

Abstract
Self-assembly of amyloid fibrils is the molecular mechanism best known for its connection with debilitating human disorders such as Alzheimer’s disease but is also associated with various functional cellular responses. There is increasing evidence that amyloid formation proceeds along two distinct assembly pathways involving either globular oligomers and protofibrils or rigid monomeric filaments. Oligomers, in particular, have been implicated as the dominant molecular species responsible for pathogenesis. Yet the molecular mechanisms regulating their self-assembly have remained elusive. Here we show that oligomers/protofibrils and monomeric filaments, formed along distinct assembly pathways, display critical differences in their ability to template amyloid growth at physiological vs denaturing temperatures. At physiological temperatures, amyloid filaments remained stable but could not seed growth of native monomers. In contrast, oligomers and protofibrils not only remained intact but were capable of self-replication using native monomers as the substrate. Kinetic data further suggested that this prion-like growth mode of oligomers/protofibrils involved two distinct activities operating orthogonal from each other: autocatalytic self-replication of oligomers from native monomers and nucleated polymerization of oligomers into protofibrils. The environmental changes to stability and templating competence of these different amyloid species in different environments are likely to be important for understanding the molecular mechanisms underlying both pathogenic and functional amyloid self-assembly.
Introduction
Self-assembly of proteins into long fibrillar aggregates with similar cross-β sheet structures is the molecular signature of amyloid diseases, including Alzheimer’s disease, tauopathies, prion diseases, type II diabetes, and rheumatoid arthritis.1 While the molecular mechanisms underlying amyloid pathogenesis remain intensely debated, the toxicity of globular oligomeric intermediates of the fibril assembly process is a prominent suspect.2−7 Yet, amyloid oligomers are considered short-lived intermediates emerging during amyloid assembly that require extrinsic stabilization for studying their properties.8 Furthermore, these oligomeric intermediates might be off-pathway from mature fibril formation and metastable against seeding with mature fibrils.9,10 This raises the question how short-lived, off-pathway intermediates of the assembly process can initiate the various pathologies associated with amyloid diseases and promote their spread throughout tissues. However, our understanding of the properties of amyloid intermediates and their ability to promote amyloid formation of monomers are typically derived from in vitro fibril growth assays often confined to a narrow range of partially denaturing solution conditions. Comparatively little is known about the behavior of distinct amyloid species close to physiological solution conditions, in particular whether they remain stable instead of dissolving or precipitating and whether they are capable of seeding growth from native monomers.11,12 Multiple experimental observations9,10,13 and theoretical considerations14−16 have suggested that changes in solution environment (pH, temperature, ionic composition, and protein concentration) could alter basic properties of amyloid aggregates, including their assembly mechanisms, their aggregation propensities, and their interactions with monomers. Investigating the aggregation propensity of different amyloid templates with native monomers also addresses recent suggestions that amyloid oligomers might assume prion-like characteristics that contribute to their spread in neurodegenerative diseases.17 We set out to investigate whether amyloid oligomers, protofibrils, and mature fibrils remained intact at physiological temperatures, and whether any of them could induce amyloid formation from lysozyme in its native fold. For these experiments we used hen egg-white lysozyme and its well-defined oligomeric and fibrillar assembly pathways as our model system.10
Hen egg-white lysozyme (hew-L) is a small enzyme of 14.3 kDa molecular weight with well-defined biochemical and physical properties. Point mutations in human lysozyme underlie non-neuropathic systemic forms of human amyloidosis, with dangerous disruptions to kidney and liver function.18 Hew-L undergoes a well-defined cooperative unfolding transition as a function of temperature, thereby permitting us to determine how different amyloid species interact with natively folded vs partially unfolded monomers. Thermal denaturation also induces spontaneous amyloid formation of lysozyme in vitro.(19,20) Fibril formation of lysozyme switches sharply from generating rigid filaments at low ionic strength to producing compact oligomeric intermediates at higher ionic strength, which subsequently nucleate curvilinear protofibrils.21 The existence of an oligomeric vs fibrillar assembly pathways has been reported for multiple amyloid proteins including amyloid-β, beta-2 microglobulin, and polyglutamine.9,22−25 For some of these systems, the oligomers and/or protofibrils are “on pathway” to mature fibril formation,24,26 while the globular oligomer and protofibrils of lysozyme and beta-2 microglobulin remain off-pathway. Furthermore, lysozyme oligomers, protofibrils, and rigid filaments all develop β-sheet peaks in their IR spectra that are considered diagnostic of amyloid structures.27 Differences in their IR peak wavenumber and morphology imply that lysozyme oligomers and protofibrils are amyloidolytic structures that are distinct from their rigid filament counterparts.28
The investigation into potential prion-like activity of lysozyme aggregates was motivated by observations that oligomer or protofibril growth at partially denaturing temperatures appeared to continue, albeit at a very slow pace, long after solutions had been returned to room temperature. We therefore tested whether oligomers, protofibrils, and mature fibrils remained intact and soluble upon return to physiological temperatures and pH, and whether they developed prion-like characteristics of self-replication from native substrate under these conditions. While we intended to confine these experiments to the physiological pH of 7, we found that thermal denaturation of hew-L at pH 7 readily produced oligomers and protofibrils but no filaments. Including pH 3 in the experiments allowed us to generate all three aggregate species by simply changing the NaCl concentration.10,28 Equally important, pH 3 preserves the native fold at 37 °C and the cooperative unfolding of lysozyme’s secondary and tertiary structure near 70 °C. At more acidic pH values, this cooperativity breaks down leading to partial loss of tertiary structure prior to secondary structure unfolding.29 In addition, pH 3 and 37 °C avoids the noticeable monomer hydrolysis at more acidic pH values.30 We also performed cross-seeding experiments by transferring filaments and oligomers/protofibrils generated at pH 3 as seeds into fresh monomer solutions at pH 7. To assess aggregate stability, aggregation kinetics, and aggregate structures during seeding experiments, we relied on a broad range of complementary experimental techniques including ThT fluorescence, static and dynamic light scattering (SLS, DLS), atomic force microscopy (AFM), and attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR).
Experimental Methods
Details for methods summarized here have been published previously.10,21,28
Protein and Chemicals
Two times recrystallized, dialyzed, and lyophilized hen egg white lysozyme (hew-L) was purchased from Worthington Biochemicals (Lakewood NJ) and used for all experiments. Ultrapure grade thioflavin T was obtained from Anaspec (Freemont, CA). All other chemicals were purchased from Fisher Scientific (Pittsburgh, PA) and were reagent grade or better.
Preparation and Isolation of Amyloid Seeds
Typically hew-L was dissolved in 20 mM HEPES at pH 7 or 10 mM of sodium citrate at pH 3 and filtered consecutively through 220 and 50 nm syringe filters.31 Lysozyme concentrations were determined from UV absorption using a280 = 2.64 mL mg–1 cm–1. For oligomer/protofibril formation at pH 7, solutions were incubated with either 40 or 80 mM NaCl at 70 °C for 1.5–6 h. At pH 3, oligomer/protofibril formation was induced by incubation lysozyme with 250 mM NaCl at 70 °C for 4–10 h. For amyloid filament growth, solutions were incubated at pH 3 with 75–100 mM NaCL for 20–30 h. Seeds were separated from lysozyme monomers using 50 kDa MW centricon filters, spun at 1400 g, and rinsed multiple times with the appropriate growth buffer. 100–150 μL of isolated seeds in buffer were collected from the top reservoir of the filter, and the seed concentration was determined from UV absorption.
Aggregate Stability and Seeded Growth Experiments
For seed growth experiments, native lysozyme solutions were mixed with 5–20% (v/v) of amyloid seed solutions and incubated for 5–14 days. For seed stability experiments, oligomers/protofibrils were added at 10–20% (v/v) to buffer and incubated up to 50 h, and up to 100 h for filaments. For experiment on monomeric dependence of seeded oligomer/protofibril growth, 10% of isolated seeds were incubated with 0.2–20 mg/mL of monomeric lysozyme solution at 37 °C and pH 7. For cross-seeding experiments, protofibrils and filaments generated at pH 3 were separated from monomers and brought to pH 3 via dialysis against pH 7 buffer. Protofibrils at various stages of growth irreversibly precipitated upon buffer exchange. Amyloid filaments also tended to precipitate but, after passing through 0.8 μm syringe filters, resolubilized. These fibrils were concentrated to 20 mg/mL right before seeding with fresh monomers at pH 7.
Static and Dynamic Light Scattering
Static and dynamic light scattering (SLS and DLS) measurements were performed using a Zetasizer Nano S (Malvern Instruments, Worchestershire, UK). For in situ measurements, autocorrelation functions of scattered light were collected using acquisition times of 3 min and converted into particle-size distributions using the “narrow modes” or “general purpose” algorithms provided with the Zetasizer Nano S. Changes in scattering intensity were derived from the count rates of the avalanche photodiode photon detector.
Atomic Force Microscopy
Amyloid aggregates were imaged by depositing samples on freshly cleaved mica and drying them with nitrogen. Images were acquired with an MFP-3D atomic-force microscope (Asylum Research, Santa Barbara, CA) using NSC36/NoAl (Mikromasch, San Jose, CA) silicon tips with nominal tip radii of 10 nm. The cantilever was driven at 60–70 kHz in alternating current mode. Images at 512 × 512 pixel resolution were acquired at a scan rate of 0.5 Hz.
Thioflavin T Fluorescence Spectroscopy
ThT fluorescence measurements were performed on a FluoroMax-4 spectrofluorometer (Horiba Jobin Yvon, Edison, NJ) using excitation and emission wavelengths of 445 and 485 nm, respectively. For ThT measurements, small aliquots of the hew-L solutions were removed and mixed 100:1 with a ThT stock solution, resulting in a final ThT concentration of 10 μM.
Fourier-Transform Infrared (FTIR) Spectroscopy of Lysozyme Solutions
Attenuated total reflectance Fourier-Transform infrared spectroscopy (ATR- FTIR) was performed on a Bruker Optik Vertex 70 (Ettlingen, Germany) spectrometer with a mid-infrared source and a pyroelectric DLATGS (deuterated l-alanine doped triglycene sulfate) room temperature detector. 30 μL of protein solution were placed on the thermostated silicon crystal of a BioATRcell II (Harrick Scientific Products, Inc.; Pleasantville, New York) attenuated total reflectance (ATR) accessory. FTIR spectra were acquired between 1000 and 4000 cm–1 wavenumbers. In general, 400–800 scans at 1 cm–1 step size were recorded and 10 such runs were averaged prior to data processing.
Results
Stability of Amyloid Oligomers, Protofibrils and Filaments at Physiological Temperatures
First, we determined how lowering the solution temperature from above the onset for monomer denaturation (70 °C) to physiological values (37 °C) affected the stability of the various amyloid species generated at either pH 3 or pH 7. Persistent solubility and lack of dissociation of aggregates near physiological temperatures is a prerequisite for their ability to propagate within tissues and seed new amyloid formation there. We used transient thermal denaturation, required to induce amyloid formation,11,12 and brought these preformed amyloid aggregates back to physiological temperature. Filaments or oligomers and protofibrils at pH 3 were formed by incubating 1.4 mM lysozyme at 70 °C with either 100 mM or 250 mM NaCl, respectively. At pH 7 only amyloid oligomer and protofibril growth was observed. Oligomers at pH 7 were generated by incubating 1.4 mM lysozyme with 40 mM NaCl at 70 °C for 1.5 h or less. After 2 h protofibril nucleation was detected by DLS, with the proportion of protofibrils in solution increasing with incubation period. In all cases amyloid growth was monitored in situ using static and dynamic light scattering (DLS). AFM images of amyloid aggregates generated at either pH 3 or pH 7 showed the characteristic morphologies associated with either amyloid filaments (Figure 1a) or oligomers and protofibrils (Figures 1b and 6a).10 They displayed the same kinetic, tinctorial, and structural features as those previously described at pH 2 and 7.21,28
Figure 1.

Stability of amyloid aggregates at physiological temperature and two pH values. AFM images of (a) lysozyme filaments and (b) protofibrils formed during incubation at 70 °C and pH 3 in 100 or 250 mM NaCl, respectively. Scale bar: 200 nm; color scale: aggregate height in nm. Images of oligomers/protofibrils at pH 7 are provided in Figure 6a. Static light scattering (Iscat) indicates that at 37 °C (c) filaments formed at pH 3 remain stable while (d) protofibrils decay very slowly. (e) At pH 7 protofibrils kept at 37 °C remain stable. Inserts: particle size distributions obtained with DLS at the beginning (---) and end (__) of the incubation period.
Figure 6.

Seeded aggregates have characteristics of amyloid oligomers/protofibrils. (a) AFM images (from left to right) of oligomeric seeds, and resulting early stage protofibrils (5 days incubation) and late stage protofibrils (3 weeks of incubation) grown via seeding of native lysozyme at pH 7 and 37 °C. (scale bar: 200 nm; color scale: height in nm). (b) Fractional increases in off-line ThT fluorescence emission (■) and light scattering intensity (●) during seeded growth of native lysozyme. (c) Normalized FTIR difference spectra within the Amide I band for late-stage amyloid aggregates at pH 7 formed after incubation at either 70 or 37 °C, following subtraction of the spectrum for lysozyme monomers.
To evaluate their thermal stability in the absence of monomers, seeds were separated from their monomer background using a 50 kDa recovery filter and subsequent resuspension in buffer. Successful separation and integrity of seeds was confirmed using DLS and AFM imaging on the separated aggregates. The stability of isolated seeds at 37 °C was determined by monitoring their scattering intensity and particle size distributions for a minimum of 24 h. Amyloid filaments, which represent the aggregate morphology most commonly associated with amyloid diseases, remained stable at 37 °C (Figure 1c). Oligomer and protofibrils at pH 3 dissociated slowly with a small monomer peak emerging in DLS after about 10 h of incubation (Figure 1 d). The rate of decay was sufficiently slow to consider these aggregates “stable” for the purpose of subsequent seeding experiments at pH 3. Protofibrils (Figure 1e) and oligomers generated at pH 7 showed no signs of decay or self-assembly at 37 °C. This stability contrasts with the rapid dissociation of both isolated fibrils or protofibrils at pH 3 and the rapid self-association of protofibrils at pH 7 upon heating near 70 °C (data not shown). The gain in thermal stability was not associated with any obvious changes to aggregate sizes (DLS), morphologies (AFM), or structures (ATR-FTIR).
Oligomers and protofibrils are considered short-lived intermediates of the assembly process. For lysozyme and beta-2 microglobulin, they form along assembly pathways distinct from that for amyloid filament assembly and are metastable against filament growth via seeding.9,10 However, upon lowering solution temperature to physiological values, oligomers and protofibrils retained their solubility and displayed either very low rates (pH 3) or no discernible signs (pH 7) of either fragmentation or dissociation into monomers. The observed thermal stability of filaments should not be considered self-evident either. This was highlighted by recent experiments on tau filaments which readily formed at high temperatures, but dissolved when returned to physiological temperatures.32 Isolated amyloid aggregates of lysozyme formed along either pathway, at a minimum, dramatically reduce their rates of dissociation, fragmentation, and self-association when lowering temperatures back into the physiological range. The stability of various amyloid species close to physiological conditions is relevant to the question how supposedly short-lived and/or metastable amyloid species can migrate within tissues or even the entire body, thereby spreading amyloid formation.
Amyloid Oligomers and Protofibrils, but not Filaments, Induce Native Lysozyme Aggregation
At denaturing temperatures used for seed growth, oligomers, protofibrils, and filaments all readily grow from partially unfolded lysozyme monomers.28 At physiological temperatures spontaneous amyloid formation from native proteins is not observed (see also Figure 4a below).12,33 We therefore tested whether the above long-lived amyloid species could induce fresh amyloid growth when added to native lysozyme maintained at physiological temperatures and pH. Initially we tested the behavior at pH 3 since it permitted inclusion of rigid filaments. Amyloid seeds of either type were diluted 10-fold into solutions of native folded lysozyme at pH 3 using the same solution composition as during seed formation, but maintaining the temperature at 37 °C throughout. The particle size distributions for fresh monomers seeded with amyloid filaments or mixtures of amyloid oligomers and protofibrils are shown in Figure 2a and d, respectively. Amyloid filaments at various stages of assembly were seeded into fresh monomers, but showed no discernible signs of growth at 37 °C over 3 days (Figure 2b, c). This indicates that monomeric filaments cannot utilize natively folded lysozyme at pH 3/37 °C as the substrate for continued growth. In stark contrast, oligomer/protofibrils at pH 3 readily induced aggregation of native lysozyme solutions. Scattering intensity doubled in as little as 18 h (Figure 2f), and the relative size of the protofibril peak grew at the expense of the monomer peak (Figure 2e). It is worth recalling that both amyloid oligomers/protofibrils and filaments at pH 3 assemble spontaneously, and continue to grow, once lysozyme is partially unfolded (T = 70 °C). Hence, both oligomers/protofibrils and filaments form suitable templates for amyloid growth from partially unfolded monomers. Upon return to 37 °C, lysozyme assumes its native fold.28 The ability of oligomers and protofibrils to seed fresh growth at 37 °C, combined with the inability of filaments to do so, implies the absence of even small percentages of partially unfolded solution states which would have supported continued growth of either template.
Figure 4.

Seeded growth requires both oligomers/protofibrils and native lysozyme, depletes native monomers. (a) Lack of changes in the hydrodynamic radius Rh (■) or light scattering intensity Iscat (○) for native lysozyme incubated at pH 7 and 37 °C for one month. (b) Changes in light scattering for lysozyme monomers incubated with 20% of either isolated protofibril (■) or monomers (○), separated from each other prior to seeding into fresh monomers. (c) Depletion of fresh lysozyme (20 mg/mL) during seeded growth at 37 °C with 15% (v/v) early stage purified protofibrils. Residual monomer concentrations in the supernatant (■) after centrifugation (2 h at 98 000 g), and light scattering intensities (○) from aliquots of seeded solution were monitored for approximately 4.5 days.
Figure 2.

Lysozyme oligomers and protofibrils, but not filaments, induce growth of native lysozyme. (a) DLS particle size distributions of monomeric filaments seeded into native lysozyme. The short and long monomeric filament peaks (S-MF, L-MF) are characteristic for this assembly pathway.10 Filament-seeded monomer solutions displayed no changes in (b) size distribution or (c) scattering intensity during 3 days of incubation. (d) Corresponding size distribution for mixtures of oligomers (merged with monomer peak) and protofibrils (40 nm peak) seeded into monomers. Within 16 h (f) the scattering intensity from oligomers/protofibril-seeded samples more than doubled. At the same time (e) the protofibril peak P grew at the expense of the monomer peak M.
To ensure that monomers retain a native fold, we repeated the seeding experiments near the physiological pH of 7. After generating either oligomers or mixtures of oligomers and protofibril at 70 °C at various stages of self-assembly, they were diluted 5- to 20-fold into 1.4 mM of fresh lysozyme at 37 °C. Seeded solutions were incubated for a period of time ranging from 4 days to 2 weeks. Solutions of native lysozyme at 37 °C seeded with oligomers only (Figure 3a) displayed kinetic growth signatures similar to those for barrier-free oligomer formation at denaturing temperatures, albeit at much reduced growth rates.10,21 This included a prominent nucleation event associated with protofibril nucleation (Figure 3b) and an accelerating increase in scattering intensity, particularly after protofibril nucleation, resulting in a 7-fold scattering increase over 2 weeks (Figure 3c). Seeding monomers with mixtures of protofibrils and oligomers (Figure 3d) resulted in noticeably faster increases in protofibril radii (Figure 3e) and scattering intensities (Figure 3f).
Figure 3.

Seeding native lysozyme with oligomers and protofibrils at pH 7. Particle size distribution derived from DLS for solutions of lysozyme monomers at pH 7 seeded with 20% (v/v) of (a) oligomers or (d) mixtures of oligomers and protofibrils. Labels indicate the (merged) monomer/oligomer peak (M/O) and protofibril peak (P). The dashed curve in (a) shows the distribution for monomers alone. Temporal evolution in the peak radii for samples seeded with (b) oligomer-seeded or (c) oligomer and protofibril. Corresponding changes in scattering intensity are displayed in panels (c) and (f). The sample seeded with oligomers nucleated protofibrils around 6 days (see panel b).
We performed a series of controls to confirm that seeded growth indeed represented induced aggregation of native monomers and required amyloid oligomers/protofibrils instead of partially denatured monomers. These control experiments were performed at pH 7 and 37 °C, because it represents the biologically most relevant situation. Incubating native lysozyme monomers without seeds, as expected, yielded no signs of aggregation for up to 3 months (Figure 4a). Isolated oligomers or protofibrils in the absence of lysozyme monomers also did not display any tendency toward self-assembly at 37 °C (Figure 1e). We also seeded fresh monomers with either transient denatured lysozyme monomers or amyloid protofibrils, separated from each other after seed growth at 70 °C and brought to identical concentrations. Only amyloid protofibrils induced aggregation of native lysozyme at 37 °C, indicating that the observed aggregation indeed required both amyloid seeds and native monomers. Moreover, we determined whether seeded aggregation did indeed consume fresh lysozyme monomers, instead of reporting on some monomer-induced self-assembly process of seeds only. Toward that end, fresh lysozyme solutions were incubated with 10% of isolated seeds. Concentrations of free monomers at various times after seeding were determined by pelleting aggregates from aliquots of growth solution via ultracentrifugation and measuring the residual monomer concentration in the supernatant. Seed-induced aggregation depleted monomers to levels far exceeding the original seed concentration (10% v/v). Monomer depletion also correlated with aggregation-induced increases in scattering intensity and particle sizes measured just prior to ultracentrifugation (Figure 4c).
The above data (Figures 2 and 3) establish that amyloid oligomers and/or protofibrils, but not filaments, were able to induce aggregation of native lysozyme at physiological temperature. The capability of lysozyme oligomers and protofibrils to seed fresh growth was preserved even at physiological pH. The observed evolution in particle size distributions (DLS) and scattering intensities (SLS) of seeded growth at physiological temperatures replicated many of the distinct kinetics features (temporal evolution of the particle size distributions; lag phase prior to protofibril nucleation; slow near-linear increase in mean protofibril radius; exponential growth in scattering intensity) associated with lysozyme fibril growth along the oligomeric pathway at denaturing temperatures.10,20,21
Cross-seeding of Filaments and Protofibrils
We attempted to cross-seed protofibrils and filaments, generated during transient denaturation at pH 3, into solutions of native lysozyme at pH 7. These experiments first indicate whether amyloid templates remain stable upon pH shifts. If successful, they also permit testing whether the capacity for growth from native protein persists with changing solution pH. Protofibrils and filaments at pH 3 were generated as described above and then either directly transferred to pH 7 or dialyzed to pH 7 and separated from monomers. Either way, oligomers and protofibrils at any stage of assembly at pH 3 irreversibly precipitated from solution at pH 7 making them unsuitable for seeding experiments. Filaments also showed signs of precipitation following dialysis, but could be resolubilized after passing them through 0.8 μm pore syringe filters. Filaments, resolubilized at pH 7, were concentrated to 20 mg/mL and then seeded into fresh lysozyme at pH 7 and 37 °C. Judged by the lack of change in scattering intensity or ThT response, filaments in the presence of monomers did not grow (Figure 5). In contrast, protofibrils generated at pH 7 and seeded into fresh monomers at the same concentration as filaments displayed robust growth.
Figure 5.

Cross-seeding of monomeric filaments. Seeds of monomeric filaments were generated at pH 3, separated from monomers, dialyzed, and resolubilized at pH 7. After concentrating seeds to 20 mg/mL, they were diluted 10-fold into either 20 mg/mL of native lysozyme or buffer only (pH 7, 40 mM NaCl) and incubated at 37 °C. Monomeric filaments (MF) did not induce growth of hew-L monomers as indicated by scattering intensity (a) or ThT fluorescence emission (b). In contrast, protofibrils (PF) generated at pH 7, concentrated (20 mg/mL), and diluted 10-fold into fresh monomers readily induced their self-replication (a). Protofibrils generated at pH 3 could not be used as seeds since they precipitated irreversibly following transfer to pH 7.
Morphological and Structural Analysis of Aggregates Derived from Seeded Growth at pH 7
To corroborate the amyloidogenic character of aggregates formed at physiological temperature and pH via seeded growth of native lysozyme we compared the aggregates’ morphological, tinctorial, and structural characteristics to those for protofibrils grown at denaturing temperatures.28 Figure 6a displays AFM images of lysozyme oligomers used for seeding and the morphologies of the resulting aggregates formed after incubation with native lysozyme at 37 °C for 5 days and 3 weeks, respectively. Following incubation, increasing numbers of highly flexible protofibrils appeared that tended to fold onto themselves upon deposition onto mica surfaces. Aggregation in seeded lysozyme solutions at 37 °C also increased thioflavin T fluorescence, a well-established amyloid indicator dye. For a given sample, fractional increases in light scattering vs ThT fluorescence intensity during incubation were of comparable magnitude (Figure 6b), replicating features of oligomer/protofibril growth at denaturing solution temperatures.13 FTIR spectra for aggregates formed at either 37 or 70 °C, after subtraction from those of native monomers, highlight aggregation-induced structural changes within the Amide-I band (Figure 6c). Aggregates emerging at the late stages of incubation at 70 °C or seeded growth at 37 °C displayed decreases in their percentage of native α-helical content (∼1660 cm–1) that correlated with their concurrent increases in β-sheet structure between 1630 and 1610 cm–1. This latter band of wavenumbers is considered diagnostic for β-sheet formation associated with amyloid fibril growth.27 We also find a much weaker peak emerging near 1695 cm–1, which has been associated with antiparallel β-sheets in Aβ amyloid oligomers.34 The above kinetic, morphological, tinctorial, and structural characteristics confirm that aggregates formed at physiological conditions via seeding native lysozyme with oligomers/protofibrils are indistinguishable from oligomers and protofibrils grown from partially denatured lysozyme.28 Therefore, lysozyme amyloid oligomers or protofibrils, when stabilized at 37 °C, do indeed induce their self-replication from native lysozyme as the growth substrate.
Seeded Oligomer/Protofibril Formation Suggests New Mode of Oligomeric Self-Assembly
There are three major models widely considered for amyloid self-assembly: templated assembly (TA),35 nucleated polymerization (NP)36 with variants accounting for secondary generation of new growth ends via fragmentation of existing seeds (NP-F)37 or via secondary surface nucleation of new seeds (NP-SN),37 and nucleated conformational conversion (NCC).26,38 These models make distinct predictions for the dependence of growth rates on both seed and monomer concentrations. Specifically, TA growth rates are reaction-limited by the number of seeds added to the system. Nucleated polymerization and nucleated conformation conversion, in turn, share characteristics of phase transitions. Consequently, seeds should decay for monomer concentrations below their solubility. The mechanisms of secondary generation incorporated into variants of nucleated polymerization both predict exponential growth rates due to the multiplicative increase of growth sites. However, fragmentation rates should be independent of monomer concentration while secondary surface nucleation rates will depend on both seed and monomer concentration. Nucleated models also presume behavior reminiscent of phase transitions, which requires monomer concentrations beyond some finite solubility limit, i.e. supersaturation.
To gain insights into what model might account for our observations of oligomer/protofibril growth under native solution conditions, we explored its dependence on seed age and monomer concentration. A fixed concentration of lysozyme monomers was seeded with oligomers only (O), early stage protofibrils (ESP), or late-stage protofibrils (LSP). Figure 6a shows a semilog plot of the increases in scattering intensity with incubation period, and linear fits through these data. The fits indicate that seeded growth from native monomers accelerates exponentially in time. Such an exponential increase implies the action of a mechanism for self-replicating growth units from those already present (dN ∝ N(t)), similar to fragmentation or secondary nucleation.37,39 The sample seeded with oligomers only also displayed a dramatic increase in the exponential growth rate near 5 days which coincided with the nucleation of protofibrils reported by DLS. Therefore, upon protofibril nucleation, this self-replication mechanism becomes more efficient or an additional mechanism is activated. The increases in growth rate with seed maturity suggest that the efficacy of this mechanism might also increase with seed age, potentially through a dependence on their contour length.
To explore the mechanisms of self-replication further, we tested the dependence of growth on monomer concentration while keeping seed concentration and age fixed. The initial slope of the temporal changes in scattering intensity (see Figure 7a) was taken as a measure of the growth rate of the seeded samples. Figure 7b is a log–log plot of the derived growth rates vs monomer concentrations for samples seeded simultaneously with 10% (v/v) of protofibrils from the same seed stock. The observed growth rate increased in nearly direct proportion (power law exponent 0.85 ± 0.15) over 2 orders of magnitude in monomer concentration. It displays no signs of a zero-point offset, indicative of a finite solubility associated with a phase transition. It also lacks signs of slowing down at high monomer concentrations as expected for a reaction-limited growth process.
Figure 7.

Dependence of growth kinetics on seed age and monomer concentration. (a) Natural logarithm of the scattering intensity (Iscat) for 20% (v/v) of amyloid oligomers (O), early stage protofibrils (ESP), or late-stage protofibrils (LSP) incubated with native lysozyme at 37 °C and pH 7. Solid lines are exponential fits with growth rate τ in days as indicated. The break in the growth rate for (O) coincided with the nucleation of protofibrils. (b) Log–log plot of the aggregate growth rate for solutions of 10% (v/v) of purified protofibrils seeded into increasing concentrations of lysozyme monomers (0.2–20 mg/mL). Aggregation rates were equated to the slope of the factional increases in scattering intensity near the onset of growth. A power law fit through the data (---) yielded an exponent of 0.85 ± 0.16.
Clearly, TA cannot account for the exponential increase in growth rates seen in Figure 7a. Furthermore, the rate-limiting step in TA is considered to be the conversion of monomers to the template structure. This would imply saturating growth rates at elevated monomer concentrations, which was not observed (Figure 7b). The strong dependence of growth rates on monomer concentration also excludes fragmentation as a mechanism for generating additional growth sites since the latter depends on aggregate but not monomer concentration.37 The absence of fragmentation is also evident from the lack of disintegration of isolated protofibrils when dissolved in buffer (Figure 1). Finally, the lack of a finite threshold to activate the self-replication process is at odds with any nucleated process since the latter requires a finite driving force (i.e., supersaturation). The inability to induce amyloid formation by native monomers at any concentration in the absence of seeds (Figure 3a), the lack of a lag phase for oligomer formation under denaturing (seed growth) conditions, and the immediate onset of growth from native monomers upon even modest seed addition, all imply that variants of nucleated polymerization and nucleated conformational conversion, by themselves, are difficult to reconcile with the growth process reported here.
Orthogonal Self-Replication and Nucleated Polymerization
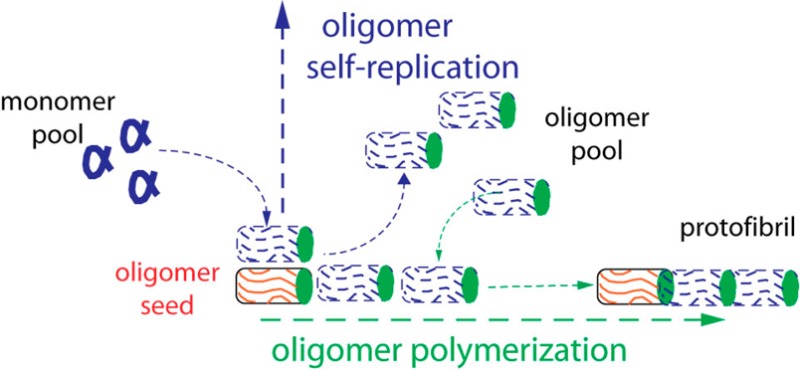
These discrepancies with existing models compel us to propose a new model for oligomeric fibril growth from native monomers. The essential features this model attempts to incorporate are a lack of spontaneous oligomer formation from native monomers, exponential growth when seeding native monomers with preformed oligomers, no discernible threshold for the onset of oligomer formation, the presence of a nucleation barrier for protofibril formation, and the previously reported morphological15 and structural similarities of oligomers and protofibrils.28 These features are consistent with two distinct “activities” of oligomers and protofibrils that operate “orthogonal” from each other: self-replication of oligomers from native monomers via a mechanism resembling a bimolecular reaction combined with nucleated polymerization of protofibrils from oligomers which resembles a nucleated phase separation. Figure 8 summarizes the aggregation behavior for oligomers/protofibrils vs those of filaments at denaturing vs native temperatures. At denaturing temperatures, amyloid filament growth proceeds via nucleated polymerization, with likely contributions from secondary generation. At physiological temperatures, these filaments are stable against dissociation or fragmentation, but can neither nucleate nor grow from native monomers. Along the oligomeric assembly pathway partially denatured monomers undergo barrier-free self-assembly into oligomers which, in turn, undergo nucleated polymerization. It should be noted that protofibril nucleation does not leave a clear signature in either ThT or SLS kinetics, but is readily apparent in DLS.21 At physiological temperatures, spontaneous formation of oligomers from monomers at even the highest concentrations is not observed, arguing further against a phase separation process. Upon addition of preformed and stabile oligomers (or protofibrils), their self-replication from native monomers emerges. Under denaturing conditions the ability for self-replication is concealed by the high rate of spontaneous oligomer assembly from denatured monomers. Similar to enzymes acting on their substrate, this autocatalytic self-replication mechanism has no discernible substrate threshold. Self-replication, in turn, accounts for the lack of a saturation threshold at high substrate concentrations. We further surmise that the subsequent polymerization of protofibrils uses oligomers as the growth substrate and proceeds at a site distinct from that for self-replication. Based on the identical cross-sectional areas of oligomers and protofibrils measured with calibrated AFM we have previously argued that oligomers, not monomers, are the growth substrate for lysozyme protofibrils.21 With oligomers as their growth substrate, protofibrils can only continue to elongate in the presence of native monomers if they provide two distinct sites, one for generating oligomers from native monomers and another for using them as a substrate for elongation. This uncoupling of activities is also supported by the observation that protofibril formation is a nucleated process while replication of oligomer from either oligomeric or protofibril seeds will proceed without a threshold.
Figure 8.

Oligomeric vs filamentous amyloid growth at denaturing vs native temperatures. Schematic of the free energy landscape for oligomeric (left) vs filamentous (right) amyloid growth at denaturing (top) vs physiological (bottom) temperatures. Solid arrows indicate processes while dashed lines represent nucleated processes. At denaturing temperatures, oligomers and protofibrils utilize denatured monomers as growth substrate. Oligomers, in turn, become the substrate for subsequent nucleation and polymerization of protofibrils. At physiological temperatures no amyloid seed can form spontaneously from native monomers. Preformed oligomers, but not filaments, can self-replicate from native monomers.
Discussion
Studies of amyloid fibril assembly under controlled in vitro conditions are critical for unraveling the molecular mechanisms underlying amyloid fibril formation in vivo. Our current understanding of molecular aggregation mechanisms, however, is mostly derived from in vitro studies under fixed solution conditions which, in the case of native proteins, require partial denaturation.11 Computations with model peptides in two and three dimensions have suggested that amyloid aggregation might proceed utilizing different conformations of monomers and aggregates, which can interact in distinct ways upon changes in solution environment (temperature, pH, ionic strength, concentration).15 This implies that mechanisms for amyloid self-assembly observed under partially denaturing conditions might not be fully representative of those active under physiological conditions. Our observations on lysozyme amyloid formation at denaturing vs physiological temperatures do indeed reveal significant changes to the thermodynamic stability, solubility, rates of fragmentation, and aggregation propensity of oligomeric vs filamentous amyloid aggregates. They particularly suggest that oligomer formation at denaturing vs physiological temperatures switches from the spontaneous self-assembly of partially denatured monomers to autocatalytic self-replication from native monomers.
Lowering solution temperatures into the physiological range resulted in either dramatic reduction of dissociation/fragmentation or self-association rates, or in complete stabilization for each of the amyloidogenic hew-L aggregates. The stability of isolated filaments might seem obvious. Yet, tau filaments generated at high temperatures readily dissociated at physiological temperatures unless they were stabilized by an extrinsic cofactor such as heparin.32 The emergence of the thermodynamic stability of oligomers at physiological pH and temperature is intriguing since oligomers are typically considered short-lived transients or metastable intermediates of the amyloid assembly process.40 Similarly, the loss of solubility of all aggregates upon increasing solution pH, while not surprising, further highlights that the properties of various templates also depend on the conditions they were generated under. Our observations suggest that the solubility and mechanical stability of all amyloid species are highly sensitive to their solution environment. The gain of stability of amyloid aggregates under physiological conditions is a property that is clearly relevant for understanding how these aggregates might spread to other cells and tissues and, potentially, promote formation of new amyloid growth there. Changes in solubility of amyloid aggregates with solution environment is equally germane for our understanding of functional amyloids.4142 For example, peptide hormones stored as amyloids in the low-pH environment of dense-core vesicles have to remain stable, but need to dissolve rapidly upon release into the bloodstream.43
Most significantly, though, is the ability for self-replication that emerged at physiological temperatures. Surprisingly, only oligomers and protofibrils were able to induce autocatalytic self-replication from natively folded lysozyme. The long rigid filaments considered most “representative” of amyloid aggregates, in contrast, remained inert at physiological temperatures. This is in stark contrast to the behavior of filaments at partially denaturing temperatures. Thermally denatured lysozyme monomers represent favorable substrates for the nucleation and growth of amyloid aggregates along either assembly pathway.10 Rapid filaments growth at denaturing temperatures vs their lack of growth at native temperatures implies that filaments require denatured monomers as a growth substrate. Conversely, the ability of oligomers and protofibrils, but not filaments, to seed growth at native temperatures highlights that seeded amyloid formation by native proteins depends not only on monomer stability and structure but on the stability and structure of amyloid templates and their interactions with their monomeric substrate. It further implies that only a subset of amyloid aggregates might have the capacity for autocatalytic self-replication required for their prion-like propagation in tissue.17,44 It is worth reiterating that there is no sign of spontaneous oligomer formation at native solution conditions (Figure 3a). Under the denaturing conditions used for seed growth, barrier-free assembly from denatured monomers is the dominant process for oligomer formation. Hence, the capacity of lysozyme amyloid oligomers and protofibrils for self-replication only becomes apparent at physiological conditions where spontaneous self-assembly is abrogated.
Are there any hints regarding how structural differences might account for the observed capacity for self-replication for oligomers and protofibrils but not filaments? We have previously shown that lysozyme filaments, oligomers, and protofibrils all develop β-sheet peaks in the “amyloid-band” of their FTIR spectra.28,45 Therefore, they all represent amyloid-like aggregates, albeit with distinguishable structural fingerprints.28 Specifically, the slight differences in the peak positions for filaments vs oligomers near 1620 cm–1 and the weak shoulder for oligomers/protofibrils near 1695 cm–1 both hint at underlying structural differences between these two aggregate species. These differences might well correlate with the β-sheet-rich but distinct structures recently reported for amyloid filaments vs oligomers reported for model peptides.2,46,47 However, we presume that not all oligomers are created equal. This is suggested by variations in the toxicity for oligomers from the N-terminal of bacterial HypF when generated at different pH’s.3 In contrast to the β-sheet enriched oligomers in this study, liquid-like disordered oligomers have been implicated to underlie nucleated conformational conversion of poly-Q.24 The ability of oligomers and their polymers for self-replication from native protein further adds to the growing list of pathogenic features ascribed to this class of amyloid aggregates.3,4,7,48,49 It also reproduces one defining characteristic of prion proteins: in vitro self-replication.35,50 The structural features that imbue lysozyme oligomers with the capacity for self-replication are not obvious, and whether this “gain of function” is limited to only certain oligomers remains to be determined.
The data on seeded amyloid growth with native lysozyme also suggest that the observed self-replication and polymerization of amyloid oligomers requires a new molecular model for oligomeric amyloid growth.51 Figure 8 summarizes how we envision the temperature-induced changes in the aggregation behavior along the filamentous and oligomeric assembly pathways seen in our experiments. As outlined above, neither templated assembly nor nucleated polymerization with inclusion of secondary mechanisms nor nucleated conformational conversion is fully consistent with the kinetics of oligomeric amyloid growth under native conditions. Our proposed model of “orthogonal autocatalytic self-replication and nucleated polymerization” shares important similarities with basic aspects of these existing models. Most prominently, self-replication provides a mechanism for spawning new aggregates similar to secondary nucleation and, thereby, generates exponential growth. In addition, we expect the nucleated polymerization of protofibrils to replicate most aspects of the current models of nucleated polymerization of filaments with a secondary nucleation pathway. It is worthwhile to also highlight the basic differences among existing models. First of all, oligomer self-replication proceeds without a discernible lower threshold concentration of monomers (saturation concentration). This implies that self-replication has the characteristics of an autocatalytic chemical reaction instead of a phase separation phenomenon (Figure 7b). The lack of saturation at high monomer concentrations, in turn, is a simple consequence of the autocatalytic feedback cycle in which the product of one replication cycle serves as the “enzyme” in the next cycle. Furthermore, the growth substrate for oligomer formation and protofibril elongation in our model are two distinct species: native monomers are the substrate for oligomer self-replication while protofibril nucleation and polymerization utilize oligomers. The separation of self-replication and elongation allows these two processes to proceed in a coordinated fashion that feeds back onto each other.
Overall, our results suggest that the process of amyloid fibril assembly, while displaying various generic aspects, might proceed through a variety of different pathways. Multiple assembly mechanisms become dominant not just by changes in monomer structure but by the type of amyloid aggregate formed and its specific propensity to interact with various monomeric conformations under various solution conditions. Such a diversity of mechanisms might help explain the wide variety of phenotypes and aggregation behavior reported for both pathological and functional amyloids.
Acknowledgments
We would like to acknowledge Tatiana Miti for support and critical input throughout this project, Garrett Matthews for generously making his AFM system available to us, and Chad Dickey, Vladimir Uversky, and Andrew Miranker for helpful comments. This work was supported, in part, by a grant from The Neuroscience Collaborative at USF and NIH Grant GM097723.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Eisenberg D.; Jucker M. Cell 2012, 148, 1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A.; Liu C.; Sawaya M. R.; Whitelegge J. P.; Park J.; Zhao M.; Pensalfini A.; Soriaga A. B.; Landau M.; Teng P. K.; Cascio D.; Glabe C.; Eisenberg D. Science 2012, 335, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campioni S.; Mannini B.; Zampagni M.; Pensalfini A.; Parrini C.; Evangelisti E.; Relini A.; Stefani M.; Dobson C. M.; Cecchi C.; Chiti F. Nat. Chem. Biol. 2010, 6, 140. [DOI] [PubMed] [Google Scholar]
- Kayed R.; Sokolov Y.; Edmonds B.; McIntire T. M.; Milton S. C.; Hall J. E.; Glabe C. G. J. Biol. Chem. 2004, 279, 46363. [DOI] [PubMed] [Google Scholar]
- Klein W. L.; Krafft G. A.; Finch C. E. Trends Neurosci. 2001, 24, 219. [DOI] [PubMed] [Google Scholar]
- Cleary J. P.; Walsh D. M.; Hofmeister J. J.; Shankar G. M.; Kuskowski M. A.; Selkoe D. J.; Ashe K. H. Nat. Neurosci. 2005, 8, 79. [DOI] [PubMed] [Google Scholar]
- Ono K.; Condron M. M.; Teplow D. B. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 14745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G. In Methods in Enzymology; Wetzel R., Kherterpal I., Eds.; Elsevier: 2006; Vol. 413, p 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosal W. S.; Morten I. J.; Hewitt E. W.; Smith D. A.; Thompson N. H.; Radford S. E. J. Mol. Biol. 2005, 351, 850. [DOI] [PubMed] [Google Scholar]
- Hill S. E.; Miti T.; Richmond T.; Muschol M. PLoS One 2011, 6, e18171/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Fernandez A.; Fink A. L. In Protein Misfolding, Aggregation and Conformational Diseases. Part A: Protein Aggregation and Conformational Diseases; Vladimir N. U., Fink A. L., Eds.; Springer: New York, 2006. [Google Scholar]
- Uversky V. N.; Fink A. L. Biochim. Biophys. Acta 2004, 1698, 131. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A.; LaBrenz S. R.; Woo L.; Serpell L. C.; Kelly J. W. J. Am. Chem. Soc. 2000, 122, 5262. [DOI] [PubMed] [Google Scholar]
- Pappu R. V.; Wang X.; Vitalis A.; Crick S. L. Arch. Biochem. Biophys. 2008, 469, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dima R. I.; Thirumalai D. Protein Sci. 2002, 11, 1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer S.; Ricchiuto P.; Kashchiev D. J. Mol. Biol. 2012, 422, 723. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B. Science 2012, 336, 1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillmore J. D.; Booth D. R.; Madhoo S.; Pepys M. B.; Hawkins P. N. Nephrol. Dial. Transplant. 1999, 14, 2639. [DOI] [PubMed] [Google Scholar]
- Krebs M. R. H.; Wilkins D. K.; Chung E. W.; Pitkeathly M. C.; Chamberlain A. K.; Zurdo J.; Robinson C. V.; Dobson C. M. J. Mol. Biol. 2000, 300, 541. [DOI] [PubMed] [Google Scholar]
- Arnaudov L. N.; de Vries R. Biophys. J. 2005, 88, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S. E.; Robinson J.; Matthews G.; Muschol M. Biophys. J. 2009, 96, 3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodali R.; Wetzel R. Curr. Opin. Struct. Biol. 2007, 17, 48. [DOI] [PubMed] [Google Scholar]
- Yong W.; Lomakin A.; Kirkitadze M. D.; Teplow D. B.; Chen S.-H.; Benedek G. B. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick S. L.; Ruff K. M.; Garai K.; Frieden C.; Pappu R. V. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 20075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis A.; Pappu R. V. Biophys. Chem. 2011, 159, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Culyba E. K.; Powers E. T.; Kelly J. W. Nat. Chem. Biol. 2011, 7, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandomeneghi G.; Krebs M. R. H.; McCammon M. G.; Fändrich M. Protein Sci. 2004, 13, 3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley J.; Hill S. E.; Miti T.; Mulaj M.; Ciesla M.; Robeel R.; Persichilli C.; Raynes R.; Westerheide S.; Muschol M. J. Chem. Phys. 2013, 139, 121901/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haezebrouck P.; Joniau M.; Van Dael H.; Hooke S. D.; Woodruff N. D.; Dobson C. M. J. Mol. Biol. 1995, 246, 382. [DOI] [PubMed] [Google Scholar]
- Mishra R.; Sörgjerd K.; Nyström S.; Nordigården A.; Yu Y.-C.; Hammarström P. J. Mol. Biol. 2007, 366, 1029. [DOI] [PubMed] [Google Scholar]
- Parmar A. S.; Gottschall P. E.; Muschol M. Biophys. Chem. 2007, 129, 224. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Dinkel P.; Yu X.; Margittai M.; Zheng J.; Nussinov R.; Wei G.; Ma B. Chem. Commun. 2013, 49, 3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth D. R.; Sunde M.; Bellotti V.; Robinson C. V.; Hutchinson W. L.; Fraser P. E.; Hawkins P. N.; Dobson C. M.; Radford S. E.; Blake C. F. F.; Pepys M. B. Nature 1997, 385, 787. [DOI] [PubMed] [Google Scholar]
- Cerf E.; Sarroukh R.; Tamamizu-Kato S.; Breydo L.; Derclaye S.; Yves F. D.; Narayanaswami V.; Goormaghtigh E.; Ruysschaert J.-M.; Raussens V. Biochem. J. 2009, 421, 415. [DOI] [PubMed] [Google Scholar]
- Griffith J. S. Nature 1967, 215, 1043. [DOI] [PubMed] [Google Scholar]
- Harper J. D.; Wong S. S.; Lieber C. M.; Landsbury P. T. J. Biochemistry 1999, 38, 8972. [DOI] [PubMed] [Google Scholar]
- Cohen S. I. A.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. J. Mol. Biol. 2012, 421, 160. [DOI] [PubMed] [Google Scholar]
- Serio T. R.; Cashikar A. G.; Kowal A. S.; Sawicki G. J.; Moslehi J. J.; Serpell L.; Arnsdorf M. F.; Lindquist S. L. Science 2000, 289, 1317. [DOI] [PubMed] [Google Scholar]
- Masel J.; Jansen V. A. A.; Nowak M. A. Biophys. Chem. 1999, 77, 139. [DOI] [PubMed] [Google Scholar]
- Bitan G.; Fradinger E. A.; Spring S. M.; Teplow D. Amyloid 2005, 12, 88. [DOI] [PubMed] [Google Scholar]
- Greenwald J.; Riek R. Structure 2010, 18, 1244. [DOI] [PubMed] [Google Scholar]
- Fowler D. M.; Koulov A. V.; Alory-Jost C.; Marks M. S.; Balch W. E.; Kelly J. W. PLoS Biol. 2005, 4, e6/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji S. K.; Perrin M. H.; Sawaya M. R.; Jessberger S.; Vadodaria K.; Rissman R. A.; Singru P. S.; Nilsson K. P. R.; Simon R.; Schubert D.; Eisenberg D.; Rivier J.; Sawchenko P.; Vale W.; Riek R. Science 2009, 325, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow C. W.; Prusiner S. B. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fändrich M.; Dobson C. M. EMBO J. 2002, 21, 5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R.; Sawaya M. R.; Balbirnie M.; Madsen A. O.; Riekel C.; Grothe R.; Eisenberg D. Nature 2005, 435, 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostol M. I.; Perry K.; Surewicz W. K. J. Am. Chem. Soc. 2013, 135, 10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malisauskas M.; Ostman J.; Darinskas A.; Zamotin V.; Liutkevicius E.; Lundgren E.; Morozova-Roche L. A. J. Biol. Chem. 2005, 280, 6269. [DOI] [PubMed] [Google Scholar]
- Mossuto M. F.; Dhulesia A.; Devlin G.; Frare E.; Kumita J. R.; de Laureto P. P.; Dumoulin M.; Fontana A.; Dobson C. M.; Salvatella X. J. Mol. Biol. 2010, 402, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr J.; Weinmann N.; Wille H.; Kaimann T.; Nagel-Steger L.; Birkmann E.; Panza G.; Prusiner S. B.; Eigen M.; Riesner D. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J. W. Nat. Struct. Biol. 2000, 7, 824. [DOI] [PubMed] [Google Scholar]