

Abstract
Purpose of Review
Macrophage activation syndrome is the rheumatic disease-associated member of a group of hyperinflammatory syndromes characterized by uncontrolled cytokine storm. In this review, we highlight recent publications related to the pathoetiology of hyperinflammatory syndromes with an emphasis on how this new knowledge will guide our diagnosis, treatment, and future research efforts to better understand these deadly conditions.
Recent findings
The heterogeneity of clinical manifestations seen in patients with hyperinflammatory syndromes continues to grow as novel genetic and immunotherapeutic triggers of cytokine storm have been identified. Recent studies characterize unique cytokine and gene expression profiles from patients with different hyperinflammatory syndromes, while novel murine models begin to define networks of immune dysregulation thought to drive excessive inflammationin cytokine storm.
Summary
Emerging evidence suggests hypercytokinemia is the driving cause of pathology and morbidity/mortality in hyperinflammatory syndromes. Therefore, approaches to block cytokine function may be fruitful in treating hyperinflammatory syndromes with less toxicity than current therapies. However, not all hyperinflammatory syndromes result in the same pathogenic cytokine profile implying a personalized approach will be required for effective use of anti-cytokine therapies in the treatment of hyperinflammatory syndromes.
Keywords: Macrophage Activation Syndrome, cytokine storm, hemophagocytosis, Hemophagocytic Lymphohistiocytosis
Introduction
Macrophage Activation Syndrome (MAS) is a life-threatening complication of acute systemic inflammation arising in the context of a variety of autoimmune and auto inflammatory conditions. More common rheumatic diseases associated with MAS include systemic juvenile idiopathic arthritis (SJIA), systemic lupus erythematosus, and Kawasaki disease, while MAS is less frequently reported to occur in patients with dermatomyositis, ankylosing spondylitis, polyarteritis nodosa, and adult-onset Still Disease. MAS is characterized by uncontrolled inflammation manifest as unremitting fevers, cytopenias, splenomegaly, hepatitis, coagulopathy, multisystem organ failure, and death in its most severe form. Evidence highlights the broad spectrum of manifestations seen in MAS from clinically unrecognized “occult” disease to the traditional fulminant and rapidly fatal disease (1).
The clinical features of MAS are shared by other cytokine storm syndromes, and are believed to represent the same end-stage pathophysiologic state reached by disparate initiating triggers of uncontrolled inflammation (2). These other cytokine storm syndromes are classified based on known underlying triggers or defects. These triggers or defects include: 1) genetic defects in cellular cytotoxicity resulting in Familial Hemophagocytic Lymphohistiocytosis (FHL), 2) immune response defects resulting in Immunodeficiency-Associated Hyperinflammatory Syndrome (IDAHS), 3) pathogenic microbial triggers resulting in Infection-Associated Hyperinflammatory Syndrome (IAHS) including severe sepsis, and 4) neoplasms resulting in Malignancy-Associated Hyperinflammatory Syndrome (MAHS) (Table 1)(3-15). However, the cellular and molecular mechanisms underpinning the pathogenesis of these disparate causes of cytokine storm are not fully understood.
Table 1.
Classification of Hyperinflammatory Syndromes.
| Disease | Gene | Protein | Defect | Trigger | Reference |
|---|---|---|---|---|---|
| Familial Hemophagocytic Lymphohistiocytosis | Infection | ||||
| FHL1 | 9q21-3-22 | Unknown | Unknown | (3) | |
| FHL2 | PRF1 | Perforin | Deficiency of cytotoxicity effector | (4) | |
| FHL3 | UNC13D | Munc13-4 | Impaired cytolytic vesicle maturation and release | (5) | |
| FHL4 | STX11 | Syntaxin 11 | Abnormal priming and release of cytolytic vesicles | (6) | |
| FHL5 | STXBP2 | Munc18-2 | Abnormal priming and release of cytolytic vesicles | (7) | |
| Immunodeficiency- Associated Hyperinflammatory | Infection | ||||
| Chédiak Higashi | LYST | LYST | Abnormal biogenesis of cytolytic vesicles | (8) | |
| Griscelli 2 | RAB27A | RAB27A | Abnormal cytolytic vesicle docking at the plasma membrane | (9) | |
| Hermansky-Pudlak 2 | AP3B1 | AP-3 | Impaired maturation and trafficking of cytotoxic granules | (10) | |
| XLP1 | SH2D1A | SAP | Defect in signaling pathway for cytotoxicity in T and NK cells | (11) | |
| XLP2 | XIAP | XIAP/Birc4 | Defect in apoptosis and immune effector signaling pathways | (12) | |
| ITK deficiency | ITK | ITK | Abnormal development, function, and/or survival of cytotoxic T cells | (13) | |
| Macrophage Activation Syndrome | Unknown | Unknown | Unknown | Rheumatic Disease Flare OR Infection | (14, 15) |
| Infection-Associated Hyperinflammatory Syndrome | Unknown | Unknown | Unknown | Infection | (14) |
| Malignancy-Associated Hyperinflammatory Syndrome | N/A | N/A | N/A | Malignancy | (14) |
In this review, we focus on recent advances in our understanding of hyperinflammatory syndromes with an emphasis on defining novel approaches to better inform the diagnosis and treatment of these conditions. We will discuss the latest advances describing cytotoxicity defects and cytokine networks thought to initiate and perpetuate systemic immunopathology in hyperinflammatory syndromes. These advances open the door to personalizing the diagnosis and treatment of hyperinflammatory syndromes, as we begin to uncover the unique pathways leading to the development of cytokine storm in individual patients.
Diagnosis
The diagnosis of hyperinflammatory syndromes is wrought with difficulty. These rapidly fatal conditions require immediate recognition to ensure urgent treatment. Secondly, tremendous clinical overlap occurs between different types of hyperinflammatory syndromes making the appropriate diagnosis challenging. Finally, appropriate diagnosis is essential, as current treatment recommendations for each individual hyperinflammatory syndrome are different.
Hemophagocytic Lymphohistiocytosis (HLH) is the only hyperinflammatory syndrome with formal diagnostic criteria, which relies on clinical criteria and testing of HLH-predisposing genetic defects to fulfill the diagnosis (Table 2)(16). Preliminary diagnostic guidelines are also available for MAS, but are restricted to patients with a known diagnosis of active SJIA (17). These guidelines may be difficult to apply to the diagnosis of MAS when it is the first manifestation of SJIA, which occurs in 0-67% of patients with SJIA complicated by MAS (17, 18). As HLH-related genetic testing can delay definitive diagnosis for days to weeks, a physician is left with HLH clinical criteria to determine whether a patient warrants toxic HLH-related therapies or not in the setting of a possible hyperinflammatory syndrome.
Table 2. 2004 Diagnostic Guidelines for HLH (16).
| The diagnosis of HLH can be established if one of the following criteria (A or B) is fulfilled. |
|
|
| (A) Molecular diagnosis consistent with known genetic susceptibility for the development of HLH (PRF1, UNC13D, STX11, STXBP2, RAB27A, SH2D1A or XIAP) |
| or |
| (B) Presence of 5 of 8 of the following clinical criteria: |
| Fever |
| Splenomegaly |
| Cytopenias affecting ≥2/3 lineages in the peripheral blood |
| Hemoglobin <90 g/L (in infants <4 weeks: hemoglobin <100 g/L) |
| Platelets <100 × 109/L |
| Neutrophils <1.0 × 109/L |
| Hypertriglyceridemia and/or hypofibrinogenemia |
| Fasting triglycerides ≥3.0 mmol/L (i.e., ≥265 mg/dL) |
| Fibrinogen ≤1.5 g/L |
| Hemophagocytosis in bone marrow, spleen, or lymph nodes |
| Low or absent natural killer cell activity (according to local laboratory reference) |
| Elevated ferritin ≥500 ug/L |
| Elevated soluble CD25 (i.e., soluble IL-2 receptor) ≥2400 U/mL |
| No evidence of malignancy |
- Absence of hemophagocytosis does not exclude a diagnosis of HLH.
- The following may provide strong supportive evidence for the diagnosis:
- spinal fluid pleocytosis (mononuclear cells) and/or elevated spinal fluid protein
- histological evidence resembling chronic persistent hepatitis by liver biopsy.
- Other abnormal clinical and laboratory findings consistent with the diagnosis: Jaundice, hepatic enzyme abnormalities, coagulopathy, lymph node enlargement, edema, skin rash, hypoproteinemia, hyponatremia, VLDL increased, HDL decreased.
The HLH clinical criteria include many non-specific findings that overlap with other diseases. For example, Ho et al. stratified 58 patients with a clinical suspicion for hyperinflammatory disease into HLH high-risk and low-risk groups, and demonstrated the amount of hemophagocytosis from bone marrow aspirates does not correlate with disease probability (19). This corroborates prior evidence showing the presence of hemophagocytosis is not sensitive or specific for hyperinflammatory syndromes (20, 21). Furthermore, Moore et al. published data on 627 patients showing a diverse range of conditions causing markedly elevated ferritin levels > 1000 μg/L (22), meaning ferritin is another nonspecific feature of HLH. In SJIA patients, the 2004 HLH criteria were shown to be an insensitive tool for the diagnosis of SJIA -related MAS, as 33% of SJIA-related MAS patients did not meet HLH diagnostic criteria (18). Therefore, it is clear the HLH diagnostic criteria should not be used to diagnose SJIA-related MAS, and should be used with caution in the diagnosis of other cytokine storm syndromes.
Alternative methods to differentiate between hyperinflammatory syndromes are needed. To this end, Lehmberg et al. recently identified absolute neutrophil count ≥1.8 × 109/L, CRP ≥90 mg/L, and sCD25 ≤7900 U/mL as cutoff values more specific for SJIA-related MAS than FHL or viral-associated HLH (18). Lehmberg et al. also demonstrated dynamic changes in standard laboratory tests, such as declining platelet and white blood cell counts, can differentiate between a flare in SJIA disease activity and full-blown MAS (18). However, they did not test whether a falling sedimentation rate or fibrinogen level would be predictive of MAS-related disease, which have been useful markers of MAS in our clinical experience. Sumegi et al. introduced another novel method for the diagnosis and differentiation of hyperinflammatory syndromes, whereby gene expression profiles of peripheral blood mononuclear cells from patients diagnosed with FHL type 2 demonstrated unique signatures compared to patients with relapsing FHL and rapidly-evolving FHL subtypes (23). It will be necessary to validate whether these cutoff values and gene expression profiles are useful in larger and more diverse cohorts of patients with cytokine storm syndromes before the full clinical benefit of these measures can be realized.
Prognostication
New insights into the basic mechanisms driving clinical heterogeneity in hyperinflammatory syndromes caused by defects in cellular cytotoxicity highlight how more informative prognoses and patient-specific treatment options may be the wave of the future. Three independent studies recently demonstrated the severity of FHL and IDAHS in genetically susceptible mice and humans correlates with the severity of the underlying cytotoxicity defect (24, 25). Jessen et al. showed patients with Syntax in 11 and LYST deficiency, conditions harboring less severe cytotoxicity defects, had a later onset of hyperinflammatory disease compared with patients with Griscelli Syndrome and FHL2, diseases with severe cytotoxicity defects (24). In a separate paper, Jessen et al. describe a mild viral-induced hyperinflammatory syndrome in pearl mice harboring a mutation in AP-3, which causes a mild defect in cytotoxicity (26). This mutation is described in Hermansky-Pudlak syndrome type 2 where the penetrance of full-blown hyperinflammatory disease is low and likely means pre-emptive bone marrow transplant is not warranted (26). Similarly, Sepulveda et al. showed the age of onset of hyperinflammatory disease in patients occurs later and a less severe viral-induced disease is seen in murine models of FHL4 compared to Griscelli Syndrome and FHL2, which correlates with the severity of the underlying cytotoxicity defects described in these diseases (25).
Recent advances in our understanding of the production, trafficking and release of cytolytic vesicles further highlight mechanisms of heterogeneity in disease severity. Interactions between Munc-18-2 and Syntaxin 11 are necessary for priming and release of cytolytic vesicles, and therefore lead to FHL4 and FHL5 when defective forms of these proteins are expressed (27). Interestingly, Hackman et al. now show how addition of interleukin (IL)-2 restores functional cytotoxicity to defective cytotoxic lymphocytes in patients with FHL4 and FHL5 by increasing levels of Syntaxin 3 and Munc 18-1, respectively (27). IL-2-induced expression of these alternative Munc18 and Syntaxin proteins complement the specific defects in Syntaxin 11 or Munc18-2 deficiencies, and highlight how IL-2-related therapies may lead to targeted interventions in patients with FHL4 and FHL5 (27).
Factors independent of defects in cytotoxicity may also regulate the severity of disease in patients with FHL. A recent study investigating a mouse model of FHL4 with a severe defect in cytotoxicity demonstrated nonfatal disease upon viral infection (28). In this model, immunopathogenic CD8 T cells had a phenotype consistent with CD8 T cell exhaustion manifest by decreased cellular activation and effector function leading to a reduction in global immunopathology (28). A better understanding of how defects in cytotoxicity affect T cell exhaustion may lead to novel therapeutic targets in hyperinflammatory diseases caused by immunopathologic CD8 T cells.
Pathogenesis
Cytokine stormsyndromes are established during self-perpetuating, feed-forward inflammatory cycles (Figure 1)(29, 30). Evidence from cytokine profiles support the notion of dysregulated cytokine production as a central driver in the propagation of systemic inflammation. Unique cytokine profiles have been identified in patients with hyperinflammatory syndromes, including an IL-18 signature in patients with SJIA-related MAS (31). More recently, Shimizu et al. described the presence of two different cytokine profiles in SJIA with an IL-6 signature in patients with arthritis-related disease manifestations and an IL-18 predominate cytokine profile in patients with MAS (32). In contrast, Wada et al. recently demonstrated IL-6, IL-18, tumor necrosis factor (TNF)α, and interferon (IFN)γ elevations in patients with active FHL, Epstein-Barr Virus (EBV)-HLH, and IDAHS (33). In a patient with SLE-related MAS, Shimizu et al. demonstrated a more striking TNFα cytokine signature compared to the cytokine profiles from patients with other forms of hyperinflammatory disease (31, 32). These data implicate unique networks of cytokines in disease pathogenesis across a variety of hyperinflammatory syndromes, and will revolutionize the diagnosis and treatment of hyperinflammatory syndromes as we begin to understand how these networks uniquely perpetuate cytokine storm in individual hyperinflammatory syndromes.
Figure 1. Self-perpetuating inflammatory cycles sustain enhanced systemic inflammation in hyperinflammatory syndromes.
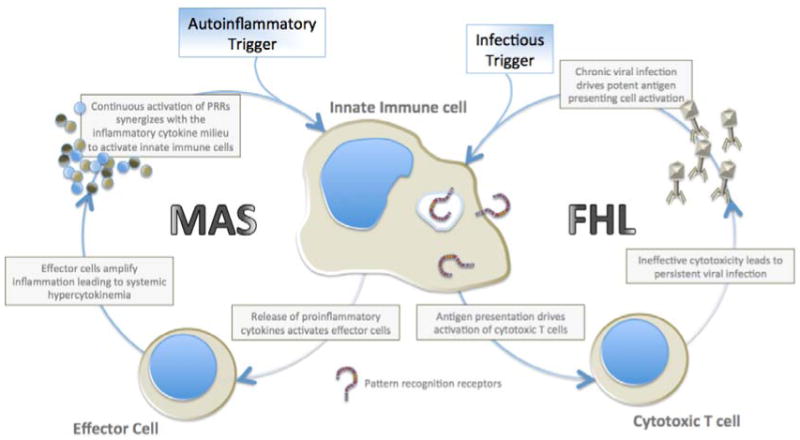
The working model of pathogenesis for hyperinflammatory diseases occurs from amplifying feed-forward inflammatory cycles leading to downstream immunopathology. In MAS, an autoinflammatory trigger likely mediated through pattern recognition receptor activation of innate immune cells leads to the release of proinflammatory mediators. Effector cells amplify this proinflammatory cytokine cascade leading to hypercytokinemia. The combination of proinflammatory cytokines and continued activation of pattern recognition receptors (PRRs) leads to synergistic activation of innate immune cells driving an enhanced immunopathologic cycle (29). In FHL, viral infections stimulate antigen-presenting cells to activate cytotoxic T cells to release tissue-damaging effector molecules. Due to defective cytotoxicity, uncontrolled viral replication propagates robust innate immune cell antigen presentation perpetuating a vicious cycle of self-sustaining and self-enhancing systemic inflammation (30).
Recent investigations using murine models of hyperinflammatory conditions have dissected the contribution of individual cytokines in the regulation and propagation of systemic inflammation. Previous reports demonstrate the central role of IL-10 as an anti-inflammatory regulator of hyperinflammatory disease (29). Interestingly, Ohyagi et al. now show an important source of IL-10 comes from hemophagocytes (34), cells once thought to be propagators of inflammation (35, 36). When hemophagocytosis is blocked or hemophagocytes are incapable of producing IL-10, viral-induced mortality is increased supporting a role for IL-10 and hemophagocytesas anti-inflammatory regulators of hyperinflammatory disease (34). Recent human and murine data support the notion that hemophagocytes are anti-inflammatory, as these cells display markers of alternatively activated macrophages, cells known to be important in the resolution phase of inflammation (37). The fact that hemophagocytosis may be reactive, not causal, may aid in understanding why its presence ultimately has a poor sensitivity and specificity for hyperinflammatory syndromes (19-21).
Previous studies have implicated IFNγ as the central mediator of the immunopathology in multiple hyperinflammatory diseases. However, two recent studies using chronic activation of innate immune cells to drive hyperinflammatory disease highlight how immunopathology can proceed in the absence of IFNγ. Canna et al. demonstrate how repeated injection of CpG, a Toll-like receptor 9 agonist, in combination with IL-10 receptor blockade leads to severe hyperinflammatory disease even in IFNγ-deficient mice (38). This study demonstrates IFNγ is dispensable for hyperinflammatory disease and implicates other inflammatory mediators as culprits of cytokine storm-related immunopathology (38). Avau et al. also describe an IFNγ-independent MAS-like disease following treatment of mice with complete Freund's adjuvant (39). This potent stimulator of innate immune cells leads to MAS-like disease in IFNγ-deficient mice with amelioration of disease following blockade of IL-12/23 or IL-17 implicating these cytokines as IFNγ-independent drivers of cytokine storm (39).
Clinically, known triggers of hyperinflammatory syndromes continue to expand with the most striking associations uncovered during the use of novel cancer immunotherapies in the treatment of specific cancers. A recent example of this is chimeric antigen receptor (CAR) T cell therapies, whereby engineered receptors graft the specificity of a monoclonal antibody onto a signaling scaffold capable of activating T cells (40). These artificial receptors can be expressed in T cells that are adoptively transferred into cancer patients to allow targeting and destruction of specific cancers (40). In patients with chemotherapy-resistant B cell acute lymphoblastic leukemia, treatment with CAR T cells has recently been shown to induce remission in patients with otherwise terminal disease (41, 42). However, this therapy has been associated with cytokine release syndrome mimicking many of the features of hyperinflammatory syndromes including massive hypercytokinemia (41, 42). Interestingly, blockade of IL-6 mitigated the effects of the cytokine release syndrome without affecting anti-leukemia efficacy (41, 42). Future studies will be necessary to determine if cytokine release syndromes are specific to the cytokine milieu produced by B cell tumor-targeted immunotherapy or whether this is specific to CAR T cell-mediated cytotoxicity. It is possible insights into the mechanism driving cytokine release syndrome may help us better understand how other hyperinflammatory diseases are triggered and regulated by specific patterns of cytokine production.
Therapeutics
Great attention has been paid to the treatment outcomes of patients with hyperinflammatory syndromes. However, treatment of hyperinflammatory syndromes remains difficult and outcomes remain poor. The best-defined treatment occurs in patients with HLH as defined by the HLH-2004 therapeutic guidelines. HLH patients are often treated with one of two different induction therapies including etoposide and dexamethasone with or without cyclosporine A or anti-thymocyte globulin and prednisone (43). Response rates and survival to hematopoietic cell transplant (HCT) approach 80% with these regimens, but complete responses are only achieved in approximately 50% of HLH patients with the majority of deaths prior to transplant accruing from uncontrolled disease (43). Adjunctive therapy for patients with refractory HLH is much less well defined. Use of infliximab, anakinra, alemtuzumab, daclizumab, and vincristine have been reported in small cases for refractory HLH, and now Jordan et al. report the use of alemtuzumab as successful salvage therapy for refractory HLH in 22 patients with 77% survival to HCT (43). While this report offers some progress in the treatment of refractory disease, the risks of infection, severe side effects, treatment-related malignancies, and death prior to transplant remain real. Promising alternative approaches to treatment of refractory disease include anti-cytokine based therapies, which are currently under investigation using therapies to neutralize the effects of IL-6 or IFNγ.
For non-HLH related hyperinflammatory syndromes, therapy is based on treatment of the underlying trigger of cytokine storm and is much less well defined than HLH-related therapies. In SJIA-related MAS, the hallmark of therapy is pulse steroids and cyclosporine A with refractory disease being treated with IVIg, rituximab (in EBV-associated MAS), alemtuzumab, daclizumabor anti-cytokine therapies including anti-IL-1 and anti-IL-6 (44, 45). For patients with IAHS, specific therapies directed at controlling infection may be sufficient for disease control (44). However, many patients with IAHS require immunosuppressive therapies to manage the hyperimmune state without clear guidelines for use of any particular immunosuppressive regimen (44). In EBV-associated hyperinflammatory syndromes, rituximab can be used in conjunction with other immunosuppressive therapies to ameliorate disease by specifically targeting B cells, which are specific targets of EBV infection (44).
Two recent reports highlight the use of recombinant thrombomodulin in ameliorating symptoms of cytokine storm in 2 patients with disease complicated by disseminated intravascular coagulation (DIC), a common complication in hyperinflammatory syndromes (46, 47). Thrombomodulin is a molecule expressed on vascular endothelium and is thought to inactivate the coagulation cascade and reduce inflammation from endogenous products of tissue damage (46). Multiple cytokines known to be elevated in hyperinflammatory syndromes down-regulate the expression of thrombomodulin and may predispose these patients to the development of DIC (47). Thrombomodulin has also shown promising results in the treatment of sepsis patients with DIC adding to the potential use of this therapeutic in the broader context of hyperinflammatory syndromes complicated by DIC (48). However, availability of this medicine may preclude its use as it is only approved in Japan.
Conclusion
New insights into the pathogenesis of hyperinflammatory syndromes continue to unravel the complex cytokine networks involved in end-organ damage and/or propagating severe inflammation. Further understanding of the immunopathogenic triggers and perpetuators of severe inflammation will likely uncover novel therapeutic targets to allow a personalized approach to the treatment and prevention of hyperinflammatory syndromes. The increasing recognition of cytokine-driven inflammation as the pathogenic mediator of disease and lack of hemophagocytes as a sensitive or specific disease marker make the name Hemophagocytic Lymphohistiocytosis obsolete. Perhaps the time is correct to rename HLH and MAS as “Hyperinflammatory” or “Hypercytokinemia” Syndromes to reflect our current understanding of the hyperinflammatory state underlying the pathogenesis of these conditions. This would provide a single term to link MAS, FHL, and other cytokine storm syndromes under a common theme, and focus clinicians on the hyperinflammatory nature of the pathology toward which therapy should be directed.
Key Points.
Clinical heterogeneity, overlap in clinical presentations, and lack of perfect diagnostic tests makes the diagnosis of hyperinflammatory syndromes difficult.
Hypercytokinemia is a common immunopathogenic state seen in hyperinflammatory syndromes.
Cytokine profiles from patients with hyperinflammatory syndromes have unique signatures and may lead to novel anti-cytokine directed therapeutic approaches.
Hemophagocytes are unlikely to be pathogenic cells, as they have poor predictive capability for disease state and appear to have regulatory roles in controlling inflammation during hyperinflammatory disease.
Acknowledgments
EMB is supported by an HHMI Early Career Physician Scientist Award, an NIH/NIAID K08-AI079396, and an NIH/NHLBI 1RO1HL112836-A1. LKW is supported by NIH T32 grants H043021 and AR007442.
Abbreviations
- FHL
Familial Hemophagocytic Lymphohistiocytosis
- MAS
Macrophage Activation Syndrome
- IDAHS
Immunodeficiency-Associated Hyperinflammatory Syndrome
- IAHS
Infection-Associated Hyperinflammatory Syndrome
- MAHS
Malignancy-Associated Hyperinflammatory Syndrome
- SJIA
systemic Juvenile Idiopathic Arthritis
- CAR
Chimeric Antigen Receptor
- HCT
Hematopoietic Cell Transplant
- DIC
Disseminated Intravascular Coagulation
- HLH
Hemophagocytic Lymphohistiocytosis
- IL
Interluekin
- IFN
Interferon
- TNF
Tumor Necrosis Factor
- EBV
Epstein Barr Virus
- PRR
Pattern Recognition Receptor
Footnotes
Conflicts of Interest: None
Bibliography
- 1.Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34(5):1133–8. Epub 2007/03/09. [PubMed] [Google Scholar]
- 2.Canna SW, Behrens EM. Making sense of the cytokine storm: a conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr Clin North Am. 2012;59(2):329–44. doi: 10.1016/j.pcl.2012.03.002. Epub 2012/05/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohadi M, Lalloz MR, Sham P, Zhao J, Dearlove AM, Shiach C, et al. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet. 1999;64(1):165–71. doi: 10.1086/302187. Epub 1999/01/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–9. doi: 10.1126/science.286.5446.1957. Epub 1999/12/03. [DOI] [PubMed] [Google Scholar]
- 5.Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115(4):461–73. doi: 10.1016/s0092-8674(03)00855-9. Epub 2003/11/19. [DOI] [PubMed] [Google Scholar]
- 6.zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14(6):827–34. doi: 10.1093/hmg/ddi076. Epub 2005/02/11. [DOI] [PubMed] [Google Scholar]
- 7.Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119(12):3765–73. doi: 10.1172/JCI40732. Epub 2009/11/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996;382(6588):262–5. doi: 10.1038/382262a0. Epub 1996/07/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25(2):173–6. doi: 10.1038/76024. Epub 2000/06/03. [DOI] [PubMed] [Google Scholar]
- 10.Clark RH, Stinchcombe JC, Day A, Blott E, Booth S, Bossi G, et al. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immunol. 2003;4(11):1111–20. doi: 10.1038/ni1000. Epub 2003/10/21. [DOI] [PubMed] [Google Scholar]
- 11.Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet. 1998;20(2):129–35. doi: 10.1038/2424. Epub 1998/10/15. [DOI] [PubMed] [Google Scholar]
- 12.Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444(7115):110–4. doi: 10.1038/nature05257. Epub 2006/11/03. [DOI] [PubMed] [Google Scholar]
- 13.Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest. 2009;119(5):1350–8. doi: 10.1172/JCI37901. Epub 2009/05/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol. 2013;161(5):609–22. doi: 10.1111/bjh.12293. Epub 2013/04/13. [DOI] [PubMed] [Google Scholar]
- 15.Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106(4):561–6. doi: 10.1016/s0022-3476(85)80072-x. Epub 1985/04/01. [DOI] [PubMed] [Google Scholar]
- 16.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31. doi: 10.1002/pbc.21039. Epub 2006/08/29. [DOI] [PubMed] [Google Scholar]
- 17.Ravelli A, Magni-Manzoni S, Pistorio A, Besana C, Foti T, Ruperto N, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146(5):598–604. doi: 10.1016/j.jpeds.2004.12.016. Epub 2005/05/05. [DOI] [PubMed] [Google Scholar]
- 18.Lehmberg K, Pink I, Eulenburg C, Beutel K, Maul-Pavicic A, Janka G. Differentiating macrophage activation syndrome in systemic juvenile idiopathic arthritis from other forms of hemophagocytic lymphohistiocytosis. J Pediatr. 2013;162(6):1245–51. doi: 10.1016/j.jpeds.2012.11.081. Epub 2013/01/22. **This study identifies cutoff values of common laboratory studies to differentiate MAS from HLH and dynamic laboratory values to differentiate MAS from SJIA flare. [DOI] [PubMed] [Google Scholar]
- 19.Ho C, Yao X, Tian L, Li FY, Podoltsev N, Xu ML. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Pathol. 2014;141(1):62–71. doi: 10.1309/AJCPMD5TJEFOOVBW. Epub 2013/12/18. [DOI] [PubMed] [Google Scholar]
- 20.Goel S, Polski JM, Imran H. Sensitivity and specificity of bone marrow hemophagocytosis in hemophagocytic lymphohistiocytosis. Ann Clin Lab Sci. 2012;42(1):21–5. Epub 2012/03/01. [PubMed] [Google Scholar]
- 21.Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;51(3):402–4. doi: 10.1002/pbc.21564. Epub 2008/06/05. [DOI] [PubMed] [Google Scholar]
- 22.Moore C, Jr, Ormseth M, Fuchs H. Causes and significance of markedly elevated serum ferritin levels in an academic medical center. J Clin Rheumatol. 2013;19(6):324–8. doi: 10.1097/RHU.0b013e31829ce01f. Epub 2013/08/24. *This study highlights the many causes of ferritin levels >1000 μg/L making it a nonspecific finding in the diagnosis of HLH. [DOI] [PubMed] [Google Scholar]
- 23.Sumegi J, Nestheide SV, Barnes MG, Villanueva J, Zhang K, Grom AA, et al. Gene-expression signatures differ between different clinical forms of familial hemophagocytic lymphohistiocytosis. Blood. 2013;121(7):e14–24. doi: 10.1182/blood-2012-05-425769. Epub 2012/12/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jessen B, Kogl T, Sepulveda FE, de Saint Basile G, Aichele P, Ehl S. Graded defects in cytotoxicity determine severity of hemophagocytic lymphohistiocytosis in humans and mice. Front Immunol. 2013;4:448. doi: 10.3389/fimmu.2013.00448. Epub 2014/01/01. *This study demonstrates the severity of the cytotoxicity defect correlates with the severity of the clinical disease in FHL and IDAHS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sepulveda FE, Debeurme F, Menasche G, Kurowska M, Cote M, Pachlopnik Schmid J, et al. Distinct severity of HLH in both human and murine mutants with complete loss of cytotoxic effector PRF1, RAB27A, and STX11. Blood. 2013;121(4):595–603. doi: 10.1182/blood-2012-07-440339. Epub 2012/11/20. *This study demonstrates the severity of the cytotoxicity defect correlates with the severity of the clinical disease in FHL and IDAHS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jessen B, Bode SF, Ammann S, Chakravorty S, Davies G, Diestelhorst J, et al. The risk of hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type 2. Blood. 2013;121(15):2943–51. doi: 10.1182/blood-2012-10-463166. Epub 2013/02/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hackmann Y, Graham SC, Ehl S, Honing S, Lehmberg K, Arico M, et al. Syntaxin binding mechanism and disease-causing mutations in Munc18-2. Proc Natl Acad Sci U S A. 2013;110(47):E4482–91. doi: 10.1073/pnas.1313474110. Epub 2013/11/07. **This study defines the molecular mechanisms underpinning the IL-2-induced restoration of cytolytic vesicle release in lymphocytes from patients with FHL4 and FHL5. This knowledge may help to direct targeted therapies for these diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kogl T, Muller J, Jessen B, Schmitt-Graeff A, Janka G, Ehl S, et al. Hemophagocytic lymphohistiocytosis in syntaxin-11-deficient mice: T-cell exhaustion limits fatal disease. Blood. 2013;121(4):604–13. doi: 10.1182/blood-2012-07-441139. Epub 2012/11/30. [DOI] [PubMed] [Google Scholar]
- 29.Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, et al. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest. 2011;121(6):2264–77. doi: 10.1172/JCI43157. Epub 2011/05/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terrell CE, Jordan MB. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+) T cells and dendritic cells. Blood. 2013;121(26):5184–91. doi: 10.1182/blood-2013-04-495309. Epub 2013/05/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology (Oxford) 2010;49(9):1645–53. doi: 10.1093/rheumatology/keq133. Epub 2010/05/18. [DOI] [PubMed] [Google Scholar]
- 32.Shimizu M, Nakagishi Y, Yachie A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine. 2013;61(2):345–8. doi: 10.1016/j.cyto.2012.11.025. Epub 2013/01/02. **This is the first paper to define unique cytokine profiles within SJIA separating patients at high-risk for MAS from those with arthritis predominant disease. [DOI] [PubMed] [Google Scholar]
- 33.Wada T, Kanegane H, Ohta K, Katoh F, Imamura T, Nakazawa Y, et al. Sustained elevation of serum interleukin-18 and its association with hemophagocytic lymphohistiocytosis in XIAP deficiency. Cytokine. 2014;65(1):74–8. doi: 10.1016/j.cyto.2013.09.007. Epub 2013/10/03. [DOI] [PubMed] [Google Scholar]
- 34.Ohyagi H, Onai N, Sato T, Yotsumoto S, Liu J, Akiba H, et al. Monocyte-derived dendritic cells perform hemophagocytosis to fine-tune excessive immune responses. Immunity. 2013;39(3):584–98. doi: 10.1016/j.immuni.2013.06.019. Epub 2013/09/17. **This paper identifies the hemophagocyte as a key producer of IL-10 in a model of hyperinflammatory disease, and highlights the critical importance of IL-10 in mitigating systemic inflammation during cytokine storm syndromes. [DOI] [PubMed] [Google Scholar]
- 35.Zoller EE, Lykens JE, Terrell CE, Aliberti J, Filipovich AH, Henson PM, et al. Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med. 2011;208(6):1203–14. doi: 10.1084/jem.20102538. Epub 2011/06/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Billiau AD, Roskams T, Van Damme-Lombaerts R, Matthys P, Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-gamma-producing lymphocytes and IL-6- and TNF-alpha-producing macrophages. Blood. 2005;105(4):1648–51. doi: 10.1182/blood-2004-08-2997. Epub 2004/10/07. [DOI] [PubMed] [Google Scholar]
- 37.Canna SW, Costa-Reis P, Bernal WE, Chu N, Sullivan KE, Paessler ME, et al. Alternative activation of laser-captured murine hemophagocytes. Arthritis Rheumatol. 2014 doi: 10.1002/art.38379. Epub 2014/01/29. *This paper supports the notion hemophagocytes are not proinflammatory cells, as they have a phenotype consistent with alternatively activated macrophages involved in the resolution phase of inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canna SW, Wrobel J, Chu N, Kreiger PA, Paessler M, Behrens EM. Interferon-gamma mediates anemia but is dispensable for fulminant toll-like receptor 9-induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum. 2013;65(7):1764–75. doi: 10.1002/art.37958. Epub 2013/04/05. *This paper is the first paper to dissociate hyperinflammatory IFNγ-driven anemia from hemophagocytosis, separating these two associated, but causally distinct processes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Avau A, Mitera T, Put S, Put K, Brisse E, Filtjens J, et al. Systemic juvenile idiopathic arthritis-like syndrome in mice following stimulation of the immune system with complete Freund's adjuvant: Regulation by IFN- gamma. Arthritis Rheumatol. 2014 doi: 10.1002/art.38359. Epub 2014/01/29. *This paper adds support to the idea IFNγ-independent hyperinflammatory disease is possible, and implicates IL-12, IL-23, and IL-17 as cytokine mediators of this process. [DOI] [PubMed] [Google Scholar]
- 40.Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol Rev. 2014;257(1):127–44. doi: 10.1111/imr.12139. Epub 2013/12/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18. doi: 10.1056/NEJMoa1215134. Epub 2013/03/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med. 2014;6(224):224ra25. doi: 10.1126/scitranslmed.3008226. Epub 2014/02/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101–9. doi: 10.1002/pbc.24188. Epub 2012/04/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandrakasan S, Filipovich AH. Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. J Pediatr. 2013;163(5):1253–9. doi: 10.1016/j.jpeds.2013.06.053. Epub 2013/08/21. [DOI] [PubMed] [Google Scholar]
- 45.Rajasekaran S, Kruse K, Kovey K, Davis AT, Hassan NE, Ndika AN, et al. Therapeutic Role of Anakinra, an Interleukin-1 Receptor Antagonist, in the Management of Secondary Hemophagocytic Lymphohistiocytosis/Sepsis/Multiple Organ Dysfunction/Macrophage Activating Syndrome in Critically Ill Children. Pediatr Crit Care Med. 2014 doi: 10.1097/PCC.0000000000000078. Epub 2014/03/04. [DOI] [PubMed] [Google Scholar]
- 46.Chi S, Ikezoe T, Takeuchi A, Takaoka M, Yokoyama A. Recombinant human soluble thrombomodulin is active against hemophagocytic lymphohistiocytosis associated with acquired immunodeficiency syndrome. Int J Hematol. 2013;98(5):615–9. doi: 10.1007/s12185-013-1450-5. Epub 2013/10/09. [DOI] [PubMed] [Google Scholar]
- 47.Yamamoto M, Hori T, Hatakeyama N, Igarashi K, Inazawa N, Tsutsumi H, et al. Use of Recombinant Thrombomodulin in Disseminated Intravascular Coagulation Complicated Hemophagocytic Lymphohistiocytosis. Indian J Pediatr. 2013 doi: 10.1007/s12098-013-1145-1. Epub 2013/08/06. [DOI] [PubMed] [Google Scholar]
- 48.Vincent JL, Ramesh MK, Ernest D, LaRosa SP, Pachl J, Aikawa N, et al. A randomized, double-blind, placebo-controlled, Phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med. 2013;41(9):2069–79. doi: 10.1097/CCM.0b013e31828e9b03. Epub 2013/08/28. [DOI] [PubMed] [Google Scholar]