

Abstract
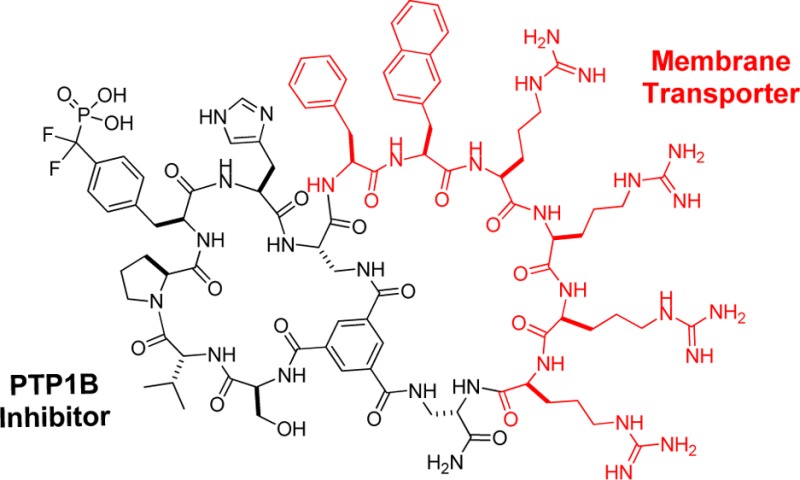
Cyclic peptides have great potential as therapeutic agents and research tools but are generally impermeable to the cell membrane. Fusion of cyclic peptides with a cyclic cell-penetrating peptide produces bicyclic peptides that are cell-permeable and retain the ability to recognize specific intracellular targets. Application of this strategy to protein tyrosine phosphatase 1B and a peptidyl-prolyl cis−trans isomerase (Pin1) isomerase resulted in potent, selective, proteolytically stable, and biologically active inhibitors against the enzymes.
Cyclic peptides (and depsipeptides) exhibit a wide range of biological activities.1 Several innovative methodologies have recently been developed to synthesize cyclic peptides, either individually2 or combinatorially,3 and screen them for biological activity. A particularly exciting application of cyclic peptides is the inhibition of protein–protein interactions (PPIs),4,5 which remain challenging targets for conventional small molecules. However, a major limitation of cyclic peptides is that they are generally impermeable to the cell membrane, precluding any application against intracellular targets, which include most of the therapeutically relevant PPIs. Although the formation of intramolecular hydrogen bonds6 or Nα-methylation of the peptide backbone7 can improve the membrane permeability of certain cyclic peptides, alternative strategies to increase the cell permeability of cyclic peptides are clearly needed.
Protein-tyrosine phosphatase 1B (PTP1B) is a prototypical member of the PTP superfamily and plays numerous roles during eukaryotic cell signaling. Because of its roles in negative regulation of insulin and leptin receptor signaling, PTP1B is a valid target for treatment of type II diabetes and obesity.8 A large number of PTP1B inhibitors have been reported,9 but none of them have succeeded in the clinic. Designing PTP inhibitors is challenging because most of the phosphotyrosine (pY) isosteres such as difluorophosphonomethyl phenylalanine (F2Pmp)10 are impermeable to the cell membrane. Additionally, because all PTPs share a similar active site, achieving selectivity for a single PTP has been difficult. In this work, we report a potentially general approach to the design of cell-permeable cyclic peptidyl inhibitors against intracellular proteins such as PTP1B.
We recently discovered cyclo(FΦRRRRQ) (cFΦR4, where Φ is l-naphthylalanine) as a novel class of cell-penetrating peptides (CPPs).11 Unlike previous CPPs, which are typically linear peptides that are entrapped in the endosome, cFΦR4 efficiently escapes from the endosome into the cytoplasm. Short peptide cargos (1–7 amino acids) can be delivered into mammalian cells by incorporating them into the cFΦR4 ring. Encouraged by this finding, we explored the possibility of developing bifunctional cyclic peptides containing both cell-penetrating and target-binding sequences as cell-permeable inhibitors against intracellular proteins. To generate specific inhibitors against PTP1B, we synthesized a one-bead two-compound library on spatially segregated ChemMatrix resin,12 in which each bead displayed a bifunctional cyclic peptide on its surface and contained the corresponding linear peptide in its interior as an encoding tag [Scheme 1 and Figure S1 in the Supporting Information (SI)]. The bifunctional cyclic peptides all featured the CPP motif FΦR4 (or its inverse sequence RRRRΦF) on one side and a random pentapeptide sequence (X1X2X3X4X5) on the other side, where X2 represents a 9:1 (mol/mol) mixture of Tyr and F2Pmp while X1 and X3–X5 are any of the 24 amino acids that included 10 proteinogenic l-amino acids (Ala, Asp, Gln, Gly, His, Ile, Pro, Ser, Tyr, Trp), five unnatural α-l-amino acids [F2Pmp, l-4-fluorophenylalanine (Fpa), l-norleucine (Nle), l-phenylglycine (Phg), l-pipecolic acid (Pip)], and nine α-d-amino acids [d-Ala, d-Asn, d-Glu, d-Leu, d-β-naphthylalanine (d-Nal), d-Phe, d-Pro, d-Thr, d-Val]. The library has a theoretical diversity of 6.6 × 105. The use of the 9:1 Tyr/F2Pmp ratio at the X2 position, together with a 5-fold reduction of the surface peptide loading, reduced the amount of F2Pmp-containing peptides at the bead surface by 50-fold, increasing the stringency of library screening.13 Screening 100 mg of the library (∼300 000 beads/compounds) against Texas red-labeled PTP1B resulted in 65 positive beads, which were individually sequenced by partial Edman degradation–mass spectrometry (PED-MS)14 to give 42 complete sequences (Table S1 in the SI).
Scheme 1. Evolution of a Cell-Permeable PTP1B Inhibitor.

Three representative hit sequences, d-Thr-d-Asn-d-Val-F2Pmp-d-Ala-Arg-Arg-Arg-Arg-Nal-Phe-Gln (inhibitor 1), Ser-d-Val-Pro-F2Pmp-His-Arg-Arg-Arg-Arg-Nal-Phe-Gln (inhibitor 2), and Ile-Pro-Phg-F2Pmp-Nle-Arg-Arg-Arg-Arg-Nal-Phe-Gln (inhibitor 3), were resynthesized and purified by HPLC. All three peptides are competitive PTP1B inhibitors, with peptide 2 being the most potent (IC50 = 31 ± 3 nM) (Table S2 and Figure S2). Unfortunately, inhibitor 2 showed no significant activity in cellular assays. Confocal microscopy analysis of human cells treated with fluorescein isothiocyanate (FITC)-labeled inhibitor 2 indicated poor cellular uptake of the peptide (Figure 1a). Although disappointing, this result was not entirely unexpected. Our previous study showed that as the size of the cargo inserted into the cFΦR4 ring increased, the cellular uptake efficiency of the cyclic peptides decreased dramatically.11 We reasoned that larger rings are more conformationally flexible and may bind less tightly to the cell-surface receptors (e.g., membrane phospholipids) during endocytosis. The negatively charged F2Pmp may also interact intramolecularly with the FΦR4 motif and interfere with its CPP function.
Figure 1.
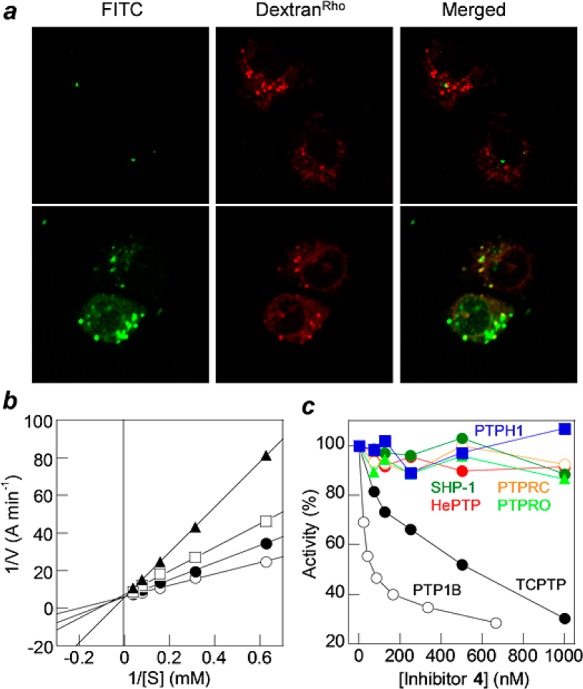
(a) Live-cell confocal microscopy images (same Z section) of A549 lung cancer cells after treatment for 2 h with 5 μM FITC-labeled inhibitor 2 (top panel) or 4 (bottom panel) and the endocytosis marker dextranRho (1.0 mg/mL). (b) Lineweaver–Burk plot showing competitive inhibition of PTP1B by inhibitor 4 at concentrations of 0 (○), 28 (●), 56 (□), and 112 nM (▲). (c) Sensitivity of various PTPs to inhibitor 4 (the y axis values are pNPP hydrolysis rates for the PTPs relative to that in the absence of inhibitor). The data shown in (b) and (c) are representative data sets.
To improve the cell permeability of inhibitor 2, we explored a bicyclic system in which the CPP motif is placed in one ring whereas the target-binding sequence constitutes the other ring (Scheme 1). The bicyclic system keeps the CPP ring to a minimal size, which according to the previously observed trend11 should result in more efficient cellular uptake. The bicyclic system should be able to accommodate cargos of any size because incorporation of the latter does not change the size of the CPP ring and therefore should not affect the delivery efficiency of the cyclic CPP. The use of a rigid scaffold (e.g., trimesic acid) may also help keep the CPP and cargo motifs away from each other and minimize any mutual interference. The smaller rings of a bicyclic peptide compared with its monocyclic counterpart should result in greater structural rigidity and improved metabolic stability.
To convert the monocyclic PTP1B inhibitor 2 into a bicyclic peptide, we replaced the Gln residue (used for attachment to the solid support and peptide cyclization) with (S)-2,3-diaminopropionic acid (Dap) and inserted a second Dap residue at the junction of the CPP and PTP1B-binding sequences (C-terminal to His) (Scheme 1). Synthesis of the bicycle was accomplished by the formation of three amide bonds between a trimesic acid and the N-terminal amine and the side chains of the two Dap residues (Scheme 2).5 Briefly, the linear peptide was synthesized on Rink amide resin using standard Fmoc chemistry and Nβ-alloxycarbonyl (Alloc)-protected Dap. After removal of the N-terminal Fmoc group, the exposed amine was acylated with trimesic acid. Removal of the Alloc groups with Pd(PPh3)4 followed by treatment with PyBOP afforded the desired bicyclic structure. To facilitate labeling with fluorescent probes, a lysine was added to the C-terminus. The bicyclic peptide (peptide 4) was deprotected using trifluoroacetic acid (TFA) and purified to homogeneity by HPLC.
Scheme 2. Solid-Phase Synthesis of Inhibitor 4.

Reagents: (a) standard Fmoc chemistry; (b) trimesic acid, HBTU; (c) Pd(PPh3)4, N-methylaniline; (d) PyBOP; (e) TFA.
Bicyclic peptide 4 acts as a competitive inhibitor of PTP1B with a KI value of 37 ± 4 nM (Figure 1b). It is highly selective for PTP1B. When assayed against PTP1B and TCPTP using p-nitrophenyl phosphate (pNPP) as substrate (500 μM), inhibitor 4 had IC50 values of 30 ± 4 and 500 ± 250 nM, respectively (Figure 1c and Table S3). It exhibited minimal inhibition of any of the other PTPs tested (≤10% inhibition of HePTP, SHP-1, PTPRC, PTPH1, or PTPRO at 1 μM inhibitor concentration). Gratifyingly, inhibitor 4 has greatly improved cell permeability over peptide 2, as detected by live-cell confocal microscopy of A549 cells treated with FITC-labeled inhibitor 4 (Figure 1a). The treated cells showed diffuse fluorescence throughout the cytoplasm and nucleus as well as fluorescence puncta, indicating that a fraction of the inhibitors reached the cytoplasm and nucleus while the rest was likely entrapped in the endosomes. Incubation of inhibitor 4 in human serum for 24 h at 37 °C resulted in ∼10% degradation, whereas 91% of inhibitor 2 was degraded under the same conditions (Figure S3). Overall, inhibitor 4 compares favorably with the small-molecule PTP1B inhibitors reported to date9 with respect to potency, selectivity over the highly similar TCPTP (17-fold), and cell permeability (Table S4).
Inhibitor 4 was next tested for its ability to perturb PTP1B function during cell signaling. Treatment of A549 cells with inhibitor 4 (0–5 μM) resulted in a dramatic and dose-dependent increase in the pY levels of a large number of proteins, consistent with the broad substrate specificity of PTP1B15 (Figure 2a). Analysis of the same samples by Coomassie blue staining showed similar amounts of proteins in all of the samples (Figure 2b), indicating that the increased pY levels reflected increased phosphorylation (or decreased PTP reaction) instead of changes in the total protein levels. Remarkably, the increase in tyrosine phosphorylation was already apparent at 8 nM inhibitor 4. Interestingly, further increases in the inhibitor concentration beyond 1 μM reversed the effect on tyrosine phosphorylation, an observation that was also made previously by Zhang and co-workers with a different PTP1B inhibitor.16 To obtain further evidence that intracellular PTP1B was inhibited by peptide 4, we monitored the pY level of insulin receptor (IR), a well-established PTP1B substrate in vivo,8 by immunoblotting with specific antibodies against the pY1162pY1163 site. Again, treatment with inhibitor 4 caused a dose-dependent increase in IR phosphorylation up to 1 μM inhibitor, and the effect leveled off at higher concentrations (Figure 2c,d). Taken together, our data indicate that bicyclic inhibitor 4 efficiently entered mammalian cells and inhibited PTP1B in vivo. The decreased phosphorylation at higher inhibitor concentrations may have been caused by nonspecific inhibition of other PTPs (which may in turn down regulate protein tyrosine kinases). It may also reflect the pleiotropic roles played by PTP1B, which can both negatively and positively regulate the activities of different protein kinases.17
Figure 2.

Inhibition of PTP1B in vivo. (a) Anti-pY immunoblot of global pY protein levels in A549 cells after treatment with 0–5 μM inhibitor 4 for 2 h. (b) SDS-PAGE analysis (Coomassie blue staining) of the same samples from (a) showing uniform sample loading in all lanes. (c) Effect of inhibitor 4 on insulin receptor phosphorylation at Tyr1162 and Tyr1163 sites. HepG2 cells were treated with the indicated concentrations of inhibitor 4 for 2 h, stimulated with insulin (100 nM) for 5 min, and analyzed by SDS-PAGE and immunoblotting with anti-IRpY1162/pY1163 antibody. (d) Quantitation of IR pY levels from (c) (data shown are means ± SD from five independent experiments).
To test the generality of the bicyclic approach, we applied it to the design of cell-permeable inhibitors against peptidyl-prolyl cis–trans isomerase (Pin1), a potential target for treatment of a variety of human diseases, including cancer,18 for which potent, selective, and biologically active inhibitors are still lacking.19 Thus, we fused cFΦR4 with the previously reported monocyclic peptide 5, which is a potent inhibitor against Pin1 in vitro (KD = 258 ± 66 nM) but membrane-impermeable20 (Scheme 3). In addition, we replaced the l-Tyr at the pThr + 3 position with Arg to improve the aqueous solubility. The resulting bicyclic peptide 6 bound Pin1 with KD = 130 ± 44 nM (Table S5 and Figure S4). Insertion of d-Ala at the pThr + 5 position to increase the separation between the Pin1-binding and cell-penetrating motifs improved the inhibitor potency by ∼2-fold (KD = 72 ± 21 nM for inhibitor 7). Inhibitor 7 competed with FITC-labeled inhibitor 5 for binding to Pin1 (Figure S5), indicating that they both bind to the Pin1 active site. Substitution of d-Thr for d-pThr of inhibitor 7 reduced its potency by ∼10-fold (KD = 620 ± 120 nM for inhibitor 8; Table S5), whereas further replacement of the pipecolyl residue with d-Ala abolished the Pin1 inhibitory activity (peptide 9). As expected, bicyclic inhibitors 7–9 are cell-permeable (Figure S6). Treatment of HeLa cells with inhibitor 7 resulted in dose-dependent inhibition of cell growth (45% inhibition after treatment for 3 days at 20 μM inhibitor 7), whereas the impermeable inhibitor 5 and inactive peptide 9 had no effect (Figure S7). Peptide 8 also inhibited cell growth, but to a lesser extent than inhibitor 7. Finally, treatment of HeLa cells with inhibitor 7 dramatically increased the cellular levels of promyelocytic leukemia protein (PML), an established Pin1 substrate destabilized by Pin1 activity (Figure S8).21
Scheme 3. Conversion of Impermeable Pin1 Inhibitor 5 into Cell-Permeable Bicyclic Inhibitor 7.

In conclusion, we have developed a potentially general approach for the design of cell-permeable bicyclic peptides against intracellular targets. Our preliminary studies show that replacement of the PTP1B-binding motif with other peptide sequences having different physicochemical properties also resulted in their efficient delivery into cultured mammalian cells.22 The availability of a general intracellular delivery method should greatly expand the utility of cyclic peptides in drug discovery and biomedical research.
Acknowledgments
This work was supported by NIH (GM062820 and CA132855).
Supporting Information Available
Experimental details and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Pomilio A. B.; Battista M. E.; Vitale A. A. Curr. Org. Chem. 2006, 10, 2075. [Google Scholar]
- For examples, see:; a Meutermans W. D. F.; Golding S. W.; Bourne G. T.; Miranda L. P.; Dooley M. J.; Alewood P. F.; Smythe M. L. J. Am. Chem. Soc. 1999, 121, 9790. [Google Scholar]; b Schafmeister C. E.; Po J.; Verdine G. L. J. Am. Chem. Soc. 2000, 122, 5891. [Google Scholar]; c Sun Y.; Lu G.; Tam J. P. Org. Lett. 2001, 3, 1681. [DOI] [PubMed] [Google Scholar]; d Kohli R. M.; Walsh C. T.; Burkart M. D. Nature 2002, 418, 658. [DOI] [PubMed] [Google Scholar]; e Qin C.; Bu X.; Zhong X.; Ng N. L. J.; Guo Z. J. Comb. Chem. 2004, 6, 398. [DOI] [PubMed] [Google Scholar]; f Turner R. A.; Oliver A. G.; Lokey R. S. Org. Lett. 2007, 9, 5011. [DOI] [PubMed] [Google Scholar]; g Hili R.; Rai V.; Yudin A. K. J. Am. Chem. Soc. 2010, 132, 2889. [DOI] [PubMed] [Google Scholar]; h Lee J.; McIntosh J.; Hathaway B. J.; Schmidt E. W. J. Am. Chem. Soc. 2009, 131, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Frost J. R.; Vitali F.; Jacob N. T.; Brown M. D.; Fasan R. ChemBioChem 2013, 14, 147. [DOI] [PubMed] [Google Scholar]
- For examples, see:; a Eichler J.; Lucka A. W.; Pinilla C.; Houghten R. A. Mol. Diversity 1996, 1, 233. [DOI] [PubMed] [Google Scholar]; b Giebel L. B.; Cass R. T.; Milligan D. L.; Young D. C.; Arze R.; Johnson C. R. Biochemistry 1995, 34, 15430. [DOI] [PubMed] [Google Scholar]; c Scott C. P.; Abel-Santos E.; Wall M.; Wahnon D. C.; Benkovic S. J. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 13638. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Millward S. W.; Takahashi T. T.; Roberts R. W. J. Am. Chem. Soc. 2005, 127, 14142. [DOI] [PubMed] [Google Scholar]; e Sako Y.; Morimoto J.; Murakami H.; Suga H. J. Am. Chem. Soc. 2008, 130, 7232. [DOI] [PubMed] [Google Scholar]; f Li S.; Marthandan N.; Bowerman D.; Garner H. R.; Kodadek T. Chem. Commun. 2005, 581. [DOI] [PubMed] [Google Scholar]; g Joo S. H.; Xiao Q.; Ling Y.; Gopishetty B.; Pei D. J. Am. Chem. Soc. 2006, 128, 13000. [DOI] [PubMed] [Google Scholar]; h Heinis C.; Rutherford T.; Freund S.; Winter G. Nat. Chem. Biol. 2009, 5, 502. [DOI] [PubMed] [Google Scholar]; i Tse B. N.; Snyder T. M.; Shen Y.; Liu D. R. J. Am. Chem. Soc. 2008, 130, 15611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples, see:; a Leduc A.-M.; Trent J. O.; Wittliff J. L.; Bramlett K. S.; Briggs S. L.; Chirgadze N. Y.; Wang Y.; Burris T. P.; Spatola A. F. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 11273. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Millward S. W.; Fiacco S.; Austin R. J.; Roberts R. W. ACS Chem. Biol. 2007, 2, 625. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tavassoli A.; Lu Q.; Gam J.; Pan H.; Benkovic S. J.; Cohen S. N. ACS Chem. Biol. 2008, 3, 757. [DOI] [PubMed] [Google Scholar]; d Wu X.; Upadhyaya P.; Villalona-Calero M. A.; Briesewitz R.; Pei D. Med. Chem. Commun. 2013, 4, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Birts C. N.; Nijjar S. K.; Mardle C. A.; Hoakwie F.; Duriez P. J.; Blaydes J. P.; Tavassoli A. Chem. Sci. 2013, 4, 3046. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Kawakami T.; Ishizawa T.; Fujino T.; Reid P. C.; Suga H.; Murakami H. ACS Chem. Biol. 2013, 8, 1205. [DOI] [PubMed] [Google Scholar]
- Lian W.; Upadhyaya P.; Rhodes C. A.; Liu Y.; Pei D. J. Am. Chem. Soc. 2013, 135, 11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai T.; Bock J. E.; Zhou M. V.; Kalyanaraman C.; Lokey R. S.; Jacobson M. P. J. Am. Chem. Soc. 2006, 128, 14073. [DOI] [PubMed] [Google Scholar]
- a Chatterjee J.; Gilon C.; Hoffman A.; Kessler H. Acc. Chem. Res. 2008, 41, 1331. [DOI] [PubMed] [Google Scholar]; b White T. R.; Renzelman C. M.; Rand A. C.; Rezai T.; McEwen C. M.; Gelev V. M.; Turner R. A.; Linington R. G.; Leung S. S. F.; Kalgutkar A. S.; Bauman J. N.; Zhang Y. Z.; Liras S.; Price D. A.; Mathiowetz A. M.; Jacobson M. P.; Lokey R. S. Nat. Chem. Biol. 2011, 7, 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Elchelby M.; Payette P.; Michaliszyn E.; Cromlish W.; Collins S.; Loy A. L.; Normandin D.; Cheng A.; Himms-Hagen J.; Chan C. C.; Ramanchandran C.; Gresser M. J.; Tremblay M. L.; Kennedy B. P. Science 1999, 283, 1544. [DOI] [PubMed] [Google Scholar]; b Zabolotny J. M.; Bence-Hanulec K. K.; Stricker-Krongrad A.; Haj F.; Wang Y.; Minokoshi Y.; Kim Y. B.; Elmquist J. K.; Tartaglia L. A.; Kahn B. B.; Neel B. G. Dev. Cell 2002, 2, 489. [DOI] [PubMed] [Google Scholar]
- He R.; Zeng L.-F.; He Y.; Zhang Z.-Y. In New Therapeutic Strategies for Type 2 Diabetes: Small Molecule Approaches; Jones R. M., Ed.; RSC Publishing: Cambridge, U.K., 2012; pp 142 ff. [Google Scholar]
- Burke T. R. Jr.; Kole H. K.; Roller P. P. Biochem. Biophys. Res. Commun. 1994, 204, 129. [DOI] [PubMed] [Google Scholar]
- Qian Z.; Liu T.; Liu Y.-Y.; Briesewitz R.; Barrios A. M.; Jhiang S. M.; Pei D. ACS Chem. Biol. 2013, 8, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Marik J.; Lam K. S. J. Am. Chem. Soc. 2002, 124, 7678. [DOI] [PubMed] [Google Scholar]
- Chen X.; Tan P. H.; Zhang Y.; Pei D. J. Comb. Chem. 2009, 11, 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar A.; Wavreille A.-S.; Pei D. Anal. Chem. 2006, 78, 5935. [DOI] [PubMed] [Google Scholar]
- Ren L.; Chen X.; Luechapanichkul R.; Selner N. G.; Meyer T. M.; Wavreille A.-S.; Chan R.; Iorio C.; Zhou X.; Neel B. G.; Pei D. Biochemistry 2011, 50, 2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L.; Lee S.-Y.; Andersen J. N.; Waters S.; Shen K.; Guo X.-L.; Moller N. P. H.; Olefsky J. M.; Lawrence D. S.; Zhang Z.-Y. Biochemistry 2003, 42, 12792. [DOI] [PubMed] [Google Scholar]
- Lessard L.; Stuible M.; Tremblay M. L. Biochim. Biophys. Acta 2010, 1804, 613. [DOI] [PubMed] [Google Scholar]
- Lu K. P.; Zhou X. Z. Nat. Rev. Mol. Cell Biol. 2007, 8, 904. [DOI] [PubMed] [Google Scholar]
- More J. D.; Potter A. Bioorg. Med. Chem. Lett. 2013, 23, 4283. [DOI] [PubMed] [Google Scholar]
- Liu T.; Liu Y.; Kao H.-Y.; Pei D. J. Med. Chem. 2010, 53, 2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke E. L.; Lam M.; Liu O.; Liu Y.; Stanya K. J.; Chang K. S.; Means A. R.; Kao H. Y. Mol. Cell. Biol. 2008, 28, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z.; LaRochelle J. R.; Jiang B.; Lian W.; Hard R. L.; Selner N. G.; Luechapanichkul R.; Barrios A. M.; Pei D. Biochemistry 2014, 53, 4034–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.