356743 |
+ |
0.64 |
23/5 |
tRNA/no |
-5.43 |
11 |
10 |
0.9996 |
0.9992 |
|
| |
1356461 |
1356575 |
- |
0.60 |
61/15 |
No/no |
-5.43 |
11 |
9 |
0.9996 |
0.9992 |
|
| t44;20 |
1531016 |
1531131 |
+ |
0.58 |
1141/437 |
ihfB/no |
-4.33 |
7 |
9 |
No |
0.9951 |
|
| 1531004 | 1531130 | - | 0.56 | 335/136 | tRNA/no | -4.33 | 7 | 9 | No | 0.9951 | ||
The genomic locations and GC-content are listed in columns 2-4. cDNA – the maximal number of observed read counts in the (+)- and (-)-library; Annotation – overlap to predicted proteins by NCBI and BacProt; RNALfold – energy in kcal/mol of locally stable RNA secondary structure predicted by RNALfold; Cons_p and Cons_t – number of species with homologous regions aligned by Pomago and TBA; RNAz – probabilities >0.5 (based on multiple sequence alignments calculated by Pomago (p) or TBA (t)). Further observations, for example that Aae-65 was described earlier in [85], are noted in the last column. A complete list of novel ncRNA candidates, and tRNAs can be found in the Supplemental Material.
Figure 10.
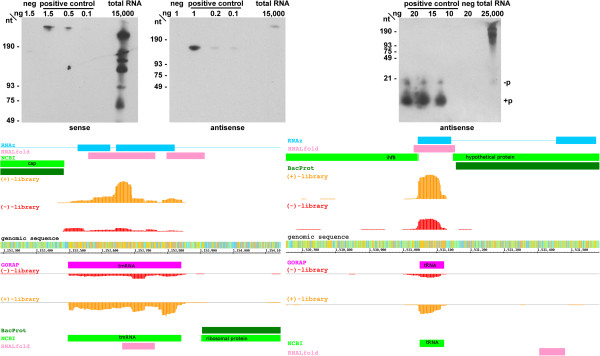
cDNA read profiles (bottom) and Northern blots (top) of selected ncRNAs. Read profiles left: tmRNA (350 counts); right: tRNA 44 (1000 counts); The upper half of each read profile represents the plus strand and the lower one the minus strand. Annotation by RNAz (blue), RNALfold (rose) and NCBI/BacProt (green), cDNA reads of the (+)-library (orange) and of the (-)-library (red) and ncRNA annotation by GORAP (pink); colors of genomic sequences represent nucleotides A (green), C (red), G (orange) and T (blue); counts - scale of read display adapted to the maximal number of detected reads (see Table 3). For Northern blots, see Methods; -p – with 5’-OH ; + p – with 5’ monophosphate.
Figure 11.
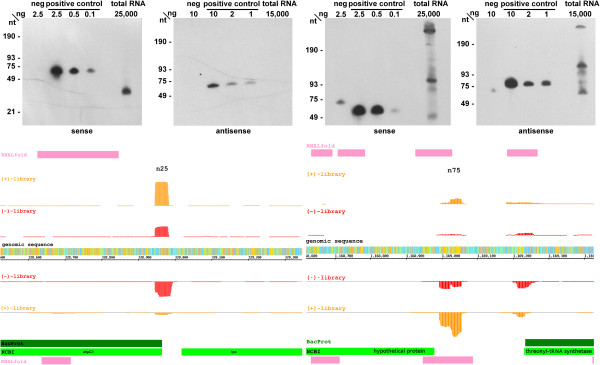
cDNA read profiles (bottom) and Northern blots (top) of selected ncRNAs. Read profiles left: n25 (600 counts); right: n75 (250 counts). Details as in Figure 10. For Northern blots, see Methods.
For comparison reasons, we also added tRNAs to our table of ncRNAs, which show the feature of sense-antisense (s/as) expression. To exclude the possibility of mapping or other artefacts, we confirmed the presence of antisense transcripts exemplarily by Northern blots for tmRNA and tRNA 44 (Pro-TGG) (Figure 10).
Furthermore, Northern blots were conducted for the loci encoding candidates n25 and n75, for which the dRNA-seq data indicated sense and antisense transcription each differing between the (+)- and (-)-library (Figure 11). For n25, we found most transcripts on the plus strand in the (+)-library (582), whereas less than half as many transcripts (250) were detected in the (-)-library. Interestingly, an inverse relation was observed for the minus strand (50/361). For n25, Northern blot detection revealed a signal somewhat shorter than the one expected from the cDNA read boundaries, whereas no signal could be detected for antisense transcripts (Figure 11, top). This finding suggests that the sense transcript is the major one. In the case of n75, both sense and antisense transcripts of comparable intensity were detected, the major signals of the Northern blot representing RNAs larger and smaller than anticipated from the read boundaries (Figure 11, bottom). Thus, the polarity of the putative ncRNA gene remains unclear.
Interestingly, very high transcription levels are found in overlapping 5’-upstream regions of two protein-coding genes located on opposing strands (Table 3). Beside these so-called head-to-head (h2h) transcripts we furthermore observed tail-to-tail overlaps (t2t, two 3’-untranslated regions overlapping on opposing strands) that are represented by very high read coverage (Supplemental Material). If these are real transcripts with a certain function or artefacts remains unclear.
Conclusion
With the advent of a growing number of Aquificales genome sequences in public databases, we have re-analyzed this group of bacteria thriving in extreme environments. The Aquificales share the feature of a small, compact genome with a reduced number of protein and ncRNA genes. The genes for tRNAs are reduced to a minimum but retain the capacity to decode all types of codons, and rRNA genes are confined to 2–3 copies each. Several classical ncRNAs are present, such as SRP RNA, tmRNA, 6S RNA, RNase P RNA (except for all Aquificaceae) and riboswitch candidates in some Aquificales. Furthermore, by combining in silico analysis with dRNA-seq data of A. aeolicus, we were able to predict nearly 100 novel ncRNA candidates, some of which might be specific to the Aquificales. Finally, CRISPR systems of bacterial immunity were identified.
Re-annotation of protein genes using BacProt revealed novel proteins with unknown function, some of which might turn out to be specific to the Aquificales as well. On average, 63 additional proteins were found that were missing in the respective original annotation.
In our cDNA libraries of A. aeolicus, we observed massive amounts of antisense reads with similar patterns (length and amount) at putative ncRNA loci and terminal regions of mRNAs. Examples of transcripts antisense to tmRNA and tRNA are illustrated in Figure 10.
We compared 40 bacterial and 2 archaeal genomes (see Supplemental Material), and the presence or absence of proteins was used to determine their position in the phylogenetic tree of bacteria. Both Archaea form a clear outgroup. Thermodesulfatator indicus branches first in the group of Bacteria, followed immediately by the Aquificales, while other bacterial branches diverge later. In an additional protein-based analysis, we took the sequences of single-copy orthologs that were present in at least 50% of all species (concatenated 57,260 aa) (see Supplemental Material). In contrast to the protein presence/absence tree, neither the Aquificales nor T. indicus were placed at a basal position here. However, the two groups are still in close vicinity to each other. This analysis not necessarily excludes the possibility of the Aquificales being a basal clade. The selection of orthologs being present in at least 50% of the species leads to a lower coverage of orthologs present in Archaea species and therefore may favor long branch attraction [99]. The idea behind selecting frequently occurring single-copy orthologs was to produce phylogenetic trees being less influenced by horizontal gene transfer. However, proteins shared by Archaea and Aquificales only are not part of the selected "50% group" of proteins and are therefore not considered in this analysis.
Both protein-based phylogenetic trees disagree with a previous study [3] where Desulfobacterium autotrophicum HRM2, a δ-proteobacterium, was added to the Desulfurobacteriaceae family based on 16S rRNA analysis. We assume that this was an artefact of the high GC-content of rRNAs due to the high environmental temperatures. Regarding their proteomes, Aquificales and D. autotrophicum are not significantly related.
The results of the 16S rRNA phylogenetic analysis did not show a clear picture. Depending on the method used for reconstruction, the Aquificales were either placed near the root of the bacterial tree (MrBayes and RAxML with GTRGAMMA substitution model) or not (NJ and RAxML with GTRCAT) (see Supplemental Material). In accordance with the results of [26], the Aquificales were always placed close to the Thermotogales and Thermales-Deinococcales, Archaea were more closely related to the Aquificales than to the Thermotogales.
We identified two 6S RNA and two tmRNA candidate genes in Hydrogenivirga sp., rather than a single one as in the other Aquificales. Likewise, Hydrogenivirga sp. has a comparatively high amount of tRNA copies and CRISPR loci and its genome is estimated to be of roughly double the size of the other Aquificales genomes. Combined, these observations support the notion that the Hydrogenivirga sp. genome assembly is erroneous or two genomes of related bacteria (one type from Hydrogenothermaceae) have entered the sequencing project, being in agreement with [32]. Based on the tmRNA tag peptides identified in the Hydrogenivirga sp. assembly, the second one (Hydrogenivirga sp.-B: IPEREIAIAA) matches the sequence exclusively found among the Hydrogenothermaceae, although Hydrogenivirga sp. belongs to the Aquificaceae (see Figure 7). This suggests that the Hydrogenivirga sp. assembly is a blend of sequences from a member of the Aquificaceae and a member of the Hydrogenothermaceae.
Endnote
aNoise cutoff is the highest observed false positive bit score for a potential gene which does not belong to the seed model.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Bioinformatical analysis: ML, SW, KR, BMB, NW, and MM. Experimental validation: AIN and RKH. Analyzed data: all. Wrote, read and approved the final manuscript: all.
Contributor Information
Marcus Lechner, Email: lechner@staff.uni-marburg.de.
Astrid I Nickel, Email: nickel@staff.uni-marburg.de.
Stefanie Wehner, Email: stefanie.wehner@uni-jena.de.
Konstantin Riege, Email: konstantin.riege@uni-jena.de.
Nicolas Wieseke, Email: wieseke@informatik.uni-leipzig.de.
Benedikt M Beckmann, Email: benedikt.beckmann@iri-lifesciences.de.
Roland K Hartmann, Email: roland.hartmann@staff.uni-marburg.de.
Manja Marz, Email: manja@uni-jena.de.
Acknowledgements
We thank Markus Fricke for tmRNA structure visualization, Brice Felden for tmRNA discussion, J. Sugahara for the SPLITS run in A. aeolicus, Jörg Vogel and Cynthia Sharma from the University of Würzburg for help with differential RNA-Sequencing. MM was funded by the Carl-Zeiss-Stiftung. This work was supported by the DFG-Graduiertenkolleg 1384 "Enzymes and multienzyme complexes acting on nucleic acids" (BMB, ML, MM, RKH, SW), and DFG project MA-5082/1 (MM, SW).
References
- Setchell WA. The upper temperature limits of life. Science. 1903;17(441):934–937. doi: 10.1126/science.17.441.934. [DOI] [PubMed] [Google Scholar]
- Reysenbach AL, Wickham GS, Pace NR. Phylogenetic analysis of the hyperthermophilic pink filament community in Octopus Spring, Yellowstone National Park. Appl Environ Microbiol. 1994;60(6):2113–2119. doi: 10.1128/aem.60.6.2113-2119.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hügler M, Huber H, Molyneaux SJ, Vetriani C, Sievert SM. Autotrophic CO2 fixation via the reductive tricarboxylic acid cycle in different lineages within the phylum Aquificae: evidence for two ways of citrate cleavage. Environ Microbiol. 2007;9:81–92. doi: 10.1111/j.1462-2920.2006.01118.x. [DOI] [PubMed] [Google Scholar]
- Reysenbach AL. Class I: Aquificae class. nov. Bergey’s Manual of Systematic Bacteriology. Edited by Garrity GM, Boone DR, Castenholz RW. New York: Springer-Verlag; 2001:359–367.
- Bonch-Osmolovskaya E. Aquificales. Chichester: Encyclopedia of Life Sciences (ELS). John Wiley & Sons, Ltd; 2008. [Google Scholar]
- Aravind L, Tatusov RL, Wolf YI, Walker DR, Koonin EV. Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet. 1998;14(11):442–444. doi: 10.1016/s0168-9525(98)01553-4. [DOI] [PubMed] [Google Scholar]
- Eder W, Huber R. New isolates and physiological properties of the Aquificales and description of Thermocrinis albus sp. nov. Extremophiles. 2002;6(4):309–318. doi: 10.1007/s00792-001-0259-y. [DOI] [PubMed] [Google Scholar]
- Santos H, Lamosa P, Borges N, Goncalves L, Pais T, Rodrigues M. Extremophiles Handbook: Organic Compatible Solutes of Prokaryotes that Thrive in Hot Environments: The Importance of Ionic Compounds for Thermostabilization. Springer Japan; 2011. [ http://dx.doi.org/10.1007/978-4-431-53898-1_23] [Google Scholar]
- Scholz S, Sonnenbichler J, Schäfer W, Hensel R. Di-myo-inositol-1,1’-phosphate: a new inositol phosphate isolated from Pyrococcus woesei. FEBS Lett. 1992;306(2–3):239–242. doi: 10.1016/0014-5793(92)81008-a. [DOI] [PubMed] [Google Scholar]
- Jorge CD, Lamosa P, Santos H. Alpha-D-mannopyranosyl-(1-2)-alpha-D-glucopyranosyl-(1-2)-glycer ate in the thermophilic bacterium Petrotoga miotherma–structure, cellular content and function. FEBS J. 2007;274(12):3120–3127. doi: 10.1111/j.1742-4658.2007.05844.x. [DOI] [PubMed] [Google Scholar]
- Brosnan CA, Voinnet O. The long and the short of noncoding RNAs. Curr Opin Cell Biol. 2009;21(3):416–425. doi: 10.1016/j.ceb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Collins LJ. The RNA infrastructure: an introduction to ncRNA networks. Adv Exp Med Biol. 2011;722:1–19. doi: 10.1007/978-1-4614-0332-6_1. [DOI] [PubMed] [Google Scholar]
- de la Fuente M, Valera S, Martínez-Guitarte JL. ncRNAs and thermoregulation: a view in prokaryotes and eukaryotes. FEBS Lett. 2012;586(23):4061–4069. doi: 10.1016/j.febslet.2012.10.018. [DOI] [PubMed] [Google Scholar]
- Marz M, Stadler PF. RNA interactions. Adv Exp Med Biol. 2011;722:20–38. doi: 10.1007/978-1-4614-0332-6_2. [DOI] [PubMed] [Google Scholar]
- Erdmann VA, Barciszewska MZ, Hochberg A, de Groot N, Barciszewski J. Regulatory RNAs. Cell Mol Life Sci. 2001;58(7):960–977. doi: 10.1007/PL00000913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R. CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip Rev RNA. 2013;4(3):267–278. doi: 10.1002/wrna.1159. [DOI] [PubMed] [Google Scholar]
- Macvanin M, Edgar R, Cui F, Trostel A, Zhurkin V, Adhya S. Noncoding RNAs binding to the nucleoid protein HU in Escherichia coli. J Bacteriol. 2012;194(22):6046–6055. doi: 10.1128/JB.00961-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier C, Boisset S, Romilly C, Masquida B, Fechter P, Geissmann T, Vandenesch F, Romby P. Staphylococcus aureus RNAIII binds to two distant regions of coa mRNA to arrest translation and promote mRNA degradation. PLoS Pathog. 2010;6(3):e1000809. doi: 10.1371/journal.ppat.1000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JB, Balasubramanian D, Vanderpool CK. Small RNA binding-site multiplicity involved in translational regulation of a polycistronic mRNA. Proc Natl Acad Sci U S A. 2012;109(40):2691–2698. doi: 10.1073/pnas.1207927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulle C, Yang Y, Marchiani M, Moore ER, Siefert JL, Aragno M, Jurtshuk P, Fox GE. Phylogenetic position of the genus Hydrogenobacter. Int J Syst Bacteriol. 1994;44(4):620–626. doi: 10.1099/00207713-44-4-620. [DOI] [PubMed] [Google Scholar]
- Brown JR, Doolittle WF. Root of the universal tree of life based on ancient aminoacyl-tRNA synthetase gene duplications. Proc Natl Acad Sci U S A. 1995;92(7):2441–2445. doi: 10.1073/pnas.92.7.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchetta M, Gribaldo S, Sanangelantoni A, Cammarano P. Phylogenetic depth of the bacterial genera Aquifex and Thermotoga inferred from analysis of ribosomal protein, elongation factor, and RNA polymerase subunit sequences. J Mol Evol. 2000;50(4):366–380. doi: 10.1007/s002399910040. [DOI] [PubMed] [Google Scholar]
- Olsen GJ, Woese CR, Overbeek R. The winds of (evolutionary) change: breathing new life into microbiology. J Bacteriol. 1994;176:1–6. doi: 10.1128/jb.176.1.1-6.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf SL, Palmer JD, Doolittle WF. The root of the universal tree and the origin of eukaryotes based on elongation factor phylogeny. Proc Natl Acad Sci U S A. 1996;93(15):7749–7754. doi: 10.1073/pnas.93.15.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmur JG, Wong DM, Ortiz B, Tong J, Reichert F, Gelfand DH. Cloning, sequencing, and expression of RecA proteins from three distantly related thermophilic eubacteria. J Biol Chem. 1994;269(41):25928–25935. [PubMed] [Google Scholar]
- Oshima K, Chiba Y, Igarashi Y, Arai H, Ishii M. Phylogenetic position of Aquificales based on the whole genome sequences of six Aquificales species. Int J Evol Biol. 2012;2012:859264–859264. doi: 10.1155/2012/859264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieseke N, Lechner M, Ludwig M, Marz M. In: Bioinformatics Research and Applications, Volume 7875 of Lecture Notes in Computer Science. Cai Z, Eulenstein O, Janies D, Schwartz D, editor. Springer Berlin Heidelberg; 2013. POMAGO: Multiple Genome-Wide Alignment Tool for Bacteria; pp. 249–260. [ http://dx.doi.org/10.1007/978-3-642-38036-5_25] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, Haussler D, Miller W. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14(4):708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose D, Hertel J, Reiche K, Stadler PF, Hackermüller J. NcDNAlign: plausible multiple alignments of non-protein-coding genomic sequences. Genomics. 2008;92:65–74. doi: 10.1016/j.ygeno.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Nakamura S, Inagaki F, Takai K, Shirai N, Sako Y. Hydrogenivirga caldilitoris gen. nov., sp. nov., a novel extremely thermophilic, hydrogen- and sulfur-oxidizing bacterium from a coastal hydrothermal field. Int J Syst Evol Microbiol. 2004;54(Pt 6):2079–2084. doi: 10.1099/ijs.0.03031-0. [DOI] [PubMed] [Google Scholar]
- Nunoura T, Miyazaki M, Suzuki Y, Takai K, Horikoshi K. Hydrogenivirga okinawensis sp. nov., a thermophilic sulfur-oxidizing chemolithoautotroph isolated from a deep-sea hydrothermal field, Southern Okinawa Trough. Int J Syst Evol Microbiol. 2008;58(Pt 3):676–681. doi: 10.1099/ijs.0.64615-0. [DOI] [PubMed] [Google Scholar]
- Gupta RS, Lali R. Molecular signatures for the phylum Aquificae and its different clades: proposal for division of the phylum Aquificae into the emended order Aquificales, containing the families Aquificaceae and Hydrogenothermaceae, and a new order Desulfurobacteriales ord. nov., containing the family Desulfurobacteriaceae. Antonie Van Leeuwenhoek. 2013;104(3):349–368. doi: 10.1007/s10482-013-9957-6. [DOI] [PubMed] [Google Scholar]
- Lechner M. Detection of Orthologs in large-scale analysis. Master’s thesis, University of Leipzig. 2009.
- Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:t24. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polard P, Prère MF, Chandler M, Fayet O. Programmed translational frameshifting and initiation at an AUU codon in gene expression of bacterial insertion sequence IS911. J Mol Biol. 1991;222(3):465–477. doi: 10.1016/0022-2836(91)90490-w. [DOI] [PubMed] [Google Scholar]
- Spiers AJ, Bergquist PL. Expression and regulation of the RepA protein of the RepFIB replicon from plasmid P307. J Bacteriol. 1992;174(23):7533–7541. doi: 10.1128/jb.174.23.7533-7541.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns N, Masters M. Expression of the Escherichia coli pcnB gene is translationally limited using an inefficient start codon: a second chromosomal example of translation initiated at AUU. Mol Microbiol. 2002;44(5):1287–98. doi: 10.1046/j.1365-2958.2002.02945.x. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara J, Yachie N, Sekine Y, Soma A, Matsui M, Tomita M, Kanai A. SPLITS: a new program for predicting split and intron-containing tRNA genes at the genome level. In Silico Biol. 2006;6(5):411–418. [PubMed] [Google Scholar]
- Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilimulati Y, Marz M, Stadler PF, Hofacker IL. Bcheck: a wrapper tool for detecting RNase P RNA genes. BMC Genomics. 2010;11:432–440. doi: 10.1186/1471-2164-11-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209–209. doi: 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35(Web Server issue):52–57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40(Database issue):71–75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, Bateman A. Rfam: updates to the RNA families database. Nucleic Acids Res. 2009;37(Database issue):136–140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuyts J, Perrière G, Van De Peer Y. The European ribosomal RNA database. Nucleic Acids Res. 2004;32(Database issue):101–103. doi: 10.1093/nar/gkh065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. RNABOB: a program to search for RNA secondary structure motifs in sequence databases. 1992–1996. [ http://selab.janelia.org/software.html]
- Hofacker IL, Fekete M, Stadler PF. Secondary structure prediction for aligned RNA sequences. J Mol Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 1994;125:167–188. [Google Scholar]
- Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008;9:474–474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto W, Will S, Backofen R. Proceedings of German Conference on Bioinformatics (GCB’2008), Volume P-136 of Lecture Notes in Informatics (LNI) Gesellschaft für Informatik (GI); 2008. Structure local multiple alignment of RNA; pp. 178–188. [ http://dblp.uni-trier.de/db/conf/gcb/gcb2008.html#OttoWB08] [Google Scholar]
- Griffiths-Jones S. RALEE—RNA ALignment editor in Emacs. Bioinformatics. 2005;21:257–259. doi: 10.1093/bioinformatics/bth489. [DOI] [PubMed] [Google Scholar]
- Federhen S. The NCBI Taxonomy database. Nucleic Acids Res. 2012;40(Database issue):D136—D143. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; 2011. Phylogenetic analysis of protein sequence data using the Randomized Axelerated Maximum Likelihood (RAXML) Program. Chapter 19 [ http://dx.doi.org/10.1002/0471142727.mb1911s96] [DOI] [PubMed] [Google Scholar]
- Subramanian AR, Kaufmann M, Morgenstern B. DIALIGN-TX: greedy and progressive approaches for segment-based multiple sequence alignment. Algorithms Mol Biol. 2008;3:6–6. doi: 10.1186/1748-7188-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SQ, Gascuel O. Phylogenetic mixture models for proteins. Mol Biol Evol. 2008;25(7):1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Liu K, Raghavan S, Nelesen S, Linder CR, Warnow T. Rapid and accurate large-scale coestimation of sequence alignments and phylogenetic trees. Science. 2009;324(5934):1561–1564. doi: 10.1126/science.1171243. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113–113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, Stadler PF, Vogel J. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464(7286):250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- Beckmann BM, Burenina OY, Hoch PG, Kubareva EA, Sharma CM, Hartmann RK. In vivo and in vitro analysis of 6S RNA-templated short transcripts in Bacillus subtilis. RNA Biol. 2011;8(5):839–849. doi: 10.4161/rna.8.5.16151. [DOI] [PubMed] [Google Scholar]
- Huber R, Wilharm T, Huber D, Trincone A, Burggraf S, Konig H, Rachel R, Rockinger I, Fricke H, Stetter KO. Aquifex pyrophilus gen.nov., sp.nov., represents a novel group of marine hyperthermophilic hydrogen-oxidizing bacteria. System Appl Microbiol. 1992;15:340–351. [Google Scholar]
- Mattatall NR, Sanderson KE. Salmonella typhimurium LT2 possesses three distinct 23S rRNA intervening sequences. J Bacteriol. 1996;178(8):2272–2278. doi: 10.1128/jb.178.8.2272-2278.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol JW, Helt GA, Blanchard SG, Raja A, Loraine AE. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25(20):2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Graham DE, Overbeek R, Snead MA, Keller M, Aujay M, Huber R, Feldman RA, Short JM, Olsen GJ, Swanson RV. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392(6674):353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermüller J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol. 2009;5(9):e1000502. doi: 10.1371/journal.pcbi.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann BM, Grünweller A, Weber MH, Hartmann RK. Northern blot detection of endogenous small RNAs (approximately 14 nt) in bacterial total RNA extracts. Nucleic Acids Res. 2010;38(14):e147. doi: 10.1093/nar/gkq437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI: Genome information by organism. [ http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi] (accessed 2013-08-05)
- Reysenbach AL, Hamamura N, Podar M, Griffiths E, Ferreira S, Hochstein R, Heidelberg J, Johnson J, Mead D, Pohorille A, Sarmiento M, Schweighofer K, Seshadri R, Voytek MA. Complete and draft genome sequences of six members of the Aquificales. J Bacteriol. 2009;191(6):1992–1993. doi: 10.1128/JB.01645-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci U S A. 2005;102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuratani M, Ishii R, Bessho Y, Fukunaga R, Sengoku T, Shirouzu M, Sekine S, Yokoyama S. Crystal structure of tRNA adenosine deaminase (TadA) from Aquifex aeolicus. J Biol Chem. 2005;280(16):16002–16008. doi: 10.1074/jbc.M414541200. [DOI] [PubMed] [Google Scholar]
- Kuratani M, Yoshikawa Y, Bessho Y, Higashijima K, Ishii T, Shibata R, Takahashi S, Yutani K, Yokoyama S. Structural basis of the initial binding of tRNA(Ile) lysidine synthetase TilS with ATP and L-lysine. Structure. 2007;15(12):1642–1653. doi: 10.1016/j.str.2007.09.020. [DOI] [PubMed] [Google Scholar]
- Soma A, Ikeuchi Y, Kanemasa S, Kobayashi K, Ogasawara N, Ote T, Kato J, Watanabe K, Sekine Y, Suzuki T. An RNA-modifying enzyme that governs both the codon and amino acid specificities of isoleucine tRNA. Mol Cell. 2003;12(3):689–698. doi: 10.1016/s1097-2765(03)00346-0. [DOI] [PubMed] [Google Scholar]
- Muramatsu T, Nishikawa K, Nemoto F, Kuchino Y, Nishimura S, Miyazawa T, Yokoyama S. Codon and amino-acid specificities of a transfer RNA are both converted by a single post-transcriptional modification. Nature. 1988;336(6195):179–181. doi: 10.1038/336179a0. [DOI] [PubMed] [Google Scholar]
- Muramatsu T, Yokoyama S, Horie N, Matsuda A, Ueda T, Yamaizumi Z, Kuchino Y, Nishimura S, Miyazawa T. A novel lysine-substituted nucleoside in the first position of the anticodon of minor isoleucine tRNA from Escherichia coli. J Biol Chem. 1988;263(19):9261–9267. doi: 10.1351/pac198961030573. [DOI] [PubMed] [Google Scholar]
- Voorhees RM, Mandal D, Neubauer C, Köhrer C, RajBhandary UL, Ramakrishnan V. The structural basis for specific decoding of AUA by isoleucine tRNA on the ribosome. Nat Struct Mol Biol. 2013;20(5):641–643. doi: 10.1038/nsmb.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalkowski M, Teune JH, Steger G, Hartmann RK, Willkomm DK. Thermostable RNase P RNAs lacking P18 identified in the Aquificales. RNA. 2006;12(11):1915–1921. doi: 10.1261/rna.242806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willkomm DK, Minnerup J, Hüttenhofer A, Hartmann RK. Experimental RNomics in Aquifex aeolicus: identification of small non-coding RNAs and the putative 6S RNA homolog. Nucleic Acids Res. 2005;33(6):1949–1960. doi: 10.1093/nar/gki334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalkowski M, Willkomm DK. Hartmann RK: 5’-end maturation of tRNA in Aquifex aeolicus. Biol Chem. 2008;389(4):395–403. doi: 10.1515/BC.2008.042. [DOI] [PubMed] [Google Scholar]
- Marszalkowski M, Willkomm DK, Hartmann RK. Structural basis of a ribozyme’s thermostability: P1-L9 interdomain interaction in RNase P RNA. RNA. 2008;14:127–133. doi: 10.1261/rna.762508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotochaud AE, Wassarman KM. A highly conserved 6S RNA structure is required for regulation of transcription. Nat Struct Mol Biol. 2005;12(4):313–319. doi: 10.1038/nsmb917. [DOI] [PubMed] [Google Scholar]
- Barrick JE, Sudarsan N, Weinberg Z, Ruzzo WL. Breaker RR: 6S RNA is a widespread regulator of eubacterial RNA polymerase that resembles an open promoter. RNA. 2005;11(5):774–784. doi: 10.1261/rna.7286705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann BM, Hoch PG, Marz M, Willkomm DK, Salas M, Hartmann RK. A pRNA-induced structural rearrangement triggers 6S-1 RNA release from RNA polymerase in Bacillus subtilis. EMBO J. 2012;31(7):1727–1738. doi: 10.1038/emboj.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman KM, Saecker RM. Synthesis-mediated release of a small RNA inhibitor of RNA polymerase. Science. 2006;314(5805):1601–1603. doi: 10.1126/science.1134830. [DOI] [PubMed] [Google Scholar]
- Neusser T, Gildehaus N, Wurm R, Wagner R. Studies on the expression of 6S RNA from E. coli: involvement of regulators important for stress and growth adaptation. Biol Chem. 2008;389(3):285–297. doi: 10.1515/BC.2008.023. [DOI] [PubMed] [Google Scholar]
- Gildehaus N, Neusser T, Wurm R, Wagner R. Studies on the function of the riboregulator 6S RNA from E. coli: RNA polymerase binding, inhibition of in vitro transcription and synthesis of RNA-directed de novo transcripts. Nucleic Acids Res. 2007;35(6):1885–1896. doi: 10.1093/nar/gkm085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karzai AW, Roche ED, Sauer RT. The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nat Struct Biol. 2000;7(6):449–455. doi: 10.1038/75843. [DOI] [PubMed] [Google Scholar]
- Tanner DR, Dewey JD, Miller MR, Buskirk AR. Genetic analysis of the structure and function of transfer messenger RNA pseudoknot 1. J Biol Chem. 2006;281(15):10561–10566. doi: 10.1074/jbc.M600167200. [DOI] [PubMed] [Google Scholar]
- Zwieb C, Gorodkin J, Knudsen B, Burks J, Wower J. tmRDB (tmRNA database) Nucleic Acids Res. 2003;31:446–447. doi: 10.1093/nar/gkg019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- Maaty WS, Ortmann AC, Dlakic̀ M, Schulstad K, Hilmer JK, Liepold L, Weidenheft B, Khayat R, Douglas T, Young MJ, Bothner B. Characterization of the archaeal thermophile Sulfolobus turreted icosahedral virus validates an evolutionary link among double-stranded DNA viruses from all domains of life. J Virol. 2006;80(15):7625–7635. doi: 10.1128/JVI.00522-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YW, Yu L, Zhang YP. Long-branch attraction artifact in phylogenetic reconstruction. Yi Chuan. 2007;29(6):659–667. doi: 10.1360/yc-007-0659. [DOI] [PubMed] [Google Scholar]