

Abstract
Prion diseases are a heterogeneous group of neurodegenerative disorders affecting various mammals including humans. Prion diseases are characterized by a misfolding of the host-encoded prion protein (PrPC) into a pathological isoform termed PrPSc. In wild-type mice, PrPC is attached to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor and PrPSc typically accumulates in diffuse nonamyloid deposits with gray matter spongiosis. By contrast, when mice lacking the GPI anchor are infected with the same prion inoculum, PrPSc accumulates in dense perivascular amyloid plaques with little or no gray matter spongiosis. In order to evaluate whether different host biochemical pathways were implicated in these two phenotypically distinct prion disease models, we utilized a proteomics approach. In both models, infected mice displayed evidence of a neuroinflammatory response and complement activation. Proteins involved in cell death and calcium homeostasis were also identified in both phenotypes. However, mitochondrial pathways of apoptosis were implicated only in the nonamyloid form, whereas metal binding and synaptic vesicle transport were more disrupted in the amyloid phenotype. Thus, following infection with a single prion strain, PrPC anchoring to the plasma membrane correlated not only with the type of PrPSc deposition but also with unique biochemical pathways associated with pathogenesis.
Keywords: LC−MS/MS, proteomics, prion disease, transmissible spongiform encephalopathies, glycosylphosphatidylinositol, amyloid, cerebral amyloid angiopathy, Alzheimer’s disease
Introduction
Prion diseases, also known as transmissible spongiform encephalopathies or TSEs, are a group of rare neurodegenerative disorders that affect humans and other mammals.1 Animal forms of prion disease include sheep scrapie, chronic wasting disease in deer and bovine spongiform encephalopathy in cattle.1 Human prion disorders include familial, sporadic and variant Creutzfeldt–Jakob disease (CJD), as well as kuru and Gerstmann–Sträussler–Scheinker (GSS) syndrome. A critical event in prion pathogenesis is the conversion of host-encoded prion protein (PrPC) into a misfolded isoform, termed PrPSc, which is widely believed to be the primary component of the infectious prion agent. Pathogenic PrPSc has the same amino acid sequence as PrPC but has significantly more β-sheet structure and is no longer monomeric.2,3 Variable disease phenotypes induced by different prion strains4−6 are often attributed to specific PrPSc conformations,7 perhaps defined by specific interactions between amino acid side-chains within individual β strands.8
Although no direct link between a specific structural motif in PrPSc and a specific in vivo prion phenotype has been made, PrPC anchoring to the plasma membrane has been shown to profoundly influence prion disease phenotypes.9−11 PrPC is normally attached to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor.12 When wild-type C57BL/10 mice are infected intracranially with the RML mouse prion strain, PrPSc accumulates in diffuse nonamyloid deposits and brain pathology is distinguished by the hallmark gray matter spongiosis for which TSEs are named. Conversely, when transgenic mice homozygous for PrPC lacking the GPI anchor (Tg44 or “anchorless” mice) are infected with the same RML strain, the incubation time is longer and PrPSc accumulates in dense perivascular amyloid plaques with little or no gray matter spongiosis.9−11 Similar amyloid PrPSc deposition and disease resulting in death has been observed in humans with an aberrant stop codon in the C-terminal portion of the PrP sequence, resulting in the generation of PrP lacking the GPI anchoring group as well as some amino acid residues.13−16 Thus, GPI-anchoring of PrPC to the plasma membrane appears to be a primary factor determining the type of PrPSc deposition and the development of brain pathology.
Despite the neuropathological differences between these two prion disease phenotypes, the mechanisms underlying these differences remain unclear. Both anchorless and wild-type mice displayed a similar cytokine release profile when infected with the 22L prion agent, suggesting a similar inflammatory response.17 When enriched PrPSc samples from RML-infected wild-type or anchorless Tg44 mouse brains were compared by infrared spectroscopy, gross differences in secondary structure were minimal.18 Furthermore, infectivity replicated to high levels in both types of mice and, when passaged back into wild-type mice, both led to prion disease typical of the original RML strain.9 Taken together, the data suggest that the dramatic phenotypic differences between these two prion diseases is determined more by the presence or absence of the GPI anchor on PrPC rather than by any changes in strain-specific β-sheet conformation within PrPSc.
Multiple studies have used microarray analysis to determine whether different pathways underlie differences in disease pathogenesis between mouse prion strains19−24 or various types of human CJD.25 A postgenomic, proteomics approach has been utilized in some studies,26−30 several of which have focused on the identification of potential prion disease biomarkers (refs (26, 31), and see ref (32) for review). However, there are no genomic or proteomic studies directly comparing two different prion diseases induced by the same prion strain. In the absence of any strain-dependent phenotypic changes, such an analysis could provide useful insights into the general pathways which underlie different prion disease phenotypes.
In the present study, wild-type C57BL/10 and anchorless Tg44 mice were infected intracranially with the RML mouse prion strain. Brain homogenates from clinically positive mice were profiled for gross differences between the nonamyloid (C57BL/10 mice) and amyloid (Tg44 mice) disease phenotypes by utilizing mass spectrometry to identify changes in protein abundance. Proteomic comparison of these two disease phenotypes allowed us to distinguish the biochemical aspects of prion pathogenesis that were likely related to a generalized host inflammatory response versus those that may be more specific to each disease model.
Experimental Procedures
Reagents and Supplies
Dithiothreitol, iodoacetamide, ammonium bicarbonate, trifluoroethanol (TFE), tributylphosphine, and membrane solubilization buffer were purchased from Sigma (St. Louis, MO, U. S. A.). Porcine trypsin (#V5111) was purchased from Promega (Madison, WI, U. S. A.). Burdick & Jackson water and acetonitrile were purchased from VWR (Pittsburgh, PA, U. S. A.). Formic acid (FA) ampules and Imperial Coomassie blue stain were purchased from Thermo-Fisher Scientific (Pittsburgh, PA, U. S. A.). SDS-PAGE was run using reagents and gels from Life Technologies-Invitrogen Corporation (Carlsbad, CA, U. S. A.), with NuPAGE Bis–Tris gels.
Histopathology and Immunohistochemistry
The protocols used were described previously.9 Briefly, PrP staining was done using an avidin–biotin complex immunoperoxidase protocol using anti-PrP antibody D13 (In-Pro Biotechnology, South San Francisco, CA, U. S. A.). Staining with a 1% Thioflavin S solution was done as previously described.11
Preparation of Mouse Brain Homogenates
The mice used in this study were housed at the Rocky Mountain Laboratories (RML) in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and the research protocols used were approved by the NIH RML Animal Care and Use Committee. Uninfected wild-type mice (C57BL/10SnJ) were obtained from Jackson Laboratories (Bar Harbor, ME, U. S. A.). Transgenic mice homozygous for PrPC without the GPI anchor (Tg44) were described previously.9
Six to eight week old mice were inoculated intracerebrally as described9 with 50 μL of a 1% brain homogenate of RML containing 0.7–1.0 × 106 ID50. Animals were then observed daily for onset and progression of TSE disease. Mice were euthanized at the time of advanced clinical disease as described previously33 and this was defined as the incubation period. Wild-type C57BL/10 mice developed clinical signs with a mean incubation time of 148 ± 9 days postinfection (dpi), whereas Tg44 mice developed clinical disease with a mean incubation time of 318 ± 16 dpi. For age-matched controls, uninoculated 188 day old C57BL/10 mice and 401 day old Tg44 mice were used. For all mice, brain samples were collected and flash frozen in liquid nitrogen then kept at −80 °C until processed. Brain tissue was homogenized as a 20% (w/v) brain homogenate in 0.01 M Tris, pH 7.2, using a mini bead beater (Biospec, Inc.), then vortexed for 1 min followed by 1 min sonication in a Misonix cuphorn sonicator and kept at −80 °C until needed.
Western Blotting
Brain homogenates analyzed without proteinase K (PK) treatment were diluted from 20% to 2.5% and boiled in Tris-buffered saline, pH 8, and sample buffer (Life Technologies) with 0.5% β-mercaptoethanol. Brain homogenates treated with PK were diluted to 6% in the presence of 0.1 M Tris, pH 8.3, 1% Triton X-100, 1% sodium deoxycholate, then digested with 125 μg/mL PK for 1 h at 37 °C and then boiled in an equal volume of sample buffer. Portions of each sample (4 μL) were separated by SDS-PAGE on a 10% Bis–Tris gel with MES running buffer (Life Technologies) and then transferred to an Immobilon PVDF-FL membrane (Millipore) by wet transfer using Towbin’s buffer. Samples were probed for PrP with mouse monoclonal antibody 6D11 (Covance Inc.) at a dilution of 1:15 000 in TBST buffer for 1 h. 6D11 recognizes residues 93–109 of mouse PrP. Antivimentin antibody (Origene #TA307358) was used at 1:2000. Anti-apoE antibody (Calbiochem #178479) was used at 1:1000 and antiactin (abcam #ab1801) was used at 1:1000. Secondary antibodies (Li-Cor Biosciences) were IRDye 800CW or IRDye 680CW anti-mouse, anti-rabbit or anti-goat, used at a 1:15 000 dilution in TBST buffer for 40 min. Imaging was performed with an Odyssey imaging system (Li-Cor) using the 800 and 680 channels.
In-Gel Trypsin Digestion
The in-gel tryptic digest procedure was conducted using 16 individual brains with 4 from each biological group: (1) RML-infected Tg44 mice, (2) age-matched uninoculated Tg44 mice, (3) RML-infected wild-type C57BL/10 mice, and (4) age-matched C57BL/10 uninoculated mice. The 20% (w/v) brain homogenates were diluted with denaturant to a final concentration of 5.3 M urea, 1.5 M thiourea, 0.8% detergent (3-(4-heptyl)phenyl-3hydroxypropyl)dimethylammoniopropanesulfonate), 2 mM tributylphosphine and 14 mM DTT, and then warmed at 37 °C for 20 min. Iodoacetamide was then added to a concentration of 50 mM and incubated at room temperature with gentle agitation in the dark for 30 min. The alkylation reaction was quenched with DTT to a final concentration of 80 mM. The mixture was then diluted with 4x sample buffer (Life Technologies), vortexed briefly, incubated at 37 °C for 20 min, and then subjected to centrifugation for 30 min at 22 000g. A 20 μL portion of the reduced and alkylated mixture corresponding to 0.5 mg brain equivalents or approximately 50 μg of total protein was loaded for SDS-PAGE onto a 10% Bis–Tris 1.5 mm thick NuPAGE gel with MES running buffer and run for 90 min at 150 V constant voltage. This was repeated for each of the 4 biological replicates from each of the 4 sample groups for a total of 16 lanes (16 brains). The gel was then stained with Imperial Coomassie blue (Thermo Fisher). Gels were subsequently destained with water, cut with a razor blade into 24 slices and then stored in individual tubes at −20 °C until needed. In-gel tryptic digestion of each gel slice was conducted overnight in a mixture of 10% trifluoroethanol in 50 mM ammonium bicarbonate, pH 8 at 37 °C for 16 h and then extracted with 70% acetonitrile as described previously.34 Each digest was then subjected to centrifugation at 22 000g (10 min) and transferred to an autosampler vial for liquid chromatography tandem mass spectrometry (LC–MS/MS) analysis.
HPLC Chip-Based Nanospraytandem Mass Spectrometry
Trypsin-digested proteins were identified by LC–MS/MS using an Agilent 1200 interfaced to an XCT Ultra Ion Trap via a microfluidic HPLC chip-cube and a nanospray source. The ion trap was calibrated externally using a tuning mix provided by Agilent specifically for this instrument. Data-dependent MS acquisition was performed with dry gas (nitrogen/air) set to 4 L/min at 350 °C, MS capillary voltage 1800 V, and a maximum accumulation time of 150 ms. The MS scan range was set to 300–1400 m/z in the Ultrascan mode. Four parent ions were selected for each MS/MS cycle with a fragmentation amplitude of 1.0 V and the SmartFrag setting on. Active exclusion for fragmentation was set after 2 spectra in order to facilitate detection of less abundant ions. The tryptic digests were loaded onto the chip (Agilent #G4240-62006, Zorbax 300SB-C18, 5 μm, 75 μm × 150 mm) with an autosampler and washed with Buffer A (3% acetonitrile/H2O and 0.1% formic acid) prior to elution at 300 nL/min by reversed-phase chromatography. The gradient was 6–10% Buffer B (90% acetonitrile and 0.1% formic acid) over 12 min, to 20% B by 65 min, to 30% B by 120 min, to 40% B by 140 min, and to 80% B by 147 min. The column was washed with 95% B until 155 min, followed by a 4 min postrun re-equilibration at 3% B with a total run time of approximately 160 min. Separate chips were used for each biological grouping of samples in order to minimize the possibility of contamination.
Protein Identification
Raw data were processed into peak lists using MASCOT Distiller v2.4.3.0. The MGF files were searched using MASCOT Daemon against a target database (www.uniprot.org) filtered for mouse taxonomy consisting of 16 315 entries. A separate reverse-sequence decoy database with the same number of entries was created from the target database using a Perl script provided by NuSep Inc. (Athens, GA, U. S. A.). The trypsin/P search parameters for MASCOT protein identification consisted of one missed tryptic cleavage allowed with a fixed carbamidomethylation (+57, Cys) and a variable oxidation (+16, Met). Mass tolerances of 2.4 and 1.0 Da were used for parent and monoisotopic fragment ions, respectively. The resulting DAT files from MASCOT were used as input files for further analysis using the ProteoIQ software bioinformatics platform (Premier Biosoft Inc.). Protein identifications with a protein false discovery rate (FDR) of ≤1% were calculated using ProteoIQ’s implementation of the ProValT algorithm35 with the constraints that only MASCOT ion scores of ≥30 and only peptides of ≥7 amino acids in length were considered in these initial calculations. The resulting 2026 protein identifications were further filtered to require at least 2 unique peptides and a protein group probability of ≥0.936 for a positive identification. Additionally, each peptide was limited to only one protein identification such that the number of protein groups equaled the number of protein identifications. The list of 1567 proteins identified according to these criteria is provided as Supporting Information Table 1.
Spectral Counting
Two data sets were derived from the original 1567 protein identifications, one from the C57BL/10 mice and the other from the Tg44 mice. For label-free quantitation,37,38 the 1567 protein identifications (Supporting Information Table 1) were normalized by dividing the spectral count (SpC) for each protein from each mouse by the mean of all proteins from that individual mouse. Once normalized, a sum was calculated for each protein for each group of mice (n = 4), and the fold change for each protein was calculated by dividing the sum for the four scrapie-infected mice by the sum from the four uninfected mice. All calculations were done using Microsoft Excel 2010. A set of ρ values was obtained by running an analysis of variance (ANOVA; Partek Genomics Suite v6.6) for the comparisons of interest, and multiple test corrected using the false discovery rate (FDR) method of Benjamini and Hochberg.39 Any proteins with a SpC average below 5 in both the infected and uninfected groups were flagged as below detection because it has been reported that mean counts of less than 5 spectra are often unreliable for quantitation.37,38
In order to identify the proteins that were most affected by prion infection in each mouse line, an ascending rank order analysis was used. Individual rankings were derived based upon the combination of greatest fold change, lowest ρ value, lowest coefficient of variation, and spectral counts from the two conditions compared (i.e., uninfected versus infected brain homogenate). All four criteria were thus given equal weight in the analysis and were used to estimate changes in relative protein abundance between RML-infected brain homogenates and their respective uninfected controls for each mouse strain. The resulting four ranks for any given protein were summed and divided by the square of their sum to produce one number from all ranks. This final number was then ranked in descending order to produce the overall reported rank. Thus, the protein with the best result across all criteria produced a rank of one, followed by the rank of two for second best and continuing for each of the 1567 proteins identified. Based on the final protein rankings, the top 100 proteins were used for subsequent IPA analysis (Supporting Information Table 2).
Ingenuity Pathway Analysis
Functional analyses and pathways were examined with Ingenuity Pathway Analysis (IPA,Ingenuity Systems, www.ingenuity.com). The top 100 ranked proteins from the Tg44 and the C57BL/10 lists, most of which had ρ values of ≤0.05, were evaluated against the Ingenuity Knowledge Base. The IPA database organizes lists of genes into graphical representations based upon text-mining of published interactions between the genes of interest and various predefined networks, disease classifications, canonical pathways or biofunctions. The scores assigned to the data sets by IPA, based upon their degree of overlap with the experimental data, are the negative exponent of the right-tailed Fisher’s exact test result. The ρ values reported estimate the probability that each disease classification or pathway assigned to the data could have been determined by random chance. For example, a ρ value of 0.01 would indicate a 1% false discovery rate for a set of protein associations with a particular classification. A random set of 100 proteins from the Swiss-Prot murine database generated IPA ρ values of 7.1 × 10–5–1.5 × 10–2 (data not shown). Therefore, we considered the association of proteins with an IPA ρ value of ≤5 × 10–5 with a particular molecular functional group to be significant and outside of the realm of random chance.
Results
Inoculation of C57BL/10 and Tg44 Mice with the RML Mouse Scrapie Strain
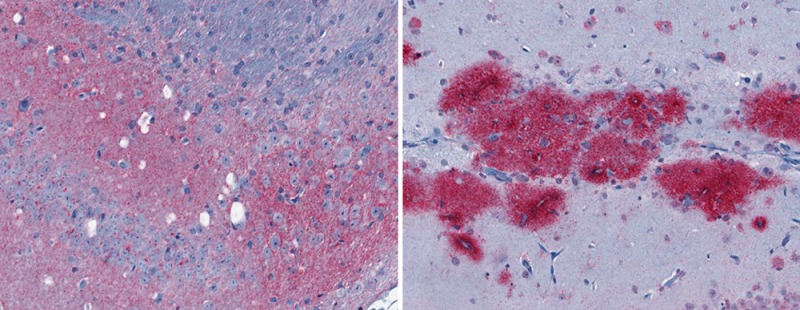
After intracranial inoculation of RML mouse scrapie, significant gray matter spongiosis was observed only in RML infected C57BL/10 mice (Figure 1A inset). C57BL/10 mice expressing GPI-anchored PrPC also exhibited diffuse PrPSc deposits in the brain (Figure 1A) and had a disease incubation time of approximately 150 days. By contrast, the same inoculum into Tg44 mice, which express anchorless PrPC, led to the deposition of PrPSc in large plaques (Figure 1B) with little or no gray matter spongiosis (Figure 1B inset) and an incubation time of approximately 320 days. When brain sections were stained with the amyloid-binding dye thioflavin S, the diffuse PrPSc deposits in C57BL/10 mice were negative (data not shown), whereas the large plaque-like PrPSc deposits in Tg44 mice were positive (Figure 1C). The disease phenotype of RML infected C57BL/10 mice will therefore be referred to as nonamyloid, whereas that of RML infected Tg44 mice will be referred to as amyloid. Consistent with previous results,9,10 these data show that the presence or absence of a GPI anchor on PrPC can induce two markedly distinct prion diseases. Moreover, the neuropathological differences observed between the two mouse models suggest that the biological pathways underlying prion pathogenesis in these mice fundamentally differ.
Figure 1.
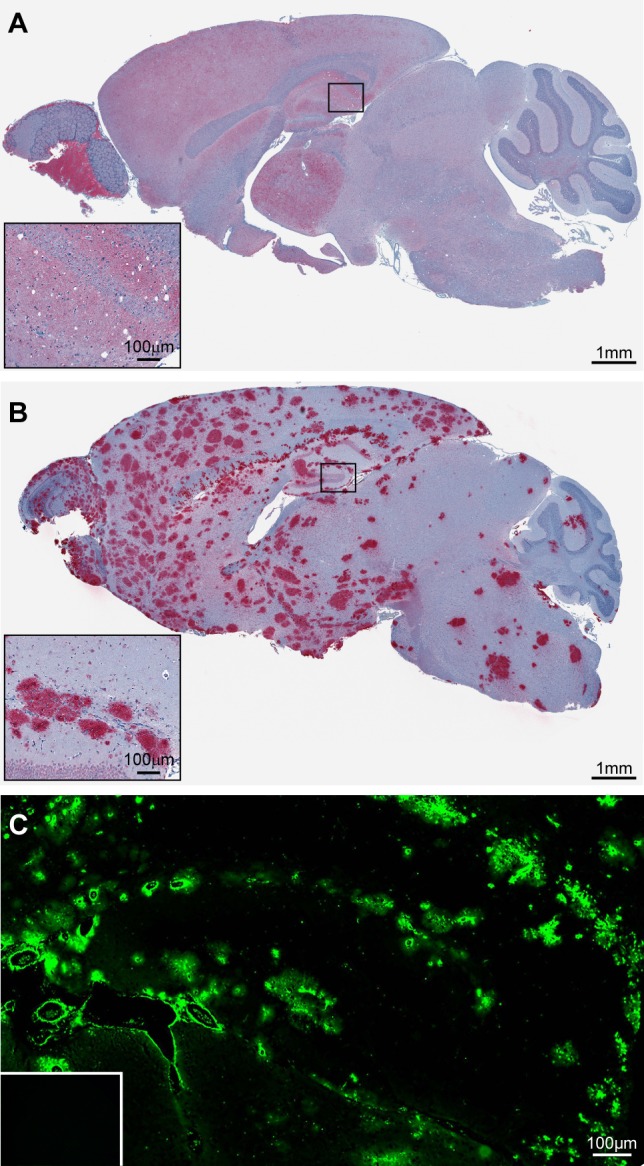
Infection of C57BL/10 and Tg44 mice with the RML prion strain yields two distinct disease phenotypes. (A) Immunostaining of RML infected C57BL/10 brain tissue with D13 prion antibody shows diffuse PrPSc deposition in most regions of the brain in addition to spongiform degeneration. (B) Immunostaining of RML infected Tg44 brain tissue with D13 prion antibody shows PrPSc deposition in the absence of spongiform degeneration. (C) Staining of the brain hippocampal region with the amyloid-specific fluorescent dye Thioflavin S confirms the presence of amyloid plaque in RML infected Tg44 brain. Thioflavin S staining is negative in uninfected Tg44 mice (inset).
Identification and Quantitation of Proteins Expressed in the Brains of Mice with the Amyloid and Nonamyloid Disease Phenotypes
In order to gain more insight into these potential differences, we adopted a proteomics approach. Brain homogenates from infected versus uninfected Tg44 and C57BL/10 mice were examined, each with four biological replicates, for a total of 16 individual brains. A representative Coomassie blue stained gel loaded with approximately 50 μg of protein for each replicate demonstrated a similar protein staining profile for each sample (Figure 2A). As outlined in Figure 2B, the initial list of proteins identified was set to a false discovery rate (FDR) threshold of ≤1%,35 resulting in 2026 nonredundant protein identifications. These data were additionally filtered so that only proteins identified by ≥2 unique peptides and a protein group probability of ≥0.9 based upon the Protein Prophet algorithm36 were selected for further analysis. The resulting protein list contained 1567 high-confidence protein identifications (Supporting Information Table 1), derived from a total of 12 834 unique peptides across all of the biological groups. The number of protein identifications was similar between groups (Figure 2C, left panel) with the vast majority of proteins common to all four groups (Figure 2C, right panel), indicating good consistency in protein sampling.
Figure 2.

Identification of proteins from prion-infected and uninfected brain homogenates. (A) Coomassie blue staining of brain homogenate from each of the 16 individual brains used for LC–MS/MS analysis. (B) Trypsin digest, LC–MS/MS data collection, processing and searching with MASCOT, and spectral counting with ProteoIQ resulted in 2026 proteins with a calculated FDR of ≤1%. Further filtering of the data resulted in 1567 high-confidence protein identifications. (C) The number of protein identifications delineated by biological group in the pie chart shows approximately 1500 identifications per group. The Venn diagram shows that 1470 proteins (94%) of all the proteins identified were common to all of the biological groups studied prior to further filtering of the data for label-free quantitation. Tg-U = uninfected Tg44 mice; Tg-Sc = RML infected Tg44 mice; C57-U = uninfected C57BL/10 mice; C57-Sc = RML infected C57BL/10 mice.
Relative Abundance of PrP Varies between Biological Groups
The relative apparent abundance of the proteins between sample groups was examined with the goal of comparing the global effects of prion infection in the amyloid and nonamyloid disease phenotypes at the end-stage of clinical disease. In order to semiquantitatively compare the relative abundance of proteins between the groups, spectral counting was utilized. Spectral counting is often used as a practical, effective approach for estimating the relative abundance of proteins between biological samples37,38,40 and is the preferred method for quantifying profiling data from ion trap mass spectrometers such as the one used in this study.41
As expected, PrP was identified as a protein with an altered relative abundance when compared to uninfected controls. Based upon spectral counts, there was an approximate 5-fold increase in PrPC in uninfected C57BL/10 mice when compared to uninfected Tg44 mice (Figure 3A). Our semiquantitative analysis using spectral counts was therefore consistent with previous findings suggesting that uninfected Tg44 mice express approximately 8-fold less PrPC relative to uninfected C57BL/10 mice.9 The amount of PrP in brain homogenates from the two prion-infected mouse lines was also significantly different. In the absence of PK treatment, total PrP levels (i.e., PrPC and PrPSc) were significantly higher in mice with the amyloid disease phenotype than in any other sample group, demonstrating an apparent 91-fold higher concentration of total PrP (Table 1) when compared to age-matched controls (Figure 3A). By contrast, a 2-fold increase in total PrP was observed with the nonamyloid disease phenotype (Table 1) when compared to their uninfected controls (Figure 3A). These differences in the total amount of PrP were confirmed by immunoblot analysis (Supporting Information Figure 1A). After PK digestion, protease-resistant PrPSc was found only in the infected samples (Figure 3B). The increased levels of total PrP in both strains of infected mice therefore appears to represent the generation and accumulation of PrPSc.
Figure 3.

Comparison of PrP in brain homogenates. (A) A spectral count comparison of PrP between sample groups shows that total PrP concentrations are lowest in the uninfected Tg44 brains (Tg-U) and highest in the RML infected Tg44 brains (Tg-Sc). Data shown are the mean and SEM of four individual mouse brains per group. (B) Brain homogenates were subjected to treatment with proteinase K and then probed with 6D11, showing that PrPSc is present only in the prion-infected samples. C57-U = uninfected C57BL/10 mice; C57-Sc = RML infected C57BL/10 mice.
Table 1. Similar Pattern of Fold Change in the Top 100 Ranked Proteins Shared between the Amyloid and Nonamyloid Prion Disease Phenotypes.
| amyloid |
nonamyloid |
|||||
|---|---|---|---|---|---|---|
| IDa | gene | shared proteins | Δ foldb | rankc | Δ fold | rank |
| P04925 | PRNP | Major prion protein (PrP) | ↑ 91.0 | 36 | ↑ 2.0 | 86 |
| Q02105 | C1QC | Complement C1q subcomponent subunit C | ↑ 17.7 | 8 | ↑ 27.6 | 4 |
| P29699 | AHSG | α-2-HS-glycoprotein | ↑ 13.7 | 20 | ↑ 26.7 | 5 |
| P20152 | VIM | Vimentin | ↑ 11.9 | 66 | ↑ 7.9 | 46 |
| P98086 | C1QA | Complement C1q subcomponent subunit A | ↑ 9.4 | 7 | ↑ 11.7 | 4 |
| P14106 | C1QB | Complement C1q subcomponent subunit B | ↑ 7.8 | 32 | ↑ 10.2 | 1 |
| Q61838 | A2M | α-2-macroglobulin | ↑ 6.0 | 28 | ↑ 2.8 | 11 |
| P03995 | GFAP | Glial fibrillary acidic protein | ↑ 5.8 | 1 | ↑ 8.9 | 44 |
| Q8BJI1 | SLC6A17 | Na and Cl-dependent neurotransmitter transporter NTT4 | ↑ 5.0 | 51 | ↑ 7.4 | 65 |
| Q99L04 | DHRS1 | Dehydrogenase/reductase SDR family member 1 | ↑ 2.3 | 78 | ↑ 3.2 | 34 |
| P26443 | GLUD1 | Glutamate dehydrogenase 1, mitochondrial | ↑ 1.6 | 99 | ↑ 1.5 | 25 |
| Q9R0P9 | UCHL1 | Ubiquitin carboxyl-terminal esterase L1 | ↓ −1.5 | 77 | ↓ −1.3 | 89 |
| P62761 | VSNL1 | Visinin-like protein 1 | ↓ −1.9 | 31 | ↓ −1.3 | 60 |
| P18760 | CFL1 | Cofilin-1 | ↓ −2.3 | 12 | ↓ −1.4 | 62 |
| Q9R0P5 | DSTN | Destrin | ↓ −2.5 | 41 | ↓ −1.6 | 94 |
| Q5PR73 | DIRAS2 | GTP-binding protein Di-Ras2 | ↓ −2.7 | 92 | ↓ −2.3 | 88 |
| Q9QZ23 | NFU1 | NFU iron–sulfur cluster scaffold homologue, mitochondrial | ↓ −3.1 | 74 | ↓ −3.5 | 67 |
Protein identifiers and gene names are from www.uniprot.org.
Fold change in abundance relative to uninfected control.
Rank out of 1567 identified proteins based upon lowest p value, highest fold change, best count combination, and lowest coefficient of variation.
Neuroinflammation and Complement Activation Are Common to Both Prion Disease Phenotypes
Each of the 1567 identified proteins was ranked for both the amyloid and nonamyloid phenotypes and the top 100 ranked proteins (Supporting Information Table 2) from each data set were compared. These proteins had the strongest association with each disease phenotype and were thus the primary focus of our subsequent data analysis. Only 17 of the top 100 proteins were common to both disease phenotypes. In every instance, proteins that showed a significant fold increase or decrease in the amyloid disease phenotype mice showed similar fold increases or decreases in the nonamyloid disease phenotype (Table 1). Consistent with the gliosis observed in both prion disease models,9,10 the astrocyte protein glial fibrillary acidic protein (GFAP) was increased 5.8 and 8.9 fold in the amyloid and nonamyloid disease phenotypes, respectively (Table 1).42 Proteins such as vimentin, α-2-macroglobulin, and α-2-HS-glycoprotein, all of which have been linked to neuroinflammation in prion as well as other neurodegenerative diseases,43−48 were increased anywhere from 2.8–26.7 fold in both disease phenotypes (Table 1). The increased levels of vimentin in prion infected mice was further confirmed by immunoblot analysis (Supporting Information Figure 1B). Finally, multiple complement factors which have been linked to neuroinflammatory responses49 also had elevated spectral counts in both prion disease models with increases in relative abundance ranging from 7.8–27.6 fold (Table 1). It was unlikely that the increased relative abundance of these proteins was due to age-related differences between C57BL/10 and Tg44 mice because they were present at comparable levels in uninfected controls from both strains of mice (Figure 4A–F). Thus, the similar expression levels of these proteins in mice with both the amyloid and nonamyloid disease phenotypes may reflect a similar response and activation of astroglia and microglia to the two different types of brain damage that result from RML prion infection in Tg44 and C57BL/10 mice.
Figure 4.

No significant age-related differences in protein abundance in C57BL/10 and Tg44 mice. Average spectral counts (SpC) for proteins associated with neuroinflammation (A–F) and lipid metabolism (G–I). In general, the relative abundance of these proteins was the same between uninfected C57BL/10 (C57-U) and Tg44 mice (Tg-U), but greater in abundance in RML infected Tg44 mice (Tg-Sc) when compared to RML infected C57BL/10 (C57-U) mice. Data shown are the mean and SEM of four individual mouse brains per group.
Increased Levels of Proteins Associated with Fatty Acid Metabolism in Both Prion Disease Mouse Models
In mice with the amyloid disease phenotype, expression levels of several key apolipoproteins (apoD, apoE, apoJ) as well as two other proteins involved in lipid transport, peroxisomal multifunctional enzyme type 2 and low density lipoprotein receptor related protein 1 (LRP1), were elevated when compared to uninfected age-matched controls (Table 2). These proteins were generally much higher ranked in the amyloid-associated data set than they were in the nonamyloid data set. The greater abundance of these proteins in mice with the amyloid phenotype was further confirmed by immunoblot for one of them, apoE (Supporting Information Figure 1C). Furthermore, the differences in relative abundance did not appear to be due to age-related differences between Tg44 and C57BL/10 mice because these proteins were present at comparable levels in uninfected controls from both strains of mice (Figure 4G–I). Thus, these data suggest that, although lipid metabolism and transport are altered in both the amyloid and nonamyloid phenotypes, this disruption is more strongly linked to the amyloid form of disease.
Table 2. Altered Levels of Apolipoproteins and Other Proteins Involved in Fatty Acid Metabolism in Prion-Infected Mice with Either the Amyloid or Nonamyloid Phenotypes.
| amyloid |
nonamyloid |
||||
|---|---|---|---|---|---|
| proteina | Δ foldb | rankc | Δ fold | rank | functional annotationd |
| Apolipoprotein D (apoD) (P51910) | ↑ 56 | 55 | ↑ 15.8 | 334 | Lipid transport and metabolism |
| Annexin A2 (P07356) | ↑ 42.7 | 14 | ↑ 3.4 | 987 | Ca2+-dependent phospholipid binding |
| Peroxisomal multifunctional enzyme type 2 (P51660) | ↑ 11 | 193 | ↑ 1.1 | 1471 | Lipid and fatty acid metabolism |
| Apolipoprotein J (apoJ or Clusterin) (Q06890) | ↑ 8.2 | 3 | ↑ 9.1 | 122 | Extracellular chaperone, lipid metabolism |
| Apolipoprotein E (apoE) (P08226) | ↑ 7.6 | 5 | ↑ 2.7 | 269 | Lipid and cholesterol transport |
| Lipoprotein receptor-related protein 1 (LRP1) (Q91ZX7) | ↑ 2.5 | 13 | ↑ 1.5 | 752 | Lipid homeostasis, intracellular signaling |
Protein identifiers and gene names are from www.uniprot.org.
Fold change in abundance relative to uninfected control.
Rank out of 1567 identified proteins based upon lowest p value, highest fold change, best count combination, and lowest coefficient of variation.
Information derived from the Ingenuity Knowledge Database.
Molecular and Cellular Functions Disrupted in Prion-Infected Mice
In order to further differentiate the two prion disease phenotypes, the top 100 ranked proteins in each data set were analyzed using Ingenuity Pathway Analysis (IPA) which maps the molecular and cellular pathways associated with those proteins. Our initial analysis showed that individual proteins were often associated with multiple functional processes, making it difficult to determine which proteins and pathways were most specifically relevant to prion disease. Therefore, to identify the potential molecular mechanisms involved in pathogenesis, we focused our initial analysis on the molecular and cellular function categories, particularly on those subcategories which contained the highest percentage of unique proteins (Table 3). Using these criteria, cell death and survival was the top scoring group in the nonamyloid phenotype, with a higher percentage of unique proteins when compared to the other groups (29% versus 3–8%, see Table 3). By contrast, in the amyloid phenotype, the highest percentage of unique proteins was associated with the molecular transport category (23%) followed by cell death and survival at 14% (Table 3). Thus, whereas cellular pathways related to cell death and survival were associated with both the amyloid and nonamyloid disease phenotypes, pathways involved in molecular transport were most strongly implicated in the amyloid disease phenotype.
Table 3. Ingenuity Pathway Analysis of the Molecular and Cellular Functions Affected in the Amyloid and Nonamyloid Prion Disease Phenotypes.
| amyloid | ||
|---|---|---|
| molecular and cellular functiona | # proteinsb | unique (%)c |
| molecular transport | 39 | 23 |
| cell death and survival | 42 | 14 |
| cellular function and maintenance | 41 | 5 |
| cellular assembly and organization | 43 | 2 |
| nonamyloid | ||
|---|---|---|
| molecular and cellular function | # proteins | unique (%) |
| cell death and survival | 48 | 29 |
| cellular assembly and organization | 39 | 8 |
| cell-to-cell signaling and interaction | 36 | 8 |
| cell morphology | 31 | 3 |
The 100 top-ranked proteins associated with the amyloid and nonamyloid disease phenotypes were subjected to IPA analysis and the top molecular and functional categories determined.
Number of proteins in the top 100 from each data set that IPA associated with that functional category.
Percentage of proteins which are uniquely associated with the specific molecular and cellular function category listed and thus do not overlap with the other three.
Proteins Associated with Metal Binding and Synaptic Vesicle Transport Are Disrupted in Mice with the Amyloid Disease Phenotype
In mice with the amyloid disease phenotype, the 39 proteins implicated in molecular transport (Table 4) were involved in the binding and transport of multiple molecules including amino acids, metals, and ADP/ATP. Interestingly, many of these proteins were associated with metal-dependent processes, many of which involved calcium (Figure 5A). Further analysis revealed another protein known to bind calcium, calcineurin subunit type B1, that had not been identified by the IPA analysis. Finally, multiple proteins involved in calcium transport, regulation, and homeostasis were also linked to molecular transport in the amyloid phenotype (Supporting Information Table 3). Thus, our data suggest that metal ion transport, and in particular calcium homeostasis, is disrupted in the amyloid form of prion infection.
Table 4. Proteins Associated with Molecular Transport in Prion-Infected Mice with the Amyloid Disease Phenotype.
| amyloid |
nonamyloid |
|||||
|---|---|---|---|---|---|---|
| IDa | gene | molecular transport proteinsb | Δ foldc | rankd | Δ fold | rank |
| P04925 | PRNP | Major prion protein (PrP) | ↑ 91.0 | 36 | ↑ 2.0 | 86 |
| P51910 | APOD | Apolipoprotein D | ↑ 56.2 | 55 | --- | --- |
| Q91 × 72 | HPX | Hemopexin | ↑ 23.1 | 2 | --- | --- |
| O35874 | SLC1A4 | Neutral amino acid transporter A | ↑ 9.2 | 3 | --- | --- |
| P08226 | APOE | Apolipoprotein E | ↑ 7.6 | 5 | --- | --- |
| P61148 | FGF1 | Heparin-binding growth factor 1 | ↑ 7.4 | 47 | --- | --- |
| Q00493 | CPE | Carboxypeptidase E | ↑ 5.2 | 38 | --- | --- |
| Q00612 | G6PD | Glucose-6-phosphate dehydrogenase X | ↑ 4.5 | 46 | --- | --- |
| Q921I1 | TF | Transferrin | ↑ 4.5 | 9 | --- | --- |
| Q9Z1Q5 | CLIC1 | Chloride intracellular channel protein 1 | ↑ 3.7 | 59 | --- | --- |
| Q01405 | SEC23A | Protein transport protein | ↑ 3.4 | 49 | --- | --- |
| Q91WC3 | ACSL6 | Long-chain-fatty-acid-CoA ligase 6 | ↑ 2.8 | 90 | --- | --- |
| P18242 | CTSD | Cathepsin D | ↑ 2.5 | 43 | --- | --- |
| O88343 | SLC4A4 | Electrogenic sodium bicarbonate cotransporter 1 | ↑ 2.1 | 97 | --- | --- |
| Q6PIC6 | ATP1A3 | Sodium/potassium-transporting ATPase subunit alpha-3 | ↓ −1.4 | 73 | --- | --- |
| P48962 | SLC25A4 | ADP/ATP translocase 1 | ↓ −1.4 | 88 | --- | --- |
| Q9CR62 | SLC25A11 | Mitochondrial 2-oxoglutarate/malate carrier protein | ↓ −1.9 | 60 | --- | --- |
| P09671 | SOD2 | Superoxide dismutase [Mn], mitochondrial | ↓ −2.0 | 58 | --- | --- |
| P01831 | THY1 | Thy-1 cell surface antigen | ↓ −2.0 | 38 | --- | --- |
| O35658 | C1QBP | Complement component 1Q, mitochondrial | ↓ −2.6 | 76 | --- | --- |
| P60766 | CDC42 | Cell division control protein 42 homologue | ↓ −3.5 | 22 | --- | --- |
| Calcium associated molecular transport proteinse | ||||||
| P07356 | ANXA2 | Annexin A2 | ↑ 42.7 | 14 | --- | --- |
| P13020 | GSN | Gelsolin | ↑ 4.9 | 45 | --- | --- |
| Q6PHS9 | CACNA2D2 | Calcium channel, voltage-dependent, α 2/δ | ↑ 3.4 | 53 | --- | --- |
| Q64331 | MYO6 | Myosin-VI | ↑ 2.9 | 62 | --- | --- |
| P08003 | PDIA4 | Protein disulfide-isomerase A4 | ↑ 2.7 | 94 | --- | --- |
| Q91ZX7 | LRP1 | Low-density lipoprotein receptor-related protein 1 | ↑ 2.5 | 13 | --- | --- |
| P61264 | STX1B | Syntaxin 1B | ↓ −1.4 | 87 | --- | --- |
| P60879 | SNAP25 | Synaptosomal-associated protein 25 | ↓ −1.5 | 75 | --- | --- |
| P63011 | RAB3A | Ras-related protein Rab-3A | ↓ −1.7 | 67 | --- | --- |
| Q60932 | VDAC1 | Voltage-dependent anion-selective channel protein 1 | ↓ −1.7 | 70 | --- | --- |
| P62761 | VSNL1 | Visinin-like protein 1 | ↓ −1.9 | 31 | ↓ −1.3 | 60 |
| Q61171 | PRDX2 | Peroxiredoxin 2 | ↓ −2.0 | 22 | --- | --- |
| O35526 | STX1A | Syntaxin-1A | ↓ −2.1 | 42 | --- | --- |
| Q9CZT8 | RAB3B | Ras-related protein Rab-3B | ↓ −2.3 | 21 | --- | --- |
| P18760 | CFL1 | Cofilin-1 | ↓ −2.3 | 12 | ↓ −1.4 | 62 |
| P49615 | CDK5 | Cyclin-dependent kinase 5 | ↓ −2.4 | 100 | --- | --- |
| P60904 | DNAJC5 | DnaJ (Hsp40) homologue, subfamily C, member 5 | ↓ −2.4 | 91 | --- | --- |
| P47708 | RPH3A | Rabphilin-3A | ↓ −2.7 | 95 | --- | --- |
Protein identifiers and gene names are from www.uniprot.org.
Proteins shown in bold are unique to molecular transport and were not found associated with any other IPA-derived molecular and cellular functions in the nonamyloid disease phenotype.
Fold change in abundance relative to uninfected control.
Rank out of 1567 identified proteins based upon lowest p value, highest fold change, best count combination, and lowest coefficient of variation. The dashed line (---) indicates the protein was not among the top 100 ranked proteins.
Proteins involved in calcium binding or calcium-mediated transport, regulation, and signaling.
Figure 5.

The majority of molecular transport proteins disrupted in prion-infected Tg44 mice were associated with metal-dependent processes. (A) Relative distribution of the metal-associated molecular transport proteins from Table 4. (B) Venn diagram illustrating the overlap between molecular transport (Table 4) and cell death and survival (Table 6) in the amyloid phenotype and their overlap with known calcium binding proteins from each category.
Interestingly, many of the proteins in the molecular transport category were also involved in synaptic vesicle transport, a process known to be regulated by calcium.50 They included synaptosomal-associated protein 25 (SNAP25) and syntaxin 1A, which are two of the three proteins present in a complex necessary for synaptic fusion and neurotransmitter release, as well as several proteins involved in synaptic vesicle exocytosis50,51 including syntaxin 1B, rabphilin 3a, and members of the Ras-related proteins (Table 4). All of these synaptic vesicle proteins were present in lower abundance when compared to noninfected controls, suggesting significant disruption to synaptic vesicle transport in prion-infected mice with the amyloid disease phenotype.
Changes in Cell Death and Survival Pathways Suggest Potentially Different Mechanisms of Cell Death in the Nonamyloid versus Amyloid Phenotypes
The highest scoring molecular function category associated with the top 100 proteins in mice with the nonamyloid disease phenotype was cell death and survival, with 48% of the 100 input proteins linked to this category (Table 5). Of those, 29% (14 out of 48) were uniquely associated with cell death and survival and were not found in any of the other categories (Table 3). The cell death and survival category proteins had diverse functions including protein folding, cytoskeletal organization, RNA processing, apoptosis, and intracellular calcium regulation. However, there was an increase in the relative levels of three proteins associated with the mitochondrial inner membrane. Two of these, the mitochondrial inner membrane protein (mitofilin) and heat shock protein HSPA9, are part of the mitochondrial inner membrane. The third is apoptosis-inducing factor 1 (AIF1), which is anchored to the mitochondrial inner membrane.52 Finally, the fold expression of several proteins associated with the ubiquitin/proteasome pathway was also increased. These included heat shock protein HSPA5 (or BiP), which interacts with proteins involved in endoplasmic reticulum associated degeneration (ERAD), as well as the protein chaperonin TCP1 (Table 5). Thus, the data are consistent with disruptions to mitochondria, ER and the ubiquitin/proteasome pathway as part of the process of cell death in the nonamyloid prion disease phenotype.
Table 5. Proteins Associated with Cell Death and Survival in Prion-Infected Mice with the Nonamyloid Disease Phenotype.
| amyloid |
nonamyloid |
|||||
|---|---|---|---|---|---|---|
| IDa | gene | cell death and survival proteinsb | Δ foldc | rankd | Δ fold | rank |
| P08905 | LYZ2 | Lysozyme C-2 | --- | --- | ↑ 23.2 | 2 |
| P98086 | C1QA | Complement C1q subcomponent subunit A | ↑ 9.4 | 7 | ↑ 11.7 | 4 |
| P03995 | GFAP | Glial fibrillary acidic protein | ↑ 5.8 | 1 | ↑ 8.9 | 44 |
| P20152 | VIM | Vimentin | ↑ 11.9 | 66 | ↑ 7.9 | 46 |
| P14733 | LMNB1 | Lamin-B1 | --- | --- | ↑ 6.4 | 33 |
| Q9Z0 × 1 | AIFM1 | Apoptosis-inducing factor 1, mitochondrial | --- | --- | ↑ 5.3 | 24 |
| Q61838 | A2M | α-2-macroglobulin | ↑ 6.0 | 28 | ↑ 2.8 | 11 |
| P56959 | FUS | RNA-binding protein FUS | --- | --- | ↑ 2.6 | 72 |
| P63158 | HMGB1 | High mobility group protein B1 | --- | --- | ↑ 2.6 | 58 |
| P40240 | CD9 | CD9 antigen | --- | --- | ↑ 2.4 | 59 |
| Q8BMS1 | HADHA | Trifunctional enzyme subunit α, mitochondrial | --- | --- | ↑ 2.3 | 76 |
| P10605 | CTSB | Cathepsin B | --- | --- | ↑ 2.2 | 65 |
| P21460 | CST3 | Cystatin-C | --- | --- | ↑ 2.0 | 51 |
| P04925 | PRNP | Major prion protein (PrP) | ↑ 91.0 | 36 | ↑ 2.0 | 86 |
| P10852 | SLC3A2 | 4F2 cell-surface antigen heavy chain | --- | --- | ↑ 2.0 | 48 |
| P62631 | EEF1A2 | Elongation factor 1-α 2 | --- | --- | ↑ 1.9 | 45 |
| P38647 | HSPA9 | Stress-70 protein, mitochondrial | --- | --- | ↑ 1.8 | 91 |
| P01942 | HBA1 | Hemoglobin subunit α | --- | --- | ↑ 1.8 | 43 |
| P10126 | EEF1A2 | Elongation factor 1-α 1 | --- | --- | ↑ 1.7 | 84 |
| P35762 | CD81 | CD81 antigen | --- | --- | ↑ 1.6 | 61 |
| P26443 | GLUD1 | Glutamate dehydrogenase 1, mitochondrial | ↑ 1.6 | 99 | ↑ 1.5 | 25 |
| P19096 | FASN | Fatty acid synthase | --- | --- | ↑ 1.5 | 95 |
| P80318 | CCT3 | Chaperonin containing TCP1, subunit 3 | --- | --- | ↑ 1.4 | 82 |
| P52480 | PKM | Pyruvate kinase | --- | --- | ↑ 1.2 | 98 |
| Q9R0P9 | UCHL1 | Ubiquitin carboxyl-terminal esterase L1 | ↓ −1.5 | 77 | ↓ −1.3 | 89 |
| P17182 | ENO1 | α-enolase | --- | --- | ↓ −1.6 | 77 |
| O08539 | BIN1 | Myc box-dependent-interacting protein 1 | --- | --- | ↓ −1.7 | 38 |
| P10637 | MAPT | Microtubule-associated protein tau | --- | --- | ↓ −2.7 | 82 |
| Q64010 | CRK | Adapter molecule crk | --- | --- | ↓ −3.0 | 96 |
| Q9Z204 | HNRNPC | Heterogeneous nuclear ribonucleoproteins C1/C2 | --- | --- | ↓ −3.1 | 57 |
| P10639 | TXN | Thioredoxin | --- | --- | ↓ −3.4 | 50 |
| P56380 | NUDT2 | Bis(5′-nucleosyl)-tetraphosphatase | --- | --- | ↓ −6.1 | 51 |
| Q9WVC2 | LYNX1 | Ly-6/neurotoxin-like protein 1 | --- | --- | ↓ −6.3 | 6 |
| Q9CY64 | BLVRA | Biliverdin reductase A | --- | --- | ↓ −6.4 | 31 |
| Q9ERK4 | CSE1L | Exportin-2 | --- | --- | ↓ −15.7 | 18 |
| Calcium associated proteinse | ||||||
| P23242 | GJA1 | Gap junction alpha-1-protein | --- | --- | ↑ 25.6 | 92 |
| Q05793 | HSPG2 | Basement membrane-heparan sulfate proteoglycan protein | --- | --- | ↑ 7.8 | 73 |
| Q62443 | NPTX1 | Neuronal pentraxin-1 | --- | --- | ↑ 5.3 | 39 |
| Q9QWI6 | SRCIN1 | SRC kinase signaling inhibitor 1 | --- | --- | ↑ 4.5 | 16 |
| P20029 | HSPA5 | Heat Shock 70 kDa Protein 5 (BiP) | --- | --- | ↑ 2.0 | 93 |
| P07724 | ALB | Serum albumin | --- | --- | ↑ 2.0 | 5 |
| Q8VDD5 | MYH9 | Myosin-9 | --- | --- | ↑ 1.8 | 46 |
| Q8CAQ8 | IMMT | Mitochondrial inner membrane protein (mitofilin) | --- | --- | ↑ 1.8 | 63 |
| P62761 | VSNL1 | Visinin-like 1 | ↓ −1.9 | 31 | ↓ −1.3 | 60 |
| P18760 | CFL1 | Cofilin 1 | ↓ −2.3 | 12 | ↓ −1.4 | 62 |
| Q9Z329 | ITPR2 | Inositol 1,4,5-trisphosphate receptor type 2 | --- | --- | ↓ −5.4 | 10 |
| Q9QZX7 | SRR | Serine racemase | --- | --- | ↓ −8.7 | 19 |
| P17809 | SLC2A1 | Solute carrier family 2, facilitated glucose transporter | --- | --- | ↓ −9.5 | 26 |
Protein identifiers and gene names are from www.uniprot.org.
Proteins shown in bold are unique to cell death and were not found associated with any other molecular and cellular functions identified by IPA in the nonamyloid disease phenotype.
Fold change in abundance relative to uninfected control.
Rank out of 1567 identified proteins based upon lowest p value, highest fold change, best count combination, and lowest coefficient of variation. The dashed line (---) indicates the protein was not among the top 100 ranked proteins.
Proteins involved in calcium binding or calcium-mediated transport, regulation, and signaling.
Cell death and survival was also the second highest scoring IPA functional category in prion-infected mice with the amyloid phenotype and contained 42 of the top 100 input proteins, 14% of which uniquely segregated into this category (Table 6). Many of these proteins, including annexin A2, gelsolin, syntaxin 1A and SNAP25, overlapped with those proteins associated with calcium binding and molecular transport (Figure 5B). This overlap suggested a strong link between perturbations in calcium homeostasis and cell death in the disease with the amyloid phenotype. Interestingly, however, none of the mitochondrial or ubiquitin/proteasome pathway proteins identified in the mice with the nonamyloid phenotype were found in the top 100 ranked proteins in mice with the amyloid disease phenotype. In fact, there was minimal overlap with the proteins associated with cell death and survival in the nonamyloid versus the amyloid disease phenotypes (9 out of 90, or 10%). Moreover, several of these 9 proteins, including GFAP, vimentin, complement, and α-2-macroglobulin have previously been associated with a strong inflammatory response in prion diseases27,49,53 and are considered global neuroinflammatory markers, indicating that they are not functionally restricted to pathways mediating cell death. Thus, our data suggest that different cell death pathways may predominate in the amyloid and nonamyloid disease phenotypes.
Table 6. Proteins Associated with Cell Death and Survival in Prion-Infected Mice with the Amyloid Disease Phenotype.
| amyloid |
nonamyloid |
|||||
|---|---|---|---|---|---|---|
| IDa | gene | cell death and survival proteinsb | Δ foldc | rankd | Δ fold | rank |
| P04925 | PRNP | Major prion protein (PrP) | ↑ 91.0 | 36 | ↑ 2.0 | 86 |
| P20152 | VIM | Vimentin | ↑ 11.9 | 66 | ↑ 7.9 | 46 |
| P07759 | SPI2 | Serine protease inhibitor A3K | ↑ 10.0 | 29 | --- | --- |
| P98086 | C1QA | Complement component 1, q, A chain | ↑ 9.4 | 7 | ↑ 11.7 | 4 |
| Q06890 | CLU | Clusterin (apolipoprotein J or apoJ) | ↑ 8.2 | 3 | --- | --- |
| P08226 | APOE | Apolipoprotein E | ↑ 7.6 | 5 | --- | --- |
| P61148 | FGF1 | Fibroblast growth factor 1 (acidic) | ↑ 7.4 | 47 | --- | --- |
| Q61838 | A2M | α-2-macroglobulin | ↑ 6.0 | 28 | ↑ 2.8 | 11 |
| P03995 | GFAP | Glial fibrillary acidic protein | ↑ 5.8 | 1 | ↑ 8.9 | 44 |
| Q8BTM8 | FLNA | Filamin A, α | ↑ 5.4 | 49 | --- | --- |
| Q78PY7 | SND1 | Staphylococcal nuclease and tudor domain | ↑ 4.7 | 30 | --- | --- |
| Q00612 | G6PD | Glucose-6-phosphate dehydrogenase | ↑ 4.5 | 46 | --- | --- |
| Q921I1 | TF | Transferrin | ↑ 4.5 | 9 | --- | --- |
| Q61292 | LAMB2 | Laminin, β 2 (laminin S) | ↑ 3.0 | 70 | --- | --- |
| P18242 | CTSD | Cathepsin D | ↑ 2.5 | 43 | --- | --- |
| Q9CZX8 | RPS19 | Ribosomal protein S19 | ↑ 2.1 | 97 | --- | --- |
| Q9DCN2 | CYB5R3 | Cytochrome b5 reductase 3 | ↑ 2.0 | 40 | --- | --- |
| P26443 | GLUD1 | Glutamate dehydrogenase 1, mitochondrial | ↑ 1.6 | 99 | ↑ 1.5 | 25 |
| Q9R0P9 | UCHL1 | Ubiquitin carboxyl-terminal esterase L1 | ↓ −1.5 | 77 | ↓ −1.3 | 89 |
| O08756 | HSD17B10 | Hydroxysteroid (17-β) dehydrogenase 10 | ↓ −1.9 | 57 | --- | --- |
| P09671 | SOD2 | Superoxide dismutase 2, mitochondrial | ↓ −2.0 | 58 | --- | --- |
| P01831 | THY1 | Thy-1 cell surface antigen | ↓ −2.0 | 38 | --- | --- |
| Q91XV3 | BASP1 | Brain, membrane attached signal protein 1 | ↓ −2.0 | 11 | --- | --- |
| P60824 | CIRBP | Cold inducible RNA binding protein | ↓ −2.4 | 89 | --- | --- |
| Q60631 | GRB2 | Growth factor receptor-bound protein 2 | ↓ −2.6 | 84 | --- | --- |
| O35658 | C1QBP | Complement component 1, q binding protein | ↓ −2.6 | 76 | --- | --- |
| P60766 | CDC42 | Cell division cycle 42 | ↓ −3.5 | 22 | --- | --- |
| Calcium associated proteinse | ||||||
| P07356 | ANXA2 | Annexin A2 | ↑ 42.7 | 14 | --- | --- |
| P13020 | GSN | Gelsolin | ↑ 4.9 | 45 | --- | --- |
| Q6PHS9 | CACNA2D | Calcium channel, voltage-dependent, α 2/Δ 2 | ↑ 3.4 | 53 | --- | --- |
| Q64331 | MYO6 | Myosin VI | ↑ 2.9 | 62 | --- | --- |
| Q91ZX7 | LRP1 | Low density lipoprotein receptor-related protein 1 | ↑ 2.5 | 13 | --- | --- |
| P48962 | SLC25A4 | ADP/ATP translocase 1 | ↓ −1.4 | 88 | --- | --- |
| P60879 | SNAP25 | Synaptosomal-associated protein, 25 kDa | ↓ −1.5 | 75 | --- | --- |
| Q60932 | VDAC1 | Voltage-dependent anion channel 1 | ↓ −1.7 | 70 | --- | --- |
| P62761 | VSNL1 | Visinin-like 1 | ↓ −1.9 | 31 | ↓ −1.3 | 60 |
| Q61171 | PRDX2 | Peroxiredoxin 2 | ↓ −1.9 | 22 | --- | --- |
| Q63810 | PPR3R1 | Calcineurin subunit B type 1 | ↓ −2.0 | 65 | --- | --- |
| O35526 | STX1A | Syntaxin 1A (brain) | ↓ −2.1 | 42 | --- | --- |
| P18760 | CFL1 | Cofilin 1 (nonmuscle) | ↓ −2.3 | 12 | ↓ −1.4 | 62 |
| P49615 | CDK5 | Cyclin-dependent kinase 5 | ↓ −2.4 | 100 | --- | --- |
| P60904 | DNAJC5 | DnaJ (Hsp40) homologue, subfamily C, member 5 | ↓ −2.4 | 91 | --- | --- |
Protein identifiers and gene names are from www.uniprot.org.
Proteins shown in bold are unique to cell death and were not found associated with any other molecular and cellular functions identified by IPA in the nonamyloid disease phenotype.
Fold change in abundance relative to uninfected control.
Rank out of 1567 identified proteins based upon lowest p value, highest fold change, best count combination, and lowest coefficient of variation. The dashed line (---) indicates the protein was not among the top 100 ranked proteins.
Proteins involved in calcium binding or calcium-mediated transport, regulation, and signaling.
Discussion
Previous genomic studies in prion-infected mice have suggested that disruptions to metal binding, calcium homeostasis, synaptic transmission, lipid metabolism, and protein folding/degradation are involved in the pathogenesis of prion diseases.19−23,53 Similarly, genomic and proteomic analysis of tissue from human CJD patients has suggested that disruptions to molecular transport, calcium homeostasis, mitochondria, synapses and glucose metabolism may be involved in the pathogenesis of CJD.25,26,29,54 Our proteomics analysis of two different models of mouse prion disease also implicated imbalances in metal binding, calcium homeostasis, mitochondria, synaptic transmission, lipid metabolism, and molecular transport as factors in the late stages of prion disease. However, unlike previous studies, our comparison of the two distinctive prion disease phenotypes induced by a single prion strain has also allowed us to distinguish which pathways were more strongly linked to amyloid or nonamyloid forms of prion disease as well as which likely represented general responses to prion infection.
Our analysis primarily focused on the 100 proteins that were linked most strongly to either the amyloid or nonamyloid disease phenotype. Of these, only 17 were common to both disease phenotypes and most had some link to neuroinflammation (Table 1). This is consistent with previous studies showing that multiple genes and proteins associated with the inflammatory response are upregulated in different types of mouse22,23,27,55 and human25,26 prion disease. Astrocytes which have been activated in response to damage or infection produce complement factors and increase the expression levels of both vimentin and GFAP,45,56 all of which are present at increased levels in our two mouse models (Table 1). The increased abundance of α-2-macroglobulin may be related to its ability to act as a carrier protein for interleukin-646 or to remove proteinases at the site of inflammation.57 Likewise, α-2-HS-glycoprotein, an acute phase protein which binds calcium and has anti-inflammatory properties, was also increased in abundance in both disease models.48 Although the magnitude of the changes in fold expression was not always identical between the two phenotypes, proteins which were increased or decreased in mice with the amyloid phenotype mice were also increased or decreased in mice with the nonamyloid phenotype (Table 1).
Interestingly, many of these same proteins are also increased or decreased in abundance in other human neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease. For example, upregulation of vimentin has recently been described in neuronal cells of AD patients as part of a damage response mechanism, whereby the neurons attempt to repair synapses and re-establish dendritic branches lost during the disease process.43 α-2-macroglobulin has been found in senile AD plaques,47 whereas α-2-HS-glycoprotein has been found, albeit at decreased levels, in the cerebrospinal fluid of Alzheimer’s patients. Complement factors (Table 1) have also been proposed to play a role in both Parkinson’s and Alzheimer’s diseases32 When taken together, the data suggest that the changes in the relative abundance of these proteins during disease may represent similar, general responses related to the damage in the brain which occurs during neurodegeneration rather than a direct response to the deposition of PrPSc as amyloid versus nonamyloid.
An increased relative abundance of several lipoproteins was also observed in both disease phenotypes (Table 2). Notably, the lipoproteins apoD, apoE and clusterin (apoJ) as well as the low density lipoprotein receptor-related protein (LRP1), were elevated in both data sets but present at higher levels in the amyloid disease phenotype (Table 2). ApoE consistently copurifies with PrPSc isolated from infected animal tissue34,58−60 and elevated levels of clusterin have been observed in the hippocampus of prion infected mice,27,28 raising the possibility that these molecules have a functional role in prion pathogenesis. The ability of clusterin to inhibit the formation of amyloid61 as well as the link between LRP1 and the apoE and α-2-macroglobulin-mediated clearance of β-amyloid62,63 may explain their elevated levels in the amyloid disease phenotype. It is possible that other clusterin functions, such as its ability to prevent apoptosis induced by the mitochondrial release of cytochrome C,64 may also be relevant in the nonamyloid disease phenotype where proteins necessary for the integrity of the mitochondrial crista (Table 5) are also elevated. Overall, our data suggest that disruption of cholesterol and lipid metabolism and alterations in the mechanisms for the clearance of misfolded proteins occur in both amyloid and nonamyloid forms of prion disease.
However, these changes are more pronounced in the mice with the amyloid disease phenotype. Thus, the presence of amyloid may exacerbate disruptions in lipid metabolism and protein clearance. Consistent with this idea, apoD and apoE have been reported as candidate biomarkers for AD and prion disease32 and are known to colocalize with amyloid plaque in both Alzheimer’s disease65,66 and cerebral amyloid angiopathy.67 Moreover, the ability of clusterin to inhibit the formation of amyloid61 as well as the link between LRP1 and the apoE and α-2-macroglobulin-mediated clearance of Alzheimer’s disease β-amyloid62,63 may explain their elevated levels in the disease phenotypes where amyloid is a prominent feature.
In addition to the perturbation of the lipid transport proteins that we observed in both phenotypes, other molecular transport proteins were also found to have significantly changed in abundance in mice with the amyloid disease phenotype (Table 4). Approximately one-third of these are known to bind metals such as iron, zinc, manganese, copper and magnesium (Figure 5A) indicating a metal imbalance that could contribute to the buildup of reactive oxygen species and reduction of superoxide dismutase activity that has been observed during prion infection.68 Interestingly, multiple proteins were involved with calcium transport, regulation, and binding (Supporting Information Table 3), suggesting a significant disruption to calcium homeostasis in mice with the amyloid disease phenotype. Divalent calcium ions are universal signaling molecules with cells maintaining an approximately 20 000-fold lower concentration of divalent calcium ions in the intracellular compartment compared to the extracellular compartment.69 Imbalances in this tightly controlled gradient can trigger a wide variety of cellular processes69 and dysregulation of calcium has been implicated in the pathogenesis of prion disease.70
Our data suggest that in addition to dysregulation of calcium homeostasis, synaptic vesicle transport may also be impaired in the mice with the amyloid phenotype. Synaptic vesicle transport and exocytosis is triggered by an influx of calcium at the synapse leading to fusion of the soluble NSF attachment protein receptor (or SNARE) complex to the membrane and the subsequent release of neurotransmitters from the synaptic vesicle.50 SNARE is essential for synaptic vesicle fusion to the membrane and syntaxin-1 and SNAP25, two critical proteins which comprise SNARE, are decreased in abundance in mice with the amyloid phenotype. Similarly, the levels of syntaxin 1B, Rab3a, Rab3b and rabphilin-3a, all of which participate in the fusion of synaptic vesicles,50,51 are also decreased (Table 4). Interestingly, altered cycling of Rab3a has recently been reported in CJD54 and alterations in synaptic vesicle fusion and recycling can lead to disruptions in neurotransmitter release and deficits in learning and memory, a phenotype which has been described in the prion-infected mice with amyloid disease.71 Thus, our data suggest that impairment of synaptic vesicle fusion and neurotransmitter release triggered by changes in calcium homeostasis may underlie the learning and memory deficits seen in mice with the amyloid disease phenotype.
SNARE proteins have also been associated with neurodegeneration in CJD,25,72 and synaptic degeneration precedes neurodegeneration in prion infected mice.73 Thus, SNARE proteins may also be involved in neurodegenerative processes. Consistent with this hypothesis, IPA analysis linked syntaxin-1 and SNAP25 to cell death and survival pathways only in mice with the amyloid disease phenotype (Table 6). However, synaptic degeneration has been observed in multiple mouse prion disease models,73,74 including the RML strain used for the current studies.75 Furthermore, a decrease in SNAP25 and syntaxin has been reported in RML-infected wild-type mice.20,75,76 Thus, contrary to the present data, synaptic transport can also be impaired in nonamyloid forms of prion disease. The reason for this discrepancy is not clear but it is possible that decreased expression of SNARE proteins in the amyloid phenotype is the result of changes in synaptic vesicle fusion not linked to SNARE-mediated neurodegeneration, perhaps because of the lack of membrane anchored PrPC in these transgenic mice. This hypothesis is consistent with the lack of significant gray matter spongiosis in the amyloid disease phenotype9−11 as well as studies showing no correlation between decreased SNAP25 and apoptosis in RML infected mice.75
In the proteins associated with cell death and survival (Tables 5 and 6), there was very little overlap between the amyloid and nonamyloid disease phenotypes even though multiple proteins linked to calcium homeostasis were identified for both disease forms. This suggested that the dominant mechanisms underlying calcium-associated cell death may differ between the two mouse models. For example, only in the nonamyloid phenotype was there an increased abundance of both chaperonin containing TCP1 and the ER protein BiP, both of which are part of a large chaperonin complex that interacts with multiple proteins linked to ERAD (Table 5). These data are consistent with the possibility that the ubiquitin-proteasome system is involved in prion pathogenesis but that it may be more disrupted in the nonamyloid disease phenotype.77,78
Mitochondrial dysfunction has been proposed as a mechanism of neurodegeneration in prion disease.79,80 Our data also implicated mitochondrial pathways of apoptosis in neurodegeneration but only in the nonamyloid disease phenotype. Mitochondria have been linked to apoptosis via the release of pro-apoptotic factors such as cytochrome C and AIF1 from the inner mitochondrial membrane.52,81,82 Disruptions in this membrane have been observed in prion infected mice in regions of synaptic degeneration.80 Mitofilin and HSPA9, two proteins associated with the mitochondrial inner membrane organizing system (MINOS) responsible for maintaining cristae morphology, were present at higher levels only in the nonamyloid phenotype. The level of AIF1, an apoptosis inducing factor anchored to the mitochondrial inner membrane, was also elevated. In response to increased cytoplasmic calcium, mitochondrial calcium also increases leading to permeabilization of the mitochondrial outer membrane, changes in cristae morphology, and the production of reactive oxygen species. The resultant cleavage of AIF1 from the inner mitochondrial membrane allows it to translocate to the nucleus where it triggers DNA fragmentation and apoptosis, primarily in neurons.52 The altered levels of mitofilin, HSPA9, AIF1 and multiple proteins involved in calcium homeostasis are all consistent with this mechanism of mitochondria-associated apoptosis in mice with the nonamyloid disease phenotype.
Overall, our data suggest that different mechanisms of cell death may predominate in amyloid versus nonamyloid forms of prion disease, with ERAD and mitochondria induced apoptosis implicated primarily in the nonamyloid disease phenotype when compared to the amyloid disease phenotype. This difference may be driven more by the presence of the GPI anchor on PrPC, either because of the possible function of PrPC in cell signaling and apoptosis83 or because of the cell-surface conversion of PrPC by PrPSc. Thus, rather than being a phenotype necessarily encoded by PrPSc, our data suggest that the spongiform change associated with prion diseases is linked to membrane-anchored PrPC and to complex calcium-regulated changes in mitochondria and the ERAD/ubiquitin-proteasome system. The lack of gray matter spongiosis in the nonamyloid phenotype mice, despite very high levels of abnormal PrPSc, is consistent with this hypothesis and further suggests that spongiform change is not necessarily driven by a loss of function of membrane anchored PrPC or by the amount of PrPSc deposited. In fact, the deposition of anchorless PrPSc as amyloid may be somewhat protective, possibly resulting in slower disease progression. This may be due to different cell death pathways being induced. Alternatively, it may simply take longer for PrPSc amyloid to alter brain homeostasis sufficiently to trigger the mechanisms that eventually lead to neurodegeneration.
Acknowledgments
The authors wish to acknowledge Dr. Michael Klingeborn and Kimberly Meade-White for technical assistance and Drs. Karin Peterson, Jay Carroll, and Katherine Taylor for critical review of the manuscript. The authors also thank Anita Mora and Austin Athman for help with the figures. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Disease.
Supporting Information Available
Supplemental Figure 1: Western blot analysis of the changes in relative abundance of selected proteins. Supplemental Table 1: List of 1567 protein identifications with normalized spectral counts. Supplemental Table 2: List of the top 100 ranked proteins for the amyloid and nonamyloid data sets based upon the lowest p value, highest fold change, best count combination, and lowest coefficient of variation. Supplemental Table 3: Proteins involved with calcium transport, regulation, and binding in the amyloid data set. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chesebro B. Introduction to the Transmissible Spongiform Encephalopathies or Prion Diseases. Br. Med. Bull. 2003, 66, 1–20. [DOI] [PubMed] [Google Scholar]
- Caughey B. W.; Dong A.; Bhat K. S.; Ernst D.; Hayes S. F.; Caughey W. S. Secondary Structure-Analysis of the Scrapie-Associated Protein Prp 27-30 in Water by Infrared-Spectroscopy. Biochemistry 1991, 30, 7672–7680. [DOI] [PubMed] [Google Scholar]
- Pan K.-M.; Baldwin M.; Nguyen J.; Gasset M.; Serban A.; Groth D.; Mehlhorn I.; Huang Z.; Fletterick R. J.; Cohen F. E.; Prusiner S. B. Conversion of Alpha-Helices into Beta-Sheets Features in the Formation of the Scrapie Prion Protein. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce M. E. TSE Strain Variation. Br. Med. Bull. 2003, 66, 99–108. [DOI] [PubMed] [Google Scholar]
- Kretzschmar H.; Tatzelt J. Prion Disease: a Tale of Folds and Strains. Brain Pathol. 2013, 23, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggiolini I.; Saverioni D.; Parchi P. Prion Protein Misfolding, Strains, and Neurotoxicity: An Update From Studies on Mammalian Prions. Int. J. Cell Biol. 2013, 2013, 910314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R. A.; Taubner L. M.; Priola S. A. Prion Protein Misfolding and Disease. Curr. Opin. Struct. Biol. 2009, 19, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltzius J. J.; Landau M.; Nelson R.; Sawaya M. R.; Apostol M. I.; Goldschmidt L.; Soriaga A. B.; Cascio D.; Rajashankar K.; Eisenberg D. Molecular Mechanisms for Protein-Encoded Inheritance. Nat. Struct. Mol. Biol. 2009, 16, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesebro B.; Race B.; Meade-White K.; LaCasse R.; Race R.; Klingeborn M.; Striebel J.; Dorward D.; McGovern G.; Jeffrey M. Fatal Transmissible Amyloid Encephalopathy: a New Type of Prion Disease Associated With Lack of Prion Protein Membrane Anchoring. PLoS Pathog. 2010, 6, e1000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingeborn M.; Race B.; Meade-White K. D.; Rosenke R.; Striebel J. F.; Chesebro B. Crucial Role for Prion Protein Membrane Anchoring in the Neuroinvasion and Neural Spread of Prion Infection. J. Virol. 2011, 85, 1484–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesebro B.; Trifilo M.; Race R.; Meade-White K.; Teng C.; LaCasse R.; Raymond L.; Favara C.; Baron G.; Priola S.; Caughey B.; Masliah E.; Oldstone M. Anchorless Prion Protein Results in Infectious Amyloid Disease Without Clinical Scrapie. Science 2005, 308, 1435–1439. [DOI] [PubMed] [Google Scholar]
- Stahl N.; Borchelt D. R.; Hsiao K.; Prusiner S. B. Scrapie Prion Protein Contains a Phosphatidylinositol Glycolipid. Cell 1987, 51, 229–240. [DOI] [PubMed] [Google Scholar]
- Ghetti B.; Piccardo P.; Spillantini M. G.; Ichimiya Y.; Porro M.; Perini F.; Kitamoto T.; Tateishi J.; Seiler C.; Frangione B.; Bugiani O.; Giaccone G.; Prelli F.; Goedert M.; Dlouhy S. R.; Tagliavini F. Vascular Variant of Prion Protein Cerebral Amyloidosis With Tau-Positive Neurofibrillary Tangles: the Phenotype of the Stop Codon 145 Mutation in PRNP. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen C.; Parchi P.; Capellari S.; Vermeij A. J.; Corrado P.; Baas F.; Strammiello R.; van Gool W. A.; van Swieten J. C.; Rozemuller A. J. Prion Protein Amyloidosis With Divergent Phenotype Associated With Two Novel Nonsense Mutations in PRNP. Acta Neuropathol. 2010, 119, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayadev S.; Nochlin D.; Poorkaj P.; Steinbart E. J.; Mastrianni J. A.; Montine T. J.; Ghetti B.; Schellenberg G. D.; Bird T. D.; Leverenz J. B. Familial Prion Disease With Alzheimer Disease-Like Tau Pathology and Clinical Phenotype. Ann. Neurol. 2011, 69, 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead S.; Gandhi S.; Beck J.; Caine D.; Gajulapalli D.; Carswell C.; Hyare H.; Joiner S.; Ayling H.; Lashley T.; Linehan J. M.; Al-Doujaily H.; Sharps B.; Revesz T.; Sandberg M. K.; Reilly M. M.; Koltzenburg M.; Forbes A.; Rudge P.; Brandner S.; Warren J. D.; Wadsworth J. D.; Wood N. W.; Holton J. L.; Collinge J. A Novel Prion Disease Associated With Diarrhea and Autonomic Neuropathy. N. Engl. J. Med. 2013, 369, 1904–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribouillard-Tanvier D.; Striebel J. F.; Peterson K. E.; Chesebro B. Analysis of Protein Levels of 24 Cytokines in Scrapie Agent-Infected Brain and Glial Cell Cultures From Mice Differing in Prion Protein Expression Levels. J. Virol. 2009, 83, 11244–11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron G. S.; Hughson A. G.; Raymond G. J.; Offerdahl D. K.; Barton K. A.; Raymond L. D.; Dorward D. W.; Caughey B. Effect of Glycans and the Glycophosphatidylinositol Anchor on Strain Dependent Conformations of Scrapie Prion Protein: Improved Purifications and Infrared Spectra. Biochemistry 2011, 50, 4479–4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen G.; Medina S.; Parchaliuk D.; Phillipson C.; Robertson C.; Booth S. A. Comprehensive Transcriptional Profiling of Prion Infection in Mouse Models Reveals Networks of Responsive Genes. BMC Genomics 2008, 9, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner P. J.; Abbassi H.; Chesebro B.; Race R. E.; Reilly C.; Haase A. T. Gene Expression Alterations in Brains of Mice Infected With Three Strains of Scrapie. BMC Genomics 2006, 7, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth S.; Bowman C.; Baumgartner R.; Sorensen G.; Robertson C.; Coulthart M.; Phillipson C.; Somorjai R. L. Identification of Central Nervous System Genes Involved in the Host Response to the Scrapie Agent During Preclinical and Clinical Infection. J. Gen. Virol. 2004, 85, 3459–3471. [DOI] [PubMed] [Google Scholar]
- Xiang W.; Windl O.; Wunsch G.; Dugas M.; Kohlmann A.; Dierkes N.; Westner I. M.; Kretzschmar H. A. Identification of Differentially Expressed Genes in Scrapie-Infected Mouse Brains by Using Global Gene Expression Technology. J. Virol. 2004, 78, 11051–11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D.; Lee I. Y.; Yoo H.; Gehlenborg N.; Cho J. H.; Petritis B.; Baxter D.; Pitstick R.; Young R.; Spicer D.; Price N. D.; Hohmann J. G.; DeArmond S. J.; Carlson G. A.; Hood L. E. A Systems Approach to Prion Disease. Mol. Syst. Biol. 2009, 5, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benetti F.; Gasperini L.; Zampieri M.; Legname G. Gene Expression Profiling to Identify Druggable Targets in Prion Diseases. Expert. Opin. Drug Discovery 2010, 5, 177–202. [DOI] [PubMed] [Google Scholar]
- Xiang W.; Windl O.; Westner I. M.; Neumann M.; Zerr I.; Lederer R. M.; Kretzschmar H. A. Cerebral Gene Expression Profiles in Sporadic Creutzfeldt-Jakob Disease. Ann. Neurol. 2005, 58, 242–257. [DOI] [PubMed] [Google Scholar]
- Gawinecka J.; Dieks J.; Asif A. R.; Carimalo J.; Heinemann U.; Streich J. H.; Dihazi H.; Schulz-Schaeffer W.; Zerr I. Codon 129 Polymorphism Specific Cerebrospinal Fluid Proteome Pattern in Sporadic Creutzfeldt-Jakob Disease and the Implication of Glycolytic Enzymes in Prion-Induced Pathology. J. Proteome. Res. 2010, 9, 5646–5657. [DOI] [PubMed] [Google Scholar]
- Brown A. R.; Webb J.; Rebus S.; Williams A.; Fazakerley J. K. Identification of Up-Regulated Genes by Array Analysis in Scrapie-Infected Mouse Brains. Neuropathol. Appl. Neurobiol. 2004, 30, 555–567. [DOI] [PubMed] [Google Scholar]
- Asuni A. A.; Gray B.; Bailey J.; Skipp P.; Perry V. H.; O’Connor V. Analysis of the Hippocampal Proteome in ME7 Prion Disease Reveals a Predominant Astrocytic Signature and Highlights the Brain-Restricted Production of Clusterin in Chronic Neurodegeneration. J. Biol. Chem. 2014, 289, 4532–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawinecka J.; Nowak M.; Carimalo J.; Cardone F.; Asif A. R.; Wemheuer W. M.; Schulz-Schaeffer W. J.; Pocchiari M.; Zerr I. Subtype-Specific Synaptic Proteome Alterations in Sporadic Creutzfeldt-Jakob Disease. J. Alzheimer’s Dis. 2013, 37, 51–61. [DOI] [PubMed] [Google Scholar]
- Campisi E.; Cardone F.; Graziano S.; Galeno R.; Pocchiari M. Role of Proteomics in Understanding Prion Infection. Expert Rev. Proteomics 2012, 9, 649–666. [DOI] [PubMed] [Google Scholar]
- Barr J. B.; Watson M.; Head M. W.; Ironside J. W.; Harris N.; Hogarth C.; Fraser J. R.; Barron R. Differential Protein Profiling As a Potential Multi-Marker Approach for TSE Diagnosis. BMC Infect. Dis. 2009, 9, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huzarewich R. L.; Siemens C. G.; Booth S. A. Application of ″Omics″ to Prion Biomarker Discovery. J. Biomed. Biotechnol. 2010, 2010, 613504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel A.; Race B.; Phillips K.; Striebel J.; Kurtz N.; Chesebro B. Distinct Patterns of Spread of Prion Infection in Brains of Mice Expressing Anchorless or Anchored Forms of Prion Protein. Acta Neuropathol. Commun. 2014, 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R. A.; Timmes A.; Wilmarth P. A.; Priola S. A. Comparative Profiling of Highly Enriched 22L and Chandler Mouse Scrapie Prion Protein Preparations. Proteomics 2010, 10, 2858–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherly D. B.; Atwood J. A. III; Minning T. A.; Cavola C.; Tarleton R. L.; Orlando R. A Heuristic Method for Assigning a False-Discovery Rate for Protein Identifications From Mascot Database Search Results. Mol. Cell Proteomics 2005, 4, 762–772. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Keller A.; Kolker E.; Aebersold R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Old W. M.; Meyer-Arendt K.; veline-Wolf L.; Pierce K. G.; Mendoza A.; Sevinsky J. R.; Resing K. A.; Ahn N. G. Comparison of Label-Free Methods for Quantifying Human Proteins by Shotgun Proteomics. Mol. Cell Proteomics 2005, 4, 1487–1502. [DOI] [PubMed] [Google Scholar]
- Lundgren D. H.; Hwang S. I.; Wu L.; Han D. K. Role of Spectral Counting in Quantitative Proteomics. Expert Rev. Proteomics 2010, 7, 39–53. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc., Ser. B (Methodological) 1995, 57, 289–300. [Google Scholar]
- Liu H.; Sadygov R. G.; Yates J. R. III A Model for Random Sampling and Estimation of Relative Protein Abundance in Shotgun Proteomics. Anal. Chem. 2004, 76, 4193–4201. [DOI] [PubMed] [Google Scholar]
- Mueller L. N.; Brusniak M. Y.; Mani D. R.; Aebersold R. An Assessment of Software Solutions for the Analysis of Mass Spectrometry Based Quantitative Proteomics Data. J. Proteome Res. 2008, 7, 51–61. [DOI] [PubMed] [Google Scholar]
- Sofroniew M. V.; Vinters H. V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin E. C.; Acharya N. K.; Sedeyn J. C.; Venkataraman V.; D’Andrea M. R.; Wang H. Y.; Nagele R. G. Neuronal Expression of Vimentin in the Alzheimer’s Disease Brain May Be Part of a Generalized Dendritic Damage-Response Mechanism. Brain Res. 2009, 1298, 194–207. [DOI] [PubMed] [Google Scholar]
- Provansal M.; Roche S.; Pastore M.; Casanova D.; Belondrade M.; Alais S.; Leblanc P.; Windl O.; Lehmann S. Proteomic Consequences of Expression and Pathological Conversion of the Prion Protein in Inducible Neuroblastoma N2a Cells. Prion 2010, 4, 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M.; Wilhelmsson U.; Bogestal Y. R.; Pekna M. The Role of Astrocytes and Complement System in Neural Plasticity. Int. Rev. Neurobiol. 2007, 82, 95–111. [DOI] [PubMed] [Google Scholar]
- Nezu T.; Hosomi N.; Aoki S.; Deguchi K.; Masugata H.; Ichihara N.; Ohyama H.; Ohtsuki T.; Kohno M.; Matsumoto M. Alpha2-Macroglobulin As a Promising Biomarker for Cerebral Small Vessel Disease in Acute Ischemic Stroke Patients. J. Neurol. 2013, 260, 2642–2649. [DOI] [PubMed] [Google Scholar]
- Van G. D.; De S. B.; Van L. F.; Triau E.; Dom R. Alpha 2-Macroglobulin Expression in Neuritic-Type Plaques in Patients With Alzheimer’s Disease. Neurobiol. Aging 1993, 14, 233–237. [DOI] [PubMed] [Google Scholar]
- Wang H.; Sama A. E. Anti-Inflammatory Role of Fetuin-A in Injury and Infection. Curr. Mol. Med. 2012, 12, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer C.; Gultner S.; Heise I.; Holtkamp N.; Baier M. Neuroinflammation in Prion Diseases: Concepts and Targets for Therapeutic Intervention. CNS Neurol. Disord. Drug Targets 2009, 8, 329–341. [DOI] [PubMed] [Google Scholar]
- Jahn R.; Fasshauer D. Molecular Machines Governing Exocytosis of Synaptic Vesicles. Nature 2012, 490, 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermeier C.; Brunger A. T. Structural Basis of Rab Effector Specificity: Crystal Structure of the Small G Protein Rab3A Complexed With the Effector Domain of Rabphilin-3A. Cell 1999, 96, 363–374. [DOI] [PubMed] [Google Scholar]
- Norberg E.; Orrenius S.; Zhivotovsky B. Mitochondrial Regulation of Cell Death: Processing of Apoptosis-Inducing Factor (AIF). Biochem. Biophys. Res. Commun. 2010, 396, 95–100. [DOI] [PubMed] [Google Scholar]
- Jang B.; Jin J. K.; Jeon Y. C.; Cho H. J.; Ishigami A.; Choi K. C.; Carp R. I.; Maruyama N.; Kim Y. S.; Choi E. K. Involvement of Peptidylarginine Deiminase-Mediated Post-Translational Citrullination in Pathogenesis of Sporadic Creutzfeldt-Jakob Disease. Acta Neuropathol. 2010, 119, 199–210. [DOI] [PubMed] [Google Scholar]
- Gawinecka J.; Cardone F.; Asif A. R.; De P. A.; Wemheuer W. M.; Schulz-Schaeffer W. J.; Pocchiari M.; Zerr I. Sporadic Creutzfeldt-Jakob Disease Subtype-Specific Alterations of the Brain Proteome: Impact on Rab3a Recycling. Proteomics 2012, 12, 3610–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer C.; Neidhold S.; Burwinkel M.; Schwarz A.; Schultz J.; Kratzschmar J.; Monning U.; Baier M. Gene Expression Profiling of Scrapie-Infected Brain Tissue. Biochem. Biophys. Res. Commun. 2004, 323, 556–564. [DOI] [PubMed] [Google Scholar]
- Gasque P.; Ischenko A.; Legoedec J.; Mauger C.; Schouft M. T.; Fontaine M. Expression of the Complement Classical Pathway by Human Glioma in Culture. A Model for Complement Expression by Nerve Cells. J. Biol. Chem. 1993, 268, 25068–25074. [PubMed] [Google Scholar]
- Fabrizi C.; Colasanti M.; Persichini T.; Businaro R.; Starace G.; Lauro G. M. Interferon Gamma Up-Regulates Alpha 2 Macroglobulin Expression in Human Astrocytoma Cells. J. Neuroimmunol. 1994, 53, 31–37. [DOI] [PubMed] [Google Scholar]
- Moore R. A.; Timmes A. G.; Wilmarth P. A.; Safronetz D.; Priola S. A. Identification and Removal of Proteins That Co-Purify With Infectious Prion Protein Improves the Analysis of Its Secondary Structure. Proteomics 2011, 11, 3853–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi A.; Di F. L.; Principe S.; Mignogna G.; Sennels L.; Mancone C.; Alonzi T.; Sbriccoli M.; De P. A.; Rappsilber J.; Cardone F.; Pocchiari M.; Maras B.; Schinina M. E. Proteomic Profiling of PrP27-30-Enriched Preparations Extracted From the Brain of Hamsters With Experimental Scrapie. Proteomics 2009, 9, 3802–3814. [DOI] [PubMed] [Google Scholar]
- Graham J. F.; Kurian D.; Agarwal S.; Toovey L.; Hunt L.; Kirby L.; Pinheiro T. J.; Banner S. J.; Gill A. C. Na+/K+-ATPase Is Present in Scrapie-Associated Fibrils, Modulates PrP Misfolding in Vitro and Links PrP Function and Dysfunction. PLoS ONE 2011, 6, e26813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerbury J. J.; Poon S.; Meehan S.; Thompson B.; Kumita J. R.; Dobson C. M.; Wilson M. R. The Extracellular Chaperone Clusterin Influences Amyloid Formation and Toxicity by Interacting With Prefibrillar Structures. FASEB J. 2007, 21, 2312–2322. [DOI] [PubMed] [Google Scholar]
- Narita M.; Holtzman D. M.; Schwartz A. L.; Bu G. Alpha2-Macroglobulin Complexes With and Mediates the Endocytosis of Beta-Amyloid Peptide Via Cell Surface Low-Density Lipoprotein Receptor-Related Protein. J. Neurochem. 1997, 69, 1904–1911. [DOI] [PubMed] [Google Scholar]
- Rolyan H.; Feike A. C.; Upadhaya A. R.; Waha A.; Van D. T.; Haass C.; Birkenmeier G.; Pietrzik C. U.; Van L. F.; Thal D. R. Amyloid-Beta Protein Modulates the Perivascular Clearance of Neuronal Apolipoprotein E in Mouse Models of Alzheimer’s Disease. J. Neural Transm. 2011, 118, 699–712. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Kim J. K.; Edwards C. A.; Xu Z.; Taichman R.; Wang C. Y. Clusterin Inhibits Apoptosis by Interacting With Activated Bax. Nat. Cell Biol. 2005, 7, 909–915. [DOI] [PubMed] [Google Scholar]
- Lashley T.; Holton J. L.; Verbeek M. M.; Rostagno A.; Bojsen-M?ller M.; David G.; van H. J.; Braendgaard H.; Plant G.; Frangione B.; Ghiso J.; Revesz T. Molecular Chaperons, Amyloid and Preamyloid Lesions in the BRI2 Gene-Related Dementias: a Morphological Study. Neuropathol. Appl. Neurobiol. 2006, 32, 492–504. [DOI] [PubMed] [Google Scholar]
- Desai P. P.; Ikonomovic M. D.; Abrahamson E. E.; Hamilton R. L.; Isanski B. A.; Hope C. E.; Klunk W. E.; DeKosky S. T.; Kamboh M. I. Apolipoprotein D Is a Component of Compact but Not Diffuse Amyloid-Beta Plaques in Alzheimer’s Disease Temporal Cortex. Neurobiol. Dis. 2005, 20, 574–582. [DOI] [PubMed] [Google Scholar]
- Attems J.; Jellinger K.; Thal D. R.; Van N. W. Review: Sporadic Cerebral Amyloid Angiopathy. Neuropathol. Appl. Neurobiol. 2011, 37, 75–93. [DOI] [PubMed] [Google Scholar]
- Wong B. S.; Brown D. R.; Pan T.; Whiteman M.; Liu T.; Bu X.; Li R.; Gambetti P.; Olesik J.; Rubenstein R.; Sy M. S. Oxidative Impairment in Scrapie-Infected Mice Is Associated With Brain Metals Perturbations and Altered Antioxidant Activities. J. Neurochem. 2001, 79, 689–698. [DOI] [PubMed] [Google Scholar]
- Clapham D. E. Calcium Signaling. Cell 2007, 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Torres M.; Castillo K.; Armisen R.; Stutzin A.; Soto C.; Hetz C. Prion Protein Misfolding Affects Calcium Homeostasis and Sensitizes Cells to Endoplasmic Reticulum Stress. PLoS ONE 2010, 5, e15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilo M. J.; Sanchez-Alavez M.; Solforosi L.; Bernard-Trifilo J.; Kunz S.; McGavern D.; Oldstone M. B. Scrapie-Induced Defects in Learning and Memory of Transgenic Mice Expressing Anchorless Prion Protein Are Associated With Alterations in the Gamma Aminobutyric Acid-Ergic Pathway. J. Virol. 2008, 82, 9890–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I.; Rivera R.; Blanco R.; Marti E. Expression of Proteins Linked to Exocytosis and Neurotransmission in Patients With Creutzfeldt-Jakob Disease. Neurobiol. Dis. 1999, 6, 92–100. [DOI] [PubMed] [Google Scholar]
- Jeffrey M.; Halliday W. G.; Bell J.; Johnston A. R.; MacLeod N. K.; Ingham C.; Sayers A. R.; Brown D. A.; Fraser J. R. Synapse Loss Associated With Abnormal PrP Precedes Neuronal Degeneration in the Scrapie-Infected Murine Hippocampus. Neuropathol. Appl. Neurobiol. 2000, 26, 41–54. [DOI] [PubMed] [Google Scholar]
- Siskova Z.; Reynolds R. A.; O’Connor V.; Perry V. H. Brain Region Specific Pre-Synaptic and Post-Synaptic Degeneration Are Early Components of Neuropathology in Prion Disease. PLoS ONE 2013, 8, e55004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siso S.; Puig B.; Varea R.; Vidal E.; Acin C.; Prinz M.; Montrasio F.; Badiola J.; Aguzzi A.; Pumarola M.; Ferrer I. Abnormal Synaptic Protein Expression and Cell Death in Murine Scrapie. Acta Neuropathol. 2002, 103, 615–626. [DOI] [PubMed] [Google Scholar]
- Moreno J. A.; Radford H.; Peretti D.; Steinert J. R.; Verity N.; Martin M. G.; Halliday M.; Morgan J.; Dinsdale D.; Ortori C. A.; Barrett D. A.; Tsaytler P.; Bertolotti A.; Willis A. E.; Bushell M.; Mallucci G. R. Sustained Translational Repression by EIF2alpha-P Mediates Prion Neurodegeneration. Nature 2012, 485, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen M.; Deriziotis P.; Dimcheff D. E.; Jackson G. S.; Ovaa H.; Naumann H.; Clarke A. R.; van Leeuwen F. W.; Menendez-Benito V.; Dantuma N. P.; Portis J. L.; Collinge J.; Tabrizi S. J. Disease-Associated Prion Protein Oligomers Inhibit the 26S Proteasome. Mol. Cell 2007, 26, 175–188. [DOI] [PubMed] [Google Scholar]
- Lin Z.; Zhao D.; Yang L. Interaction Between Misfolded PrP and the Ubiquitin-Proteasome System in Prion-Mediated Neurodegeneration. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 477–484. [DOI] [PubMed] [Google Scholar]
- Choi S. I.; Ju W. K.; Choi E. K.; Kim J.; Lea H. Z.; Carp R. I.; Wisniewski H. M.; Kim Y. S. Mitochondrial Dysfunction Induced by Oxidative Stress in the Brains of Hamsters Infected With the 263 K Scrapie Agent. Acta Neuropathol. 1998, 96, 279–286. [DOI] [PubMed] [Google Scholar]
- Siskova Z.; Mahad D. J.; Pudney C.; Campbell G.; Cadogan M.; Asuni A.; O’Connor V.; Perry V. H. Morphological and Functional Abnormalities in Mitochondria Associated With Synaptic Degeneration in Prion Disease. Am. J. Pathol. 2010, 177, 1411–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L.; Ashiya M.; Buttle K.; Weiler S.; Oakes S. A.; Mannella C. A.; Korsmeyer S. J. A Distinct Pathway Remodels Mitochondrial Cristae and Mobilizes Cytochrome c During Apoptosis. Dev. Cell 2002, 2, 55–67. [DOI] [PubMed] [Google Scholar]
- Polster B. M.; Fiskum G. Mitochondrial Mechanisms of Neural Cell Apoptosis. J. Neurochem. 2004, 90, 1281–1289. [DOI] [PubMed] [Google Scholar]
- Linden R.; Martins V. R.; Prado M. A.; Cammarota M.; Izquierdo I.; Brentani R. R. Physiology of the Prion Protein. Physiol. Rev. 2008, 88, 673–728. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.