

Abstract
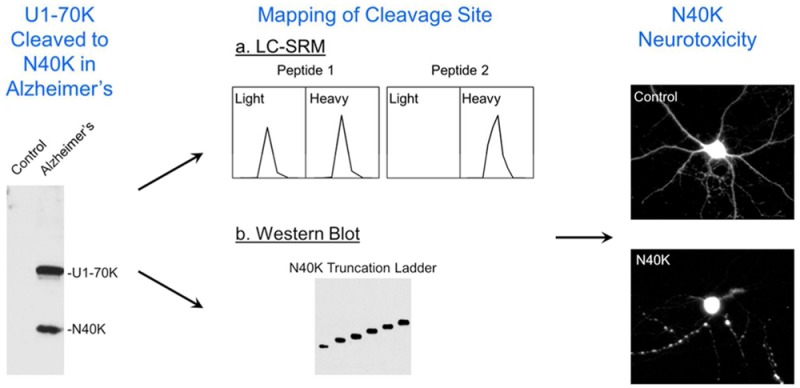
The accumulation of pathologic protein fragments is common in neurodegenerative disorders. We have recently identified in Alzheimer’s disease (AD) the aggregation of the U1-70K splicing factor and abnormal RNA processing. Here, we present that U1-70K can be cleaved into an N-terminal truncation (N40K) in ∼50% of AD cases, and the N40K abundance is inversely proportional to the total level of U1-70K. To map the cleavage site, we compared tryptic peptides of N40K and stable isotope labeled U1-70K by liquid chromatography–tandem mass spectrometry (MS), revealing that the proteolysis site is located in a highly repetitive and hydrophilic domain of U1-70K. We then adapted Western blotting to map the cleavage site in two steps: (i) mass spectrometric analysis revealing that U1-70K and N40K share the same N-termini and contain no major modifications; (ii) matching N40K with a series of six recombinant U1-70K truncations to define the cleavage site within a small region (Arg300 ± 6 residues). Finally, N40K expression led to substantial degeneration of rat primary hippocampal neurons. In summary, we combined multiple approaches to identify the U1-70K proteolytic site and found that the N40K fragment might contribute to neuronal toxicity in Alzheimer’s disease.
Keywords: mass spectrometry, LC−MS/MS, proteomics, neurodegenerative disease, Alzheimer’s disease, stable isotope labeling, proteolytic cleavage, U1 snRNP, U1-70K, RNA splicing
Introduction
Alzheimer’s disease (AD) is the most common form of dementia and the sixth-leading cause of death in the U. S., affecting ∼5 million people in the U. S., with a care cost of ∼$200 billion in 2012.1 However, no treatment is available to slow memory decline in AD. Analysis of familial early onset AD patients identified the association of three disease genes,2 including amyloid β (Aβ) precursor protein (APP), presenilin 1, and presenilin 2, the mutations of which are thought to alter the cleavage of APP to accelerate the production of neurotoxic Aβ peptides. Genetic susceptibility, such as ApoE-ε43 and a TREM2 variant,4,5 and environmental factors also influence the onset and progression of AD. Pathologically, AD is manifested by amyloid plaques and neurofibrillary tangles mainly composed of Aβ and tau, respectively.6−9 The resulting amyloid10 and tau hypotheses11 have dominated AD research. However, the precise disease mechanism is still not fully understood.12
The advent of proteomics technology13−15 provides an unprecedented opportunity to characterize human AD brain tissues. Highly sensitive liquid chromatography–tandem mass spectrometry (LC–MS/MS) has become the mainstream platform in proteomics.16 We have been using this advanced platform to analyze AD specimens,17−25 and recently discovered the aggregation of U1 small nuclear ribonucleoprotein complex (snRNP),26,27 a spliceosome complex consisting of U1-70K, U1A, U1C, Sm proteins, and U1 snRNA.28 Both U1-70K and U1A form cytoplasmic tangle-like structures in AD but not in other neurodegenerative disorders. Deep RNA sequencing revealed global dysregulation of RNA splicing in AD brains. Moreover, U1-70K knockdown or antisense oligonucleotide inhibition of U1 snRNP increases the levels of APP and Aβ in cellular models. Importantly, U1-70K aggregates are present in cases of mild cognitive impairment, a prodromal stage of AD, suggesting that U1 snRNP alteration occurs early in AD development. These results support specific U1 snRNP pathology and implicate dysfunctional RNA processing in AD pathogenesis.26,27
During our analysis of the aggregation-enriched proteome in AD by 1D SDS gel coupled with LC–MS/MS, U1-70K peptides were detected in two gel regions (∼70 kDa and ∼40 kDa), corresponding to the full length protein and a truncation form, respectively. The truncation contained only N-terminal peptides and was thus named N40K.26 The N-terminal sequence in N40K was further confirmed by immunoblotting using antibodies specific to either the N-terminus or C-terminus of U1-70K. As the N40K truncation cannot be explained by the alternative splicing isoforms of U1-70K, the data indicate that U1-70K is internally cleaved to produce the N40K fragment in AD.
Protein cleavage is common in neurodegeneration, with the resulting proteolytic fragments forming aggregates and contributing to pathogenesis,6,29 we sought to fully characterize the N40K fragment in AD. In the present study, we evaluated the N40K occurrence in AD cases and mapped the cleavage site of N40K by combining quantitative MS and Western blotting. Finally, the neuronal toxicity of the N40K fragments was investigated in cultured primary neuronal cells.
Materials and Methods
AD Sample Analysis and Protein Preparation
Cortical Brain Tissues
Human frozen tissues from prefrontal cortical regions were provided by the brain bank of AD Research Center at Emory University. The cases (Supporting Information Table S1) were clinically and pathologically characterized in accordance with established criteria.30
Sequential Protein Extraction
The brain samples were sequentially extracted with increasingly stringent detergents.26 Briefly, the cortical tissues were homogenized in a low salt buffer (10 mM Tris–HCl, pH 7.5, 5 mM EDTA, 1 mM DTT, 10% sucrose, and Sigma protease inhibitor cocktail), with 10mL buffer per gram of tissue, yielding the “total homogenate”. After centrifugation at 180 000g for 30 min, the pellet was re-extracted with the same volume of the low salt buffer with the addition of 1% Triton X-100 and sonication. The extraction was further performed with the low salt buffer plus 1% sarkosyl (N-lauroylsarcosine) twice. Finally, the pellet was dissolved in 8 M urea with 2% SDS, generating the “detergent insoluble fraction”.
Western Blotting
Protein samples were analyzed by NuPAGE Bis–Tris gel (Invitrogen) polyacrylamide gel electrophoresis and then transferred to nitrocellulose membrane. The membrane was blocked with 5% dry milk, incubated with antibodies, and followed by chemiluminescent detection (Thermo Fisher Scientific). The specificity of antibodies used in this study was previously characterized.26 The band intensities of selected proteins were quantified by the ImageJ program (National Institutes of Health).
Generation of N40K Truncations and Neuronal Toxicity Analysis
DNA Plasmids and HEK293 Cell Transfection
Full-length U1-70K and its truncations were cloned from a commercial plasmid (U1-70K-myc-DDK, pCMV6, Origene, RC201713). The PCR products were inserted into the mammalian expression vector “pcDNA3.1(+)” between BamHI and EcoRI sites using “Quick Ligation” kit (New England Biolabs). All DNA plasmids were validated by restrictive enzymatic digestion patterns and DNA sequencing. To express the recombinant proteins in HEK293 cells, the plasmids were transfected by the phospholipid-based transfection reagent FuGENE HD (Promega).
Recombinant U1-70K Expression and Purification
Full-length U1-70K-myc-DDK (FLAG tag) was overexpressed in HEK293 cells for 48 h after transfection. The cells were harvested and lysed in the Tris-buffered saline (TBS, 50 mM Tris–HCl, pH 7.5, and 100 mM NaCl) plus 1% Triton X-100 and Sigma protease inhibitor cocktail. After centrifugation at ∼20 000g for 3 min, the pellet was dissolved with sonication in the radioimmunoprecipitation assay buffer (RIPA, 50 mM Tris–HCl, pH 7.4, 100 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and Sigma protease inhibitor cocktail). FLAG-tagged U1-70K was purified by M2 anti-FLAG beads (Sigma), and confirmed by Western blotting and mass spectrometry.
Rat Primary Hippocampal Neuron Culture, Transfection, and Toxicity Assays
Hippocampal neurons were isolated from Sprague–Dawley rat embryos at E18, and cultured in completed Neurobasal A (Life Technologies) supplemented with B27 complex and GlutaMAX. Transfection was carried out using the calcium phosphate method (CalPhos, Clontech) in the complete medium Hibernate E for 3 h without CO2 in the incubator. The morphology of neurons was imaged after 2 day transfection. Transfection efficiency was optimized by using high quality plasmids (EndoFree Plasmid Maxi Kit, Qiagen) and adjusting the ratio of plasmids to calcium phosphate. Approximately 80 neurons were transfected per well in one 12-well plate for analyzing the degree of neuronal degeneration. Neuronal toxicity was also measured by the MTT assay (Thermo Fisher Scientific) upon 12 day Lentiviral infection (the multiplicity of infection of 10), in which mitochondrial activity in living cells converts the tetrazolium dye MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to insoluble formazan crystals for detection.
Quantitative Mass Spectrometry
Metabolic Labeling of U1-70K and Purification
Full-length U1-70K-myc-DDK (FLAG tag) was expressed in HEK293 cells labeled with heavy isotopes: [13C615N4] arginine (+10.0083 Da) and [13C6] lysine (+6.0201 Da) from Cambridge Isotope Laboratories, following our previous protocol.24 The U1-70K was affinity purified and validated by Western blotting.
Targeted LC–SRM Analysis
The purified labeled U1-70K was analyzed on an SDS gel, together with N40K isolated from AD brain. The corresponding gel bands were excised for standard in-gel digestion (12.5 ng/μL trypsin in 50 mM NH4HCO3 overnight). Small peptide aliquots of heavy isotope labeled U1-70K and native N40K were mixed at different ratios to examine their relative amounts. Then the two samples were mixed at an equal molar ratio for LC–SRM analysis, in which a number of peptide pairs were coeluted by nanoscale reverse phase LC–MS. During selected reaction monitoring (SRM), the peptide ion pairs were selected for fragmentation and quantified according to the signal ratio of coeluting product ion pairs. The analysis was performed on an LTQ-Velos Orbitrap (Thermo Fisher Scientific). The optimized parameters for the peptide pairs are included in Supporting Information (Supporting Information Table S2).
LC–MS/MS Analysis of Recombinant U1-70K to Confirm the N-Terminal Acetylated Peptide
Purified recombinant U1-70K sample was resolved on an SDS gel followed by in-gel digestion. The resulting peptides were analyzed on our optimized platform,31 which includes an LTQ-Velos Orbitrap (Thermo Fisher Scientific), Nano Acquity UPLC (Waters), and a C18 reverse phase column (75 μm ID × 20 cm, 2.7 μm HALO beads, Michrom Biosources). The buffer system consisted of buffer A (0.1% formic acid) and buffer B (0.1% formic acid plus 70% AcN). The peptides were eluted by a gradient of 15–50% buffer B in 35 min at 0.3 μL per min. The MS settings included one survey scan (60 000 resolution in the Orbitrap, 106 automatic gain control (AGC), and 50 ms maximal ion time), and top 10 low resolution (linear ion trap) MS/MS scans (5000 AGC, 250 ms maximal ion time, 3 m/z isolation window, default collision-induced dissociation, and 15 s dynamic exclusion). Charge state screening was enabled to exclude precursor ions of singly charged or unassigned charge states.
Acquired MS/MS raw files were searched against Uniprot human databases by the Sequest algorithm (v28, revision 13) with the target–decoy strategy to analyze false discovery rate (FDR).32,33 Spectra were matched with a mass tolerance of ± 10 ppm for precursor ions and ± 0.5 Da for product ions, with partially tryptic restriction and three maximal missed cleavages. Dynamic mass shift parameters were oxidized Met (+15.9949) and N-terminal acetylation (+42.0106), and four maximal modification sites. Only b and y ions were scored. Matched peptides were first refined by mass accuracy (±4 standard deviations, defined by all empirical good matches of doubly charged peptides with Xcorr of at least 2.5). These good matches were also utilized for mass recalibration. The refined matches were classified by precursor ion charge state and further filtered by Xcorr and ΔCn values until a protein FDR below 1% was reached. When peptides were matched to multiple proteins, these peptides were assigned to the proteins with the highest spectral counts based on the rule of parsimony.
Results
N40K, a Proteolytic Product of U1-70K, Occurs Frequently in AD
To probe AD disease mechanism, we performed a comprehensive GeLC–MS/MS analysis of detergent insoluble proteome and found unique U1-70K pathology in AD.26,27 In the analysis, U1-70K was identified by a large number of spectral counts and distributed in two main gel areas shown in a MS-based virtual Western blot image34 (Figure 1A and Supporting Information Figure S1A), including 10 spectral counts in the ∼40 kDa region and 33 spectral counts near its full-length 70 kDa region. Western blot demonstrated the presence of detergent insoluble U1-70K and the fragment (N40K) only in the diseased sample, whereas the nonspecific actin was found at an equal level in the control and AD samples (Figure 1B).
Figure 1.

N40K occurrence is independent of the postmortem interval and correlates with the decrease of the U1-70K level in AD. (A) Identification of N40K in the detergent insoluble fraction of AD brain by 1D SDS gel and LC–MS/MS. Ctl: control case. (B) Validation of N40K only in AD (case 8 in Supporting Information Table S1) by Western blotting. (C) No statistically significant difference in postmortem intervals between the N40K negative and positive cases. (D) Western blotting to show reduced U1-70K level in the total homogenate in N40K positive cases. The numbers above the gel indicate the corresponding case numbers in Supporting Information Table S1. (E) Quantitation of the Western blotting intensity in panel D to indicate an inverse correlation of U1-70K in total homogenate with insoluble N40K. Relative intensities of U1-70K and N40K were normalized by setting the maximal values to 100, respectively.
We further examined the prevalence of N40K in AD cases (n = 17, disease onset age of 58.7 ± 7.8 years old, age of death of 67.7 ± 8.6 years old, postmortem interval of 10.1 ± 5.7 h, Supporting Information Table S1). Approximately half of the cases (9/17, ∼52.9%) exhibited N40K, and there was no significant difference between N40K negative (N40K–) and N40K positive (N40K+) AD cases when comparing the age of disease onset (N40K–: 58.5 ± 7.7 y; N40K+: 58.9 ± 8.4 y) or the age of death (N40K–: 66.8 ± 9.7 y; N40K+: 68.5 ± 8.1 y). Although the postmortem interval of the two AD groups appears to display a difference (N40K–: 8.1 ± 5.3 h; N40K+: 12 ± 5.8 h), it is not statistically significant (p = 0.165, Student’s t-test, Figure 1C). The results suggest that the generation of N40K is unlikely caused by prolonged postmortem interval during the collection of brain tissues.
When extracting proteins from AD brain tissues, we observed that the U1-70K level was low in total homogenate of some cases.26 Interestingly, the occurrence of N40K in the detergent insoluble fraction was concomitant with the reduction of total U1-70K (Figure 1D and Supporting Information Figure S1B), and the two protein abundances showed an inverse correlation (Figure 1E).
We then sought to determine whether N40K is an alternative splicing isoform or a cleaved fragment of full-length U1-70K. In the UniProt/Swiss-Prot database, U1-70K has four different splicing isoforms of 437 (full length, Figure 2), 428, 341, and 166 residues. None of these four isoforms fit the size of N40K. To explore potential new U1-70K splicing isoforms in AD, we analyzed RNA-seq data sets of multiple AD brains26 but did not identify any additional isoforms. All exon reads of U1-70K were evenly distributed along the transcript, consistent with only one major RNA form of the full length U1-70K. In addition, U1 SnRNP deficiency may cause premature cleavage and polyadenylation in AD,26 leading to shortened RNA transcripts. However, we did not find any premature polyadenylation in the exons of U1-70K. Therefore, N40K is most likely produced by the proteolysis of full length U1-70K.
Figure 2.

Identification of U1-70K and N40K peptides in AD. These peptides are shown in different colors in the protein sequence, together with peptides monitored in LC–SRM.
Analysis of N40K Termini by Mass Spectrometry
We attempted to determine both termini of N40K protein by mass spectrometry. First, shotgun MS analysis26 identified seven peptides in N40K in the region from Asp17 to Arg200 (Figure 2), but N40K is clearly larger than a protein of 200 amino acids. Additional database search for protein modifications did not uncover any modified peptides. Some N40K peptides may be missed in the analysis, possibly due to the low level of N40K, undersampling of shotgun proteomics, or the incompatibility with LC–MS/MS.
To address the undersampling problem of shotgun MS, we then performed a systematic LC–SRM screening of N40K tryptic peptides using stable isotope labeled, recombinant U1-70K protein as an internal standard (Figure 3A). Compared to precursor ion quantification on survey MS scans that may be interfered by coeluted isobaric ions, the SRM enables targeted analysis of product ions from precursor ion fragmentation (Supporting Information Table S2), typically showing better specificity and sensitivity. Theoretically, when U1-70K peptides are equally mixed with N40K peptides, shared peptides have a 1:1 ratio and unique peptides have a 1:0 or 0:1 ratio. On the basis of this principle, we purified heavy Lys and Arg labeled U1-70K from HEK293 cells and validated by immunoblotting. The sample was resolved on an SDS gel, and the U1-70K-containing band was excised for in-gel trypsin digestion (Figure 3B). Meanwhile, N40K extracted from the AD insoluble fraction was similarly processed (Figure 3C). The two samples were mixed at a nearly 1:1 ratio for LC–SRM, in which four selected peptides were monitored according to MS/MS spectra of purified U1-70K (Supporting Information Figure S2–S5). Two peptides (residues 17–27 and 219–231) showed roughly 1:1 ratio, whereas the other two peptides (residues 311–346 and 394∼411) were not detected in the light forms, suggesting that the C-terminus of N40K is between amino acids 231 and 345 (Figure 3D).
Figure 3.

Analysis of the N40K C-terminus by LC–SRM. (A) Work flow of the method. (B) Identification and size confirmation of purified recombinant U1-70K by Western blotting, followed by band excision and in-gel digestion. (C) Identification of a N40K–containing gel band by Western blotting. The 10 kDa ladder was also used for precise alignment of immunoblotting images with stained gel. (D) The intensity ratios of light/heavy (L/H) of U1-70K peptide pairs, leading to the predicted cleavage region.
Additional attempts to analyze putative peptides between residues 231 and 345 were futile, as the region is highly repetitive and hydrophilic, enriched in Lys, Arg, Asp, and Glu residues (Figure 2). Any peptides digested by available proteases are either too small or too hydrophilic for LC–MS/MS analysis using standard methodology. Thus, we decided to use Western blotting to narrow down the range of N40K C-terminus. Prior to the Western blotting analysis, we needed to ensure that full length U1-70K and N40K have the same N-terminus (Figure 2) and lack other post-translational modifications that may affect protein migration.
As the N-terminal amino acid of proteins is usually modified,35 we tested multiple modification possibilities and found that U1-70K and N40K have the first Met removed and the second Thr acetylated (Figure 4A). The MS/MS pattern of the resulting tryptic peptide (residues 2–16) was detected for both U1-70K and N40K in AD (Figure 4A and B). Importantly, the MS/MS pattern was fully recapitulated in the analysis of recombinant U1-70K purified from HEK293 cells (Figure 4C). Moreover, the precursor ions in these three independent LC–MS/MS runs showed highly similar isotopic patterns and LC retention properties (i.e., eluted by ∼33% acetonitrile, Supporting Information Figure S6). These data strongly support that N40K has the identical N-terminus as the full length U1-70K.
Figure 4.

Analysis of the N40K N-terminus by MS. (A–C) MS/MS spectra for identifying the N-terminal peptide with methionine removal and threonine acetylation in U1-70K, N40K, and purified U1-70K in HEK293 cells, respectively. Matched ions were assigned by arrows indicating specific b or y ions.
N40K C-Terminus Is Around Arg300 Based on Western Blotting
Protein migration during Western blotting is determined by its amino acid sequence and post-translational modifications. Our MS analysis did not identify any major post-translational modification sites with high occupancy in N40K. RNA-seq analysis did not reveal any alternative splicing isoforms similar to N40K. To ensure the comparison of proteins with the same amino acid composition, we constructed serial U1-70K N-terminal truncations (N272, N282, N289, N300, N306, and N316, without any tag) based on the MS-mapped C-terminal range. When expressed in HEK293 cells, these proteins showed a ladder on Western blot using U1-70K polyclonal antibodies26 that recognize a peptide antigen of residues 99–120 (Figure 5A). After multiple rounds of analyses, the cleavage site was determined to be Arg300 [±0.8 kDa (six residues)] (Figure 5B; only the three closest truncations are shown). As most proteases exhibit residue specificity, we generated six N-terminal truncations including all possible C-terminal residues for subsequent functional studies (Figure 5C-5D).
Figure 5.
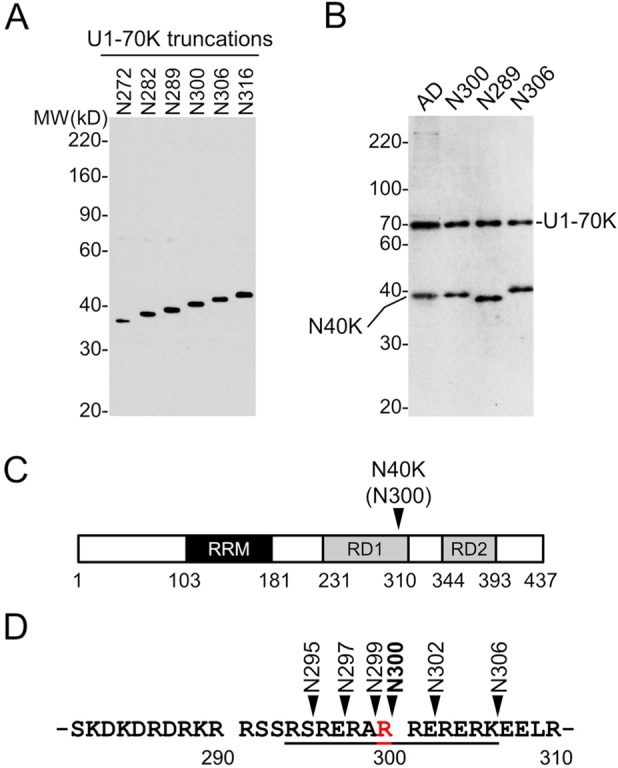
Determination of N40K C-terminus by Western blotting. (A) Expression of U1-70K N-terminal truncations in HEK293 cells. (B) Analysis of N40K in AD and three recombinant truncations. (C) Diagram of U1-70K domains and predicted N40K cleavage region. (D) N40K truncations covering all possible cleavage products with different C-terminal residues.
N40K Fragment Expression Induces Neuronal Toxicity
As proteolytic cleavage may result in neurotoxic fragments in neurodegeneration,6,29 we examined the toxicity of the full-length U1-70K and six N40K truncations (N295, N297, N299, N300, N302, and N306) in cultured primary neurons. To test the expression efficiency of these recombinant proteins, we transfected equal amounts of DNA plasmids (Figure 6A) in HEK293 cells and detected highly similar levels of recombinant proteins (Figure 6B). These plasmids were then cotransfected into rat primary hippocampal neurons that were cultured for 14 days in vitro, together with a vector expressing red fluorescent protein (RFP) to monitor neuronal morphology. Interestingly, at two days after transfection, all six truncations caused neuritic beading and dystrophy, reminiscent of neuronal morphologies observed in degeneration.36−38 In contrast, the effects of mock vector and expression of enhanced green fluorescent protein (EGFP) were mild, and the expression of U1-70K showed a moderate toxicity (Figure 6C). This conclusion was further confirmed by statistical analysis of quantitative data (i.e., counting morphologically altered neurons, Figure 6D).
Figure 6.

Toxicity analysis of N40K in rat hippocampal primary neurons. (A) Restriction enzyme digestion at the cloning sites by BamHI and EcoRI to show equal levels of the plasmids of U1-70K and six N40K truncations. (B) Western blotting to show similar protein expression in HEK293 cells. (C) Co-transfection of nine plasmids each with RFP by the calcium phosphate method. (D) Quantitative analysis by counting degenerating neurons. Error bars represent standard errors of mean (SEM) of analyzed neurons in duplicated plates of the same batch of neuronal culture. The experiments were repeated three times with different batches of culture, all showing similar results. The asterisks indicate p < 0.05 by the ANOVA and Tukey range test. (E) The MTT assay of neuronal viability upon Lentiviral N40K transfection. Error bars represent SEM. The double asterisks indicate p < 0.01 by Student’s t-test.
As all of the six N40K truncations showed almost identical effect during the morphology analysis, we selected a representative construct N300 and U1-70K to perform the MTT cell viability assay,39 in which mitochondrial activity in living cells converts MTT to formazan crystals for detection. Dying cells have less mitochondrial activity and, thus, produce weaker signals. Because this biochemical assay requires a large number of neurons expressing targeted proteins, we generated recombinant Lentivirus to infect hippocampal neurons. The U1-70K expression exhibited a moderate neurotoxity to decrease cellular viability by ∼35%, whereas the N300 expression resulted in ∼50% decrease compared to the control virus (Figure 6E). These results strongly support that N40K expression in cultured neurons induces cellular death.
Conclusions and Discussion
In this study, we have demonstrated that N40K, a cleaved fragment of U1-70K, occurs frequently in Alzheimer’s disease. Whereas the N-terminus of N40K is identical to that of U1-70K, N40K C-terminus is approximately Arg300 ± 6 residues. Extremely repetitive, hydrophilic residues in the C-terminal area raise a challenge to precisely define the cleavage site. All putative N40K truncations exhibit similar molecular properties, with respect to protein expression level and neuronal toxicity, suggesting that the accumulation of N40K in human brain may contribute to neuronal degeneration in Alzheimer’s disease.
Interestingly, we have found that the N40K level is inversely correlated with the total level of full length U1-70K, which might result in the loss of function of U1-70K in addition to the toxic gain of function of N40K. It is notable that expression of C-terminal fragment of TDP-43,40 another RNA processing factor involved in the pathogenesis of ALS, also caused similar suppression of endogenous full-length protein.41 Both N40K and the TDP-43 fragments contain RNA recognition motifs, suggesting a common self-regulation mechanism of RNA binding proteins. Indeed, another U1 snRNP subunit U1A binds to U1 snRNA to maintain its expression level.42 It is also possible that the reduction of U1-70K level is primarily caused by proteolytic cleavage because numerous proteases are activated in AD, such as caspases, calpains, and cathepsins.43−45 Moreover, it is known that U1-70K is cleaved by caspase-3, granzyme B, and Cu/H2O2 oxidation reactions.46−48 However, these protease-derived fragments are larger than N40K. Further investigation of the protease responsible for N40K cleavage is important to elucidate its involvement in AD.
In addition, we have used several approaches for mapping N40K proteolytic site. N40K was first identified by virtual Western blotting reconstructed from GeLC–MS/MS and then validated by traditional Western blotting (Figure 1A and B). It needs to be mentioned that the virtual Western blot image showed a more complex pattern than conventional Western blot, partially because of ambiguity during gel excision. In addition, the GeLC–MS/MS detects proteins in a large dynamic range (∼10 000), whereas Western blot only displays proteins in a narrow dynamic range (<100). These features contribute to the different images from the two semiquantitative methods.
To overcome the undersampling problem for proteins of low abundance in shotgun LC–MS/MS analysis, we have developed an LC–SRM method to determine the N40K C-terminus. The direct monitoring of U1-70K peptides based on pre-existing recombinant protein data greatly enhanced the sensitivity of detection, which was critical for the analysis of N40K in a limited amount of human samples. Because of irregular amino acid composition of N40K C-terminus, no suitable peptides are compatible with standard LC–MS/MS settings. We fully explored the usage of Western blotting using a ladder of recombinant proteins, which restricted the C-terminus in a small region.
We have further tested a series of putative N40K truncations in HEK293 cells and primary neuronal culture. All N40K proteins had no obvious influence on HEK293 cells but clearly demonstrated toxicity in rat primary neurons. Although full length U1-70K also led to a moderate toxicity, reminiscent of cell death caused by the expression of the RNA processing factor TDP-43,49 U1-70K had less effect than the N40K fragments. Apoptosis is a fundamental process essential for tissue homeostasis and its dysfunction is involved in AD pathogenesis.50 Neuronal apoptosis results in neuritic degeneration manifested by beading, thinning, and breakage, as well as the destruction of mitochondria.36−38 N40K-expressing neurons displayed these morphological changes reported in apoptotic neurons and showed mitochondrial deficit in the MTT assay, suggesting that the toxicity in cell culture might be mediated by apoptotic events, although other alternative cell death mechanisms cannot be excluded.
In summary, we have identified that the N40K fragment in AD not only is associated with the reduction of full length U1-70K but also directly exerts toxic effects in cultured neurons, providing lines of evidence to support that the U1-70K cleavage event occurs in human AD brain and might contribute to the neurodegeneration in AD.
Acknowledgments
The authors thank all of the lab members for helpful discussion. This work was partially supported by NIH grants P50AG005136, and American Academy of Neurology Foundation Clinical Research Training Fellowship to C.M.H. J.P. is supported by ALSAC (American Lebanese Syrian Associated Charities). The MS analysis was performed in the St. Jude Children’s Research Hospital Proteomics Facility, which is partially supported by NIH Cancer Center Support Grant (P30CA021765).
Glossary
Abbreviations
- LC
liquid chromatography
- MS
mass spectrometer
- U1 snRNP
U1 small nuclear ribonucleoprotein complex
- AD
Alzheimer’s disease
Supporting Information Available
Demographic information, LC–SRM analysis, Western blots, MS/MS spectra, and retention time data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- 2012 Alzheimer’s disease facts and figures. Alzheimer's Dementia 2012, 8 (2), 131–68. [DOI] [PubMed] [Google Scholar]
- Tanzi R. E., The genetics of Alzheimer disease. Cold Spring Harbor Perspect. Med. 2012, 2 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Kanekiyo T.; Xu H.; Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 92106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T.; Stefansson H.; Steinberg S.; Jonsdottir I.; Jonsson P. V.; Snaedal J.; Bjornsson S.; Huttenlocher J.; Levey A. I.; Lah J. J.; Rujescu D.; Hampel H.; Giegling I.; Andreassen O. A.; Engedal K.; Ulstein I.; Djurovic S.; Ibrahim-Verbaas C.; Hofman A.; Ikram M. A.; van Duijn C. M.; Thorsteinsdottir U.; Kong A.; Stefansson K. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 3682107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H.; Daly M. J. Variant TREM2 as risk factor for Alzheimer’s disease. N. Engl. J. Med. 2013, 3682182–4. [DOI] [PubMed] [Google Scholar]
- Taylor J. P.; Hardy J.; Fischbeck K. H. Toxic proteins in neurodegenerative disease. Science 2002, 29655751991–5. [DOI] [PubMed] [Google Scholar]
- Glenner G. G.; Wong C. W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 1203885–90. [DOI] [PubMed] [Google Scholar]
- Masters C. L.; Simms G.; Weinman N. A.; Multhaup G.; McDonald B. L.; Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U. S. A. 1985, 82124245–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee V. M.; Balin B. J.; Otvos L. Jr.; Trojanowski J. Q. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991, 2514994675–8. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002, 2975580353–6. [DOI] [PubMed] [Google Scholar]
- Ballatore C.; Lee V. M.; Trojanowski J. Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 89663–72. [DOI] [PubMed] [Google Scholar]
- Pimplikar S. W.; Nixon R. A.; Robakis N. K.; Shen J.; Tsai L. H. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J. Neurosci. 2010, 304514946–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass spectrometry-based proteomics. Nature 2003, 4226928198–207. [DOI] [PubMed] [Google Scholar]
- Peng J.; Gygi S. P. Proteomics: the move to mixtures. J. Mass Spectrom. 2001, 36101083–91. [DOI] [PubMed] [Google Scholar]
- Cravatt B. F.; Simon G. M.; Yates J. R. 3rd The biological impact of mass-spectrometry-based proteomics. Nature 2007, 4507172991–1000. [DOI] [PubMed] [Google Scholar]
- Mann M.; Kulak N. A.; Nagaraj N.; Cox J. The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 2013, 494583–90. [DOI] [PubMed] [Google Scholar]
- Zhou J. Y.; Jones D. R.; Duong D. M.; Levey A. I.; Lah J. J.; Peng J. Proteomic Analysis of Postsynaptic Density in Alzheimer Disease. Clin. Chim. Acta 2013, 420, 62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal Y. M.; Duong D. M.; Gearing M.; Cheng D.; Hanfelt J. J.; Funderburk C.; Peng J.; Lah J. J.; Levey A. I. Proteomics analysis reveals novel components in the detergent-insoluble subproteome in Alzheimer’s disease. J. Proteome Res. 2009, 8115069–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal Y. M.; Dammer E. B.; Duong D. M.; Cheng D.; Gearing M.; Rees H. D.; Peng J.; Lah J. J.; Levey A. I. Proteomic analysis of hippocampal dentate granule cells in frontotemporal lobar degeneration: application of laser capture technology. Front. Neurol. 2011, 2, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal Y. M.; Seyfried N. T.; Gearing M.; Glass J. D.; Heilman C. J.; Wuu J.; Duong D. M.; Cheng D.; Xia Q.; Rees H. D.; Fritz J. J.; Cooper D. S.; Peng J.; Levey A. I.; Lah J. J. Aberrant septin 11 is associated with sporadic frontotemporal lobar degeneration. Mol. Neurodegener. 2011, 6, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammer E. B.; Na C. H.; Xu P.; Seyfried N. T.; Duong D. M.; Cheng D.; Gearing M.; Rees H.; Lah J. J.; Levey A. I.; Rush J.; Peng J. Polyubiquitin linkage profiles in three models of proteolytic stress suggest the etiology of Alzheimer disease. J. Biol. Chem. 2011, 2861210457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q.; Cheng D.; Duong D. M.; Gearing M.; Lah J. J.; Levey A. I.; Peng J. Phosphoproteomic analysis of human brain by calcium phosphate precipitation and mass spectrometry. J. Proteome Res. 2008, 772845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L.; Cheng D.; Wang J.; Duong D. M.; Losik T. G.; Gearing M.; Rees H. D.; Lah J. J.; Levey A. I.; Peng J. Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J. Biol. Chem. 2004, 2793537061–8. [DOI] [PubMed] [Google Scholar]
- Seyfried N. T.; Gozal Y. M.; Dammer E. B.; Xia Q.; Duong D. M.; Cheng D.; Lah J. J.; Levey A. I.; Peng J. Multiplex SILAC analysis of a cellular TDP-43 proteinopathy model reveals protein inclusions associated with SUMOylation and diverse polyubiquitin chains. Mol. Cell. Proteomics 2010, 94705–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. Y.; Afjehi-Sadat L.; Asress S.; Duong D. M.; Cudkowicz M.; Glass J. D.; Peng J. Galectin-3 Is a Candidate Biomarker for Amyotrophic Lateral Sclerosis: Discovery by a Proteomics Approach. J. Proteome Res. 2010, 9105133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B.; Hales C. M.; Chen P. C.; Gozal Y.; Dammer E. B.; Fritz J. J.; Wang X.; Xia Q.; Duong D. M.; Street C.; Cantero G.; Cheng D.; Jones D. R.; Wu Z.; Li Y.; Diner I.; Heilman C. J.; Rees H. D.; Wu H.; Lin L.; Szulwach K. E.; Gearing M.; Mufson E. J.; Bennett D. A.; Montine T. J.; Seyfried N. T.; Wingo T. S.; Sun Y. E.; Jin P.; Hanfelt J.; Willcock D. M.; Levey A.; Lah J. J.; Peng J. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 2013, 1104116562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales C. M.; Dammer E. B.; Diner I.; Yi H.; Seyfried N. T.; Gearing M.; Glass J. D.; Montine T. J.; Levey A. I.; Lah J. J. Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain Pathol. 2014, 10.1111/bpa.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley J. P.; Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 1998, 923315–26. [DOI] [PubMed] [Google Scholar]
- Ross C. A.; Poirier M. A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10SupplS10–7. [DOI] [PubMed] [Google Scholar]
- Hyman B. T.; Trojanowski J. Q. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J. Neuropathol. Exp. Neurol. 1997, 56101095–7. [DOI] [PubMed] [Google Scholar]
- Xu P.; Duong D. M.; Peng J. Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome Res. 2009, 883944–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J.; Elias J. E.; Thoreen C. C.; Licklider L. J.; Gygi S. P. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC–MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2003, 2, 43–50. [DOI] [PubMed] [Google Scholar]
- Elias J. E.; Gygi S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 43207–14. [DOI] [PubMed] [Google Scholar]
- Seyfried N. T.; Xu P.; Duong D. M.; Cheng D.; Hanfelt J.; Peng J. Systematic approach for validating the ubiquitinated proteome. Anal. Chem. 2008, 80114161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonissone S.; Gupta N.; Romine M.; Bradshaw R. A.; Pevzner P. A. N-terminal protein processing: a comparative proteogenomic analysis. Mol. Cell. Proteomics 2013, 12114–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla K. V.; Goldman S. T.; Lattarulo C. R.; Wu H. Y.; Hyman B. T.; Bacskai B. J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 592214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M.; Shepardson N.; Yang T.; Chen G.; Walsh D.; Selkoe D. J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U. S. A. 2011, 108145819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A.; McLaughlin T.; O’Leary D. D.; Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 4577232981–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lobner D. Comparison of the LDH and MTT assays for quantifying cell death: validity for neuronal apoptosis?. J. Neurosci. Methods 2000, 962147–52. [DOI] [PubMed] [Google Scholar]
- Neumann M.; Sampathu D. M.; Kwong L. K.; Truax A. C.; Micsenyi M. C.; Chou T. T.; Bruce J.; Schuck T.; Grossman M.; Clark C. M.; McCluskey L. F.; Miller B. L.; Masliah E.; Mackenzie I. R.; Feldman H.; Feiden W.; Kretzschmar H. A.; Trojanowski J. Q.; Lee V. M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 3145796130–3. [DOI] [PubMed] [Google Scholar]
- Nonaka T.; Kametani F.; Arai T.; Akiyama H.; Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 2009, 18183353–64. [DOI] [PubMed] [Google Scholar]
- Kambach C.; Mattaj I. W. Intracellular distribution of the U1A protein depends on active transport and nuclear binding to U1 snRNA. J. Cell Biol. 1992, 118111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais F. G.; Xu D.; Robertson G. S.; Vaillancourt J. P.; Zhu Y.; Huang J.; LeBlanc A.; Smith D.; Rigby M.; Shearman M. S.; Clarke E. E.; Zheng H.; Van Der Ploeg L. H.; Ruffolo S. C.; Thornberry N. A.; Xanthoudakis S.; Zamboni R. J.; Roy S.; Nicholson D. W. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999, 973395–406. [DOI] [PubMed] [Google Scholar]
- Saito K.; Elce J. S.; Hamos J. E.; Nixon R. A. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc. Natl. Acad. Sci. U. S. A. 1993, 9072628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A. M.; Nixon R. A. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc. Natl. Acad. Sci. U. S. A. 1990, 87103861–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L.; Nicholson D. W.; Chong T.; Rowan K. R.; Thornberry N. A.; Miller D. K.; Rosen A. Apopain/CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J. Exp. Med. 1996, 18351957–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L.; Andrade F.; Ulanet D.; Wong W. B.; Rosen A. Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J. Exp. Med. 1999, 1906815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L.; Wigley F.; Rosen A. Scleroderma autoantigens are uniquely fragmented by metal-catalyzed oxidation reactions: implications for pathogenesis. J. Exp. Med. 1997, 185171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. B.; Lee V. M.; Trojanowski J. Q. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2012, 13138–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J.; Yankner B. A. Apoptosis in the nervous system. Nature 2000, 4076805802–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.