

Abstract

We report the late-stage functionalization of multisubstituted pyridines and diazines at the position α to nitrogen. By this process, a series of functional groups and substituents bound to the ring through nitrogen, oxygen, sulfur, or carbon are installed. This functionalization is accomplished by a combination of fluorination and nucleophilic aromatic substitution of the installed fluoride. A diverse array of functionalities can be installed because of the mild reaction conditions revealed for nucleophilic aromatic substitutions (SNAr) of the 2-fluoroheteroarenes. An evaluation of the rates for substitution versus the rates for competitive processes provides a framework for planning this functionalization sequence. This process is illustrated by the modification of a series of medicinally important compounds, as well as the increase in efficiency of synthesis of several existing pharmaceuticals.
Introduction
Pyridines and diazines are among the most prevalent heterocycles in biologically active compounds. They are found in 2 of the 5 top-selling pharmaceuticals1 and 6 of the 23 small molecules approved by the FDA in 2013.2 Of course, the groups appended to the heteroarene and the heteroarene core together affect the activity of the molecule, not just the heteroarene itself. Thus, studies of the structure–activity relationships (SAR) require derivatives containing a range of substituents attached to pyridines and diazines. One potential method to create these derivatives is C–H bond functionalization of the heteroarene. Yet, few C–H bond functionalization reactions are amenable to late-stage functionalization of heteroarenes. Instead, multistep syntheses are typically conducted to study the effects of substituents on the heteroarene units.
Some C–H bond functionalization reactions of heteroarenes have been developed that exploit a directing group, but methods for site-selective functionalization without the influence of such groups would allow modifications at different positions and without the need to install and remove such groups.
Three major classes of reactions lead to the functionalization of C–H bonds in pyridines and diazines without the influence of directing groups. The first class comprises Minisci-type reactions, which occur by the addition of carbon-centered radicals to heteroarenes (Scheme 1A).3,4 These reactions are versatile, but mixtures of isomers are formed in most cases, and the regioselectivity depends on the steric and electronic properties of both the heteroarene and the radical partner.3a,5
Scheme 1. Strategies for C–H Functionalization of Pyridines.

A second class of heteroarene C–H bond functionalization reactions includes additions of nucleophiles to pyridine N-oxides in combination with a reagent to activate and dehydrate the pyridine N-oxide (Scheme 1B). Reactions of this type have been developed to install Br, Cl, CN, amino, phenoxy, and nucleophiles derived from reagents containing acidic C–H and N–H bonds (pKa range of ∼10–20).6 In most cases, the scope of the pyridine N-oxides that undergo these reactions is limited to those containing ether, ester, and nitrile substituents or halides at positions not reactive toward SNAr. In general, functionalization occurs at the 2-position, but competing functionalization at the 4-position or on pendant alkyl groups is often observed. In addition to these issues of functional group compatibility and site selectivity, the need to prepare and isolate the N-oxides limits the use of these reactions.
Most closely related to the work reported here, C–H functionalization reactions at the 2-position of pyridines and pyridine N-oxides have been developed with transition metal catalysts.7 However, these reactions are limited to the formation of C–C bonds, and the reaction conditions and scope suggest that these reactions are not suitable for complex substrates.8 Other functionalizations of heteroaryl C–H bonds involve cross-coupling at the most acidic C–H bond; these methods occur most commonly with five-membered ring heteroarenes.7a−7c
Finally, C–H borylation reactions catalyzed by transition metal complexes provide a means to install substituents onto heteroarenes by conversion of the heteroarene to the corresponding heteroarylboronate ester and subsequent transformations of the C–B bond.9 However, borylations of pyridines occur predominantly at the 3- and 4-positions and, therefore, are complementary to the reactions at the 2-position described here.
We aimed to develop a method for the late-stage functionalization of pyridines and diazines that would address the limitations of the regioselectivity and scope of C–H functionalizations of heteroarenes. Our approach was inspired by the value of borylation reactions developed in the authors’ laboratory to create synthetic intermediates that can be converted to a variety of functionalized products.9b We considered that the C–H fluorination of pyridines and diazines10 at the position α to nitrogen with AgF2 we developed recently could be used for the late-stage functionalization of medicinally relevant compounds because pyridines and diazines are contained in many such compounds and the 2-fluoro group could be replaced with a wide range of nucleophiles.
Herein, we report mild conditions for the SNAr reaction of fluoroheteroarenes, an assessment of the potential of the fluorination and SNAr reactions to be conducted with complex structures, and the application of these findings to the development of late-stage functionalizations of complex heterocyclic compounds by the combination of C–H fluorination and SNAr reactions (Scheme 1C). In addition to revealing the potential of this reaction for the functionalization of complex heteroarenes, we demonstrate how the combination of C−H bond fluorination and SNAr creates routes to several active pharmaceutical ingredients that occur in higher yields and fewer steps than previously reported syntheses of these molecules.
Results and Discussion
Conditions for the Nucleophilic Aromatic Substitution of 2-Fluoropyridines
SNAr reactions of 2- or 4-halopyridines comprise a site-specific method to synthesize substituted pyridines.11 However, this approach requires initial synthesis and isolation of halogenated pyridines that are typically prepared from pyridine N-oxides or hydroxypyridines with neat POX3 (X = Br, Cl) at high temperatures.
The majority of SNAr reactions with halopyridines have been performed with chloropyridines. Chloropyridines are more available commercially than other halopyridines, but the reactions of fluoropyridines are likely to be faster than those of chloropyridines. As for SNAr reactions of arenes, the SNAr reactions of pyridines and diazines are likely to be accelerated by the high electronegativity of fluorine. Indeed, the reaction of 2-fluoropyridine with NaOEt in EtOH is 320 times faster than the reaction of 2-chloropyridine.12 This higher reactivity of fluoropyridines could allow SNAr reactions to occur under conditions that are mild enough to allow this class of reaction to occur on complex molecules; however, a more detailed assessment of the rates and yields for the SNAr reactions of fluoropyridines and diazines would be needed to predict the scope of the SNAr process and a method to create the 2-fluoropyridines and diazines that could be conducted in a typical laboratory on complex pyridines and diazines would be required.
Thus, to develop a sequence of C–H bond fluorination and SNAr, we first evaluated conditions to conduct SNAr reactions of 2-fluoropyridines. Although SNAr reactions of electron-deficient fluoroarenes and chloropyridines are commonplace,11 few studies have been performed on SNAr reactions of fluoropyridines.13 The majority of the published reactions of 2-fluoropyridines have been conducted with unsubstituted 2-fluoropyridine; a few examples were conducted with 2-fluoropyridines containing a single bromide substituent. Moreover, these reactions were performed under conditions involving strong nucleophiles and bases at high temperatures (up to 130 °C), neat reagents, microwave heating, strong reducing agents (LiBH3NR2), or toxic solvents (HMPA), and these conditions are unlikely to tolerate the functional groups found in complex molecules relevant to medicinal chemistry. Several SNAr reactions of 2-fluoropyridines have been reported in the patent literature on both simple and complex substrates. However, the reactions described in these documents for a given class of nucleophile occur under varying conditions. Therefore, the scope of each set of reaction conditions is unclear, and the SNAr reactions were performed with activated 2-fluoropyridines in most cases. From this body of work, it was unclear whether mild and general SNAr reactions with fluoroheteroarenes could be developed and if selectivity could be obtained for substitution of fluoride over other halides. In fact, previous reports showed that the same forcing reaction conditions lead to substitution of F, Cl, Br, and I.
To identify conditions for the SNAr reaction that would tolerate common functionality, we studied reactions of unactivated 2-fluoropyridines with nucleophiles derived from alcohols, phenols, amines, amides, N-heterocycles, cyanide, and thiols under relatively mild conditions. The reaction conditions we identified afforded quantitative conversion to the substitution products, as indicated by GC/MS and TLC (Table 1). Reactions with KCN proceeded in approximately 80% yield. Variation of the cyanide source, stoichiometry, temperature, and solvent did not increase the yield, but the initial conditions did form the cyanopyridine product in a synthetically useful yield. Having developed a set of SNAr reactions that occur under mild conditions, we explored the tandem fluorination–substitution process.
Table 1. Reaction Conditions for the SNAr of 2-Fluoropyridines.

| NuH | base | solvent | temp (°C) | time (h) | conv (%) |
|---|---|---|---|---|---|
| 1, 2, or 3° alcohol | KOtBu | THF | 50 | 3 | 100 |
| ArOH | KOtBu | DMF | 80 | 6 | 100 |
| 1 or 2° amine | iPr2NEt | DMSO | 120 | 18 | 100 |
| amide (N–H) | NaH | DMF | 100 | 3 | 100 |
| N-heterocycle | NaH | DMF | 100 | 1–3 | 100 |
| KCN (3 equiv) | DMSO | 120 | 18 | ∼80 | |
| NaSR | THF | 50 | 3 | 100 |
For the SNAr reactions to occur in tandem with C–H fluorination, the MeCN and silver salts from the fluorination reaction needed to be removed. Filtering the fluorination reactions through a short silica-filled pipet and evaporating the solvent was sufficient to perform the subsequent SNAr reactions. Yields of the SNAr reactions conducted after filtering the fluorination reaction through Celite were low, due to the presence of soluble Ag salts.
Convenient Protocols for Performing the Fluorination of Pyridines and Diazines
For the fluorination–SNAr sequence to be used broadly, procedures for conducting the reaction without specialized equipment for excluding air and moisture are needed. AgF2 is a hygroscopic solid that decomposes in the presence of water. Therefore, during our initial study, the fluorination reactions were assembled in a glovebox with rigorously dried MeCN.10 However, despite the water sensitivity of AgF2, simple procedures can be followed for conducting the reactions without rigorous exclusion of air or moisture.
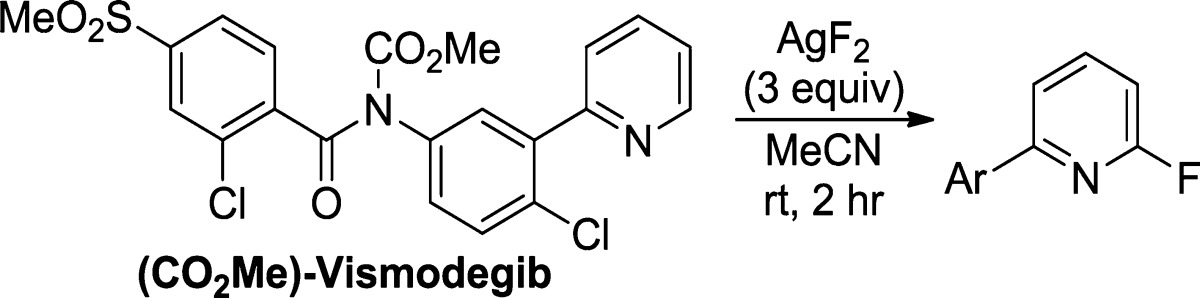
To assess the impact of water and oxygen on the yield of the fluorination reaction of complex molecules, we performed a series of experiments with (CO2Me)-vismodegib, a drug recently approved for the treatment of basal-cell carcinoma (Table 2). Assembling the fluorination reaction in a glovebox (oxygen and water content <1 ppm), in an oven-dried vial, with MeCN that had been rigorously dried over CaH2, afforded the fluorinated vismodegib derivative in 99% yield by 19F NMR spectroscopy.
Table 2. Assessing the Impact of Water and Air on the Fluorination Reaction with AgF2a.
| method of drying MeCN | rxn vial dried | solids weighed | atmosphere | yield (%) (NMR) |
|---|---|---|---|---|
| CaH2 | yes | in glovebox | N2 | 99 |
| CaH2 | yes | in air | N2 | 84 |
| CaH2 | no | in air | air | 79 |
| molecular sieves | no | in air | air | 85 |
| none | no | in air | air | 65 |
Reactions were performed with 0.2 mmol of (CO2Me)-vismodegib with 2 mL of MeCN in 4 mL vials.
Since the use of a glovebox is not practical for all chemists, we studied the effects of weighing the solid reagents in air and assembling the reaction using standard air-free techniques. A reaction was assembled by adding the pyridine substrate to a dry vial in air and adding MeCN that had been dried over CaH2. AgF2 was weighed quickly in air, added to the pyridine solution, and the vial was sealed under an atmosphere of N2. The reaction assembled in this manner afforded the 2-fluoropyridine product in 84% yield, only a slight decrease in yield compared to the reaction assembled entirely in the glovebox. Performing the reaction in a similar manner with a nondried vial and sealing the reaction under an atmosphere of air (2 mL of MeCN in a 4 mL vial; 2 mL headspace of air) resulted in a similar yield of the fluoropyridine product (79%).
Acetonitrile dried with 5 wt % of 3 Å molecular sieves for 24 h was a suitable solvent; the reaction of (CO2Me)-vismodegib with AgF2 assembled by weighing the solid reagents in air in a nondried vial sealed under an atmosphere of air afforded the fluorinated product in 85% yield. The water content in MeCN dried over 5 wt % molecular sieves for 24 h is near 4 ppm, and the water content further decreases with time.14 The water content of commercial “anhydrous” MeCN is below 10 ppm water and should be equally suitable for this reaction.
Finally, the same reaction was assembled in air with ACS grade MeCN directly from a commercial bottle that had been opened and used over the course of a year; a noticeable decrease in yield to 65% was observed, but a substantial amount of product was still formed. Together, these results demonstrate that these fluorination reactions can be conveniently assembled completely in air, without the use of a glovebox or air-free techniques, and with MeCN dried over molecular sieves, even though AgF2 is sensitive to water and should be stored under an inert atmosphere. Reactions performed on the benchtop occur in yields that are comparable to those performed under rigorously anhydrous reaction conditions.
AgF2 is supplied as a black, microcrystalline solid, which should be stored under an inert atmosphere. Because AgF2 undergoes decomposition with moisture, a noticeable color change from black to yellow or brown is observed when it is stored in air. For all reactions reported here, and in our previous paper,10 AgF2 was used as received from Alfa Aesar and stored under a nitrogen atmosphere in a plastic bottle. In our experience, reactions with AgF2 supplied from Strem occurred in comparable rates and yields to those performed with AgF2 puchased from Alfa Aesar.
Scope of the Tandem C–H Fluorination and Nucleophilic Aromatic Substitution of Pyridines
Having identified convenient methods for conducting both the fluorination and SNAr reactions and having developed a protocol to conduct the two reactions in sequence, we investigated the scope of the C−H bond fluorination–SNAr process. Representative examples illustrating the scope of the combined reactions are shown in Tables 3 and 4. Yields given are for isolated products starting from the heteroarene. The yields for the fluorination step are also shown to illustrate how the values for the two-step process compare to those of the first step.
Table 3. Tandem C–H Fluorination and SNAra.

Isolated yields from the two-step sequence for reactions performed with 0.2 mmol of heteroarene. Yields in parentheses were determined by 19F NMR spectroscopy for the independent fluorination reaction.
The isolated product contained ∼5% of an inseparable impurity.
The Boc group was cleaved during the SNAr reaction.
Table 4. Tandem C–H Fluorination and SNAr of Medicinally Important Compoundsa.
Isolated yields from the two-step sequence for reactions performed with 0.2 mmol of heteroarene. Yields in parentheses were determined by 19F NMR spectroscopy for the independent fluorination reaction.
The isolated product contained ∼5% of an inseparable impurity.
A variety of pyridines that are sterically hindered (2, 4, 11, 12, 14, 15) and/or electronically deactivated (2, 9, 13, 15) toward SNAr reactions afforded the substitution products in good yields. Substrates containing alkyl groups in the 2-position reacted selectively at the 6-position (2, 4, 9, 14), while analogous reactions with pyridine N-oxides are known to result in substitution of a C–H bond on the alkyl group (Scheme 1B). A wide range of functional groups were tolerated, including ethers, halides, ketones, acetals, esters, amides, ethyl and t-butyl carbamates, nitriles, and sulfones. It is notable that the azetidine in 11 (Table 4) did not undergo ring opening, a competing reaction observed under acidic conditions.15
The reactions of chloropyridines 7 and 11 revealed a high selectivity for substitution of a fluoride over a chloride under conditions of the SNAr reactions shown in Table 1. This high selectivity, along with the high functional group compatibility, is attributed to the milder reaction conditions we developed for the SNAr reaction at the 2-fluoro position, relative to the conditions typically used to conduct substitutions with 2-fluoropyridines. In sum, this work shows that fluoropyridines undergo substitution reactions under conditions much milder than previously reported and can be performed in the presence of a wide range of functional groups, including those that are electrophilic.
Tandem C–H Fluorination and Nucleophilic Aromatic Substitution of Diazines
Six-membered heteroarenes containing two nitrogen atoms (diazines) are prevalent subunits in medicinal chemistry. Radical addition reactions to diazines are commonplace3 (Scheme 1A), but a single example of C−H bond functionalization by nucleophilic addition of a heteroatom to a diazine N-oxide (Scheme 1B) has been demonstrated.6c Like pyridines, diazines react with AgF2 with exclusive selectivity for fluorination adjacent to nitrogen.10 Thus, we considered that the combination of C–H bond fluorination and SNAr reactions could be conducted with these heterocycles to form functionalized diazine products.
Indeed, pyrimidines (5, 6) and pyrazines (8) reacted in the two-step sequence following the standard conditions we developed for the fluorination and SNAr reactions reported in Table 1. This sequence allowed several polysubstituted diazines to be prepared through C–H functionalization. Because the conditions for both the fluorination and the SNAr reactions with diazines are the same as those for pyridines, the tolerance of the reactions toward functional groups on pyridines and diazines should be comparable. This C–H fluorination–SNAr sequence was also applied to the synthesis of a reverse transcriptase inhibitor containing a tetra-substituted pyrimidine (vide infra).
Late-Stage Functionalization of Complex Molecules via Tandem C–H Fluorination and Nucleophilic Aromatic Substitution
With conditions established for the fluorination and SNAr reactions of pyridines and diazines, we evaluated this sequence for the late-stage functionalization of more complex molecules in medicinal chemistry. First, we used our tandem sequence to prepare several 2-substituted derivatives of (Boc-protected) betahistine (9), a histamine agonist used in the treatment of Ménière’s disease. Reaction of 9 with AgF2 formed the corresponding 2-fluoropyridine in nearly quantitative yield (98%). This electronically deactivated fluoropyridine intermediate reacted with nucleophiles derived from butanol, morpholine, and indole to provide several 2-substituted analogues of betahistine. Although betahistine is a relatively simple compound that can be prepared in one step from 2-vinylpyridine, the synthesis of derivatives that are similar to those we report here would require 2-substituted 6-vinylpyridines. Few such pyridines are commercially available.16 Thus, our C−H bond fluorination−SNAr strategy for late-stage functionalization avoids lengthy synthetic sequences to prepare derivatives of betahistine.
We also conducted the fluorination of compound 10, the direct precursor to loratadine (Claritin) and desloratadine (Clarinex), two common antihistamines. Compound 10 is prepared in two steps from 3-methylpicolinic acid under relatively harsh reaction conditions (nBuLi, KOtBu for the first step; SOCl2, AlCl3 for the second step).17 Therefore, the synthesis of 2-substituted derivatives of 10 would require access to the appropriately substituted 3-methylpicolinic acid, which could require several steps to prepare, and an additional two-step sequence to construct the tricycle for each derivative. We prepared various analogues of 10 more directly through fluorination and SNAr reactions to form the corresponding 2-alkoxy-, 2-amino-, and 2-pyrazolyl-substituted derivatives. It is worthy to note that the substituents we installed in 10a–10c would be unlikely to tolerate the conditions of a de novo synthesis of similar analogues of 10.
Our C–H functionalization method also gave access to a series of (Boc-protected) derivatives of tebanicline (11), a potent non-opiod analgesic that is structurally related to several nicotinic acetylcholine receptor agonists. As mentioned above, no ring opening of the azetidine or substitution of the chloride was observed. De novo syntheses of compounds similar to 11a–11c would require access to 2-substituted 3-hydroxy-6-chloropyridine substrates and an additional C–O bond-forming reaction to complete the synthesis of each derivative.
The sequence of C–H bond fluorination and SNAr also led to a convenient synthesis of 2-alkoxy and 2-amino analogues of roflumilast (12). Roflumilast is a recently approved PDE-4 inhibitor used in the treatment of chronic obstructive pulmonary disease. The reported syntheses of this compound involve amide bond formation with 3,5-dichloro-4-aminopyridine. Thus, the syntheses of the 2-substituted analogues we report would require access to 2-alkoxy or 2-amino-3,5-dichloro-4-aminopyridine, for which none are commercially available. Therefore, preparing derivatives of roflumilast would mandate multistep syntheses of the appropriate pyridine, in addition to performing the subsequent amide bond formation for each derivative. In contrast, the C−H fluorination−SNAr strategy we report allows rapid access to analogues that would otherwise require several synthetic steps to prepare.
In a similar manner, analogues of the precursor to pirenzepine (13), a benzodiazepine-based M1-selective antagonist used for the treatment of ulcers, were prepared. The sequence was used to install alkoxy, thio, and aryloxy substituents at the 2-position in good overall yields. Competing reactions at the electrophilic ethyl carbamate were not observed. The synthesis of the core of 13 requires three steps from 2-chloro-3-aminopyridine.
Relative Rates for the Fluorination of Pyridines and Diazines Having Different Electronic Properties
Because many medicinally important compounds contain multiple heteroaryl rings, it would be valuable if the fluorination was selective for the functionalization of one type of ring system over another. The proposed mechanism10 for the fluorination of pyridines and diazines with AgF2 (eq 1) is initiated by coordination of the basic nitrogen to silver. This coordination could cause a more basic heterocycle to be more reactive than a less basic heterocycle. However, the second step in the proposed mechanism is addition of fluoride to the π system, which would be favored for a more electron-deficient heteroarene. Finally, the third step, a formal oxidation of the heterocycle through hydrogen atom abstraction, would likely be favored for a more electron-rich substrate. Our previous studies of the selectivity between pyridine and pyridine-d5 demonstrated that coordination of AgF2 to pyridine is reversible and that cleavage of the C–H bond is irreversible.10
 |
1 |
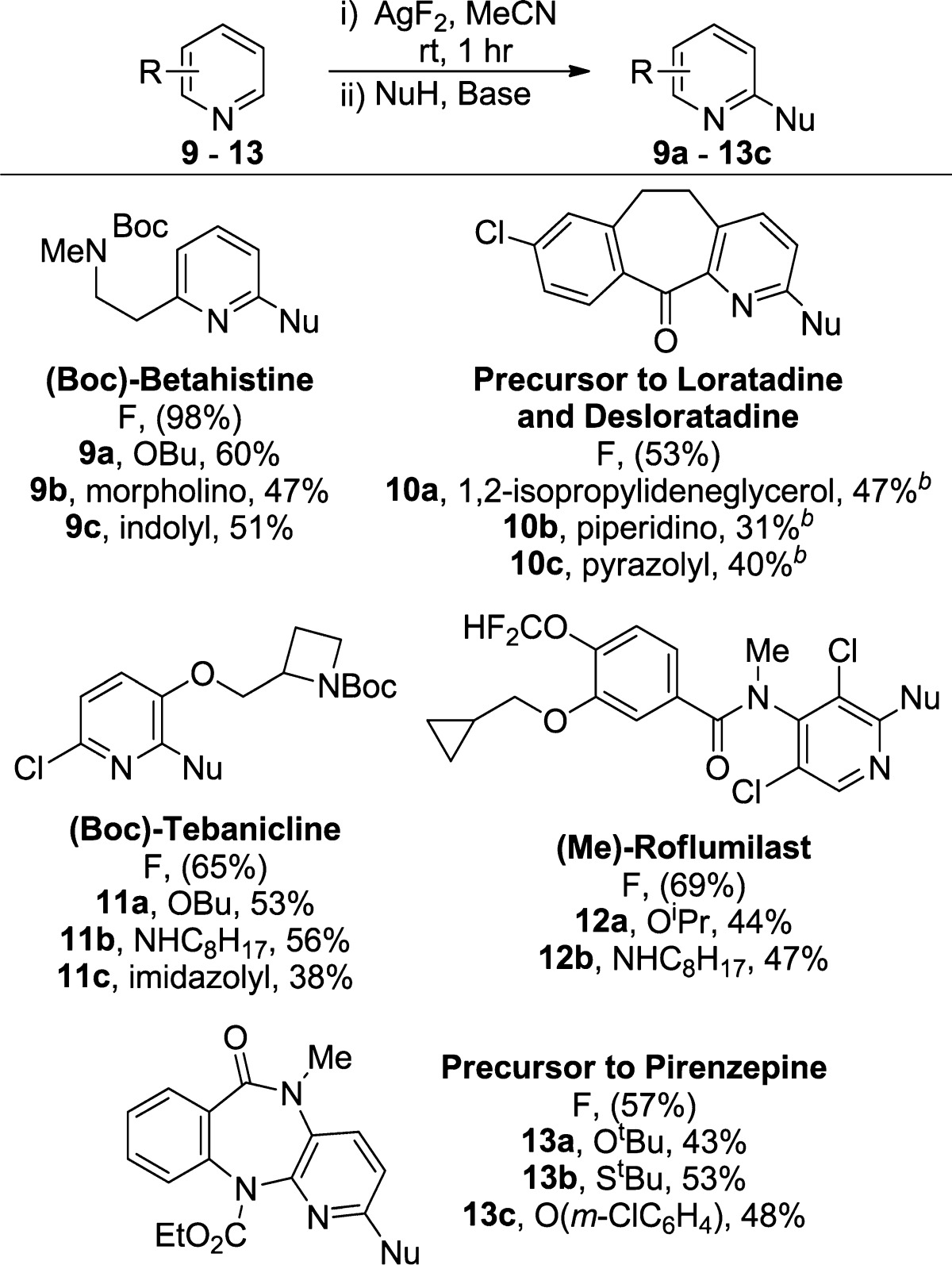
To determine how the electronic properties of the heteroarene influence the relative rates of fluorination, we conducted a series of competition experiments. In these experiments, AgF2 was allowed to react with a 1:1 mixture of two different pyridines and diazines. Because the yield of the fluorination reactions conducted with a large excess of pyridine, relative to AgF2, is low, reactions containing 1 equiv of each heteroarene (0.1 mmol each) and 2 equiv of AgF2 (0.2 mmol) were run, and the reactions were quenched after 15 min so that the selectivities were being measured at low conversion (25 ± 2%). Competition experiments between 2-ethyl, 2-methoxy, and 2-chloropyridine were conducted; the steric properties of these substrates are similar to each other, but the basicity of the heterocycles differ incrementally from each other by ∼2.6 pKa units. Competition experiments were also conducted between alkyl-substituted pyridines, pyrimidines, and pyrazines containing two available C–H bonds for fluorination. The results of these competition experiments are shown in Table 5. These data show that more Lewis basic pyridines undergo the C–H fluorination reactions in preference to less Lewis basic pyridines. Moreover, exclusive selectivity for fluorination of a 4-alkylpyridine over two alkyl-substituted diazines was observed; the competition between 2-methylpyrimidine and 2,3-dimethylpyrazine showed that the pyrimidine was the more reactive diazine by a factor of 3.3.
Table 5. Competition Experiments between Electronically Different Pyridines and Diazines with AgF2a.
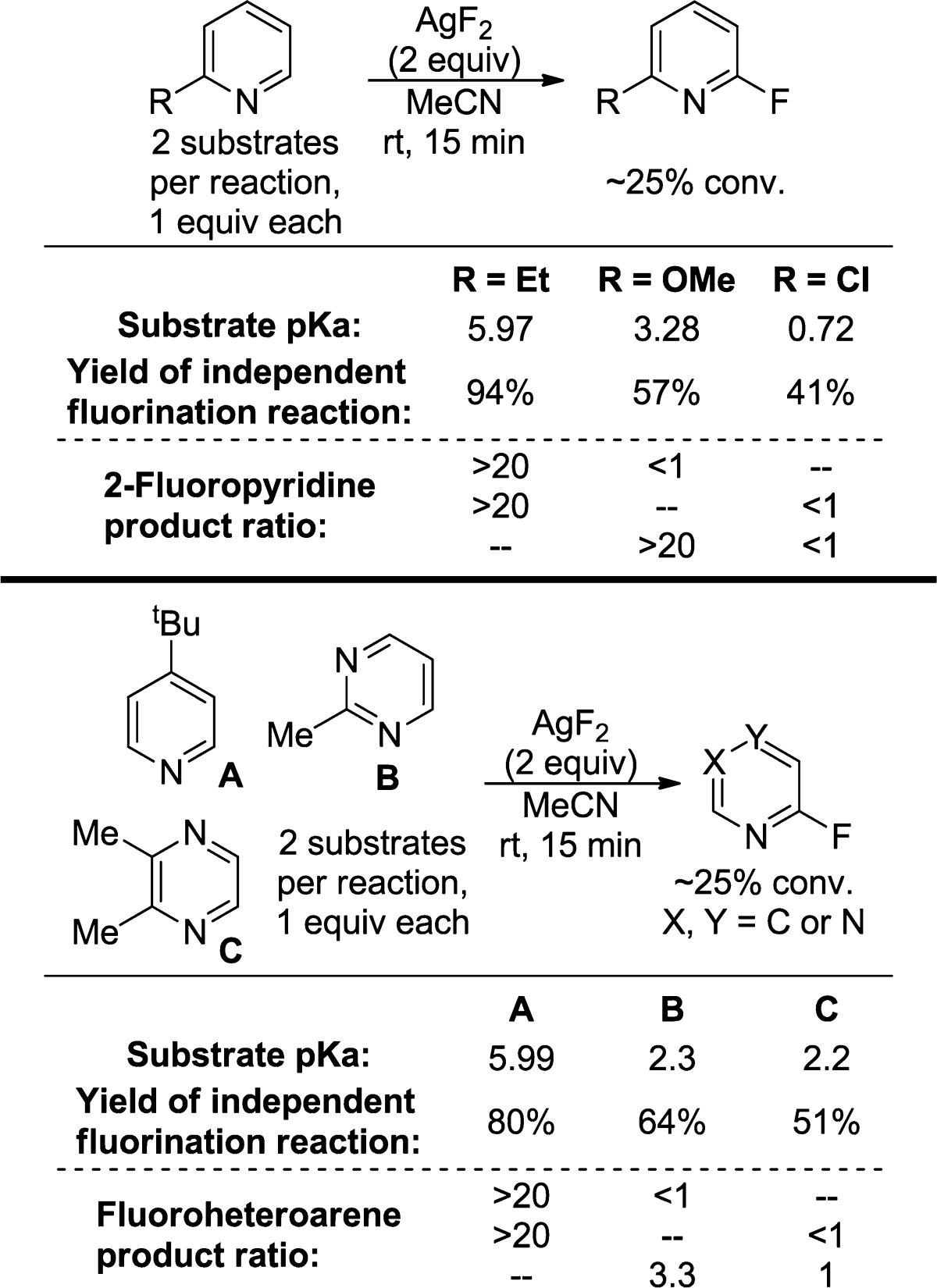
Product ratios were determined by 19F NMR spectroscopy of the crude reaction mixture after quenching with aqueous NaHCO3.
The results of these competition experiments contrast with what would be predicted based only on the relative rates of independent reactions between each substrate and AgF2. 2-Ethylpyridine reacts with 2 equiv of AgF2 to give 38% yield of the 6-fluoropyridine after 15 min. Similarly, 2-methoxypyridine reacts in 36% yield, and 2-chloropyridine reacts in 9% yield after 15 min. Because the rates for fluorination of 2-ethylpyridine and 2-methoxypyridine are comparable, and because AgF2 is present in excess, little selectivity for 2-ethylpyridine over 2-methoxypyridine would be expected based on these data alone. However, AgF2 has negligible solubility in MeCN; therefore, competitive binding of the two substrates to the limited amount of available AgF2 likely results in fluorination of the more basic pyridine.
 |
2 |
To assess the relative reactivity of multiple pyridines in the context of a medicinally important compound, we conducted the fluorination and SNAr reaction of etoricoxib (eq 2), a selective COX-2 inhibitor used in the treatment of arthritis. This compound contains two different pyridine rings. The more electron-rich ring contains methyl and 2-pyridyl substituents, and the less electron-rich ring contains chloro, aryl, and 3-pyridyl substituents. This molecule reacted with AgF2 with complete selectivity for fluorination of the more basic pyridine system, as predicted from the results in Table 5. No product resulting from fluorination of the 3-chloropyridine was observed. Following this site-selective fluorination, several derivatives of etoricoxib containing pendant alkoxy, amino, cyano, and pyrazolyl units were prepared.
Site Selectivity for the Fluorination of 3,5-Disubstituted Pyridines
Many medicinally active pyridines contain two inequivalent C–H bonds that could undergo fluorination with AgF2. We previously demonstrated10 that several 3-substituted pyridines undergo fluorination with exclusive selectivity to form the 2-fluoro-3-substituted pyridine product. The 3-substituted pyridines that react selectively at the 2-position include those containing 3-halo, alkoxy, cyano, or CF3 groups. 3-Substituted pyridines that give a mixture of 2-fluoropyridine isomers include those containing 3-alkyl, 3-CO2R, and 3-C(O)NR2 substituents. It was unclear from these results if 3,5-disubstituted pyridines would undergo the fluorination selectively. A set of 3,5-disubstituted pyridines containing phenyl, cyano, benzyloxy, bromo, methyl, and CF3 substituents was prepared. The fluorination of the 15 unsymmetrical pyridines containing these substituents occurred with poor site selectivity (from 1:1 up to 6:1), with the exception of the benzyloxy-substituted pyridines. The 3-benzyloxy-substituted pyridines containing various substituents in the 5-position reacted with AgF2 with modest to high selectivity (4.2:1 to 20:1) for fluorination adjacent to the ether substituent (Table 6).
Table 6. Fluorination of 3,5-Disubstituted Pyridines.
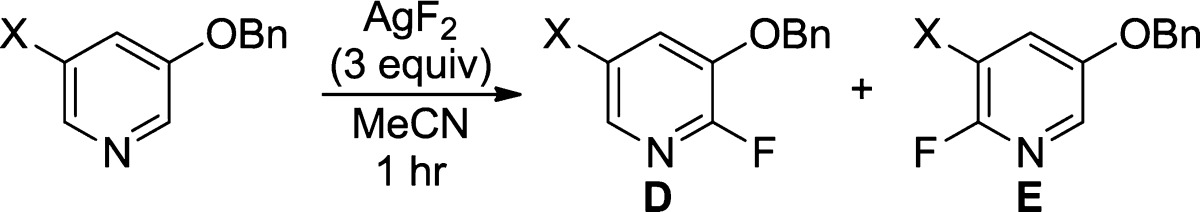
| X | temp (°C) | concentration (M) |
ratio (D/E) |
combined yield (%) (D+E) |
|---|---|---|---|---|
| Br | 50 | 0.050 | 15:1 | 49 |
| CI | 50 | 0.025 | 8.1:1 | 64 |
| Me | 50 | 0.025 | >20:1 | 62 |
| Ph | 50 | 0.050 | 4.2:1 | 68 |
| CN | 50 | 0.025 | 12:1 | 67 |
| CF3 | rt | 0.050 | 20:1 | 85 |
Having shown that an alkoxy group can lead to selective fluorination of a 3,5-disubstituted pyridine, we exploited this selectivity to conduct the late-stage fluorination of a medicinally relevant, 3,5-disubstituted pyridine. The core of crizotinib (eq 3), a drug used for the treatment of metastatic non-small cell lung cancer, contains such a heteroaromatic unit. Reaction of 15 with AgF2 gave products reflecting a 7.2:1 selectivity for fluorination adjacent to oxygen. This selectivity is lower than that observed for the fluorination of 3-bromo-5-benzyloxypyridine (Table 6), likely due to the steric hindrance of the arylethoxy substituent. To determine if the lower selectivity of this substrate is due to the greater steric hindrance of the benzyloxy group of 15 than that of a simple benzyloxy substrate, the fluorination reaction was also performed with a 2,6-dimethylphenyl arylethoxy substrate. This compound reacted in 62% yield with similar 5.9:1 selectivity for fluorination adjacent to the ether substituent. Thus, the selectivity is lower for reactions of pyridines containing more hindered benzyloxy groups but is still significant. The steric hindrance of the arylethoxy substrates disfavors both the second and third steps in our proposed mechanism (eq 1)
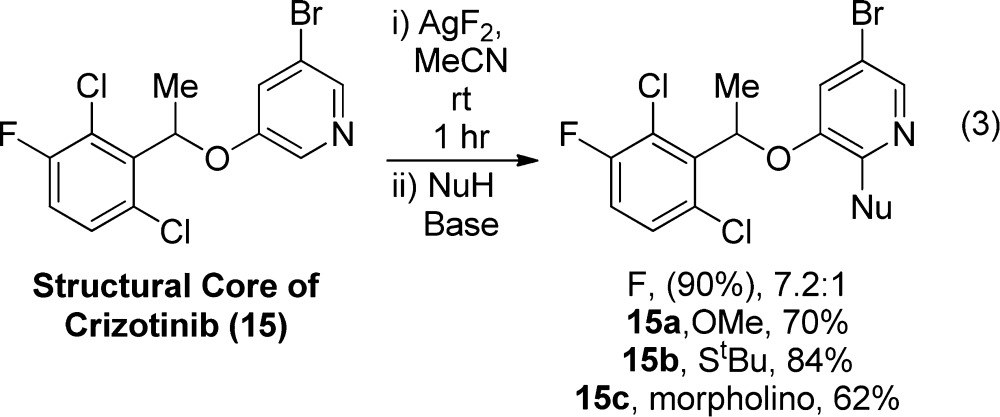 |
3 |
leading to an increase in the relative amount of product from fluorination adjacent to the bromide.
Even though the 2-fluoro-3-benzyloxypydridine intermediate is both sterically and electronically deactivated toward SNAr, several 2-substituted derivatives were prepared under the standard conditions for the SNAr step shown in Table 1. Even a secondary amine reacted with the 2-fluoropyridine to form the sterically congested product 15c. The isomeric products prepared from 15 after C–H bond fluorination–SNAr reactions were not separable by standard silica gel chromatography, even though the 2-fluoropyridine isomers could be separated by HPLC and GC.
Limitations of the C–H Fluorination and SNAr Reactions
Although we have demonstrated that the C–H fluorination and SNAr reactions occur with broad scope and can be conducted on complex molecules, there are some limitations. As we reported previously,10 the fluorination reaction is not compatible with free amines or alcohols, carboxylic acids, aldehydes, or electron-rich five-membered heterocycles; however, several protected derivatives of these groups are tolerated by AgF2. In addition, we have found that pyridines or diazines containing multiple electron-withdrawing substituents undergo the fluorination reaction in lower yields than those containing electron-neutral or electron-donating groups. Examples of substrates that reacted with AgF2 in low yields (0–30%) are shown in Table 7. Although the Boc-protected derivative of HG-10-102-01 reacted with AgF2 in low yield, a similar tetra-substituted pyrimidine reacted in high yield for the synthesis of etravirine (vide infra).
Table 7. Substrates that Reacted in Low Yields for the C–H Fluorination or SNAr Reactions.
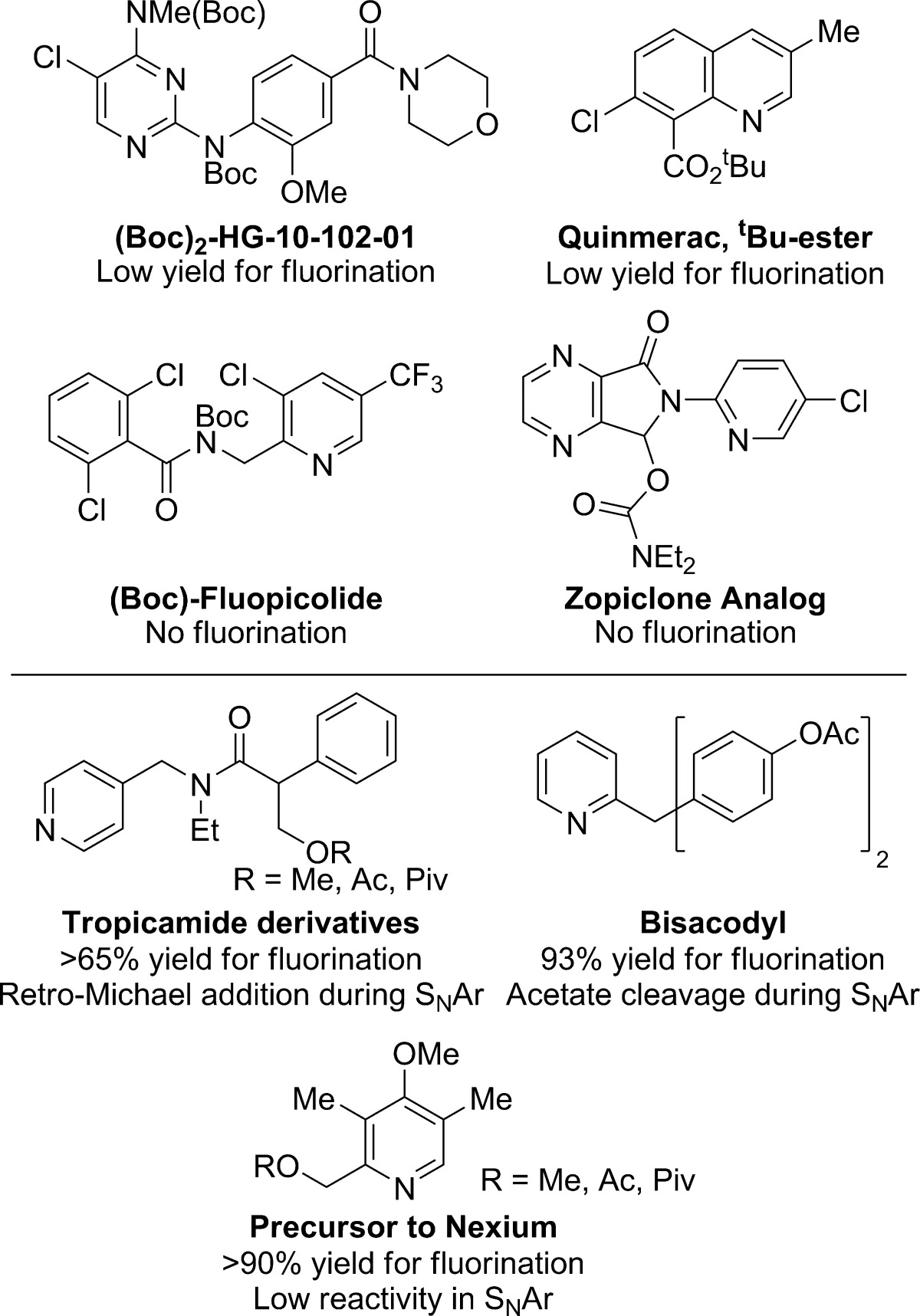
We have demonstrated that a simple set of SNAr reaction conditions can be employed for substitution reactions on a variety of 2-fluoroheteroarenes. However, we did find substrates that underwent competing side reactions faster than they underwent SNAr (Table 7). Fluorinated analogues of tropicamide underwent retro-Michael addition under the basic reaction conditions. Although we demonstrated that tert-butyl esters remain intact during the SNAr reactions (Table 3, substrate 3), we found that aryl acetates are cleaved faster than substitution of fluoride. This cleavage was observed when attempting SNAr reactions of bisacodyl. Finally, very electron-rich and sterically hindered pyridines, such as the precursor to Nexium, underwent the SNAr step in low yields; more forcing conditions resulted in the formation of side products.
Applications of C–H Fluorination and SNAr for the Synthesis of Medicinal Compounds
Having demonstrated the potential of the C–H fluorination–SNAr sequence for the late-stage derivatization of medicinally important compounds, we sought to evaluate whether the same strategy could create shorter and higher-yielding synthetic routes to the same types of compounds. Although this chemistry requires stoichiometric silver and is not designed for process-scale chemistry, the strategy of C–H fluorination and SNAr can be useful in discovery chemistry. One example of a synthesis that could be simplified by the fluorination and substitution process is the synthesis of the simple compound 6-(methylamino)-2-pyridineethanol (Scheme 2, 16). Compound 16 is a precursor to several important compounds, including the integrin inhibitor SB-273005 (Scheme 2C). Although 16 is structurally simple, the two reported syntheses of 16 by medicinal chemists were conducted in five and seven steps from 2-amino-6-methylpyridine (Scheme 2A). We considered that the 2-methylamino group could be installed through C–H bond fluorination and SNAr from a derivative of 2-pyridineethanol.
Scheme 2. Synthesis of 6-(Methylamino)-2-pyridineethanol through C–H Fluorination.
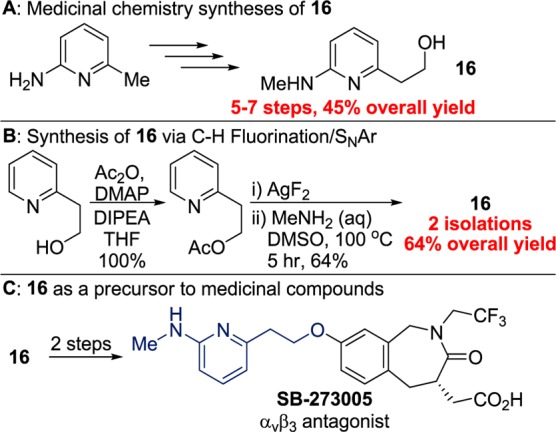
Fluorination did not occur on the free alcohol, but it did occur after protecting the alcohol as an ester. An acetyl protecting group was chosen because it could be cleaved in concert with the SNAr reaction. The fluorination of acetylated 2-pyridineethanol occurred in 88% yield (by 19F NMR spectroscopy). Treatment of the crude mixture from the fluorination step with aqueous MeNH2 in DMSO led to formation of the methylaminopyridine unit and cleaveage of the acetyl group to reveal the free alcohol. This route gave 16 with only two isolations in 64% overall yield, a significantly shorter and higher-yielding approach than the reported syntheses requiring five or seven steps. Although our route requires stoichiometric silver, it is worthy to note that an improved process used to prepare 30 g of 16 still required four steps and occurred in only 39% overall yield.18
The combination of C–H bond fluorination and SNAr also shortened the synthesis of PF-1247324, a potent and selective NaV1.8 inhibitor (Scheme 3).19 The medicinal chemistry route to this compound comprised six steps and occurred in less than 1% yield; an advanced intermediate was outsourced for the kilogram-scale synthesis of PF-1247324.20 Our route to this compound began with the fluorination of methyl 5-bromopicolinate. The fluoropyridine intermediate contains two electrophilic sites for nucleophilic substitution: a methyl ester and a 2-fluoropyridine. We demonstrated (Tables 3 and 4) that substitution at the fluorine of a 2-fluoropyridine can occur in the presence of auxiliary electrophilic functional groups, including a tert-butyl ester. However, we considered that conditions could be identified to transform the methyl ester to the N-methyl amide without substitution of the fluoride. The reaction between the fluoropyridine methyl ester with aqueous MeNH2 in THF formed a mixture of products resulting from substitution of both methoxide and fluoride. However, simply changing the reaction solvent from THF to MeOH allowed for the selective transformation of the methyl ester to the N-methyl amide without substitution of the fluoride.
Scheme 3. Synthesis of PF-1247324 through C–H Fluorination.
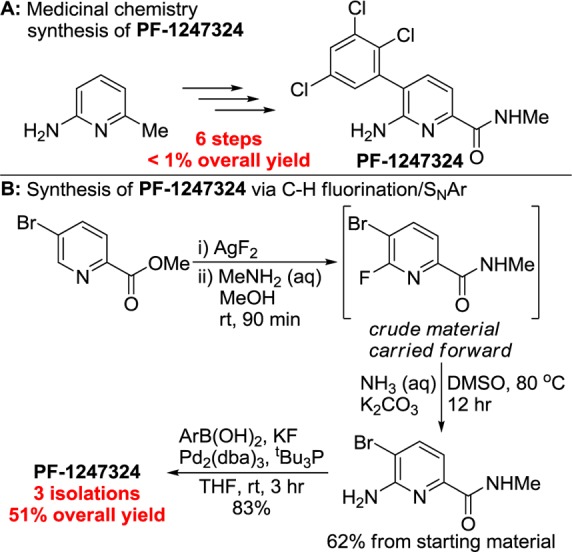
After aqueous workup, the crude reaction mixture was treated with ammonium hydroxide in DMSO to substitute an NH2 group for the fluoride. Finally, the 3-bromopyridine was subjected to the reported Suzuki cross-coupling reaction conditions20 to provide the title compound. By our route, the synthesis of PF-1247324 involving C–H fluorination was completed with just three total isolations in 51% overall yield, a major improvement in yield and step count over the published synthesis. Furthermore, the route performed by medicinal chemists required 120 h of total reaction time, while the route we report here required less than 18 h.
Finally, we used our chemistry to prepare the non-nucleoside reverse transcriptase inhibitor Intelence (etravirine, Scheme 4), a compound used in the treatment of HIV. The route to this compound developed by medicinal chemists occurred in five steps and 9% yield from N-(4-cyanophenyl)guanidine hydrochloride.21 An alternative route used to prepare etravirine on kilogram scale required four steps and occurred in 30% yield.22
Scheme 4. Synthesis of Intelence (Etravirine) through C–H Fluorination.
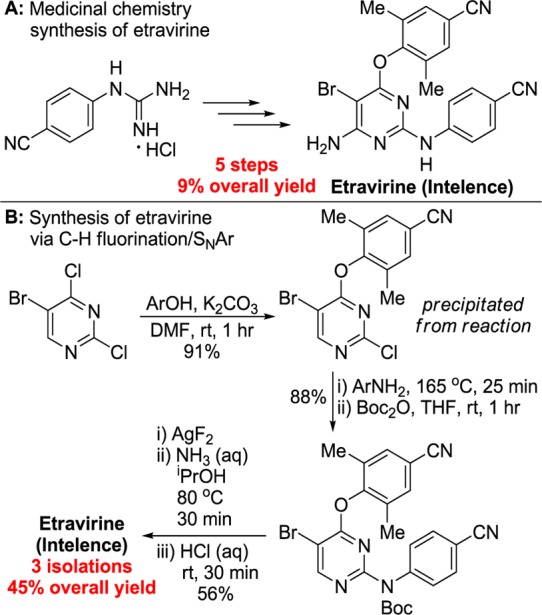
Our synthesis of etravirine began with substitution of the 4-chloro substituent in 2,4-dichloro-5-bromopyrimidine with 2,6-dimethyl-4-cyanophenol, from which the solid product was isolated after the addition of water. Next, substitution with 4-aminobenzontirile and Boc protection in situ was conducted, and the product was isolated in 88% yield. The installation of a protecting group for the N–H bond was necessary for the subsequent fluorination reaction to proceed. The synthesis of etravirine was then completed by C–H fluorination of the pyrimidine (71% yield by 19F NMR spectroscopy), substitution with aqueous ammonia, and addition of HCl(aq) to cleave the Boc group. Through this route, etravirine was prepared with three total isolations in 45% overall yield in under 6 h of total reaction time.
Summary
In summary, we have developed a broadly applicable strategy for the diverse, site-selective C–H functionalization of pyridines and diazines. The reaction sequence occurs to provide alkoxy-, aryloxy-, amino-, amido-, heteroaryl-, thio-, and cyano-substituted heterocycles that can be difficult to access through traditional methods. This tandem sequence is attractive for the direct diversification of heteroarenes, due to the exquisite site selectivity for C–H functionalization and the mild conditions for the SNAr reaction. In addition, high site selectivity for the fluorination of substrates containing more than one heteroarene or more than one reactive C–H bond is possible using this chemistry. Finally, the process of fluorination and SNAr can allow medicinal compounds containing substituted pyridines and diazines to be prepared by short synthetic routes. We anticipate that these reactions will find immediate use for both late-stage functionalization and efficient syntheses of complex molecules.
Acknowledgments
We thank the NIH-NIGMS (R29-55382) for support of this work, and the ACS Division of Organic Chemistry for a fellowship to P.S.F.
Supporting Information Available
Experimental procedures and characterization of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- http://www.drugs.com/stats/top100/2013/sales.
- http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM381803.pdf.
- a Minisci F.; Vismara E.; Fontana F. Heterocycles 1989, 28, 489. [Google Scholar]; b Duncton M. A. J. MedChemComm 2011, 2, 1135. [Google Scholar]; c Fujiwara Y.; Dixon J. A.; O’Hara F.; Funder E. D.; Dixon D. D.; Rodriguez R. A.; Baxter R. D.; Herle B.; Sach N.; Collins M. R.; Ishihara Y.; Baran P. S. Nature 2012, 492, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a single report on the addition of nitrogen radicals to limiting amounts of heteroarenes, see:Foo K.; Sella E.; Thomé I.; Eastgate M. D.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara F.; Blackmond D. G.; Baran P. S. J. Am. Chem. Soc. 2013, 135, 12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bull J. A.; Mousseau J. J.; Pelletier G.; Charette A. B. Chem. Rev. 2012, 112, 2642. [DOI] [PubMed] [Google Scholar]; b Farrell R. P.; Elipe M. V. S.; Bartberger M. D.; Tedrow J. S.; Vounatsos F. Org. Lett. 2013, 15, 168. [DOI] [PubMed] [Google Scholar]; c Keith J. M. J. Org. Chem. 2008, 73, 327. [DOI] [PubMed] [Google Scholar]; d Yin J. J.; Xiang B. P.; Huffman M. A.; Raab C. E.; Davies I. W. J. Org. Chem. 2007, 72, 4554. [DOI] [PubMed] [Google Scholar]; e Londregan A. T.; Jennings S.; Wei L. Q. Org. Lett. 2011, 13, 1840. [DOI] [PubMed] [Google Scholar]; f Wengryniuk S. E.; Weickgenannt A.; Reiher C.; Strotman N. A.; Chen K.; Eastgate M. D.; Baran P. S. Org. Lett. 2013, 15, 792. [DOI] [PubMed] [Google Scholar]
- a Alberico D.; Scott M. E.; Lautens M. Chem. Rev. 2007, 107, 174. [DOI] [PubMed] [Google Scholar]; b Seregin I. V.; Gevorgyan V. Chem. Soc. Rev. 2007, 36, 1173. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McGlacken G. P.; Bateman L. M. Chem. Soc. Rev. 2009, 38, 2447. [DOI] [PubMed] [Google Scholar]; d Liu B.; Huang Y. M.; Lan J. B.; Song F. J.; You J. S. Chem. Sci. 2013, 4, 2163. [Google Scholar]; e Nakao Y.; Kanyiva K. S.; Hiyama T. J. Am. Chem. Soc. 2008, 130, 2448. [DOI] [PubMed] [Google Scholar]; f Wen J.; Qin S.; Ma L. F.; Dong L. A.; Zhang J.; Liu S. S.; Duan Y. S.; Chen S. Y.; Hu C. W.; Yu X. Q. Org. Lett. 2010, 12, 2694. [DOI] [PubMed] [Google Scholar]; g Lewis J. C.; Bergman R. G.; Ellman J. A. J. Am. Chem. Soc. 2007, 129, 5332. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Wen P.; Li Y. M.; Zhou K.; Ma C.; Lan X. B.; Ma C. W.; Huang G. S. Adv. Synth. Catal. 2012, 354, 2135. [Google Scholar]
- Xiao B.; Liu Z. J.; Liu L.; Fu Y. J. Am. Chem. Soc. 2013, 135, 616. [DOI] [PubMed] [Google Scholar]
- a Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. Chem. Rev. 2010, 110, 890. [DOI] [PubMed] [Google Scholar]; b Hartwig J. F. Acc. Chem. Res. 2012, 45, 864. [DOI] [PubMed] [Google Scholar]
- Fier P. S.; Hartwig J. F. Science 2013, 342, 956. [DOI] [PubMed] [Google Scholar]
- Terrier F.Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Schlosser M.; Rausis T. Helv. Chim. Acta 2005, 88, 1240. [Google Scholar]
- a Loupy A.; Philippon N.; Pigeon P.; Galons H. Heterocycles 1991, 32, 1947. [Google Scholar]; b Cherng Y. H. Tetrahedron 2002, 58, 4931. [Google Scholar]; c Thomas S.; Roberts S.; Pasumansky L.; Gamsey S.; Singaram B. Org. Lett. 2003, 5, 3867. [DOI] [PubMed] [Google Scholar]; d Klapars A.; Waldman J. H.; Campos K. R.; Jensen M. S.; McLaughlin M.; Chung J. Y. L.; Cvetovich R. J.; Chen C. Y. J. Org. Chem. 2005, 70, 10186. [DOI] [PubMed] [Google Scholar]; e Seki K.; Ohkura K.; Terashima M.; Kanaoka Y. Heterocycles 1994, 37, 993. [Google Scholar]
- Bradley D.; Williams G.; Lawton M. J. Org. Chem. 2010, 75, 8351. [DOI] [PubMed] [Google Scholar]
- Lynch J. K.; Holladay M. W.; Ryther K. B.; Bai H.; Hsiao C. N.; Morton H. E.; Dickman D. A.; Arnold W.; King S. A. Tetrahedron: Asymmetry 1998, 9, 2791. [Google Scholar]
- 2-Bromo-6-vinylpyridine is listed at $360 for 250 mg from Matrix Scientific.
- Doran H. J.; O’Neill P. M.; Williams R. P. U.S. Patent US2003144314, 2003.
- Conde J. J.; Goldfinger L. L.; Mcguire M. A.; Shilcrat S. C.; Wallace M. D.; Yu M. S. Patent WO/2004/089890, 2004.
- Lane C. A. L.; Maw G. N.; Rawson D. J.; Thompson L. R. Patent WO/2006/011050, 2006.
- Fray M. J.; Gillmore A. T.; Glossop M. S.; McManus D. J.; Moses I. B.; Praquin C. F. B.; Keeves K. A.; Thompson L. R. Org. Process Res. Dev. 2010, 14, 263. [Google Scholar]
- Ludovici D. W.; De Corte B. L.; Kukla M. J.; Ye H.; Ho C. Y.; Lichtenstein M. A.; Kavash R. W.; Andries K.; de Bethune M. P.; Azijn H.; Pauwels R.; Lewi P. J.; Heeres J.; Koymans L. M. H.; de Jonge M. R.; Van Aken K. J. A.; Daeyaert F. F. D.; Das K.; Arnold E.; Janssen P. A. J. Bioorg. Med. Chem. Lett. 2001, 11, 2235. [DOI] [PubMed] [Google Scholar]
- Joshi S.; Maikap G. C.; Titirmare S.; Chaudhari A.; Gurjar M. K. Org. Process Res. Dev. 2010, 14, 657. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.