

Abstract
Mevalonate diphosphate decarboxylase (MDD) catalyzes the final step of the mevalonate pathway, the Mg++-ATP dependent decarboxylation of mevalonate 5-diphosphate (MVAPP), producing isopentenyl diphosphate (IPP). Synthesis of IPP, an isoprenoid precursor molecule that is a critical intermediate in peptidoglycan and polyisoprenoid biosynthesis, is essential in Gram-positive bacteria (e.g. Staphylococcus, Streptococcus and Enterococcus spp.) and thus the enzymes of the mevalonate pathway are ideal antimicrobial targets. MDD belongs to the GHMP superfamily of small molecule (i.e. metabolite) kinases that have been extensively studied for the past 50 years, yet the crystallization of GHMP kinase ternary complexes has proven difficult. To further our understanding of the catalytic mechanism of GHMP kinases with the purpose of developing broad spectrum antimicrobial agents that target the substrate and nucleotide binding sites, we report the crystal structures of wild-type and mutant (S192A and D283A) ternary complexes of Staphylococcus epidermidis MDD. Comparison of apo-, MVAPP-bound and ternary complexed wild-type MDD provides structural information on the mode of substrate binding and the catalytic mechanism. Structural characterization of ternary complexes of catalytically deficient MDD S192A and D283A (decreased kcat of 103-fold and 105-fold, respectively) provides insight into MDD function. The carboxylate side chain of invariant Asp283 functions as a catalytic base and is essential to the proper orientation of the MVAPP C3-hydroxyl group within the active site funnel. Several MDD amino acids within the conserved phosphate binding loop (‘P-loop’) provide key interactions, stabilizing the nucleotide triphosphoryl moiety. The crystal structures presented here provide a useful foundation for structure-based drug design.
Synthesis of isopentenyl 5-diphosphate (IPP1) is critical in eubacteria, where it serves as an essential intermediate in the synthesis of diverse polyisoprenoid compounds. Whereas Gram-negative bacteria utilize the methylerythritol phosphate pathway for the creation of IPP (1), certain species of pathogenic Gram-positive organisms (such as Staphylococcus aureus and Staphylococcus epidermidis) utilize the mevalonate pathway (2). In total, three molecules of acetyl-CoA are used to produce one molecule of IPP via the mevalonate pathway through six enzymatic steps. Within this sequence, there are three consecutive ATP-dependent reactions needed to produce IPP from mevalonic acid in a manner that is essential for bacterial viability (2). The last of these reactions is catalyzed by the enzyme mevalonate diphosphate decarboxylase (MDD). MDD (EC 4.1.1.33) catalyzes the divalent cation-dependent (3), irreversible decarboxylation of mevalonate 5-diphosphate (MVAPP) into IPP, along with the concurrent hydrolysis of ATP to ADP and formation of CO2 and inorganic phosphate (4). The reaction proceeds as follows:
The mevalonate pathway is operative in higher order eukaryotes, plants, some protists and many Gram-positive prokaryotes, and so the extensive characterization of MDD from yeast (4, 5), avian (6), and mammalian sources (6-8) has elucidated many details on the MDD mechanism2. Deprotonation of the acceptor MVAPP C3-hydroxyl occurs via the general base Asp283, which leads to a phosphorylation to form the putative metastable 3-phospho-MVAPP adduct (9). Creation of a carbocation intermediate at the C3 position results from the departure of the newly transferred phosphoryl. Decarboxylation proceeds after the formation of this intermediate via the interaction of Arg144 with the C1 carboxylate of MVAPP, and thereby yields the product, IPP. Mutagenesis studies on both of these side chains revealed upwards of a 103-fold decrease in catalytic activity, which confirmed the function of these residues’ side chains in catalysis (5, 10).
MDD is a member of the GHMP kinase (galactokinase; homoserine kinase; mevalonate kinase; phosphomevalonate kinase) family (11). These enzymes are characterized by a cone-shaped fold with a deep active site cleft that houses the previously mentioned catalytic Asp and Arg residues, while the β-grasp-like N-terminal region packs against the C-terminal five α-helical bundle (12). Although previous work resulted in the determination of a number of different MDD crystal structures (10-12), none of these studies succeeded in crystallizing the enzyme in a metabolite or ligand-bound form. More recently, however, our group reported the high-resolution co-crystal structures of S. epidermidis MDD bound to the inhibitory substrate analogs diphosphoglycolyl proline (DPGP) and 6-fluoromevalonate diphosphate (FMVAPP) (13). Both FMVAPP and DPGP are competitive inhibitors, though their Ki values (50 nM and 4.3 μM, respectively) differ considerably. Comparison of their respective crystal structures when bound to MDD provided a physical explanation for this difference (13). Inspection of these structures also identified the side chain of invariant Ser192 as making potential contributions to catalysis. Indeed, loss of Ser192 resulted in a nearly 103-fold decrease in kcat when compared to wild-type MDD.
Unfortunately, one limitation of these recent structures was that they lacked a bound nucleotide within the MDD active site. This restricted any mechanistic conclusions to those features which concerned the phosphoryl acceptor, MVAPP. Thus, to characterize the MDD structure, active site and catalytic mechanism more completely, we report here a series of seven additional high-resolution X-ray co-crystal structures of S. epidermidis MDD bound to both inhibitory substrate analogs as well as to the nucleotide analog, ATPγS. Included in this cohort are ternary complexes of two previously characterized catalytically deficient MDD mutants, S192A (13) and D283A (5), bound to inhibitory substrate analogs as well as the nucleotide analog, ATPγS. Analysis of these mutant MDD ternary complexes, along with that of wild-type MDD bound to FMVAPP and ATPγS has revealed significant insight into the conformational changes that occur upon nucleotide binding, and thereby significantly augments our collection of structural “snapshots” of the reaction cycle.
With antibiotic resistant strains of gram-positive bacterial organisms becoming an increasingly serious healthcare issue (14, 15), focus on identification of novel classes of antimicrobial drugs is now of paramount importance. The apparently essential nature of the mevalonate pathway ATP-dependent kinases for bacterial cell viability strongly suggests that these enzymes are excellent targets for antimicrobial compounds (2). In this regard, it is worth noting that recent investigation of small molecule inhibitors that selectively target the nucleotide binding region within ATP-dependent enzymes has met with great success (16). Thus, we believe that development of selective inhibitors that target the substrate and nucleotide binding regions of bacterial MDDs will be greatly aided by the structure/function studies presented and discussed here.
Experimental Procedures
Materials
Unless specified, all chemical and biological products used in these studies were reagent grade materials purchased from Fisher Scientific or Sigma-Aldrich.
Substrates and Analogs
The synthesis of mevalonate 5-diphosphate (MVAPP) has been previously reported (17) and briefly summarized elsewhere (13). Preparation of 6-fluoromevalonate diphosphate (FMVAPP) employed the method described by Voynova et al. (10) and was also described previously (13). The competitive inhibitor diphosphoglycolylproline (DPGP) was synthesized by the strategy of Vlattas et al. (18) using methodology described by Krepkiy and Miziorko (9). ATPγS was purchased from Calbiochem.
Cloning, Overexpression and Purification of of MDD
Expression and purification of wild-type and S192A S. epidermidis MDD was described previously (13). An analogous expression vector for the Asp283→Ala mutant was constructed using standard molecular biology techniques (19). Expression and purification of this mutant enzyme was performed in an equivalent manner to the wild-type protein.
Crystallization
Crystallization of S. epidermidis MDD was described previously (13). Briefly, 1 μl of protein solution (5 mg/ml in 10 mM Tris-HCl (pH 7.5), 50 mM NaCl) was mixed with 1 μl of reservoir solution containing 0.25 M sodium formate and 16% (w/v) PEG 3350 that had been previously diluted in an equal volume of ddH2O, and equilibrated over 500 μl of reservoir solution. Crystals of apo-S192A MDD were obtained in an analogous manner. Co-crystallization of MDD (wild-type of mutant) with a molar excess (∼0.5 mM) of ligands (50mM MgCl2 for ternary co-crystallization) was achieved in a similar fashion. Co-crystallization through the substitution of magnesium formate in place of sodium formate for all ternary experiments was also performed in an attempt to obtain Mg++-bound MDD. All crystals were flash cooled in a cryoprotectant solution consisting of reservoir buffer with an additional 15% (v/v) glycerol. Ternary complexed co-crystals were flash cooled in a cryoprotectant containing an additional 50mM MgCl2.
Diffraction Data Collection, Structure Determination, Refinement and Analysis
Monochromatic X-ray diffraction data were collected from single crystals at 100 K using beamlines 22-ID and 22-BM of the Advanced Photon Source, Argonne National Laboratory (Table 1). Following data collection, individual reflections were indexed, integrated, merged and scaled using HKL2000 (20). Initial phase information was obtained for all MDD structures by maximum-likelihood molecular replacement using PHASER (21). Chain A of PDB entry 3QT5 (S. epidermidis MDD) was used as a search model. The single most highly scored solution for each molecular replacement search contained two copies of MDD in the asymmetric unit for crystals of the space group C2221 or C2 while crystals within the space group P212121 contained 8 copies of MDD within the asymmetric unit.
Table 1. Diffraction Data Collection and Structure Refinement Statistics3.
| Diffraction Dataa | ||||||||
| Protein | S192A MDD | S192A MDD | S192A MDD | MDD | D283A MDD | MDD | D283A MDD | |
| Ligands | DPGP | FMVAPP | FMVAPP | MevPP | MevPP | DPGP | ||
| ATPγS | ATPγS | ATPγS | ||||||
| Beamline | APS 22-ID | APS 22-ID | APS 22-ID | APS 22-ID | APS 22-ID | APS 22-BM | APS 22-BM | |
| Space Group | C2221 | C2221 | C2221 | C2 | P212121 | C2221 | C2221 | |
| Molecules in asu | 2 | 2 | 2 | 2 | 8 | 2 | 2 | |
| Unit-cell parameters | ||||||||
| a (Å) | 82.922 | 82.678 | 82.732 | 102.251 | 96.459 | 82.592 | 82.966 | |
| b (Å) | 102.273 | 101.733 | 103.14 | 82.46 | 99.445 | 101.32 | 102.05 | |
| c (Å) | 155.942 | 155.498 | 156.749 | 93.649 | 314.418 | 154.932 | 155.143 | |
| β (°) | 122.34 | |||||||
| Wavelength (Å) | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.0000 | |
| Resolution (Å) | 50.0 - 1.95 | 50.0 - 2.15 | 50.0 - 1.90 | 50.0 - 2.19 | 50.0 - 2.60 | 50.0 - 2.20 | 50.0 - 2.10 | |
| Completeness (%) | 100.0 (100.0) | 91.7 (82.0) | 99.7 (99.6) | 99.9 (99.4) | 98.2 (99.9) | 99.8 (99.1) | 95.0 (88.1) | |
| Total Reflections | 355,071 | 195,781 | 539,192 | 183,389 | 699,223 | 306,240 | 232,548 | |
| Unique Reflections | 48,719 | 33,395 | 52,862 | 33,632 | 91,579 | 33,287 | 36,836 | |
| Redundancy | 7.3x | 5.9x | 10.2x | 5.5x | 7.6x | 9.2x | 6.3x | |
| Rmerge (%)b | 8.3 (38.2) | 7.6 (32.0) | 8.0 (58.4) | 7.5 (49.7) | 20.3 (59.2) | 11.7 (55.7) | 9.3 (53.8) | |
| <I>/<σI> | 19.3 (5.83) | 15.7 (5.29) | 20.5 (3.2) | 19.7 (3.31) | 7.57 (3.75) | 19.8 (3.04) | 19.3 (3.58) | |
| PDB | 4DPX | 4DPY | 4DPU | 4DPT | 4DPW | 4DU7 | 4DU8 | |
| Refinement | ||||||||
| Rwork/Rfreec | 16.24/20.01 | 16.75/22.62 | 17.91/22.37 | 18.62/24.07 | 19.87/25.08 | 19.15/23.88 | 18.63/23.87 | |
| RMSD | ||||||||
| Bond Length (Å) | 0.007 | 0.008 | 0.007 | 0.010 | 0.009 | 0.007 | 0.007 | |
| Bond Angle (°) | 1.005 | 1.017 | 1.058 | 1.192 | 1.121 | 0.971 | 1.001 | |
| Ramachandran | ||||||||
| Favored (%) | 98.6 | 98.0 | 98.5 | 96.5 | 97.7 | 98.6 | 98.0 | |
| Allowed (%) | 1.4 | 2.0 | 1.4 | 3.3 | 2.3 | 1.4 | 2.0 | |
| Outliers (%) | 0.0 | 0.0 | 0.2 | 0.2 | 0.0 | 0.0 | 0.0 | |
| Number of Atoms | ||||||||
| Protein | 5102 | 5102 | 5125 | 5017 | 20,386 | 5055 | 5074 | |
| Solven | ||||||||
| t | 587 | 399 | 488 | 256 | 266 | 275 | 293 | |
| Ligand | N/A | 40 | 100 | 50 | 392 | 36 | 40 | |
| B factor (ÅΛ2) | ||||||||
| Protein | 23.2 | 26.5 | 27.9 | 37.7 | 50.9 | 35.0 | 27.7 | |
| Solven | ||||||||
| t | 30.6 | 29.3 | 31.8 | 37.6 | 51.0 | 34.6 | 28.9 | |
| Ligand | N/A | 23.5 | 27.9 | 55.3 | 45.7 | 30.9 | 29.3 | |
| Density Galleryd | ||||||||
|

|
|
|
|
|
Numbers in parentheses are for the highest-resolution shell.
Rmerge = ΣhΣi|Ii(h)-<I(h)>|/ΣhΣiIi(h), where Ii(h) is the ith measurement of reflection h and <I(h)> is a weighted mean of all measurements of h.
R = Σh|Fobs(h)-Fcalc(h)|/ Σh|Fobs|. Rcryst and Rfree were calculated from the working and test reflection sets, respectively. The test set constituted 5% of the total reflections not used in refinement.
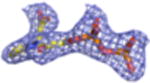
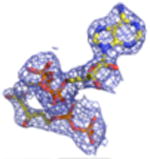
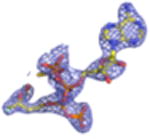
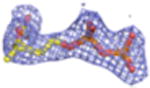
2Fo-Fc electron density maps (blue mesh contoured at 1.0σ) of modeled ligands.
Structure refinement was carried out using the protocols implemented in phenix.refine (22). One round of individual coordinate and isotropic atomic-displacement factor refinement were conducted, and the refined model was used to calculate both 2Fo-Fc and Fo-Fc difference maps. These maps were used to iteratively improve the model by manual building in Coot (23, 24), followed by additional coordinate and atom-displacement factor refinement. Ordered solvent molecules were added to all structures according to the default criteria of phenix.refine, and inspected manually using Coot prior to model completion. Additional information and refinement statistics for all seven structures are presented in Table 1.
Regions of poor electron density prevented the complete modeling of each polypeptide in the asymmetric unit within several of the structures described here. Typically these regions of low map quality were between residues 185-191. Further details regarding the residues that could not be modeled accurately may be found in the corresponding PDB files.
Ligand Fitting
Models for each ligand were generated using the PRODRG server (25), and restraint files were generated using phenix.elbow (22). Inspection of the initial Fo-Fc maps described above revealed unmodeled contiguous density that corresponded to ordered ligands in the active site of all copies of MDD found within the asymmetric unit, with a single exception. Only a single active site within the co-crystal structure of wild-type MDD bound to FMVAPP and ATPγS was found to be occupied, thus the second copy of MDD within the asymmetric unit was modeled in the apo-conformation. Phenix.ligandfit (22) was subsequently used to fit and model an inhibitor, substrate and/or nucleotide molecule in each active site. Refinement of complexed MDD structures was carried out as described above, with the exception that constrained group occupancy refinement was used to estimate the fraction of ligand bound at each site independently (STable 1).
Miscellaneous
Multiple sequence alignments were carried out using CLUSTALW (26) and aligned with secondary structure elements using ESPRIPT (27). MDD sequences used in alignments, along with their respective GenBank accession numbers, were as follows: Staphylococcus epidermidis, 27467280; Staphylococcus aureus, 14246359; Legionella pneumophila subsp. pneumophila, 52842257; Streptococcus pyogenes, 5093120; Homo sapiens, 4505289; Saccharomyces cerevisiae, 1706682; Trypanosoma brucei, 149241992; Mus musculus, 13539580; Listeria monocytogenes, 217965923; Enterococcus faecalis, 315577315; Xenopus (Silurana) tropicalis, 39645379; Bos taurus, 115495513; Arabidopsis thaliana, 18410026. Three-dimensional structures were superimposed using the Local-Global Alignment method (LGA) (28). Representations of all structures were generated using PyMol (29). The coordinates for ligand AMPPNP were obtained from the Streptococcus pneumoniae PDB structure 3GON (30).
Results
The Co-crystal Structure of a Staphylococcus epidermidis MDD Ternary Complex Illustrates the Conserved Nature of the Nucleotide Binding Site.
We undertook co-crystallization trials of S. epidermidis enzyme in the presence of the competitive inhibitor FMVAPP and various nucleotide analogs (ATPγS, AMPPCP and AMPPNP) in order to identify and investigate the ATP binding site of MDD. Large, birefringent crystals were obtained only with the combination of FMVAPP and ATPγS, and diffracted synchrotron X-rays to 2.19 Å resolution (Table 1). Intriguingly, this crystal belonged to the space group C2, while both apo- and inhibitor-only bound S. epidermidis MDD crystals belonged to the space group C2221 (13). The structure of this crystal was solved by molecular replacement using a single copy of unliganded S. epidermidis MDD as a search model. Analysis of the highest scoring solution revealed two copies of MDD within the C2 asymmetric unit, as was previously described for the MDD crystals in C2221; however, the change to a lower symmetry lattice indicated that the crystal packing interactions had been altered by the presence of the nucleotide analog ATPγS. Following initial modeling of both MDD copies, analysis of the Fo-Fc difference maps indeed revealed two regions of strong, contiguous density housed within a single MDD active site cleft (SFig.1a). A single copy of both FMVAPP and ATPγS was modeled within this active site and refined to final occupancy values of 78% and 79%. Overall, the final structure is in good agreement with the observed diffraction data, as judged by Rwork and Rfree values of 18.6 and 24.1%, respectively (Table 1, STable 1, SFig. 2a).
Numerous amino acid backbone atoms and residue side chains (invariant across all species of MDD) are involved in the extensive interaction between MDD and ATPγS (Fig. 1a,b). Conserved amino acids Ser94 and Asn96 contribute three side chain hydrogen bonding pairs to the nucleotide purine ring, while the only non-conserved side chain interaction between MDD and ATPγS is found between the side chain of Glu69 and the ribose 2′ hydroxyl of the nucleotide. We recently documented a catalytic role for Ser192 (13) by demonstrating that a S192A mutant of MDD has a greater than 103-fold diminution in catalytic activity (kcat). Interestingly, this mutant did not affect the location or KM of substrate binding (near wild-type values). The side chain and backbone nitrogen of Ser192 are within appropriate hydrogen bonding distances to the α-phosphoryl of ATPγS (STable 2), which suggests that the catalytic roles of this amino acid include positioning the nucleotide triphosphoryl moiety. The remainder of the MDD-ATPγS interaction sites are located on the opposite side of the active site cleft from Ser192, and fall primarily within the consensus GHMP kinase phosphate binding loop (residues P-Xaaa-GLASSAA) (31-33).
Figure 1.
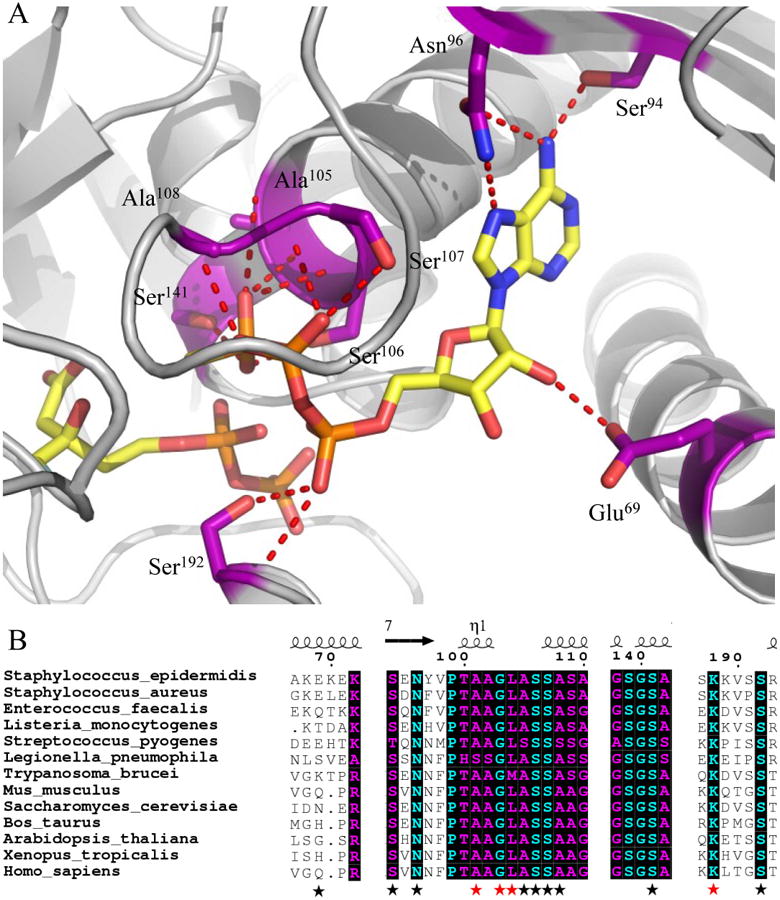
Co-crystal structure (2.19 Å) of MDD from Staphylococcus epidermidis bound to inhibitor FMVAPP and nucleotide analog ATPγS. (A) Co-crystal structure of S. epidermidis MDD in cartoon format (gray). Inhibitor FMVAPP and nucleotide analog ATPγS are represented as ball and stick (yellow). Active site side chains within interaction distance (2.5-3.4 Å) of ATPγS are depicted in ball and stick (purple) with red dashes. Hydrogen bonding distances can be found in Supplemental Table 3. (B) Limited structure-based sequence alignment of prokaryotic and eukaryotic MDD proteins colored according to residue conservation (cyan = absolute, purple = similar). Alignment was generated using ClustalW. Numbers above the sequences correspond to S. epidermidis MDD. Black stars below the sequences correspond to WT MDD amino acid side chains involved in ATPγS interaction, while red stars correspond to additional ATPγS interactions in the D283A mutant. Sequences from the following organisms were used in alignment: S. epidermidis; S.aureus; L. pneumophila; S. pyogenes; H. sapiens; S. cerevisiae; T. brucei; M. musculus; L. monocytogenes; E. faecalis; X. tropicalis; B. taurus; and A. thaliana.
Despite the fact that 50mM MgCl2 was used in the buffer during ternary wild-type MDD crystallization trials, no interpretable electron density was observed to suggest a stably coordinated Mg++ atom. Indeed, the distance between the C3-hydroxyl of FMVAPP and the γ-phosphoryl of ATPγS is nearly 5.5 Å. This relatively large distance may very well be due to the absence of a coordinated Mg++ ion which is expected to be required to detect a catalytically productive conformation for this complex.
Conformational Changes within MDD upon Nucleotide Binding
Even in the crystalline state, wild-type enzymes are known to be quite catalytically efficient (34). This typically precludes the simultaneous use of bona fide substrate (i.e. MVAPP) and nucleotide (i.e. ATP) compounds in co-crystallography studies. To circumvent this problem, our initial work described above employed both FMVAPP and ATPγS, which are inhibitory analogs of their MDD substrate and nucleotide targets, respectively. However, the use of these analogs may have contributed as well to the excessive distance between the y-phosphoryl of ATPγS and the C3-hydroxyl of FMVAPP. In order to discern whether binding of FMVAPP rather than MVAPP within the MDD active site resulted in any conformational changes, we obtained a co-crystal structure of the MVAPP-bound enzyme at 2.20 Å limiting resolution (Table 1). Nearly identical binding modes and active site contacts are observed in both MVAPP- (Fig. 2a) and FMVAPP-bound (13) wild-type MDD. Additionally, superposition of the main chain atoms from both MDD co-crystal structures reveals that 322/324 MVAPP-bound Cα atoms align within 5.0 Å distance with those of the FMVAPP-bound enzyme at an overall RMSD of 0.40 Å (STable 3). Thus, the nearly indistinguishable active site and tertiary structures in both co-crystal structures strongly suggest that binding of neither the MVAPP substrate nor the FMVAPP inhibitor results in conformational differences within MDD.
Figure 2.
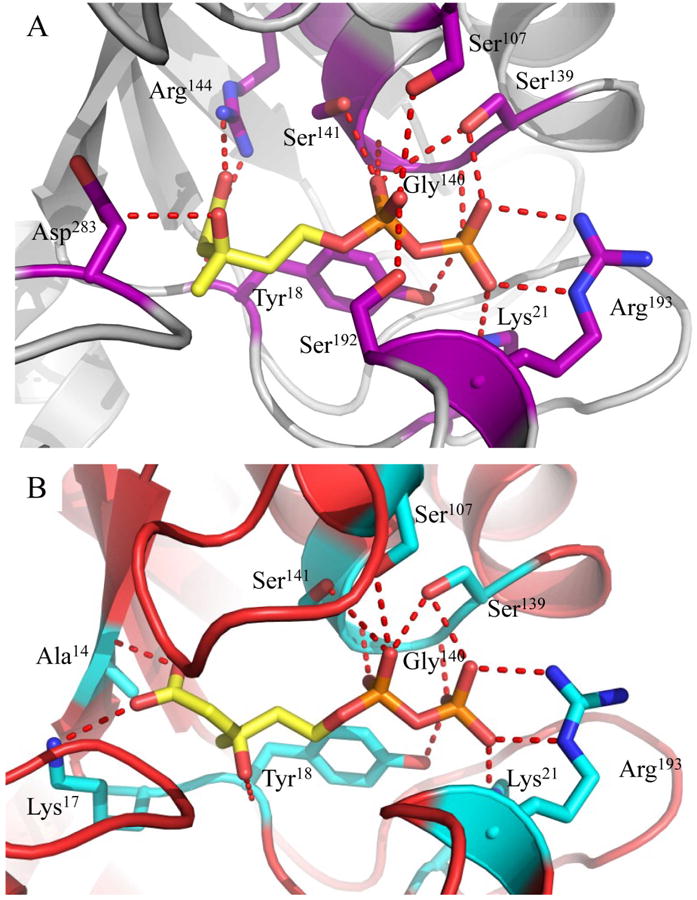
Co-crystal structures of WT and D283A MDD bound to the substrate MVAPP. (A) 2.20 Å co-crystal structure of S. epidermidis WT MDD in cartoon format (gray). Substrate MVAPP is represented as ball and stick (yellow). Active site side chains within interaction distance of MVAPP are depicted in ball and stick (purple). (B) 2.10 Å co-crystal structure of S. epidermidis D283A MDD in cartoon format (red). Substrate MVAPP is represented as ball and stick (yellow). Active site side chains within interaction distance of MVAPP are depicted in ball and stick (cyan). Hydrogen bonding distances can be found in Supplemental Table 4.
By contrast, structural superposition of apo-, MVAPP-bound and FMVAPP/ATPγS-bound MDD reveals several conformational changes within MDD that arise only upon nucleotide binding (Fig. 3). The conserved phosphate binding loop (‘P-loop’) clamps down around the triphosphoryl group of ATPγS, and culminates in a movement that spans ∼7.9 Å as measured from the Cα of Ala within each enzyme. Similar conformational changes within the ‘P-loop’ have been previously documented for other GHMP kinase family members upon nucleotide binding (35, 36). For example, within the ternary complex of homoserine kinase the ‘upper lip’ (residues 181-189) moves ∼3.5 Å in order to interact with the P-phosphoryl of ATP, while Arg187 swings around 180° and entraps homoserine within the active site cleft (37, 38). Arg within S. epidermidis MDD accomplishes a similar effect, and locks MVAPP into the active site funnel (13). Separately, within the rat mevalonate kinase-ATP binary complex, the active site was deemed too large to snugly fit the mevalonic acid substrate; this prompted the authors to suggest that side chain reorientations and loop movements would likely need to occur in a ternary complex (39). Indeed, an additional 2.0 Å movement within helix 1 (residues 69-83) further constricts the active site cleft upon formation of the MDD ternary complex presented here.
Figure 3.
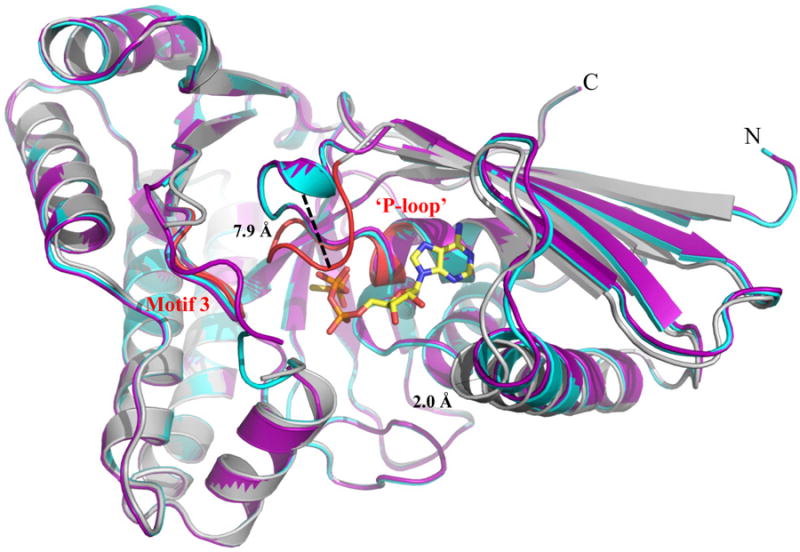
Structural alignment of WT MDD crystal structures. Cartoon ribbon diagram of the structural alignment of apo-MDD (purple), MDD bound to the substrate MVAPP (cyan) and MDD bound to the inhibitor FMVAPP and nucleotide analog ATPγS (gray). Only ATPγS is shown for clarity in ball and stick format (yellow). Conserved GHMP kinase family motifs 2 (‘P-loop’) and 3 are colored red within the ternary MDD co-crystal structure. A 7.9 Å shift within the ‘P-loop’, as measured from the Cα of Ala110 on both the apo-MDD and ternary MDD crystal structures, is highlighted for clarity. RMSD values for the structural alignment of all three structures can be found in Supplemental Table 1.
With these conformational changes now documented, superpositioning of the apo-, MVAPP-bound and ternary S. epidermidis MDD crystal structures can be used to provide informative visual cues into the sequential binding of the MVAPP acceptor and ATP donor substrates into the enzyme active site (Fig. 4). To begin, the MVAPP substrate must first enter the unoccupied active site. MVAPP binds in the interior-most region of this funnel-shaped cavity, in close proximity to the catalytic residues Arg144 and Asp283 (13). No apparent structural changes are required to allow or result from acceptor binding. Next, the phosphoryl donor ATP binds at the active site periphery, atop of the already positioned MVAPP. Upon assuming this ternary state, however, the MDD ‘P-loop’ must clamp down upon the nucleotide triphosphoryl group. This rearrangement allows for the phosphoryl transfer and subsequent decarboxylation to proceed, at which time the structural changes can reverse to allow release of the product ADP and IPP.
Figure 4.
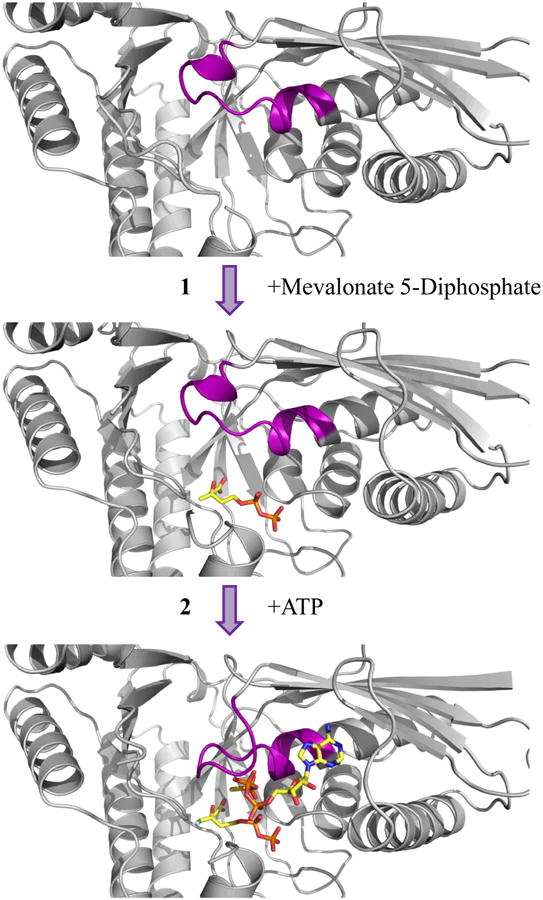
Sequential binding of substrate and nucleotide within the active site of MDD. Top, Cartoon ribbon diagram of apo-MDD (gray). Middle, Cartoon ribbon diagram of MDD (gray) in complex with the substrate MVAPP (ball and stick, yellow). Bottom, Cartoon ribbon diagram of MDD (gray) in complex with the inhibitor FMVAPP and nucleotide analog ATPγS (ball and stick, yellow). Within all three panels, the conserved ‘P-loop’ sequence (PTXaaaGLASSAS) is colored purple. The conformational change within the ‘P-loop’ occurs after both substrate and nucleotide have bound the enzyme active site.
Structural Basis for Catalytic Deficiencies in MDD Mutants
Previous structure/function studies on various MDDs have identified several residues that serve crucial roles in enzyme catalysis (5, 9, 10, 13). Among these, kinetic evaluations of mutants of two such residues (Ser192 and Asp283) are especially intriguing. The S192A mutant exhibits a thousand-fold decrease in catalytic rate (kcat = 0.0058 s-1), despite its near wild-type KM for MVAPP (5.0 μM) and its slightly decreased KM for ATP (70 μM) (13). Curiously, however, the active site side chain interactions from the previously described S192A MDD-FMVAPP complex agree well with those obtained for the wild-type enzyme (13). To characterize the S192A MDD mutant more fully, we obtained ternary co-crystals of FMVAPP/ATPγS bound to S192A MDD that diffracted X-rays to 1.90 Å resolution (Table 1). Interestingly, this co-crystal belongs to the space group C2221 and has unit cell dimensions that are nearly identical to either apo- or FMVAPP-bound MDD (13). Following structure solution and initial refinement, inspection of Fo-Fc difference maps revealed regions of contiguous, well-defined electron density visible in both enzyme active sites (SFig. 1b). This allowed modeling of both FMVAPP and ATPγS at high occupancy (>70%) prior to final refinement to Rwork and Rfree values of 17.9 and 22.4%, respectively. Several active site side chain and backbone interactions (Fig. 5a) are present that compare favorably with the WT ternary complex. However, the absence of Ser192 appears to adversely affect the localization of the triphosphoryl group of ATPγS. Most significantly, the ‘P-loop’ remains locked in the apo-conformation, as superposition of this ternary structure with apo-S192A MDD show that 330/330 Cα atoms align within 5.0 Å with an RMSD of 0.49 Å (STable 3). Thus, even though both substrate analogs are present, a lack of conformational change in the ‘P-loop’ region accounts for the fact that these crystals maintain the same space group as both the apo- and FMVAPP-bound forms (13).
Figure 5.
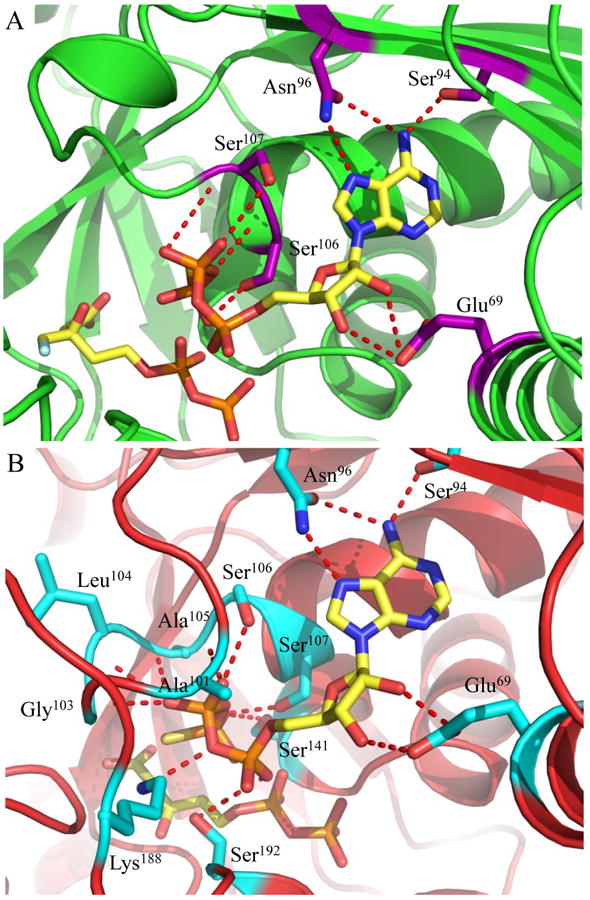
Active sites of ternary co-crystal structures from mutant forms of MDD. (A) 1.90 Å co-crystal structure of S. epidermidis S192A MDD in cartoon format (green). Inhibitor FMVAPP and nucleotide analog ATPγS are represented as ball and stick (yellow). Active site side chains within interaction distance of ATPγS are depicted in ball and stick (purple). (B) 2.60 Å co-crystal structure of S. epidermidis D283A MDD in cartoon format (red). Substrate MVAPP and nucleotide analog ATPγS are represented as ball and stick (yellow). Active site side chains within interaction distance of ATPγS are depicted in ball and stick (cyan). Hydrogen bonding distances can be found in Supplemental Table 3.
Studies on the catalytic base Asp283 revealed that a D283A mutant in Sacchromyces cerevisae MDD displays a 105-fold decrease in catalytic rate ( kcat=1.64 × 10−5 s-1) and a three fold inflation in the KM for MVAPP (397 μM), but a KM for ATP could not be directly determined due to the low activity of this mutant at subsaturating ATP levels, as well as a low intrinsic ATPase activity (5). To structurally examine the ATP binding behavior this mutant, we co-crystallized the D283A variant of S. epidermidis MDD as a ternary complex with MVAPP and ATPγS. These crystals diffracted X-rays to 2.60 Å limiting resolution, belonged to the space group P212121, and had vastly larger cell dimensions than any of those so far obtained for S. epidermidis MDD (Table 1). Following structure solution and initial refinement, contiguous regions of high-quality density were visible in each MDD active site within all 8 copies in the asymmetric unit, upon inspection of the Fo-Fc difference maps (SFig. 1c). This allowed modeling of both MVAPP and ATPγS at high occupancy (>88%) prior to final refinement to Rwork and Rfree values of 19.9 and 25.1%, respectively.
The D283A ternary complex active site reveals numerous additional MDD-ATPγS interactions (Fig. 5b). In particular, several novel backbone atom interactions are present within the ‘P-loop’ (Ala101, Gly103 and Leu104) while the loop region connecting beta sheet 9 and helix 5 (residues 182-192) has swung into a position that allows conserved Lys188 to interact with the γ-phosphoryl of ATPγS. Furthermore, the acceptor substrate binding site appears to be the same as wild-type within the D283A mutant, with the exception of a notable 120° rotation of the carboxylate and C3-hydroxyl groups of MVAPP (compare Fig. 2a vs. 2b). The absence of Asp283, which has largely been confirmed as the general base responsible for deprotonation (5) and is believed to aid in the orientation of the MVAPP C3-hydroxyl (9), provides a reasonable explanation for this misorientation.
Superposition of all three ternary MDD complexes sheds light on the structural alterations and mechanistic impact key mutations have on MDD catalysis (Fig. 6a). Conformational changes within the ‘P-loop’ appear very similar for both wild-type and D283A MDD, while the loop region in S192A appears unchanged. Indeed, both loops reposition by ∼8.1 Å (as measured from wild-type or D283A Ala101 Cα to S192A Ala101 Cα) to enclose the triphosphoryl moiety of ATPγS. Large scale variances in the position of the beta sheet 9 and helix 5 connecting loop are also seen, as a ∼9.0 Å shift away from the active site (as measured from D283A Gln186 Cα to S192A Gln186 Cα) is observed for the S192A complex relative to the D283A complex. Analysis of the active site funnel dimensions (Fig. 6b-d) further corroborates the functional studies in site directed mutants (distances were measured from equivalent amino acid Cα atoms of each MDD active site within PyMol (40)). The wild-type funnel area measures ∼145 Å2, whereas the S192A funnel is over two-fold larger at ∼300 Å2. The large S192A funnel size, resulting from the lack of ‘P-loop’ movement and translation of the sheet9/helix5 loop away from the active site, accounts for the significantly reduced kcat of S192A MDD. Separately, the D283A funnel is ∼50% smaller than wild-type at ∼99 Å2, and is primarily due to novel movements of the sheet9/helix5 loop. Thus, it seems that the apparently tighter binding of ATP in the D283A mutant can be rationalized by both increased enzyme surface contacts as well as the more occluded state of the active site funnel that follows nucleotide binding.
Figure 6.
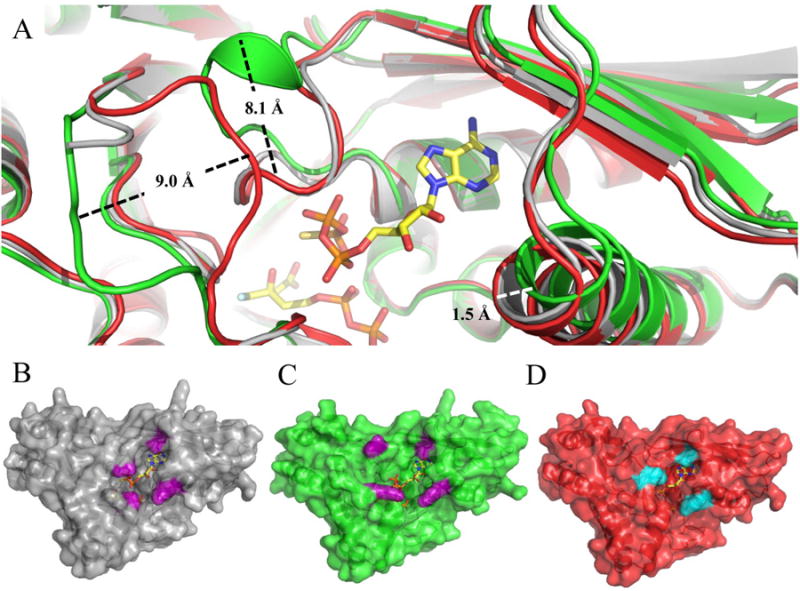
Structural alignment of ternary wild type and mutant MDD co-crystal structures. (A) Cartoon ribbon diagram of the structural alignment of WT MDD (gray) in complex with the inhibitor FMVAPP and nucleotide analog ATPγS (ball and stick, yellow), S192A MDD (green) in complex with the inhibitor FMVAPP and nucleotide analog ATPγS (not shown for clarity) and D283A MDD (red) in complex with the substrate MVAPP and nucleotide analog ATPγS (not shown for clarity). Measured conformational changes between the S192A and D283A MDD mutants are depicted; distances were measured from equivalent amino acid Cα atoms. (B) Surface representation of ternary WT MDD co-crystal structure (gray). Area of active site ‘funnel’ (∼145 Å2) measured from Ala101 Cβ to Lys68 Cs (10.5 Å) and Ser191 Cα to Leu60 CD2 (13.8 Å), side chains colored purple. (C) Surface representation of ternary S192A MDD co-crystal structure (green). Area of active site ‘funnel’ (∼300 Å2) measured from Ala101 Cβ to Lys68 NZ (19.3 Å) and Lys188 NZ to Leu60 CD2 (15.7 Å), side chains colored purple. (D) Surface representation of ternary D283A MDD co-crystal structure (red). Area of active site ‘funnel (∼99 Å2) measured from Ala101 Cβ to Lys68 NZ (10.5 Å) and Ser187 OG to Leu60 CD2 (9.4 Å), side chains colored cyan.
Discussion
MDD belongs to a large group of small molecule kinases that is known as the GHMP kinase superfamily (33). Each of the members in this group catalyzes a sequential reaction, whereby the γ-phosphoryl group of ATP is transferred to an enzyme-specific acceptor, thereby yielding ADP and a phosphorylated product. While there exists a great deal of similarity among these proteins at the structural level (Cα RMSD values from 2.6 – 4.0 Å) (41), they share relatively low levels of sequence identity (10-20%) overall. This considerable divergence, perhaps reflective of their different acceptor substrates, has given rise to unique combinations of both active site residues and catalytic mechanisms. In addition, the order of substrate binding varies greatly between GHMP kinase family members, with acceptor binding first in some (e.g. mevalonate kinase (39) and MDD (42)), ATP binding first in others (e.g. liver galactokinase (43), Salmonella enterica threonine kinase (44)), and still others incorporating either substrate randomly (e.g. phosphomevalonate kinase (45)). We recently published an article (13) that described the first crystal structures of an MDD bound to two acceptor substrate-like inhibitory analogs. This work demonstrated that the active site of MDD resembles a funnel, with the acceptor substrate-binding site and catalytic residues located at the bottom. Given this restriction, it seemed very likely that the MVAPP acceptor-binding site would be occluded if the ATP nucleotide donor bound first. The ternary co-crystal structures presented here now demonstrate this point conclusively. In this regard, our structural analyses presented here are consistent with those of Jabalquinto and Cardemil, whose kinetic characterization of avian liver MDD suggested a sequential substrate binding order over two decades ago (42).
The binding site of ATPγS within the ternary MDD crystal structure places the γ -phosphoryl ∼5.5 Å away from the C3-hydroxyl of MVAPP. Such a large distance most likely signifies that this complex has been trapped in a catalytically incompetent state, and could have resulted from crystallization with inhibitory mimics of both acceptor (FMVAPP) and nucleotide (ATPγS) rather than bona fide substrates. In addition to this, the triphosphoryl group of ATPγS appears to adopt a ‘kinked’ conformation; this may be due to the lack of an ordered Mg++ in any of our ternary co-crystal structures. Nevertheless, a model for a catalytically competent complex can be readily derived by superimposing the AMPPNP nucleotide analog found in the Streptococcus pneumoniae phosphomevalonate kinase ternary complex (PDB ID: 3GON) onto the ATPγS nucleotide in the wild-type MDD ternary complex presented here (Fig. 7). In this case, the γ-phosphoryl of AMPPNP is now only 3.4 Å from the C3-hydroxyl of MVAPP and is correctly positioned for in-line phosphoryl transfer (5). Both mutagenesis (5) and structural studies (13) have implicated Asp283 as the catalytic base responsible for deprotonation of the MVAPP C3-hydroxyl. Indeed, the crystal structure of D283A MDD bound to MVAPP (Fig. 2b) reveals that mutagenesis of this side chain eliminates the optimal alignment of the C3-hydroxyl of MVAPP. When both the D283A-MVAPP and WT MDD ternary complex are considered together (along with previous mutagenesis studies) it appears that the role of Asp283 must involve both the deprotonation and optimal orientation of the C3-hydroxyl group of MVAPP.
Figure 7.
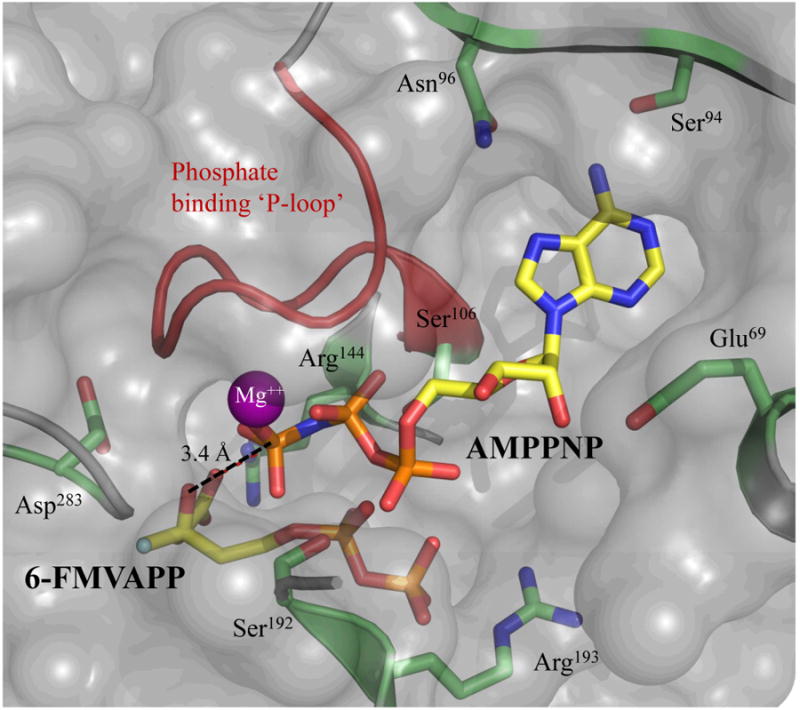
Molecular surface of the substrate and nucleotide binding sites of MDD. An AMPPNP-Mg++ molecule (yellow, ball and stick) from phosphomevalonate kinase (PDB code 3GON) was manually aligned (within COOT (23, 24)) onto the ATPγS molecule present in the WT MDD ternary co-crystal structure. The surface is rendered semitransparent (gray) with the phosphate binding ‘P-loop’ (red) and relevant active site side chains (lime) represented as cartoon ribbon and ball and stick, respectively. The AMPPNP γ-phosphoryl is within 3.4 Å of the C3-hydroxyl of FMVAPP (yellow, ball and stick).
Considering the structures presented here and elsewhere (13), along with an array of mutagenesis studies (5, 9, 10, 13), we can now propose a detailed schematic for the catalytic mechanism of MDD (Fig. 8). Asp283 correctly positions the MVAPP acceptor substrate, while functioning as a catalytic base to abstract the acidic proton found on its C3-hydroxyl. This deprotonation facilitates an in-line transfer of the γ -phosphoryl from the ATP donor (5). In addition to the previously discussed catalytic effects of Asp283, several amino acids within the ‘P-loop’ are involved in extensive interactions with the triphosphoryl group of ATP. These residues are conserved across GHMP kinase family members and include Ala105, Ser106, Ser107 and Ala108. Previous mutagenesis of Ser (9) revealed only modest changes in the Kd for ATP-Mg and the KM for MVAPP despite a >42,000 fold reduction in kcat. This side chain is appropriately positioned to hydrogen bond with both the β- and γ-phosphoryls of ATPγS within the MDD ternary structure, which strongly suggests that its role in MDD catalysis involves properly positioning the ATP phosphoryls. Ser106 and Ser192 each interact with the α-phosphoryl of ATPγS and FMVAPP, which further suggests that their roles involve appropriately aligning both substrate and nucleotide within the active site funnel. Separately, two arginine side chains (Arg144 and Arg193) flank either side of the MVAPP acceptor. The guanidinium group of Arg144 aids in catalysis by influencing the position of the C1 carboxyl group of MVAPP, and promotes decarboxylation. This interaction was disrupted in the D283A ternary complex. Arg193, found only in prokaryotes (13), interacts with the phosphoryl groups of MVAPP, and locks the acceptor in position within the active site. In summary, the reaction mechanism of MDD appears to be quite similar to that of mevalonate kinase (39), and provides further evidence that the GHMP kinases involved in the mevalonate pathway hold many features in common with one another.
Figure 8.

Proposed scheme for the catalytic mechanism of mevalonate diphosphate decarboxylase. Abstraction of the C3-hydroxyl proton from MVAPP is aided by the carboxyl group of Asp283 which functions as the catalytic base in the MDD reaction. Several MDD amino acid side chains within the ‘P-loop’ interact with the ATP phosphoryl groups. Ser107 and Ser141 play prominent roles in stabilizing the γ -phosphoryl group. The guanidinium group of Arg144 interacts with the MVAPP carboxylate group assisting in decarboxylation after formation of the carbocation intermediate. Not all ligand interactions within the ternary crystal structure are depicted. Hydrogen bonds are shown as dashed lines.
The human genome encodes for over 500 different kinases, which is perhaps not surprising since phosphoryl transfer events are found in a multitude of biochemical and signal transduction pathways. Since many of these enzymes (e.g. protein kinases in particular) have well-defined roles in serious diseases such as cancer, kinases as a whole have become an extremely popular target of small molecule inhibition studies in recent years (46). Intriguingly, the development of highly specific small molecule inhibitors has been quite successful despite a high degree of conservation within the respective ATP binding sites of these enzymes (47). Because it is possible to design truly specific kinase inhibitors for therapeutic use, it seems all the more important that a detailed understanding of both the structure and mechanism of the kinase target be in place prior to beginning this work. Thus, the crystal structures presented within these studies and elsewhere (13) constitute a valuable platform for rational drug design. By employing high-throughput screening with biochemical assays and structure-guided design (through crystallographic data), novel compounds that specifically target mevalonate pathway enzymes may be developed into antimicrobials effective against significant Gram-positive pathogens such as Staphylococcus aureus, Staphylococcus epidermidis, and Enterococcus faecalis. In this regard, the fact that the mevalonate pathway enzymes have not yet been targeted for antibiotic purposes strongly suggests that any novel inhibitors are likely to be active against deadly bacterial strains already resistant to current therapeutic agents.
Supplementary Material
Acknowledgments
The authors acknowledge generous technical assistance of SER-CAT staff during X-ray diffraction data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) beamlines at the Advanced Photon Source, Argonne National Laboratory. A list of supporting member institutions may be found at www.ser-cat.org/members.html.
Footnotes
This work was supported by National Institutes of Health Grants AI071028, AI090149, and the Marion-Merrell-Dow Foundation.
Abbreviations used are: IPP, isopentenyl diphosphate; MDD, mevalonate diphosphate decarboxylase; DPGP, diphosphoglycolyl proline; MVAPP, mevalonate 5-diphosphate; FMVAPP, 6-fluoromevalonate 5-diphosphate; GHMP, galactokinase, homoserine kinase, mevalonate kinase, phosphomevalonate kinase; ATPγS, adenosine 5′-O-(thiotriphosphate); AMP-PNP, adenosine 5′-(β,γ-imidotriphosphate); PDB, Protein Data Bank; RMSD, root-mean-square deviation; P-loop, phosphate binding loop.
Numbering of all residues in this manuscript reflects their position in the Staphylococcus epidermidis MDD sequence.
The refined coordinates and structure factors (codes 4DPT-U, 4DPW-Y and 4DU7-8) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rscb.org/).
Supporting Information Available: Four supplemental figures, their corresponding legends, and five supplemental tables accompany this manuscript. This material is available free of charge via the Internet at http://pubs.acs.org/
References
- 1.Eisenreich W, Bacher A, Arigoni D, Rohdich F. Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell Mol Life Sci. 2004;61:1401–1426. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilding EI, Brown JR, Bryant AP, Chalker AF, Holmes DJ, Ingraham KA, Iordanescu S, So CY, Rosenberg M, Gwynn MN. Identification, evolution, and essentiality of the mevalonate pathway for isopentenyl diphosphate biosynthesis in gram-positive cocci. J Bacteriol. 2000;182:4319–4327. doi: 10.1128/jb.182.15.4319-4327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jabalquinto AM, Cardemil E. Kinetic effects of ATP, divalent metal ions and pH on chicken liver mevalonate 5-diphosphate decarboxylase. Biochim Biophys Acta. 1987;916:172–178. doi: 10.1016/0167-4838(87)90105-1. [DOI] [PubMed] [Google Scholar]
- 4.Bloch K, Chaykin S, Phillips AH, De Waard A. Mevalonic acid pyrophosphate and isopentenylpyrophosphate. J Biol Chem. 1959;234:2595–2604. [PubMed] [Google Scholar]
- 5.Krepkiy D, Miziorko HM. Identification of active site residues in mevalonate diphosphate decarboxylase: implications for a family of phosphotransferases. Protein Sci. 2004;13:1875–1881. doi: 10.1110/ps.04725204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvear M, Jabalquinto AM, Eyzaguirre J, Cardemil E. Purification and characterization of avian liver mevalonate-5-pyrophosphate decarboxylase. Biochemistry. 1982;21:4646–4650. doi: 10.1021/bi00262a020. [DOI] [PubMed] [Google Scholar]
- 7.Michihara A, Sawamura M, Nara Y, Ikeda K, Yamori Y. Purification and characterization of two mevalonate pyrophosphate decarboxylases from rat liver: a novel molecular species of 37 kDa. J Biochem. 1997;122:647–654. doi: 10.1093/oxfordjournals.jbchem.a021802. [DOI] [PubMed] [Google Scholar]
- 8.Chiew YE, O'Sullivan WJ, Lee CS. Studies on pig liver mevalonate-5-diphosphate decarboxylase. Biochim Biophys Acta. 1987;916:271–278. doi: 10.1016/0167-4838(87)90170-1. [DOI] [PubMed] [Google Scholar]
- 9.Krepkiy DV, Miziorko HM. Investigation of the functional contributions of invariant serine residues in yeast mevalonate diphosphate decarboxylase. Biochemistry. 2005;44:2671–2677. doi: 10.1021/bi0484217. [DOI] [PubMed] [Google Scholar]
- 10.Voynova NE, Fu Z, Battaile KP, Herdendorf TJ, Kim JJ, Miziorko HM. Human mevalonate diphosphate decarboxylase: characterization, investigation of the mevalonate diphosphate binding site, and crystal structure. Arch Biochem Biophys. 2008;480:58–67. doi: 10.1016/j.abb.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonanno JB, Edo C, Eswar N, Pieper U, Romanowski MJ, Ilyin V, Gerchman SE, Kycia H, Studier FW, Sali A, Burley SK. Structural genomics of enzymes involved in sterol/isoprenoid biosynthesis. Proc Natl Acad Sci U S A. 2001;98:12896–12901. doi: 10.1073/pnas.181466998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byres E, Alphey MS, Smith TK, Hunter WN. Crystal structures of Trypanosoma brucei and Staphylococcus aureus mevalonate diphosphate decarboxylase inform on the determinants of specificity and reactivity. J Mol Biol. 2007;371:540–553. doi: 10.1016/j.jmb.2007.05.094. [DOI] [PubMed] [Google Scholar]
- 13.Barta ML, Skaff DA, McWhorter WJ, Herdendorf TJ, Miziorko HM, Geisbrecht BV. Crystal structures of Staphylococcus epidermidis mevalonate diphosphate decarboxylase bound to inhibitory analogs reveal new insight into substrate binding and catalysis. J Biol Chem. 2011;286:23900–23910. doi: 10.1074/jbc.M111.242016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zetola N, Francis JS, Nuermberger EL, BIshai WR. Community-acquired meicillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect Dis. 2005;5:275–286. doi: 10.1016/S1473-3099(05)70112-2. [DOI] [PubMed] [Google Scholar]
- 15.Tenover FC, Biddle JW, Lancaster MV. Increasing resistance to vancomycin and other glycopeptides in Staphylococcus aureus. Emerging Infect Dis. 2001;7:327–332. doi: 10.3201/eid0702.010237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 17.Reardon JE, Abeles RH. Inhibition of cholesterol biosynthesis by fluorinated mevalonate analogues. Biochemistry. 1987;26:4717–4722. doi: 10.1021/bi00389a018. [DOI] [PubMed] [Google Scholar]
- 18.Vlattas I, Dellureficio J, Ku E, Bohacek R, Zhang X. Inhibition of mevalonate 5-pyrophosphate decarbarboxylase by a proline-containing transition state analog. Bioorg Med Chem Lett. 1996;6:2091–2096. [Google Scholar]
- 19.Brøns-Poulsen J, Nøhr J, Larsen L. Megaprimer method for polymerase chain reaction-mediated generation of specific mutations in DNA. Methods Mol Biol. 2002;182:71–76. doi: 10.1385/1-59259-194-9:071. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z, M W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.McCoy A, Grosse-Kunstleve R, Storoni L, Read R. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 22.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 23.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 26.Thompson J, Higgins D, Gibson T. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gouet P, Courcelle E, Stuart D, Métoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 28.Zemla A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeLano W. The PyMOL Molecular Graphics System. 2002 In http://www.pymol.org.
- 30.Andreassi JL, Vetting MW, Bilder PW, Roderick SL, Leyh TS. Structure of the Ternary Complex of Phosphomevalonate Kinase: the Enzyme and its Family. Biochemistry. 2009;48:6461–6468. doi: 10.1021/bi900537u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saraste M, Sibbald PR, Wittinghofer A. The P-loop--a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- 32.Tsay YH, Robinson GW. Cloning and characterization of ERG8, an essential gene of Saccharomyces cerevisiae that encodes phosphomevalonate kinase. Mol Cell Biol. 1991;11:620–631. doi: 10.1128/mcb.11.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bork P, Sander C, Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993;2:31–40. doi: 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mozzarelli A, Rossi GL. Protein function in the crystal. Annu Rev Biophys Biomol Struct. 1996;25:343–365. doi: 10.1146/annurev.bb.25.060196.002015. [DOI] [PubMed] [Google Scholar]
- 35.Thoden JB, Timson DJ, Reece RJ, Holden HM. Molecular structure of human galactokinase: implications for type II galactosemia. J Biol Chem. 2005;280:9662–9670. doi: 10.1074/jbc.M412916200. [DOI] [PubMed] [Google Scholar]
- 36.Miallau L, Alphey MS, Kemp LE, Leonard GA, McSweeney SM, Hecht S, Bacher A, Eisenreich W, Rohdich F, Hunter WN. Biosynthesis of isoprenoids: crystal structure of 4-diphosphocytidyl-2C-methyl-D-erythritol kinase. Proc Natl Acad Sci U S A. 2003;100:9173–9178. doi: 10.1073/pnas.1533425100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou T, Daugherty M, Grishin NV, Osterman AL, Zhang H. Structure and mechanism of homoserine kinase: prototype for the GHMP kinase superfamily. Structure. 2000;8:1247–1257. doi: 10.1016/s0969-2126(00)00533-5. [DOI] [PubMed] [Google Scholar]
- 38.Krishna SS, Zhou T, Daugherty M, Osterman A, Zhang H. Structural basis for the catalysis and substrate specificity of homoserine kinase. Biochemistry. 2001;40:10810–10818. doi: 10.1021/bi010851z. [DOI] [PubMed] [Google Scholar]
- 39.Fu Z, Wang M, Potter D, Miziorko HM, Kim JJ. The structure of a binary complex between a mammalian mevalonate kinase and ATP: insights into the reaction mechanism and human inherited disease. J Biol Chem. 2002;277:18134–18142. doi: 10.1074/jbc.M200912200. [DOI] [PubMed] [Google Scholar]
- 40.DeLano WL. The PyMOL Molecular Graphics System. 2002;2009 http://www.pymol.org. [Google Scholar]
- 41.Andreassi JL, Leyh TS. Molecular functions of conserved aspects of the GHMP kinase family. Biochemistry. 2004;43:14594–14601. doi: 10.1021/bi048963o. [DOI] [PubMed] [Google Scholar]
- 42.Jabalquinto AM, Cardemil E. Substrate binding order in mevalonate 5-diphosphate decarboxylase from chicken liver. Biochim Biophys Acta. 1989;996:257–259. doi: 10.1016/0167-4838(89)90256-2. [DOI] [PubMed] [Google Scholar]
- 43.Ballard FJ. Purification and properties of galactokinase from pig liver. Biochem J. 1966;98:347–352. doi: 10.1042/bj0980347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan C, Fromm HJ, Bobik TA. Kinetic and Functional Analysis of L-threonine Kinase, the PduX Enzyme of Salmonella enterica. J Biol Chem. 2009;284:20240–20248. doi: 10.1074/jbc.M109.027425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pilloff D, Dabovic K, Romanowski MJ, Bonanno JB, Doherty M, Burley SK, Leyh TS. The kinetic mechanism of phosphomevalonate kinase. J Biol Chem. 2003;278:4510–4515. doi: 10.1074/jbc.M210551200. [DOI] [PubMed] [Google Scholar]
- 46.Cohen P. Protein kinases--the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 47.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.