

Abstract
Kras-induced non-small-cell lung adenocarcinoma is the major subtype of lung cancers and is associated with poor prognosis. Using a lung cancer mouse model that expresses a cre-mediated KrasG12D mutant, we identified a critical role for the cell surface molecule CD44 in mediating cell proliferation downstream of oncogenic Kras signaling. The deletion of CD44 attenuates lung adenocarcinoma formation and prolongs the survival of these mice. Mechanistically, CD44 is required for the activation of Krasmediated signaling through the mitogen-activated protein kinase (MAPK) pathway and thus promotes tumor cell proliferation. Together, these results reveal an unrecognized role for CD44 in oncogenic Kras-induced lung adenocarcinoma and suggest that targeting CD44 could be an effective strategy for halting Kras-dependent carcinomas.
Keywords: CD44, Kras, lung adenocarcinoma
Introduction
Lung cancer is the leading cause of cancer death worldwide. Most patients are diagnosed in advanced stages, and despite the development of new chemotherapy and radiation protocols, the overall 5-year survival rate is approximately 14%. Recent studies have identified several genetic abnormalities in human lung cancer.1 Among these, mutations in the Kras gene are detected in 20–30% of human lung adenocarcinomas.2 Determining the pathways that mediate Kras-dependent lung adenocarcinoma is thus critical for the development of more effective therapeutic agents.
The cell adhesion molecule CD44 is a transmembrane glycoprotein that mediates responses to the microenvironment. CD44 is thought to perform multiple cellular functions including regulating cell proliferation, adhesion, migration and invasion.3 Recently, CD44 has been used as a marker for the enrichment of cancer stem cells.4–10 Through extensive alternative splicing, a large number of CD44 isoforms are generated, including CD44 variants (CD44v), which consist of various combinations of CD44 variable exons, and CD44 standard (CD44s), which is devoid of all variable exons. We have previously shown that the variable exon 6 containing variants regulate ras/mitogen-activated protein kinase (MAPK) signaling through a positive feedback loop, resulting in sustained ras/MAPK signaling and cell cycle progression, which are critical for the transition from normal to transformed phenotypes.11 These findings illustrate the importance of CD44 isoforms in maintaining ras/MAPK signaling. However, the in vivo role of CD44 in ras-mediated carcinogenesis has not been investigated. In this study, we used a mouse model of Kras-induced lung adenocarcinoma,12 and genetically ablated CD44 to evaluate the role of CD44 in ras-dependent signaling and tumorigenesis. Combining this genetic approach with molecular analysis using the human lung cancer cells, our results reveal that CD44 significantly contributes to Kras-mediated lung adenocarcinoma.
Results and Discussion
Expression of CD44 is upregulated during Kras-induced lung adenocarcinoma
The goal of this study was to investigate the role of CD44 in Kras-dependent carcinogenesis in vivo. Thus, we used the conditional LSL-KrasG12D mouse model of lung cancer,12 in which expression of the oncogenic KrasG12D allele is controlled by a removable transcriptional termination element that is flanked by two Lox P sites. When Cre-recombinase is introduced by intranasal administration of an adenoviral vector (Ad-Cre), oncogenic Kras is expressed at physiological levels in the lungs. Characterization of this mouse model indicates that oncogenic Kras expression in the lungs results in multifocal hyperplasia followed by adenoma and adenocarcinoma.12
To investigate the possible involvement of CD44 in Kras-induced lung adenocarcinoma, we first examined whether CD44 expression is altered in these tumors. Normal mouse lung tissue and lung tumors from the KrasG12D mice were recovered and subjected to quantitative reverse transcriptase–PCR analysis to assess mRNA levels of CD44. Interestingly, there was a statistically significant threefold increase (P<0.0001) in CD44 mRNA expression in tumors as compared with the normal lung tissue (Figure 1a). These findings were confirmed by immunohistochemistry analysis using the lung sections from normal lung and Kras-induced tumor tissue (Figure 1b), demonstrating that the levels of CD44 are increased in tumors. Thus, the expression of CD44 is upregulated in Kras-induced lung adenocarcinomas.
Figure 1.
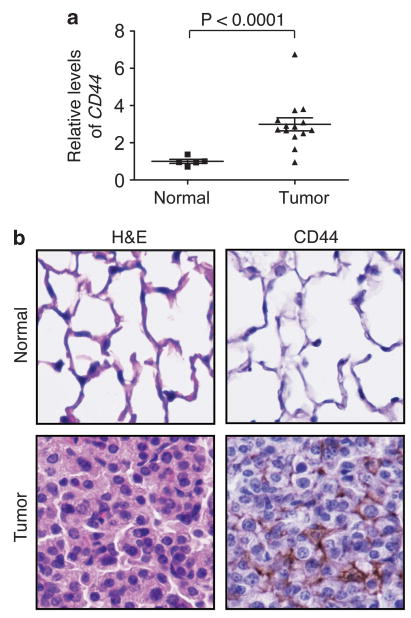
CD44 expression is increased in Kras-dependent lung adenocarcinoma. (a) The relative mRNA levels of CD44 in individual normal mouse lung tissue and KrasG12D-induced tumor samples were compared by quantitative reverse transcriptase-PCR. P values were calculated using a two-tailed Student's t test. TATA-binding protein (TBP) served as a loading control. Primer sequences used were: CD44 forward: 5′-TATGACACATATTGCTTCAATGC-3′, CD44 reverse: 5′-GTGTACCATCACGGTTGACA-3′, TBP forward: 5′-TGCACAGGAGCCAAGAGTGA-3′; TBP reverse: 5′-AGCTGGGAAGCCCAACTTCT-3′. (b) Images (original magnification, × 40) of immunohistochemistry staining of CD44 in normal mouse lung and Kras-induced tumor. Hematoxylin and eosin (H&E) stains on the left panels show normal and tumor lung epithelial cells.
Deletion of CD44 increases survival of mice with Kras-induced lung adenocarcinomas
To directly examine the role of CD44 in Kras-dependent lung adenocarcinoma, we used a CD44 knockout mouse strain and evaluated whether CD44 is required for Kras-induced lung tumor formation. As the CD44 null mice are in a B6/129 mixed background while the LSL-KrasG12D mice are in a B6 background, we generated cohorts of LSL-Kras;CD44-/- mice and their littermate-control LSL-Kras;CD44+/- mice for comparison, in order to minimize the potential influence of strain background. Following intranasal administration of Ad-Cre (1 × 108 plaque-forming unit),12 mice from these cohorts were monitored for survival. Control LSL-KrasG12D;CD44+/- mice developed multiple tumors and had a median survival of 101.5 days (average of 13 mice, Figure 2a). Strikingly, LSL-KrasG12D;CD44-/- mice, despite also developing lung tumors, had a significantly longer median survival of 144 days (average of 14 mice). These results indicate that deletion of CD44 confers a survival advantage to mice expressing oncogenic KrasG12D in the lung.
Figure 2.
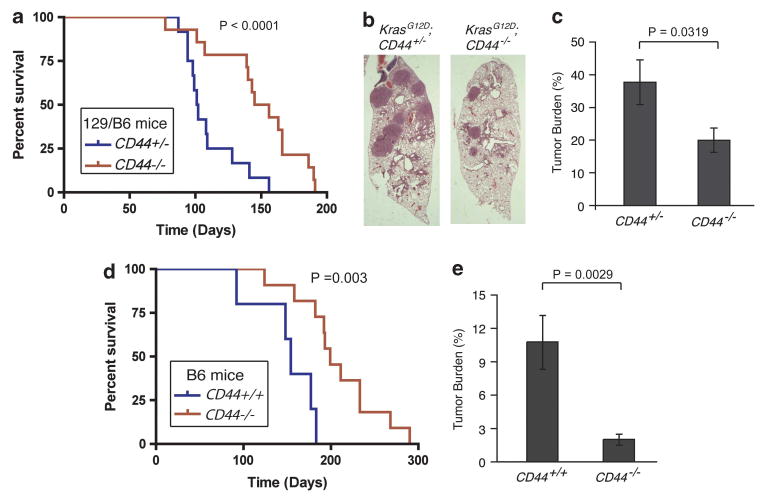
CD44 promotes Kras-dependent lung adenocarcinoma in mice. (a) Survival rate of B6/129 mixed-background mice infected with Ad-Cre. 13 LSL-KrasG12D CD44+/- mice and 14 LSL-KrasG12D CD44-/- mice were infected with Ad-Cre at the same age, and survival was followed over time. Data are presented as a Kaplan-Meier plot. P<0.0001. (b) Representative H&E-stained sections of KrasG12D;CD44+/- and KrasG12D;CD44-/- lungs (original magnification, × 1.25). (c) Tumor burden at a 12-week time point of mixed-background mouse lungs from 17 KrasG12D;CD44+/- mice and 14 Kras 12D;CD44-/- mice, s.e.m. n⩾14. H&E-stained lung sections were used for tumor size quantification with Image J software (NIH,Bethesda, MD, USA, http://imagej.nih.gov/ij/). (d) Prolonged survival of KrasG12D;CD44-/- B6 mice that express KrasG12D in the lung. Data are presented as a Kaplan-Meier plot from five KrasG D;CD44+/+ mice and 11 KrasG12D;CD44-/- mice. P = 0.003. (e) Tumor burden at a 15-week time point of seven KrasG12D;CD44+/+ and ten KrasG12D;CD44-/- mice in B6 background, s.e.m. n⩾7.
CD44 null mice develop fewer lesions
The above results suggest that CD44 could have a critical role in Kras-induced lung adenocarcinoma. To further test this hypothesis, time course experiments were performed to examine tumor development in mice as gauged by tumor burden following the initiation of oncogenic KrasG12D expression via intranasal introduction of Ad-Cre. Atypical hyperplasia formed in the mouse lungs 6 weeks after Ad-Cre intranasal administration. Analysis of the lungs in a cohort of control and LSL-KrasG12D;CD44-/- mice showed that the area of hyperplasia in the CD44-/- group was similar to that in the CD44+/- group, suggesting that tumor initiation is not significantly affected in the CD44 null background (Supplementary Figure S1). In contrast, analysis of tumors arising 12-weeks after infection indicated that CD44-null mice had fewer tumor lesions. Aproximately 38% of the lung area was comprised of tumor lesions in control mice, while tumors occupied less than 20% of the lung area in the CD44-null mice (Figures 2b and c, P = 0.0319). These results suggest that deletion of CD44 impairs the progression of oncogenic Kras-induced lung adenocarcinomas.
As the above observations were obtained in mouse strains with mixed genetic backgrounds, a possible alternative explanation for the delay in tumor formation was that unidentified modifiers adjacent to the CD44 gene, rather than the CD44 gene itself, contribute to tumor formation in mice. To address this concern, we next used a CD44 deletion strain in a pure B6 background, which became available during the course of this study, and generated cohort of B6 compound mice that are either LSL-KrasG12D;CD44+/+ or LSL-KrasG12D;CD44-/-. Consistent with the results obtained from the mixed background mice, survival of the B6 cohort mice following Ad-Cre induction of KrasG12D expression showed that deletion of CD44 in the B6 background caused a significant increase in survival. The median survival in the control mice was 154 days, whereas the median life span was extended to 199 days in the CD44-null mice (P = 0.003, Figure 2d). The observed extended life span in the above B6 strains as compared with the B6/129 mixed background is consistent with previous reports that show that the B6 strain has a longer tumor latency than those with mixed background.13 Further analysis of tumor volume from each group of mice 15 weeks after Ad-Cre intranasal administration revealed that LSL-Kras;CD44-/- mice exhibited a five-fold decrease in tumor burden relative to LSL-Kras mice (P = 0.0029, Figure 2e). Together, these multiple approaches clearly indicate that CD44 deletion attenuates Kras-induced lung adenocarcinoma formation in mice.
CD44v is significantly upregulated in Kras-induced lung tumors CD44 undergoes extensive alternative splicing that generates a family of CD44 splice isoforms with varying functions. We and others have shown that activation of ras/MAPK signaling stimulates CD44 alternative splicing, resulting in the production of CD44v.11,14,15 As the oncogenic event driving lung tumor formation is dependent on ras signaling, these observations promoted us to examine whether CD44v expression is preferentially upregulated in the Kras-mediated lung adenocarcinomas in mice. As shown in Figure 3, analysis of CD44 isoform expression in normal lung tissue and lung tumors from the LSL-KrasG12D mice showed that the expression of CD44v, as assayed for the presence of v5 and v6 exons (CD44v5/v6) or v8 and v9 exons (CD44v8/v9), is upregulated five- to six-fold in tumors (P = 0.0002 and P< 0.0001, respectively, Figures 3a and b), while the CD44s isoform only showed a marginal 1.3-fold increase and it was not statistically significant (Supplementary Figure S2). These results suggest that the increase in CD44v is the major event accounting for the upregulation of the CD44 gene in mouse lung adenocarcinomas and may also be responsible for the signaling events mediated by oncogenic Kras.
Figure 3.
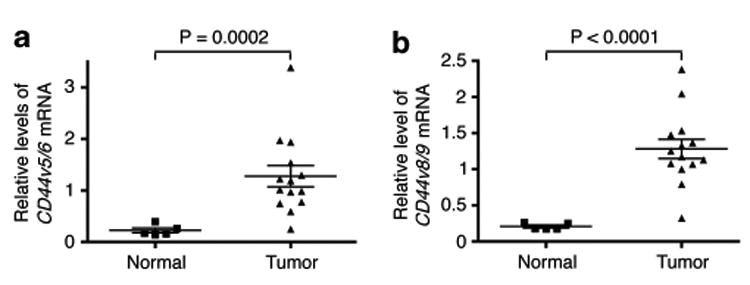
CD44v is upregulated in Kras-dependent lung adenocarcinoma. Expression levels of CD44v5/v6 (a) and CD44v8/v9 (b) isoforms in normal and Kras-driven lung tumors. P values were calculated using a two-tailed Student's t test. TBP served as a loading control. Primer sequences used were: CD44v5/v6 forward: 5′ -TATAGACA GAATCAGCACCA-3′, reverse: 5′-GCTGCTTCTGCTGTACTATTAG-3′; CD44v8/v9 forward: 5′-CCAGTCATAGTACAACCCTT-3′, reverse: 5′-CCAT GTAATGTAGAGAAGTTCTG-3′.
CD44 promotes proliferation and Erk activation in human lung adenocarcinoma cells
To investigate the mechanism by which CD44 could promote tumorigenesis in Kras-induced lung adenocarcinoma, signaling cascades downstream of Kras that are modulated by CD44 were examined. Mutant-activated Kras activates the Raf/Mek/Erk pathway, which is important in mitogenic signaling for cell proliferation. Thus, we examined whether CD44 is important for MAPK activation in a human lung adenocarcinoma cell line H358, which carries the KrasG12C mutation. H358 cells predominantly express CD44v, and introducing a human CD44 small hairpin RNA (shRNA)16 efficiently knocked down CD44 to an undetectable level (Figure 4a). Quantitative reverse transcriptase–PCR analysis confirmed that the CD44 shRNA efficiently knocks down CD44v mRNAs by greater than 10-fold (Supplementary Figure S3). To detect the role of CD44 in MAPK activity, H358 cells expressing control or CD44 shRNAs were starved for 24 h followed by stimulation with epidermal growth factor (EGF) (0.5 ng/ml). Cells were collected at different time intervals for a period of 4 h and the levels of Erk phosphorylation were measured as a read of MAPK activation. In control cells, EGF treatment resulted in increased Erk phosphorylation that peaked at 10 min and then declined with time (Figure 4b). Analysis of cells in which CD44 had been silenced showed that the degree of Erk phosphorylation was impaired in all timepoints in which Erk phosphorylation is detactable (Figure 4b and Supplementary Figure S4A for longer exposure at 4 h). Similarly, results using a higher dosage of EGF (5 ng/ml) to stimulate control and CD44 shRNA-expressing cells also showed that CD44 knockdown inhibited Erk activity (Supplementary Figure S4B). In addition to using EGF as a stimulus for Erk activation, we have also treated cells with 5% serum. While both control and CD44 knockdown cells showed much lower degrees of Erk activation in response to serum addition compared with EGF stimulation, CD44-silenced cells consistently showed a slight decrease in the levels of phosphorylated Erk (Supplementary Figure S4C). Hence, these data demonstrate that CD44 is a critical component mediating MAPK signaling downstream of Kras.
Figure 4.
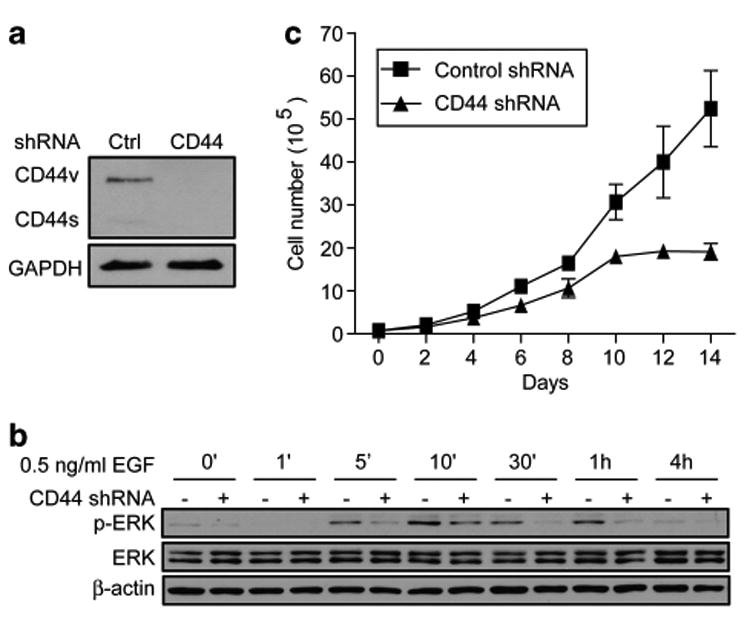
Depletion of CD44 in human Kras-mediated lung adenocarcinoma cells inhibits MAPK activation and cell proliferation. (a) Immunoblot analysis of CD44 (antibody from R&D, Minneapolis, MN, USA) in H358 cells expressing control or CD44 shRNAs. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH, antibody from Millipore, Billerica, MA, USA) serves as a loading control. (b) Immunoblot analysis of phospho-Erk (antibody from Cell Signaling, Danvers, MA, USA) in control and CD44 shRNA-expressing H358 cells. Cells were serum starved and then stimulated with EGF (0.5 ng/ml) at different time intervals as indicated. b-Actin (antibody from Sigma-Aldrich, St Louis, MO, USA) serves as a loading control. (c) Cell proliferation assay of H358 cells that express control or CD44 shRNAs.
As MAPK activity drives cell proliferation, we next assessed whether silencing CD44 by shRNA affects H358 tumor cell growth. H358 cells expressing control or CD44 shRNA were cultured in growth media containing 5% serum and counted every other day for a consecutive 14 days. Control cells grow exponentially in culture. However, the CD44 shRNA-expressing cells proliferate at a much slower rate as compared with control cells (Figure 4c), suggesting that CD44 promotes cell proliferation in the Kras-expressing lung adenocarcinoma H358 cells.
To confirm the generality of CD44-mediated MAPK signaling, we have also used LKR13 tumor cells17,18 that were derived from mouse lung tumors of the KrasG12D mice and examined whether CD44 is important for MAPK activation in these cells. As shown in Supplementary Figure S5, EGF-induced Erk phosphorylation was impared in cells in which CD44 had been silenced. Furthermore, these CD44-silenced cells displayed decreased potential in forming colonies in soft agar (Supplementary Figure S5C). Thus, these observations show that CD44 mediates Kras-dependent MAPK activation and cell proliferation, supporting our findings that CD44 deletion inhibits Kras-driven lung adenocarcinomas in vivo.
Conclusions
We describe here a critical role for CD44 in Kras-mediated lung adenocarcinoma. Our data demonstrate that genetic deletion of CD44 inhibits Kras-mediated lung adenocarcinoma development in mice, and extends the survival of these animals. Functionally, we show that CD44 promotes ras-mediated MAPK signaling and cell proliferation. The mechanism by which CD44 activates rasmediated effectors is currently unclear. As CD44 is a cell surface molecule, we speculate that CD44 may act as a sensor/coreceptor for growth factors and cytokines thereby sustaining ras effector activity through mechanisms such as a positive feedback loop. As CD44 has been previously suggested to mediate signaling of the epidermal growth factor receptor (EGFR) family proteins,19–21 we have also examined levels of phosphorylated EGFR. Under conditions that impaired Erk actvity was observed in CD44 knockdown cells, we have not detected impaired EGFR activation in these CD44-silenced cells (data not shown). This suggests that CD44 is involved in promoting MAPK signaling downstream of EGFR activation. Future studies investigating the molecular connection between CD44 and MAPK are necessary to better understand the mechanisms governing CD44-mediated activation of ras effectors.
Using genetic approaches, we observed that deletion of CD44 inhibits formation of Kras-mediated lung adenocarcinoma. Of note, the CD44 null mouse strain used in this study ablates CD44 in all tissues. In addition to the fact that CD44 functions autonomously to promote lung adenocarcinoma, potential contributions of CD44 from non-lung cells may also exist. One such example would include the possible role of CD44 in immune cells that are recruited to the microenvironment of the lung adenocarcinoma to accelerate lung carcinogenesis. Developing a conditional mouse strain of CD44 deletion will be necessary to address the non-autonomous effect of CD44 in lung adenocarcinoma.
The role of CD44 in tumorigenesis has been studied in other mouse models. We have previously shown that expression of CD44v isoforms increases in primary breast tumors in an oncogenic Neu-induced breast cancer mouse model. Interestingly, recurrent tumors that develop after regression of the primary tumors in this mouse model show high levels of CD44s, but not CD44v, suggesting that CD44 isoform expression is stage-specific during tumor progression.22 Our current data showing that CD44v is more significantly increased in lung primary tumors are consistent with this previous finding that CD44v is highly expressed in the breast primary tumors. Interestingly, CD44 is upregulated in non-small cell lung tumors but not in small cell lung tumors.23 Immunohistological analysis also showed that the expression of variable exon 6 containing variants in stage I non-small cell lung carcinomas inversely correlates with the patient's 5-year survival rate.24 Together with our data, these results suggest that CD44 has an important role in promoting tumor development and progression. Future studies examining the expression of CD44 isoforms in different stages of human lung tumors will be essential for deciphering the role of CD44 isoforms in lung cancer progression.
In summary, our results show that CD44 deletion attenuates Kras-mediated lung adenocarcimonas. CD44 thus emerges as a critical factor regulating Kras-expressing lung adenocarcinomas and represents potential therapeutic target for the treatment of Kras-dependent carcinomas.
Supplementary Material
Acknowledgments
We thank Dr Tyler Jacks for providing the LSL-K-RasG12D mouse strain and Kim Mercer for technical assistance. We also thank members in the Cheng laboratory for discussions and suggestions.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Girard L, Zochbauer-Muller S, Virmani AK, Gazdar AF, Minna JD. Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res. 2000;60:4894–4906. [PubMed] [Google Scholar]
- 2.Slebos RJ, Kibbelaar RE, Dalesio O, Kooistra A, Stam J, Meijer CJ, et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N Engl J Med. 1990;323:561–565. doi: 10.1056/NEJM199008303230902. [DOI] [PubMed] [Google Scholar]
- 3.Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 4.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fillmore C, Kuperwasser C. Human breast cancer stem cell markers CD44 and CD24: enriching for cells with functional properties in mice or in man? Breast Cancer Res. 2007;9:303. doi: 10.1186/bcr1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 8.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 9.Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 10.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng C, Yaffe MB, Sharp PA. A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev. 2006;20:1715–1720. doi: 10.1101/gad.1430906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davie SA, Maglione JE, Manner CK, Young D, Cardiff RD, MacLeod CL, et al. Effects of FVB/NJ and C57Bl/6J strain backgrounds on mammary tumor phenotype in inducible nitric oxide synthase deficient mice. Transgenic Res. 2007;16:193–201. doi: 10.1007/s11248-006-9056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng C, Sharp PA. Regulation of CD44 alternative splicing by SRm160 and its potential role in tumor cell invasion. Mol Cell Biol. 2006;26:362–370. doi: 10.1128/MCB.26.1.362-370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weg-Remers S, Ponta H, Herrlich P, Konig H. Regulation of alternative pre-mRNA splicing by the ERK MAP-kinase pathway. Embo J. 2001;20:4194–4203. doi: 10.1093/emboj/20.15.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, et al. Growth-inhibitory and tumor-suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 18.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 19.Bourguignon LY, Zhu H, Chu A, Iida N, Zhang L, Hung MC. Interaction between the adhesion receptor, CD44, and the oncogene product, p185HER2, promotes human ovarian tumor cell activation. J Biol Chem. 1997;272:27913–27918. doi: 10.1074/jbc.272.44.27913. [DOI] [PubMed] [Google Scholar]
- 20.Sherman LS, Rizvi TA, Karyala S, Ratner N. CD44 enhances neuregulin signaling by Schwann cells. J Cell Biol. 2000;150:1071–1084. doi: 10.1083/jcb.150.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu WH, Woessner JF, Jr, McNeish JD, Stamenkovic I. CD44 anchors the assembly of matrilysin/MMP-7 with heparin-binding epidermal growth factor precursor and ErbB4 and regulates female reproductive organ remodeling. Genes Dev. 2002;16:307–323. doi: 10.1101/gad.925702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121:1064–1074. doi: 10.1172/JCI44540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penno MB, August JT, Baylin SB, Mabry M, Linnoila RI, Lee VS, et al. Expression of CD44 in human lung tumors. Cancer Res. 1994;54:1381–1387. [PubMed] [Google Scholar]
- 24.Hirata T, Fukuse T, Naiki H, Hitomi S, Wada H. Expression of CD44 variant exon 6 in stage I non-small cell lung carcinoma as a prognostic factor. Cancer Res. 1998;58:1108–1110. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.