

Abstract
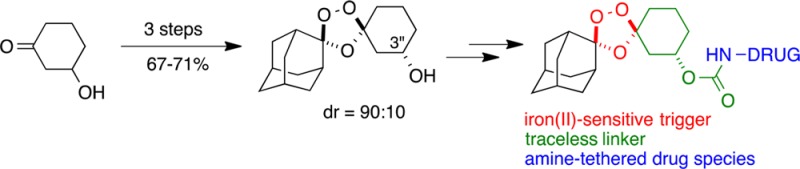
Ferrous iron-promoted reduction of a hindered peroxide bond underlies the antimalarial action of the 1,2,4-trioxane artemisinin and the 1,2,4-trioxolane arterolane. In appropriately designed systems, a 1,2,4-trioxolane ring can serve as a trigger to realize ferrous iron-dependent and parasite-selective drug delivery, both in vitro and in vivo. A stereocontrolled, expeditious (three steps), and efficient (67–71% overall yield) synthesis of 1,2,4-trioxolanes possessing the requisite 3″ substitution pattern that enables ferrous iron-dependent drug delivery is reported. The key synthetic step involves a diastereoselective Griesbaum co-ozonolysis reaction to afford primarily products with a trans relationship between the 3″ substituent and the peroxide bridge, as confirmed by X-ray structural analysis of a 3″-substituted 4-nitrobenzoate analogue.
Combination therapy with the sesquiterpene endoperoxide artemisinin is the current standard of care for treating uncomplicated malaria. A variety of synthetic molecules unrelated to artemisinin except for the presence of a hindered peroxide bond have been shown to also exhibit potent antimalarial properties. The 1,2,4-trioxolane arterolane1−3 (1, Figure 1) was the first such synthetic peroxide to be approved for clinical use (in India), while a related analogue with improved properties (OZ439)4 is currently progressing through human clinical trials. While their mechanism of antimalarial action is still studied and debated,5−16 the prevailing view is that Fenton-type reduction by ferrous iron sources within the parasite is an essential, activating chemical event. Various in vitro studies7,11,13,17 have confirmed the intermediacy of carbon-centered radicals following exposure of arterolane-like model systems to inorganic iron(II) salts or iron(II) heme. The concomitant production of ketone products in these reactions suggested to us the possibility of exploiting trioxolane fragmentation chemistry for ferrous iron-dependent and parasite-selective drug delivery.
Figure 1.
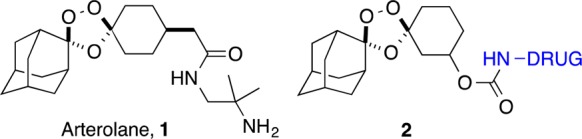
Structure of arterolane (1) and arterolane-inspired trioxolane–drug conjugate 2. Free drug is released from 2 in the presence of ferrous iron.
To realize iron(II)-dependent drug delivery, we designed 1,2,4-trioxolanes in which the liberated ketone species is a substrate for subsequent retro-Michael reaction.18−20 Once revealed, this ketone intermediate undergoes spontaneous β-elimination and decarboxylation to release a drug species attached at the β-position. Thus, iron(II)-dependent drug delivery is achieved by coupling trioxolane fragmentation and β-elimination chemistries. In previous work, we demonstrated a proof-of-concept for this approach in cultured P. falciparum parasites18,19 and in a mouse model of malaria.20 We found that trioxolane-mediated delivery of a protease inhibitor led to more sustained inhibition of the desired parasite protease and reduced inhibition of mammalian off-target proteases in vivo.
Most recently, we described21 a new generation of molecules 2 in which drug is released via β-elimination from cyclohexanone intermediate 3 (Scheme 1). This design requires conjugation of drug species at the 3″ position of the cyclohexane ring in 2. As with our previous systems, release of drug is “traceless” and can occur only after initial unveiling of the ketone function in 3 by iron(II)-promoted trioxolane scission in 2. A conjugate bearing the aminonucleoside antibiotic puromycin was used to confirm drug release from 2 in live Plasmodium falciparum parasites. Thus, α-puromycin antibodies were employed to follow the incorporation of released puromycin in the parasite proteome. Significantly, treatment with nonperoxidic dioxolane control 4 did not lead to puromycin incorporation in parasite proteins, confirming the peroxide dependence of drug release from 2.
Scheme 1. Mechanism of Ferrous Iron-Promoted Drug Release from Trioxolane 2.
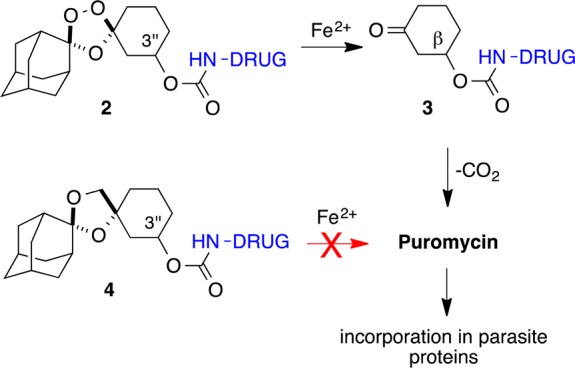
Release of a drug species (e.g., puromycin) was detected in parasites treated with 2 but not in those treated with dioxolane control 4(21).
To further explore the therapeutic potential of trioxolane conjugates 2, we sought to identify an efficient and stereocontrolled synthetic approach to these systems. A second objective was to prepare analogues of 2 with 3″-vinyl substitution, which we expected should exhibit enhanced rates of retro-Michael reaction. Ready access to drug substance is of particular importance in the case of antimalarial therapies, which must be produced at low cost for use in malaria-endemic countries. As noted above, realizing drug delivery from trioxolane 2 requires conjugation of drug at the 3″ position of the cyclohexane ring since this position represents the β position in the corresponding retro-Michael intermediate 3 (Scheme 1). However, a key synthetic challenge presented by 3″ substitution is the consequent desymmetrization of the molecule, resulting in four possible stereoisomers. By contrast, symmetrical 4″-substituted trioxolanes like 1 are achiral, existing as cis or trans diastereomers. Increased stereochemical complexity may explain why 3″-substituted trioxolanes have been scarcely reported22,23 in the literature, an exception being the ester 5(23) (Scheme 2).
Scheme 2. Reduction and Addition Reactions of Ketone 6.

Our initial synthetic efforts focused on nucleophilic additions to the racemic ketone 6,21 which we hoped would allow access to the desired 3″-vinylic alcohol intermediate (Scheme 2). While this proved a viable approach, we found that the intrinsic diastereofacial selectivity of ketone 6 was modest. Thus, reduction of 6 with sodium borohydride proceeded with little selectivity, and 1H NMR analysis of the corresponding methyl ether 7 revealed a 40:60 diastereomeric ratio (dr). Addition of vinylmagnesium bromide to 6 was more selective but complicated by competing enolization of the substrate, leading to poor yields. In an attempt to circumvent this issue, we examined additions of the less basic vinyl organocerium species. This approach afforded modest yields of 8 as a separable mixture of diastereomers (45:55 dr). While intermediate 8 proved useful for studies of retro-Michael release, we found that conjugation of drug species at this sterically encumbered tertiary alcohol was often challenging. Thus, we returned to focus on unsubstituted systems (secondary alcohols) with the goal of achieving improved stereocontrol.
One attractive strategy involved setting relative stereochemistry in the Griesbaum co-ozonolysis24 reaction used to install the trioxolane ring. This remarkable reaction proceeds through multiple steps, the final one involving a [3 + 2] cycloaddition between a carbonyl oxide and ketone. In their studies of 4″-substituted trioxolanes, Vennerstrom and co-workers concluded that the stereochemistry-defining [3 + 2] cycloaddition step occurs with a preference for axial addition to 4-substituted cyclohexanone substrates.25 Axial addition to 4-cyclohexanone substrates affords trioxolane product with an equatorial 4″ substituent and a cis relationship to the axial peroxide function (as in 1). By analogy, axial attack of the carbonyl oxide intermediate on a 3-substituted cyclohexanone should produce products with an equatorial 3″ substituent and a trans relationship with the axial peroxide (Scheme 3, top).
Scheme 3. Stereocontrolled Synthesis of Key Alcohol Intermediate 13.

To explore this possibility, we prepared the tert-butyldiphenylsilyl ether 10 as a substrate for Griesbaum co-ozonolysis with oxime 11 (Scheme 3). In the event, 10 reacted smoothly with 2.5 equiv of 11 and ozone in CCl4 to afford the desired product 12 in high yield. Silyl ether 12 was immediately subjected to reaction with TBAF in THF to afford alcohol 13 in three steps and 67–71% overall yield from 9. The 1H NMR spectrum of 12 and 13 suggested that the Griesbaum reaction had proceeded with good diastereocontrol. This was confirmed following efficient conversion by an established method26 to the methyl ether 7, in which diastereomeric ratios could be readily determined (Scheme 3). Integration of the −OMe resonances in the 1H NMR of 7 indicated a 90:10 dr. This compares to a 40:60 dr for 7 prepared via the reduction of ketone 6 (Scheme 2).
To further explore the diastereoselectivity of this reaction, we explored additional cyclohexanone substrates bearing 3-substituents with varying steric bulk (e.g., −OAc, −OSiMe3, −OSi(i-Pr)3). Following Griesbaum reaction with 11, protecting groups were removed and the resulting alcohols 13 converted to methyl ethers 7 for determination of diastereomeric ratios. Interestingly, we observed a similar ∼90:10 dr in each of these reactions, regardless of the ketone substrate employed. The only previously described Griesbaum co-ozonolysis involving a similar 3-substituted substrate was that leading to 5, which was reported23 to proceed with “high” diastereoselectivity, consistent with our results. The trans stereochemistry assigned to 5 was based on the predicted mode of axial addition, however, and was not rigorously established.
We found that the high proportion of overlapping aliphatic resonances in the 1H NMR spectra of 13 and its analogues made an unambiguous stereochemical assignment challenging. We therefore prepared a number of analogues that we expected might yield diffraction quality crystals. Ultimately, a suitable crystal of the 4-nitrobenzoate analogue 14 was obtained from a saturated toluene/ethanol/methanol solution by vapor diffusion of hexane. A complete, high-quality X-ray diffraction data set was collected on a single crystal of 14, and the resulting structure confirms the expected axial disposition of the peroxide bridge with a trans relationship to the equatorially positioned 3″-substituent (Figure 2). A 1H NMR spectrum of the recovered crystal used for the X-ray diffraction experiment showed the material to be solely the major diastereomeric product (Supporting Information). Thus, the X-ray structure of 14 establishes unambiguously for the first time the stereochemical preference of Griesbaum co-ozonolysis reactions involving 3-substituted cyclohexanones.
Figure 2.
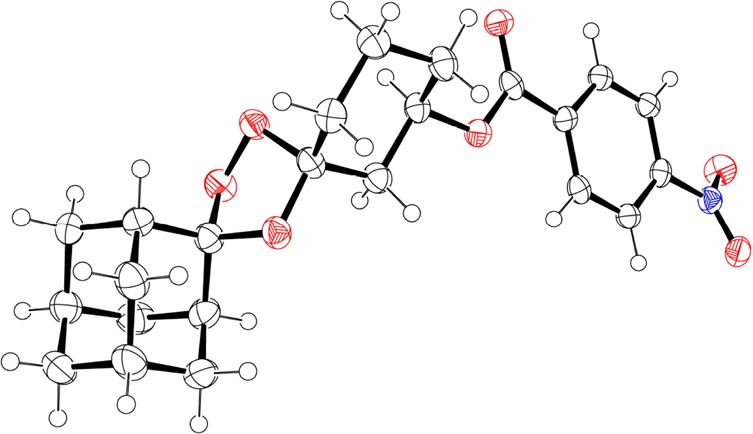
ORTEP of trans-14 derived from single-crystal X-ray diffraction data (ellipsoids at 50% probability). Note the axial position of the peroxide moiety and the equatorially disposed 3″-substituent.
Scheme 4. Synthesis of 4-Nitrobenzoate Analogue 14.

Upon re-crystallization of 14, the pure trans diastereomer was obtained.
The conjugation of amine-bearing drugs to alcohol 13 can be achieved in high yield via the corresponding nitrophenyl carbonate intermediate 15. To illustrate this, we introduced the diamino side chain of arterolane via a carbamate linkage at the 3″ position, yielding 16 (Scheme 5). Note that the trans stereochemistry of this “pseudoarterolane” analogue (16) is conformationally analogous to the cis stereochemistry of arterolane in that the side chain in both 1 and 16 is equatorially disposed (Scheme 5). Thus, the trans stereochemistry afforded by the chemistry described herein is also the “correct” stereochemistry for drug delivery conjugates 2, in that it is most similar to clinical compounds like arterolane and OZ439. Indeed, we have found that 16 exhibits in vitro antiplasmodial activity indistinguishable from that of 1 (Gut, J., personal communication). We are currently exploring the in vitro and in vivo properties of various drug conjugates of 13, which will be reported in due course.
Scheme 5. Synthesis of Pseudoarterolane Analogue 16.

In this report, we have described a short, efficient, and stereocontrolled synthesis of antimalarial 1,2,4-trioxolanes with a hitherto underexplored 3″-substitution pattern. Carbamate-conjugated, 3″-substituted trioxolanes of this type are uniquely capable of realizing ferrous iron-dependent drug delivery to the malaria parasite.21 The Griesbaum reaction between ketone 10 and oxime 11 proceeds in a stereocontrolled fashion to provide primarily products with a trans relationship between the peroxide bridge and the 3″-substituent. In our hands, this chemistry has provided ready access to gram quantities of 13, and no obvious barriers should bar the preparation of much larger quantities of this key intermediate. Finally, the use of nonracemic ketone substrates in this process should enable access to single enantiomers of 3″-substituted 1,2,4-trioxolanes and their corresponding ferrous iron-reactive drug conjugates.
Acknowledgments
We thank B. Spangler (UCSF) for helpful discussions and experimental assistance. A.R.R. acknowledges the support of the US National Institutes of Health (AI105106). A.G.D. acknowledges the support of the US NIH Shared Instrumentation Grant No. S10-RR027172 for the purchase of the diffractometer used in this research.
Supporting Information Available
Supplementary figures, detailed experimental procedures and compound characterization data, crystallographic procedures and data, and CIF/PLATON report. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Vennerstrom J. L.; Arbe-Barnes S.; Brun R.; Charman S. A.; Chiu F. C. K.; Chollet J.; Dong Y.; Dorn A.; Hunziker D.; Matile H.; McIntosh K.; Padmanilayam M.; Santo Tomas J.; Scheurer C.; Scorneaux B.; Tang Y.; Urwyler H.; Wittlin S.; Charman W. N. Nature 2004, 430, 900–904. [DOI] [PubMed] [Google Scholar]
- Valecha N.; Looareesuwan S.; Martensson A.; Abdulla S. M.; Krudsood S.; Tangpukdee N.; Mohanty S.; Mishra S. K.; Tyagi P. K.; Sharma S. K.; Moehrle J.; Gautam A.; Roy A.; Paliwal J. K.; Kothari M.; Saha N.; Dash A. P.; Björkman A. Clin. Infect. Dis. 2010, 51, 684–691. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Wittlin S.; Sriraghavan K.; Chollet J.; Charman S. A.; Charman W. N.; Scheurer C.; Urwyler H.; Santo Tomas J.; Snyder C.; Creek D. J.; Morizzi J.; Koltun M.; Matile H.; Wang X.; Padmanilayam M.; Tang Y.; Dorn A.; Brun R.; Vennerstrom J. L. J. Med. Chem. 2010, 53, 481–491. [DOI] [PubMed] [Google Scholar]
- Charman S. A.; Arbe-Barnes S.; Bathurst I. C.; Brun R.; Campbell M.; Charman W. N.; Chiu F. C. K.; Chollet J.; Craft J. C.; Creek D. J.; Dong Y.; Matile H.; Maurer M.; Morizzi J.; Nguyen T.; Papastogiannidis P.; Scheurer C.; Shackleford D. M.; Sriraghavan K.; Stingelin L.; Tang Y.; Urwyler H.; Wang X.; White K. L.; Wittlin S.; Zhou L.; Vennerstrom J. L. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 4400–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein-Ludwig U.; Webb R. J.; Van Goethem I. D. A.; East J. M.; Lee A. G.; Kimura M.; O’Neill P. M.; Bray P. G.; Ward S. A.; Krishna S. Nature 2003, 424, 957–961. [DOI] [PubMed] [Google Scholar]
- O’Neill P. M.; Posner G. H. J. Med. Chem. 2004, 47, 2945–2964. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Dong Y.; Wang X.; Sriraghavan K.; Wood J. K.; Vennerstrom J. L. J. Org. Chem. 2005, 70, 5103–5110. [DOI] [PubMed] [Google Scholar]
- Creek D.; Charman W.; Chiu F. C. K.; Prankerd R. J.; Mccullough K. J.; Dong Y.; Vennerstrom J. L.; Charman S. A. J. Pharm. Sci. 2007, 96, 2945–2956. [DOI] [PubMed] [Google Scholar]
- Stocks P. A.; Bray P. G.; Barton V. E.; Al-Helal M.; Jones M.; Araujo N. C.; Gibbons P.; Ward S. A.; Hughes R. H.; Biagini G. A.; Davies J.; Amewu R.; Mercer A. E.; Ellis G.; O’Neill P. M. Angew. Chem., Int. Ed. 2007, 46, 6278–6283. [DOI] [PubMed] [Google Scholar]
- Haynes R. K.; Chan W. C.; Lung C.-M.; Uhlemann A.-C.; Eckstein U.; Taramelli D.; Parapini S.; Monti D.; Krishna S. ChemMedChem 2007, 2, 1480–1497. [DOI] [PubMed] [Google Scholar]
- Creek D. J.; Charman W. N.; Chiu F. C. K.; Prankerd R. J.; Dong Y.; Vennerstrom J. L.; Charman S. A. Antimicrob. Agents Chemother. 2008, 52, 1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garah F. B.-E.; Stigliani J.-L.; Coslédan F.; Meunier B.; Robert A. ChemMedChem 2009, 4, 1469–1479. [DOI] [PubMed] [Google Scholar]
- Fügi M. a; Wittlin S.; Dong Y.; Vennerstrom J. L. Antimicrob. Agents Chemother. 2010, 54, 1042–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill P. M.; Barton V. E.; Ward S. A. Molecules 2010, 15, 1705–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes R. K.; Cheu K.-W.; Chan H.-W.; Wong H.-N.; Li K.-Y.; Tang M. M.-K.; Chen M.-J.; Guo Z.-F.; Guo Z.-H.; Sinniah K.; Witte A. B.; Coghi P.; Monti D. ChemMedChem 2012, 7, 2204–2226. [DOI] [PubMed] [Google Scholar]
- Robert A.; Benoit-Vical F.; Claparols C.; Meunier B. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 13676–13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousejra-El Garah F.; Wong M. H.-L.; Amewu R. K.; Muangnoicharoen S.; Maggs J. L.; Stigliani J.-L.; Park B. K.; Chadwick J.; Ward S. A.; O’Neill P. M. J. Med. Chem. 2011, 54, 6443–6455. [DOI] [PubMed] [Google Scholar]
- Mahajan S. S.; Deu E.; Lauterwasser E. M. W.; Leyva M. J.; Ellman J. A.; Bogyo M.; Renslo A. R. ChemMedChem 2011, 6, 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan S. S.; Gut J.; Rosenthal P. J.; Renslo A. R. Future Med. Chem. 2012, 4, 2241–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deu E.; Chen I.; Lauterwasser E. M. W.; Valderramos J.; Li H.; Edgington L.; Renslo A. R.; Bogyo M. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 18244–18249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine S. D.; Spangler B.; Gut J.; Lauterwasser E. M. W.; Rosenthal P. J.; Renslo A. R. ChemMedChem 2014, 10.1002/cmdc.201402362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y.; Chollet J.; Matile H.; Charman S. A.; Chiu F. C. K.; Charman W. N.; Scorneaux B.; Urwyler H.; Santo Tomas J.; Scheurer C.; Snyder C.; Dorn A.; Wang X.; Karle J. M.; Tang Y.; Wittlin S.; Brun R.; Vennerstrom J. L. J. Med. Chem. 2005, 48, 4953–4961. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Vargas M.; Dong Y.; Zhou L.; Wang X.; Sriraghavan K.; Keiser J.; Vennerstrom J. L. J. Med. Chem. 2010, 53, 4223–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbaum K.; Övez B.; Huh T. S.; Dong Y. Liebigs Ann. 1995, 1995, 1571–1574. [Google Scholar]
- Tang Y.; Dong Y.; Karle J. M.; DiTusa C. A.; Vennerstrom J. L. J. Org. Chem. 2004, 69, 6470–6473. [DOI] [PubMed] [Google Scholar]
- Johnstone R. A. W.; Rose M. E. Tetrahedron 1979, 35, 2169–2173. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.