

Abstract
Gemcitabine is a modified cytidine analog having two fluorine atoms at the 2′-position of the ribose ring. It has been proposed that gemcitabine inhibits RNR activity by producing a C3′• intermediate via direct H3′-atom abstraction followed by loss of HF to yield a C2′• with 3′-keto moiety. Direct detection of C3′• and C2′• during RNR inactivation by gemcitabine still remains elusive. To test the influence of 2′- substitution on radical site formation, electron spin resonance (ESR) studies are carried out on one-electron oxidized gemcitabine and other 2′-modified analogs, i.e., 2′-deoxy-2′-fluoro-2′-C-methylcytidine (MeFdC) and 2′-fluoro-2′-deoxycytidine (2′-FdC). ESR line components from two anisotropic β-2′-F-atom hyperfine couplings identify the C3′• formation in one-electron oxidized gemcitabine, but no further reaction to C2′• is found. One-electron oxidized 2′-FdC is unreactive toward C3′• or C2′• formation. In one-electron oxidized MeFdC, ESR studies show C2′• production presumably from a very unstable C3′• precursor. The experimentally observed hyperfine couplings for C2′• and C3′• match well with the theoretically predicted ones. C3′• to C2′• conversion in one-electron oxidized gemcitabine and MeFdC has theoretically been modeled by first considering the C3′• and H3O+ formation via H3′-proton deprotonation and the subsequent C2′• formation via HF loss induced by this proximate H3O+. Theoretical calculations show that in gemcitabine, C3′• to C2′• conversion in the presence of a proximate H3O+ has a barrier in agreement with the experimentally observed lack of C3′• to C2′• conversion. In contrast, in MeFdC, the loss of HF from C3′• in the presence of a proximate H3O+ is barrierless resulting in C2′• formation which agrees with the experimentally observed rapid C2′• formation.
Introduction
Gemcitabine is a modified cytidine analog having two fluorine atoms at the 2′-position in the deoxyribose sugar moiety (Scheme 1). For nearly 20 years, it has been widely used to treat specifically pancreatic cancer.1−4 It has been proposed that gemcitabine inhibits ribonucleotide reductase (RNR) activity5−7 as well as acting as a replication stop,8,9 thereby affecting DNA synthesis and elongation.
Scheme 1. Structural Formula of Gemcitabine Including the Standard Numbering Convention for Atoms According to IUPAC Nomenclature.

The two highly electronegative F-atoms at C2′ substantially increase the acidity of H3′ through the inductive effect.
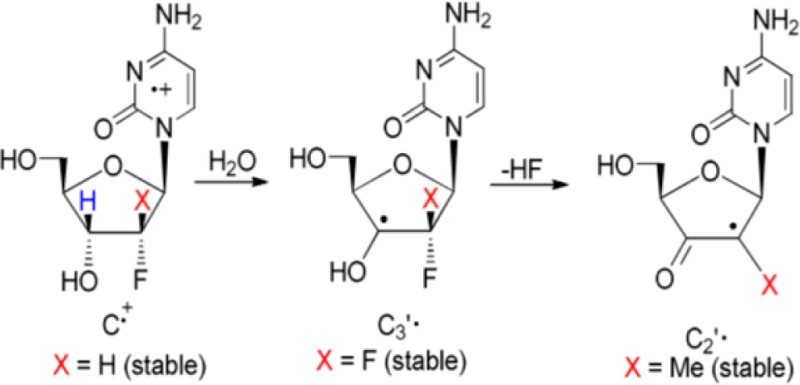
It is well established that in the absence of oxygen, thiyl radicals are able to abstract hydrogen (H) atoms to form neutral C-centered radicals. For example, H-atom abstraction by thiyl radicals has been shown to induce isomerization of cis-2,5-dimethyltetrahydrofuran to the trans-2,5-dimethyltetrahydrofuran.10,11 On this basis, Stubbe et al.,5−7 in their enzymatic and electron spin resonance (ESR) studies with gemcitabine, have proposed that inhibition of the RNR activity by gemcitabine should occur via radical formation at the C3′ site by direct H-atom abstraction to produce a C3′• via a enzymatic thiyl radical (reaction 1). Subsequently, the C3′• intermediate has been proposed to form 3′-keto C2′• via HF loss. This C2′• is stabilized by an oxy radical resonance contribution (reaction 2). C2′• and its immediate precursor C3′• (reactions 1 and 2) play a key role in the RNR inactivation.5−7
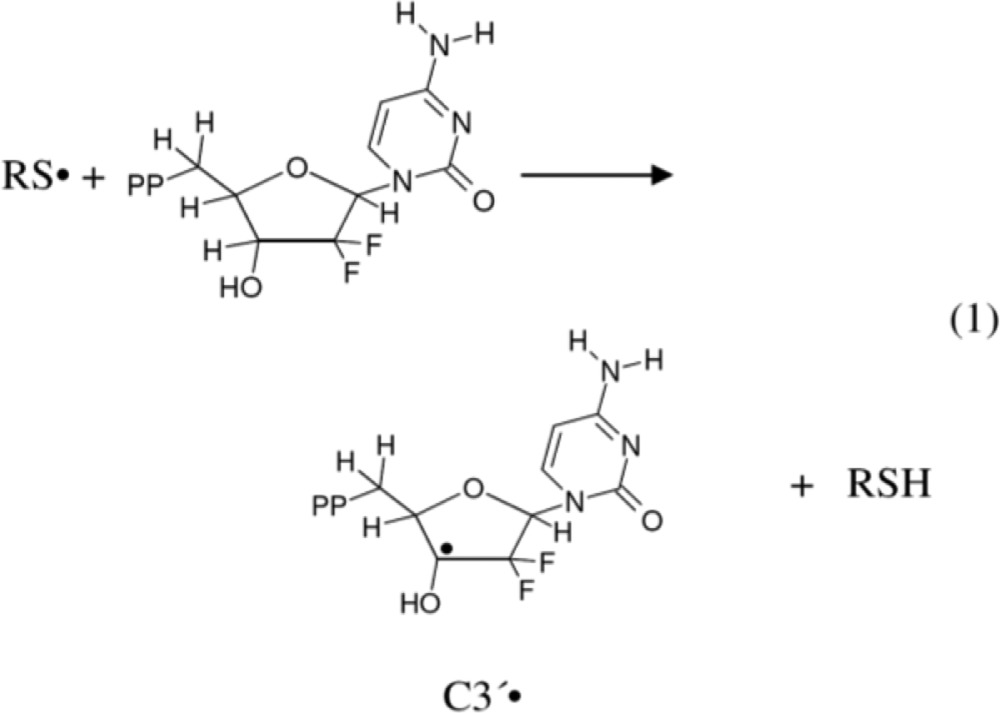 |
1 |
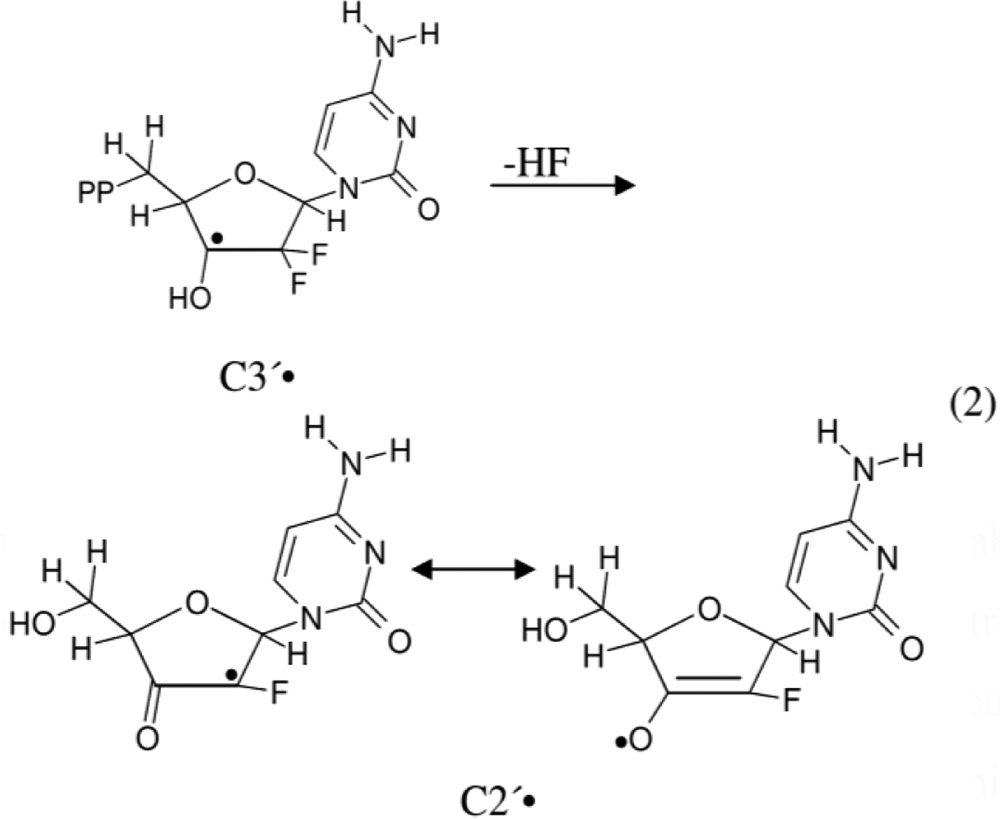 |
2 |
The H-atom abstraction reaction (reaction 1) is not only important in RNR activation but also plays a very key role toward stable product formation in other biologically damage processes in DNA, such as oxidative intrastrand cross-link formation12 and in the formation of sugar radicals that are strand break precursors.13,14
Theoretical modeling,15,16 along with chemical biomimetic studies by Giese et al.,17 by Robins et al.18,19 as well as McCarthy’s 2′-deoxy-2′-fluoromethylenecytidine20,21 support the mechanism shown in reactions 1 and 2. It is noteworthy that pulse radiolysis experiments with a 1,4-anhydro-5-deoxy-6-thio-d-ribo-hexofuranitol detected the formation of ribosyl-based carbon-centered radical(s) after H-atom abstraction by thiyl radicals.22 These studies are supportive of reactions 1 and 2 but unequivocal, and direct detection of C3′• and C2′• employing ESR or pulse radiolysis during RNR-catalyzed deoxygenation of the natural substrates23−25 or during inactivation by gemcitabine still remains elusive.
In this work, we report the formation of C2′• from a likely C3′• in a gemcitabine analog which mimics the mechanism proposed above. From the structural formula of gemcitabine (Scheme 1), it is expected that the negative inductive effect (−I) of two highly electronegative F-atoms at C2′ should increase the acidity of H3′. From our previous work on nucleoside cation radicals,26−38 the gemcitabine cation radical formed upon one-electron oxidation is expected to produce C3′• after deprotonation of the acidic proton H3′. In this work, ESR spectroscopy has been employed to investigate one-electron oxidation of gemcitabine and other 2′-modified derivatives, for example, 2′-deoxy-2′-fluoro-2′-C-methylcytidine (MeFdC (PSI-6130); Scheme 2)39−42 and 2′-fluoro-2′-deoxycytidine (2′-FdC, Scheme 2), in order to test the influence of 2′- substituent on radical site formation. It is noteworthy that MeFdC is a well-known clinically efficacious inhibitor of hepatitis C virus.39,42 The ESR results clearly identify the C3′• formation in one-electron oxidized gemcitabine and the production of C2′• in one-electron oxidized MeFdC. These ESR studies are supported by density functional theory (DFT) calculations. These calculations show that in the case of one-electron oxidized MeFdC, the lowest energy path is the rapid formation of C2′• from C3′• via F– loss. This F– loss is a barrierless reaction between the 2′-F-atom and the proximate H3O+ which was formed via deprotonation of H3′ in the cation radical.
Scheme 2. Structural Formulae of the Compounds (Apart from Gemcitabine (Scheme 1)) Used in This Work.

Materials and Methods
Compounds
Gemcitabine (Scheme 1) and 2′-FdC (Scheme 2) were obtained from Carbosynth Ltd. (Berkshire, UK). MeFdC (Scheme 2) was prepared as described39 or purchased from ADooQ Bioscience (Irvine, CA).
Lithium chloride (LiCl) (ultra dry, 99.995% (metals basis)) was obtained from Alfa Aesar (Ward Hill, MA, USA). 2′-Deoxycytidine (2′-dC) was obtained from Sigma Chemical Company (St Louis, MO, USA). Deuterium oxide (D2O) (99.9 atom % D) was purchased from Aldrich Chemical Co. Inc. (Milwaukee, WI, USA). Potassium persulfate (K2S2O8) was procured from Mallinckrodt, Inc. (Paris, KY, USA). Cytidine-5,6-d2 ([5,6-D,D]-Cyd, 99 atom % D) was purchased from CDN Isotopes (Quebec, Canada). All compounds were used without further purification.
Preparations of Samples
Preparation of Homogeneous Solutions
Homogeneous solutions of gemcitabine were prepared by dissolving 2–10 mg/mL either in 7.5 M LiCl in D2O or in H2O. Solutions of other compounds (2′-dC, 2′-F-dC, MeFdC, and [5,6-D,D]-Cyd) were prepared by dissolving ca. 2–3 mg/mL in 7.5 M LiCl in D2O. K2S2O8 (6–16 mg/mL) was added as an electron scavenger so that only the formation of the one-electron oxidized species and its subsequent reactions can be followed by employing ESR spectroscopy. The above-mentioned procedure for preparation of solutions is according to our ongoing studies on various model systems of DNA and RNA.26−38
pH Adjustments
The pH of gemcitabine in 7.5 M LiCl/D2O was adjusted to the range of ca. 8–12 depending on the experiment. The pH of gemcitabine in 7.5 M LiCl/H2O and the pH of other compounds (2′-dC, 2′-F-dC, MeFdC, and [5,6-D,D]-Cyd) in 7.5 M LiCl/D2O was adjusted at pH ca. 10. These pH adjustments were performed by adding μL amounts 1 M NaOH as per our previous efforts.26−30,32,38 These solutions have high ionic strength (7.5 M LiCl); therefore, the pH meters would not provide accurate pH measurements of these solutions. Instead, as per our previous works,26−30,32,38 pH values reported in this work were obtained using pH papers and are approximate measurements.
Preparation of Glassy Samples and Their Storage
As per our previous works,26−30,32,38 these pH-adjusted homogeneous solutions were thoroughly bubbled with nitrogen to remove the dissolved oxygen. Immediately, these solutions were drawn into 4 mm Suprasil quartz tubes (Catalog no. 734-PQ-8, WILMAD Glass Co., Inc., Buena, NJ, USA) and were rapidly cooled in liquid nitrogen (77 K). The rapid cooling of these homogeneous liquid solutions at 77 K leads to the formation of transparent homogeneous glassy solutions. These glassy solutions were later used for the irradiation and subsequent progressive annealing experiments. All glassy samples were stored at 77 K in Teflon containers in the dark.
Irradiation and Storage of γ-Irradiated Glassy Samples
All samples were γ (60Co)-irradiated (absorbed dose =1.4 kGy) at 77 K and stored at 77 K in Teflon containers in dark following our previous efforts.26−30,32,38
Annealing of Glassy Samples
A variable-temperature assembly was employed which passed liquid nitrogen cooled nitrogen gas past a thermister and over the sample as described in our earlier studies.27 The glassy samples have been annealed anywhere from (140–170) K for 15 min. Annealing leads to one-electron oxidation of the solute by the matrix radical Cl2•– thus, forming only the cation radical of the solute, e.g., gemcitabine.
Electron Spin Resonance
Following our earlier studies,26−30,32,38 immediately after γ-irradiation of the glassy sample at 77 K, the ESR spectrum was recorded at 77 K. Also, immediately after each annealing step, the sample was cooled to 77 K by immersing in liquid nitrogen (77 K), and the ESR spectrum was recorded at 77 K which maximizes signal height and allows for comparison of signal intensities. A Varian Century Series X-band (9.3 GHz) ESR spectrometer with an E-4531 dual cavity, 9 in. magnet, and a 200 mW Klystron was used, and Fremy’s salt (gcenter = 2.0056, A(N) = 13.09 G) was employed for the field calibration. All ESR spectra have been recorded at 77 K and at 40 dB (20 μ W).
Anisotropic simulations of ESR spectra have been performed using the WIN-EPR and SimFonia programs of Bruker as per our previous works.26,28−38,43 The simulated spectra thus obtained were compared to experimental spectra, and ESR parameters were adjusted for the best fit26,28−38,43 (also Supporting Information Figure S3).
Calculations Based on DFT
Theoretical calculations were performed using the Gaussian 09 program.44a GaussView44b and JMOL44c programs were used to plot the spin densities and molecular structures. The geometries of all the radicals considered in the present study were fully optimized using the ωb97x functional45 and 6-31G(d) basis set. We note here that ωb97x functional was developed by the group of Head–Gordon and found to be very successful for the calculations of various properties of molecules in their different spin states.45,46 The hyperfine coupling constants (HFCCs) of the radicals were calculated using the same method and basis set, i.e., ωb97x/6-31G(d) in the gas phase. In order to treat the effect of solvent on HF loss from the C3′• in gemcitabine and in MeFdC, we employ the integral equation formalism polarized continuum model47 (IEF-PCM) as implemented in Gaussian 09. In addition to PCM, for C3′• in both systems a H3O+ is placed in the vicinity of the C3′-OH bond for C3′• in gemcitabine and for C3′• in MeFdC and have optimized the structures. The electronic energy profile of F– dissociation from C2′ site of C3′• in MeFdC as well as the electronic energy profile of F– dissociation for each of the two F-atoms from C2′ site of C3′• in gemcitabine were obtained in the presence of a single water molecule at the same level of theory (Supporting Information Figure S4C,D). Furthermore, employing the wb97x/6-31++G(d,p) method along with the IEF-PCM model for the solvent effect, the pKa of the C3′-OH group for the C3′• of gemcitabine and also of the C3′-OH group for the C3′• of 2′-dC was calculated.
Results and Discussion
Experimental Section
C3′• Formation via One-Electron Oxidation of Gemcitabine in the pH Range ca. 7–12
In Figure 1A, we show the experimentally recorded (77 K) ESR spectrum (green) of one-electron oxidized gemcitabine at pH (pD) ca. 7 in a homogeneous glassy 7.5 M LiCl/D2O solution. The one-electron oxidation of gemcitabine was induced by Cl2•– attack after annealing at 155 K in the dark. The computer simulated spectrum is shown in blue.
Figure 1.
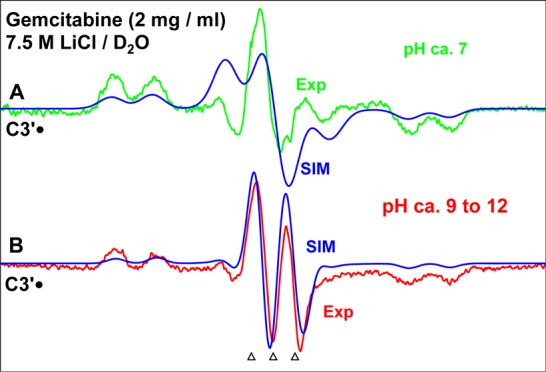
ESR spectra obtained from matched gemcitabine samples [concentration of gemcitabine in each sample = 2 mg/mL in 7.5 M LiCl/D2O] in the presence of the electron scavenger K2S2O8 (8 mg/mL in each sample). Each sample has been γ-irradiated (absorbed dose = 1.4 kGy at 77 K), subsequently annealed to 155 K for 15 min in the dark at various pHs (A) pH ca. 7 (green) and (B) pH ranging ca. 9–12 (pink). Here the spectrum recorded at pH ca. 10 is shown. The blue spectra that are superimposed on the experimentally recorded spectra are the simulated spectra of C3′•. See text for the details of simulation. All ESR spectra are recorded at 77 K. The three reference markers (open triangles) in this figure and in the subsequent figures show the position of Fremy’s salt resonance with the central marker at g = 2.0056. The spacing separating the markers is 13.09 G.
Matched samples of gemcitabine at pDs ranging from ca. 9–12 showed identical spectra after one-electron oxidation of gemcitabine by Cl2•– on annealing at 150–155 K. Thus, only the spectrum obtained from the gemcitabine sample at pD ca. 10 is presented in Figure 1B along with the simulated spectrum in blue. It is evident from Figure 1A,B, the line shape, total hyperfine splitting, and the center of the simulated spectra match with those of the experimentally recorded spectra quite well.
Each of the spectra in Figure 1A,B show two anisotropic β-F-atom hyperfine couplings and a β-H-atom hyperfine coupling. The β-H-atom hyperfine coupling creates the doublet splitting in the line components in Figure 1A,B.
Figure 1A is best matched with a simulation employing the two different anisotropic β-F-atom (nuclear spin = 1/2) HFCC values of (15.0, 15.0, 105) G and (15.0, 15.0, 69.0) G, one β-H HFCC as (15.0, 15.0, 24.0) G, gxx, gyy, gzz (2.0080, 2.0050, 2.0020) along with a mixed (Lorentzian/Gaussian (1:1)) line-width of 14 G. The simulated spectrum in blue is superimposed on the experimentally recorded spectrum in Figure 1A. On the other hand, the best fit for Figure 1B is obtained employing the two identical anisotropic β-F-atom (nuclear spin = 1/2) HFCC as (17.0, 17.0, 86.0) G, one β-H HFCC as (15.0, 15.0, 24.0) G, gxx, gyy, gzz (2.0060, 2.0050, 2.0020) along with a mixed (Lorentzian/Gaussian (1:1)) line-width of 10 G.
Following our work on the radicals produced in monomers of DNA and RNA,28−38,43 the A∥ (i.e., the Azz) component of each of the two anisotropic β-F-atoms (see Table 1) as well as the A∥ of the β-H are directly measured from the width of the experimentally recorded spectra with an uncertainty of ±2 G (see Supporting Information Figure S3). On the other hand, the theoretically obtained values of Axx and Ayy components of each of the two anisotropic β-F-atoms and of the β-H-atom in Table 1 were adjusted to fit the experimentally recorded spectra with estimated uncertainty of ±4 G (see Supporting Information Figure S3).
Table 1. Comparison of the Experimentally Obtained HFCCs Values of C3′• and C2′• in Gauss (G) with Those Obtained by Calculation Using DFT/ωb97x/6-31G(d) Method.
| HFCC (G) |
|||||||
|---|---|---|---|---|---|---|---|
| theory |
expa,b | ||||||
| molecule | radical | atoms | AIso | AAniso | A(total)a | A(total)a,b | |
| gemcitabine | C3′• pH ca. 7 | two β-F-atoms (C2′) | 37.24 | Axx | –14.61 | 22.63 | 15.0 |
| Ayy | –13.95 | 23.29 | 15.0 | ||||
| Azz | 28.56 | 65.8 | 69.0 | ||||
| 69.55 | –29.79 | 39.76 | 15.0 | ||||
| –27.76 | 41.79 | 15.0 | |||||
| 57.55 | 127.1 | 105.0 | |||||
| β-H-atom (C4′) | 28.61 | –1.73 | 26.88 | 15.0 | |||
| –1.03 | 27.58 | 15.0 | |||||
| 2.76 | 31.37 | 24.0 | |||||
| C3′• pH ca. 9–12 | two β-F-atoms (C2′) | 17.0 | |||||
| 17.0 | |||||||
| 86.0 | |||||||
| β-H-atom (C4′) | 15.0 | ||||||
| 15.0 | |||||||
| 24.0 | |||||||
| MeFdCc | C2′• | three β-H-atoms CH3 group | 17.5d (average) | 21.5 | |||
| one β-H-atom (C1′-H) | 23.31 | –1.77 | 21.54 | 25.5 | |||
| –0.73 | 22.77 | ||||||
| 2.51 | 25.82 | ||||||
A(total) = AIso + AAniso.
Experiments give the magnitude but not the sign of the couplings. Estimated errors are of ±2 G for Azz and ±4 G for Axx and also for Ayy. See Supporting Information Figure S3 for details.
Calculated in the presence of one water molecule
Only isotropic HFCC values have been considered.
Thus, the one-electron oxidized gemcitabine spectrum at pH ca. 7 show two nonequivalent anisotropic β-F-atom HFCCs, whereas, the one-electron oxidized gemcitabine spectrum at pH ca. 10 shows two equivalent anisotropic β-F-atom HFCCs. The β-H-atom HFCC does not show any observable change in the one-electron oxidized gemcitabine spectrum throughout the pH range ca. 7–12.
The coupling to two β-F-atoms (C2′) and one β-H-atom (C4′) is clear evidence for the generation of C3′• after one electron oxidation of gemcitabine at 150–155 K. The electron-withdrawing effect of the two electronegative F-atoms at C2′ increases the acidity of H3′, which leads to deprotonation (see Supporting Information Table T2) and prevents observation of the initially formed cytosine base π-cation radical (C•+) in gemcitabine as indicated in reaction 3. Therefore, the mechanism of C3′• formation due to one-electron oxidation of gemcitabine is proposed as follows: one-electron oxidation of gemcitabine results in the formation of metastable C•+, which is unstable even at ca. 155 K. The metastable C•+ quickly deprotonates at C3′ in the sugar moiety producing C3′• (reaction 3) via a proton-coupled electron-transfer (PCET) mechanism.
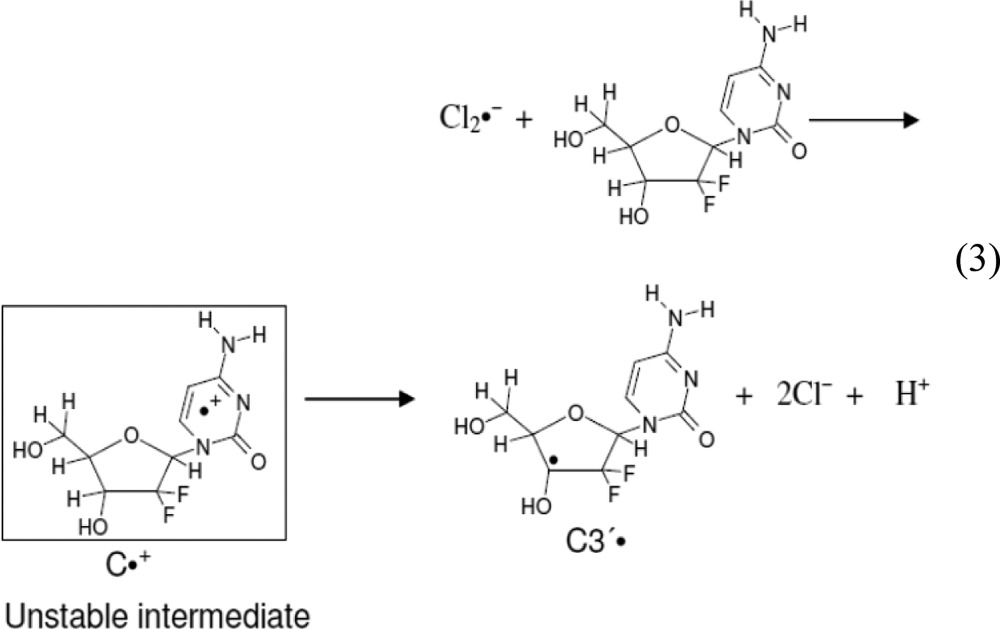 |
3 |
Origin of the pH Effect
As is evident from the HFCC values of the spectra shown in Figure 1 and also in Table 1, the pH of the solution clearly affects the individual β-F-atom anisotropic HFCC but not the sum of the two β-F-atom anisotropic HFCC along with the C4′ β-proton HFCC (see section above). At pH ca. 9–12, the two β-F-atom anisotropic HFCCs are equivalent; whereas, at pH ca. 7, the sum of the two β-F-atom anisotropic HFCCs remain the same, but they individually differ.
The presence of two 2′-F-atoms in gemcitabine will lower the pKa value of both H3′ and C3′-OH hydrogens. For example, the OH in 2,2-difluoroethanol has its pKa lowered by 3.5 units in comparison with ethanol.48 Further, radical formation has also been shown to lower the pKa of the alcoholic OH group (e.g., pKa (CH3)2CHOH = 17.1, pKa (CH3)2C·OH = 12.03).49 Based on these factors, the pKa value of the C3′-OH group for C3′• in gemcitabine is estimated to be in the range of 7–9. Employing DFT/ωb97x/6-31++G(d,p) method along with the IEF-PCM model for the solvent effect, the pKa value of the C3′-OH group for C3′• in 2′-dC has been calculated as 14.2. The same level of calculation for the C3′-OH group of C3′• in gemcitabine which replaces each of the two hydrogens at C2′ with fluorine predicts a pKa of 6.8 (see Supporting Information pages S8, S9). Therefore, the two 2′-F-atoms are predicted to lower the pKa of the C3′-OH group in C3′• by 7 full units. Considering the sensitivity of the calculations for predicting pKa to the small changes in free energy,37 these results are very reasonable. Furthermore, the theoretically calculated pKa value 6.8 of the C3′-OH group for C3′• in gemcitabine is in good agreement with its experimentally estimated value (7–9).
The spectrum in Figure 1A (pH ca. 7) should therefore be for C3′• in gemcitabine with a C3′-OH group and the spectrum in Figure 1B (pH ca. 9–12) should be for C3′• with the deprotonated group, i.e., C3′-O– (reaction 4). Thus, we attribute the variation of the two β-F-atom anisotropic HFCC to the deprotonation of the C3′-OH group at higher pHs.
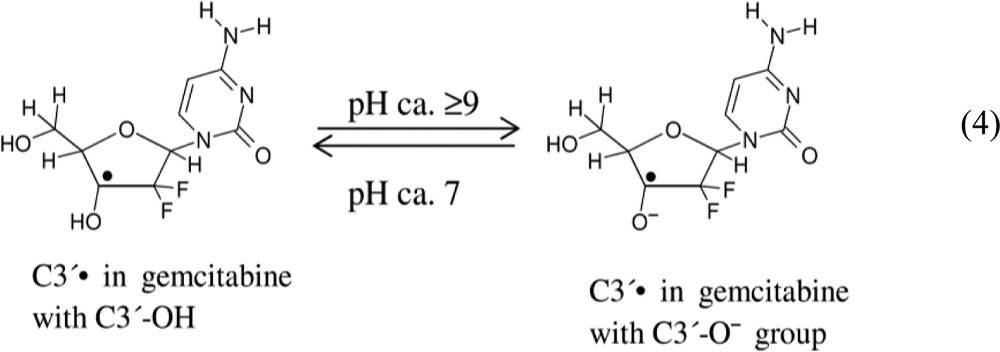 |
4 |
Influence of the Solvent (D2O vs H2O) on One-Electron Oxidation of Gemcitabine
ESR spectral studies of one-electron oxidation of gemcitabine in H2O glasses (7.5 M LiCl/H2O) were performed and compared with the results found in D2O glasses (7.5 M LiCl/D2O). These results are shown in Supporting Information Figure S1. No observable difference in spectra other than a small line broadening was observed on formation of C3′• in H2O glasses versus in D2O glasses. Since C3′• only has the C3′-OH as an exchangeable proton at pH 7 and this does not contribute to a significant hyperfine coupling, the ESR spectrum found on formation of C3′• is not altered by a change of the solvent from D2O to H2O.
C2′• Formation in One-Electron Oxidized, 2′-Deoxy-2′-fluoro-2′-C-methylcytidine (MeFdC)
Similar experiments to those performed for gemcitabine were carried out for the methyl/fluoro analog MeFdC (Scheme 2). Using MeFdC, we investigated whether the formation of C3′• observed in gemcitabine bearing geminal difluoro unit at C2′ (Figure 1) is affected by the substitution of one of the F-atoms (−I) with a methyl (Me) group (+I). The results are presented in Figure 2.
Figure 2.
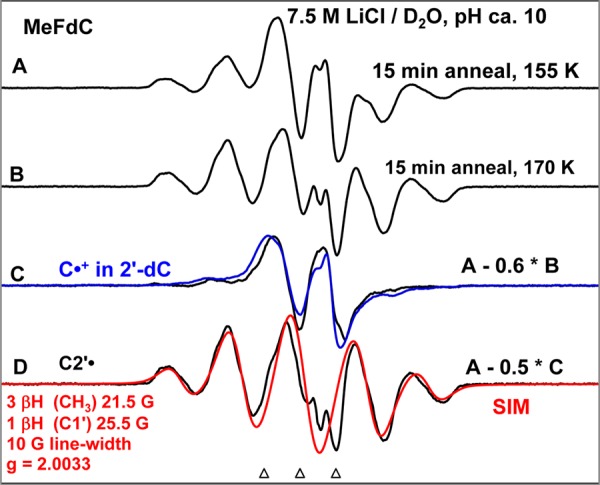
(A) ESR spectrum (black) obtained from MeFdC [concentration = 2 mg/mL in 7.5 M LiCl/D2O] in the presence of the electron scavenger K2S2O8 (8 mg/mL), pH ca. 10, γ-irradiated to a dose of 1.4 kGy at 77 K and subsequently annealed to 155 K for 15 min. (B) Annealed to 170 K for 15 min. (C) Spectrum (black) obtained after subtraction of 60% of spectrum (B) from spectrum (A). For comparison, the C•+ spectrum (blue) in 2′-dC (Supporting Information Figure S2 and pp S3–S5) is superimposed. (D) Spectrum (black) assigned to C2′• is obtained after subtraction of 50% of spectrum (C) from spectrum (A). The simulated C2′• spectrum (for simulation parameters see Figure 2 and text) is superimposed on the experimentally isolated spectrum for comparison. All the spectra are recorded at 77 K.
Shown in Figure 2A is the ESR spectrum (black) of a matched sample of MeFdC that has been γ-irradiated (absorbed dose = 1.4 kGy), subsequently annealed to 155 K for 15 min in the dark, and recorded at 77 K.
Figure 2B was obtained by annealing this sample for 15 min to 170 K. Comparison of spectrum 2A with spectrum 2B shows clearly that a central doublet decreases along with an increase of the other line components upon annealing. Therefore, the central doublet (black) shown in Figure 2C is isolated by subtraction of 60% of the spectrum 2B from spectrum 2A. The doublet due to C•+ spectrum (blue) in 2′-dC (see Supporting Information Figure S2 and its discussion (pp S3–S5)) is superimposed on it for comparison. From the spectral similarities of both doublets, the doublet in black shown in Figure 2C is assigned to C•+ in MeFdC.
Subtraction of 50% C•+ spectrum 2C (black) from spectrum 2A results in the black spectrum shown in Figure 2D. This overall quintet spectrum arises from 4 isotropic β-proton couplings: three methyl β-protons (ca. 21.5 G each) and an isotropic splitting of ca. 25.5 G due to a β-proton assigned to the C1′-H (vide infra). The experimental (black) spectrum is simulated using the above-mentioned HFCC values along with a 10 G line-width and g-value = 2.0033 (this g-value is typical for C-centered sugar radicals).28,32,33,35,36,38,50−55 The simulated spectrum (red) in Figure 2D matches the overall line components of the experimental spectrum well. Since the C2′• (reaction 5) is the only likely radical structure that would explain the large hyperfine coupling to a methyl group and the additional β-proton hyperfine coupling (assigned to C1′), the experimental spectrum in Figure 2D has been assigned to C2′• (reaction 5).
The spectra 2A and 2B are a composite of C•+ (black, Figure 2C) and C2′• (black, Figure 2D) in different amounts. Under the same constant gain and constant microwave power and upon gradual and stepwise annealing of the sample from 155 K (spectrum 2A) to 170 K (spectrum 2B), no loss of spectral intensity was observed, and our analyses show an additional (ca. 20%) conversion of the C•+ to C2′•.
Consideration of the results presented in Figures 1 and 2 suggest that for MeFdC, C•+ is produced first and on annealing converts to a transient C3′•, which is not observed. In contrast, the line shape, line width, and the overall hyperfine splitting of the C3′• spectrum in gemcitabine do not change upon annealing to ca. 165 K (i.e., within the temperature range 155–165 K). Thus, unlike the rapid conversion of C3′• to C2′• found in one-electron oxidized MeFdC, a similar conversion of C3′• to C2′• is not observed for one-electron oxidized gemcitabine at these low temperatures. The lack of observation of the transient C3′• in one-electron oxidized MeFdC and the low temperatures employed in these experiments implies a very low activation barrier for the conversion of C3′• to C2′•; whereas, the activation barrier for the conversion of C3′• to C2′• for one-electron oxidized gemcitabine should be ≥4 kcal/mol. Calculations suggest that the proximity of the lost H3′ proton as H3O+ to the 2′-F-atom quite likely provides the driving force for this rapid unimolecular reaction (see Figure 3). Therefore, we propose that C3′• in one-electron oxidized MeFdC readily converts to C2′• via a barrierless F– loss (see reaction 5, Figure 3, and Supporting Information S5).
Figure 3.

PCM-ωb97x/6-31G(d) calculated structures of C3′• in (A) gemcitabine and in (B) MeFdC. As indicated in (A) the C3′• in gemcitabine involves a barrier of 5 kcal/mol (detailed electronic energy profiles are provided in Supporting Information Figure S4C,D) to HF loss in the presence of H3O+. In (B), the C3′• in MeFdC is unstable in the presence of H3O+ and reacts without a barrier to form C2′• via HF loss. The animations (movies) of the optimization steps for reaction involving H3O+ for C3′• in both gemcitabine (A) and MeFdC (B) are provided in the Supporting Information (S5).
Theoretical
Comparison of the Theoretically Calculated HFCC Values of Radicals with Their Experimentally Obtained HFCC Values
The ωb97x/6-31G(d) calculated HFCCs of C3′• and C2′• found in gemcitabine and in MeFdC along with experimental HFCCs (in Gauss) are presented in Table 1. It is evident from Table 1 that experimental and theoretically calculated HFCCs are in reasonably good agreement.
Mechanism of C3′• to C2′• Conversion in MeFdC
To explore the reaction mechanism of C2′• formation from C3′• in one-electron oxidized MeFdC and gemcitabine, using the DFT ωb97x/6-31G(d) method, we considered three possible reaction paths: (i) HF loss due to deprotonation of 3′-hydroxyl group,5−7 (ii) HF loss due to fluorine dissociation,5−7 and additionally, (iii) HF loss in the presence of a hydronium ion (H3O+).
The electronic energy profile of HF loss from C3′• in MeFdC and also from C3′• in gemcitabine in the presence of a water molecule is shown in Figure S4 in the Supporting Information. From Supporting Information Figure S4A, it is evident that for C3′• of MeFdC, the formation of C2′• via HF loss with deprotonation of 3′-OH has a significant barrier of ca. 18 kcal/mol. For C3′• in gemcitabine, the HF loss associated with deprotonation of 3′-hydroxyl group was calculated to be ca. 27 kcal/mol (Supporting Information, Figure S4B).
We have also considered the dissociation of fluorine in C3′• of MeFdC and in C3′• of gemcitabine and found that stretching the C2′-F bond up to 1.8 Å needs ca. 9 kcal/mol for C3′• of MeFdC and ca. 17 kcal/mol for C3′• of gemcitabine. This shows that dissociation of fluorine for C3′• in MeFdC occurs at lower energy than dissociation of fluorine for C3′• in gemcitabine (Supporting Information Figure S4).
Alternatively, we consider the fact that deprotonation of H3′ will form H3O+ initially in close proximity of the C2′-C3′ bond which may then induce HF loss (reaction 5). Employing the ωb97x/6-31G(d) method and considering the full solvent effect through the polarized continuum model (PCM), this mechanism have been modeled by placing a H3O+ in the vicinity of the C3′-OH bond for C3′• in gemcitabine and for C3′• in MeFdC and have optimized the structures. From our calculations, we have observed that for C3′• in gemcitabine, a minimum structure exists in the electronic energy profile in which the H3O+ stabilizes the F″-atom through forming a hydrogen bond (1.57 Å) (see Figure 3A and Supporting Information Figure S4C). A second minimum structure was also found in the electronic energy profile in which the H3O+ stabilizes the F′-atom through forming a hydrogen bond of identical length, 1.57 Å (see Supporting Information Figure S4D). A barrier of 5 kcal/mol and the overall reaction energy of −7 to −8 kcal/mol were found for HF formation in both cases (see Figure 3A and Supporting Information Figures S4C,D). However, for C3′• in MeFdC, the H3O+ reacts with the 2′-F-atom without a barrier and forms HF and C2′• (see Figure 3B). The bond distances of C3′-O3′ and O3′-H bonds for C3′• in gemcitabine are calculated as 1.34 Å (primarily C–O single bond character)56 and 0.97 Å, respectively. The values of the corresponding bond distances of C3′-O3′ and O3′-H bonds for C3′• in MeFdC are obtained as 1.28 Å (mainly double bond character56 and 1.0 Å respectively).
Thus, these calculations show that the HF loss from C3′• in gemcitabine has a ca. 5 kcal/mol barrier (Supporting Information Figure S4C,D) while for C3′• in MeFdC, the loss of HF is barrierless, and the C2′• production is exothermic in nature as shown in Figure 3. These findings support our experimental observations that in MeFdC, C3′• is too unstable to be observed, and only C2′• is found. In contrast, in gemcitabine only C3′• formation is observed without any conversion to C2′• in the same temperature range.
Conclusion
Our work has the following two salient findings: (i) One electron oxidation leads to cytosine base π-cation radical (C•+) in 2′-dC and 2′-F-dC but to C3′• in gemcitabine. As expected from the one-electron redox potentials of the bases and the backbone,27−30,57−59 one-electron oxidation of 2′-dC and 2′-F-dC leads to C•+ formation as evidenced by the ca. 16 G doublet that is characteristic of C•+. However, gemcitabine (Scheme 1) leads to the formation of C3′• on one-electron oxidation. The two highly electronegative F-atoms at 2′-position, through their negative inductive effect, lead to a substantial increase in the acidity of H3′. Therefore, C•+ in gemcitabine is highly unstable toward the loss of H3′ as deprotonation at 150–155 K. This is evidenced by the free energy changes of the cation radical for the loss of H3′ as deprotonation to the surrounding solvent (see Supporting Information Table T2); this deprotonation shifts the unpaired spin from the cytosine base of metastable C•+ in gemcitabine to sugar at C3′• via a PCET process.
(ii) C2′• formation does not occur in gemcitabine but does in its analog MeFdC. It has been proposed in the literature that in gemcitabine both C3′• and C2′• (reactions 1 and 2) play an important role in the RNR inactivation.5−7 Conversion of C3′• to C2′• takes place via an irreversible F– loss from C2′ during RNR inactivation by gemcitabine.5−7 However, experimental and theoretical results shown in this work have clearly demonstrated that in our system (supercooled homogeneous glassy solutions), C3′• in gemcitabine does not convert to C2′• on annealing up to 170 K owing to theoretically predicted barrier of greater than 5 kcal/mol. Theoretically, DFT calculations support the mechanism involving a H3O+ induced barrierless conversion of C3′• to C2′• in one-electron oxidized MeFdC. Experimentally, C2′• is observed in one-electron oxidized MeFdC upon annealing to ca. 160–170 K. Thus, our study in one-electron oxidized MeFdC provides the first evidence of formation of C2′• (via the unstable intermediate C3′• (reaction 5)) in a nonenzymatic system even at low temperature.
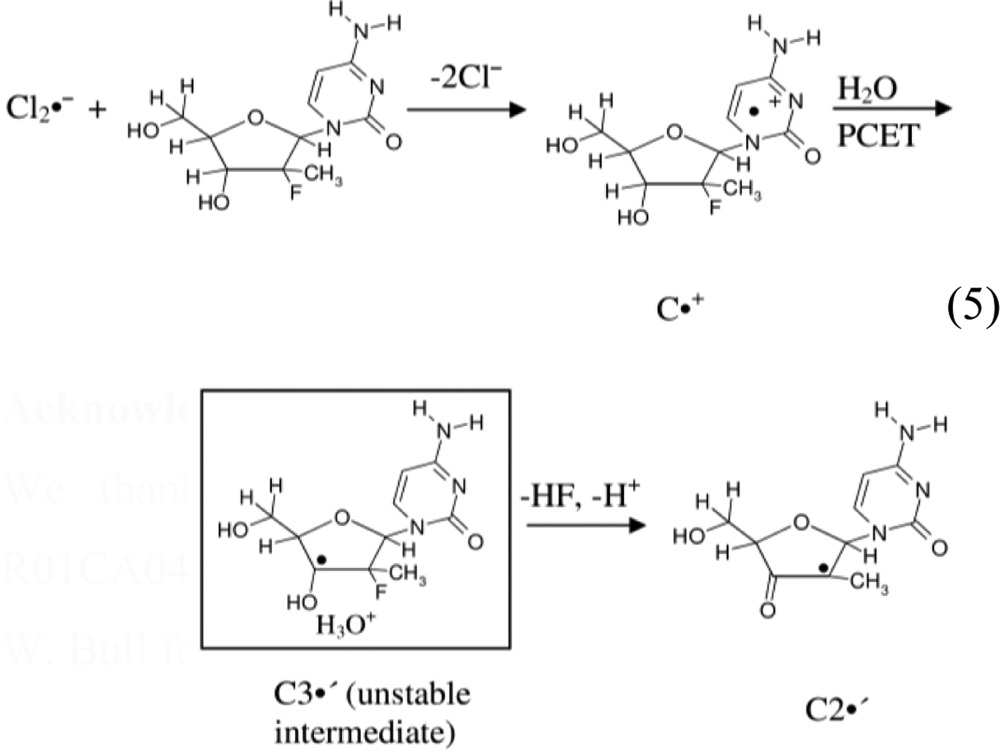 |
5 |
Acknowledgments
We thank the National Cancer Institute of the National Institutes of Health (Grants R01CA045424 for MDS and SC1CA138176 for SFW) for support. The authors thank Prof. A. W. Bull for helpful suggestions and critical review of the manuscript.
Supporting Information Available
Supporting Information contains the following: (i) C3′• formation in one-electron oxidized gemcitabine in D2O and in H2O; (ii) formation of cytosine cation radical via one-electron oxidation of 2′-dC, 2′-F-dC, and of [5, 6-D,D-Cyd]; (iii) estimated uncertainties (errors) in HFCC values; (iv) prediction of pKa values of the C3′-OH group of C3′• in gemcitabine and also of the C3′-OH group of C3′• in 2′-dC; (v) calculated free energy changes for deprotonation of H3′ in cation radicals; (vi) electronic energy profile of HF loss from C3′• in MeFdC and from C3′• in gemcitabine; (vii) animations (movies) of the optimization steps for reaction involving H3O+ for C3′• in both gemcitabine and MeFdC, and (viii) wb97x/6-31G(d) optimized structures of various radicals considered in this work and their isotropic and anisotropic HFCC values. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Burris H. A. III; Moore M. J.; Andersen J.; Green M. R.; Rothenberg M. L.; Modiano M. R.; Cripps M. C.; Portenoy R. K.; Storniolo A. M.; Tarassoff P.; Nelson R.; Dorr F. A.; Stephens C. D.; Von Hoff D. D. J. Clin. Oncol. 1997, 15, 2403–2413. [DOI] [PubMed] [Google Scholar]
- Von Hoff D. D.; Ervin T.; Arena F. P.; Chiorean E. G.; Infante J.; Moore M.; Seay T.; Tjulandin S. A.; Ma W. W.; Saleh M. N.; Harris M.; Reni M.; Dowden S.; Laheru D.; Bahary N.; Ramanathan R. K.; Tabernero J.; Hidalgo M.; Goldstein D.; Van Cutsem E.; Wei X.; Iglesias J.; Renschler M. F. New Engl. J. Med. 2013, 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thota R.; Pauff J. M.; Berlin J. D. Oncology 2014, 28, 70–74. [PubMed] [Google Scholar]
- Kleger A.; Perkhofer L.; Seufferlein T. Ann. Oncol. 2014, 25, 1260–1270. [DOI] [PubMed] [Google Scholar]
- Wang J.; Lohman G. J. S.; Stubbe J. Biochemistry 2009, 48, 11612–11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman G. J. S.; Stubbe J. Biochemistry 2010, 49, 1404–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artin E.; Wang J.; Lohman G. J.; Yokoyama K.; Yu G.; Griffin R. G.; Bar G.; Stubbe J. Biochemistry 2009, 48, 11622–11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi V.; Legha J.; Chen F.; Hertel L. W.; Plunkett W. Cancer Res. 1996, 56, 4453–4459. [PubMed] [Google Scholar]
- Plunkett W.; Huang P.; Xu Y. Z.; Heinemann V.; Grunewald R.; Gandhi V. Semin. Oncol. 1995, 22, 3–10. [PubMed] [Google Scholar]
- Akhlaq M. S.; Schuchmann H. P.; Von Sonntag C. Int. J. Radiat. Biol. 1987, 51, 91–102. [DOI] [PubMed] [Google Scholar]
- Wardman P.; von Sonntag C. Methods Enzymol. 1995, 251, 31–45. [DOI] [PubMed] [Google Scholar]
- Cerón-Carrasco J. P.; Jacquemin D.; Dumont E. J. Phys. Chem. B 2013, 117, 16397–16404. [DOI] [PubMed] [Google Scholar]
- von Sonntag C.Free-radical-induced DNA Damage and Its Repair; Springer-Verlag: Berlin, Heidelberg, 2006; pp 288–299. [Google Scholar]
- Chatgilialoglu C. In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry; Greenberg M. M., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2009; pp 99–133. [Google Scholar]
- Pereira S.; Fernandes P. A.; Ramos M. J. J. Comput. Chem. 2004, 25, 1286–1294. [DOI] [PubMed] [Google Scholar]
- Cerqueira N. M. F. S. A.; Fernandes P. A.; Ramos M. J. Chem.—Eur. J. 2007, 13, 8507–8515. [DOI] [PubMed] [Google Scholar]
- Lenz R.; Giese B. J. Am. Chem. Soc. 1997, 119, 2784–2794. [Google Scholar]
- Robins M. J.; Guo Z.; Samano M. C.; Wnuk S. F. J. Am. Chem. Soc. 1999, 121, 1425–1433. [Google Scholar]
- Robins M. J.; Ewing G. J. J. Am. Chem. Soc. 1999, 121, 5823–5824. [Google Scholar]
- van der Donk W. A.; Yu G.; Silva D. J.; Stubbe J.; McCarthy J. R.; Jarvi E. T.; Matthews D. P.; Resvick R. J.; Wagner E. Biochemistry 1996, 35, 8381–8391. [DOI] [PubMed] [Google Scholar]
- Gerfen G. J.; van der Donk W. A.; Yu G.; McCarthy J. R.; Jarvi E. T.; Matthews D. P.; Farrar C.; Griffin R. G.; Stubbe J. J. Am. Chem. Soc. 1998, 120, 3823–3835. [Google Scholar]
- Wnuk S. F.; Penjarla J. A. K.; Dang T. P.; Mebel A. M.; Nauser T.; Schöneich C. Collect. Czech. Chem. Commun. 2011, 76, 1223–1238. [Google Scholar]
- Persson A. L.; Sahlin M.; Sjoberg B.-M. J. Biol. Chem. 1998, 273, 31016–31020. [DOI] [PubMed] [Google Scholar]
- Lawrence C. C.; Bennati M.; Obias H. V.; Bar G.; Griffin R. G.; Stubbe J. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipse H.; Artin E.; Wnuk S.; Lohman G. J. S.; Martino D.; Griffin R. G.; Kacprzak S.; Kaupp M.; Hoffman B.; Bennati M.; Stubbe J.; Lees N. J. Am. Chem. Soc. 2009, 131, 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Adhikary A.; Khanduri D.; Sevilla M. D. J. Am. Chem. Soc. 2009, 131, 8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Adhikary A.; Sevilla M. D. J. Phys. Chem. B 2011, 115, 8947–8948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanduri D.; Adhikary A.; Sevilla M. D. J. Am. Chem. Soc. 2011, 133, 4527–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Becker D.; Palmer B. J.; Heizer A. N.; Sevilla M. D. J. Phys. Chem. B 2012, 116, 5900–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Kumar A.; Heizer A. N.; Palmer B. J.; Pottiboyina V.; Liang Y.; Wnuk S. F.; Sevilla M. D. J. Am. Chem. Soc. 2013, 135, 3121–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Kumar A.; Palmer B. J.; Todd A. D.; Sevilla M. D. J. Am. Chem. Soc. 2013, 135, 12827–12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Kumar A.; Munafo S. A.; Khanduri D.; Sevilla M. D. Phys. Chem. Chem. Phys. 2010, 12, 5353–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Malkhasian A. Y. S.; Collins S.; Koppen J.; Becker D.; Sevilla M. D. Nucleic Acids Res. 2005, 33, 5553–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Becker D.; Collins S.; Koppen J.; Sevilla M. D. Nucleic Acids Res. 2006, 34, 1501–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Kumar A.; Becker D.; Sevilla M. D. J. Phys. Chem. B 2006, 110, 24170–24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanduri D.; Collins S.; Kumar A.; Adhikary A.; Sevilla M. D. J. Phys. Chem. B 2008, 112, 2168–2178. [DOI] [PubMed] [Google Scholar]
- Adhikary A.; Khanduri D.; Kumar A.; Sevilla M. D. J. Phys. Chem. B 2008, 112, 15844–15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Kumar A.; Khanduri D.; Sevilla M. D. J. Am. Chem. Soc. 2008, 130, 10282–10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A.; Kumar A.; Palmer B. J.; Todd A. D.; Heizer A. N.; Sevilla M. D. Int. J. Radiat. Biol. 2014, 90, 433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J. L.; Hollecker L.; Mason J. C.; Stuyver L. J.; Tharnish P. M.; Lostia S.; McBrayer T. R.; Schinazi R. F.; Watanabe K. A.; Otto M. J.; Furman P. A.; Stec W. J.; Patterson S. E.; Pankiewicz K. W. J. Med. Chem. 2005, 48, 5504–5508. [DOI] [PubMed] [Google Scholar]
- Wang P.; Chun B.-K.; Rachakonda S.; Du J.; Khan N.; Shi J.; Stec W.; Cleary D.; Ross B. S.; Sofia M. J. J. Org. Chem. 2009, 74, 6819–6824. [DOI] [PubMed] [Google Scholar]
- Peifer M.; Berger R.; Shurtleff V. W.; Conrad J. C.; MacMillan D. W. C. J. Am. Chem. Soc. 2014, 136, 5900–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia M. J.; Chang W.; Furman P. A.; Mosley R. T.; Ross B. S. J. Med. Chem. 2012, 55, 2481–2581. [DOI] [PubMed] [Google Scholar]
- Adhikary A.; Khanduri D.; Pottiboyina V.; Rice C. T.; Sevilla M. D. J. Phys. Chem. B 2010, 114, 9289–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009.; b GaussView; Gaussian, Inc.: Pittsburgh, PA, 2003.; c Jmol: An open-source Java viewer for chemical structures in 3D; Jmol Development Team, An Open-Science Project, 2004; http://jmol.sourceforge.net.
- Chai J.-D.; Head-Gordon M. J. Chem. Phys. 2008, 128, 084106. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Sevilla M. D. J. Phys. Chem. B 2014, 118, 5453–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Chem. Rev. 2005, 105, 2999–3094. [DOI] [PubMed] [Google Scholar]
- Engbersen J. F. J.; Broos J.; Verboom W.; Reinhoudt D. N. Pure Appl. Chem. 1996, 68, 2171–2178. [Google Scholar]
- Laroff G. P.; Fessenden R. W. J. Phys. Chem. 1973, 77, 1283–1288. [Google Scholar]
- De Cooman H.; Vanhaelewyn G.; Pauwels E.; Sagstuen E.; Waroquier M.; Callens F. J. Phys. Chem. B 2008, 112, 15045–15053. [DOI] [PubMed] [Google Scholar]
- Pauwels E.; De Cooman H.; Vanhaelewyn G.; Sagstuen E.; Callens F.; Waroquier M. J. Phys. Chem. B 2008, 112, 15054–15063. [DOI] [PubMed] [Google Scholar]
- Tarpan M. A.; Pauwels E.; Vrielinck H.; Waroquier M.; Callens F. J. Phys. Chem. A 2010, 114, 12417–12426. [DOI] [PubMed] [Google Scholar]
- Close D. M. Radiat. Res. 1997, 147, 663–673. [PubMed] [Google Scholar]
- Close D. M. In Radiation Induced Molecular Phenomena in Nucleic Acid: A Comprehensive Theoretical and Experimental Analysis; Shukla M. K., Leszczynski J., Eds.; Springer-Verlag: Berlin, Heidelberg, 2008; pp 493–529. [Google Scholar]
- Bernhard W. A.; Close D. M. In Charged Particle and Photon Interactions with Matter Chemical, Physicochemical and Biological Consequences with Applications; Mozumdar A., Hatano Y., Eds.; Marcel Dekkar, Inc.: New York, 2004; pp 431–470. [Google Scholar]
- Glockler G. J. Phys. Chem. 1958, 62, 1049–1054. [Google Scholar]
- Steenken S. Chem. Rev. 1989, 89, 503–520. [Google Scholar]
- Steenken S. Free Radical Res. Commun. 1992, 16, 349–379. [DOI] [PubMed] [Google Scholar]
- Steenken S. Biol. Chem. 1997, 378, 1293–1297. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.