

Abstract
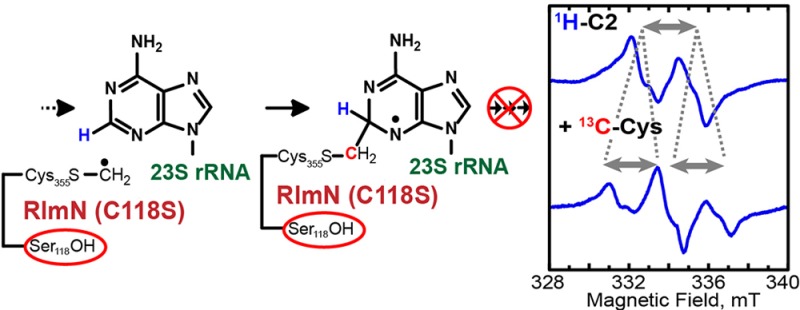
RlmN and Cfr are methyltransferases/methylsynthases that belong to the radical S-adenosylmethionine superfamily of enzymes. RlmN catalyzes C2 methylation of adenosine 2503 (A2503) of 23S rRNA, while Cfr catalyzes C8 methylation of the exact same nucleotide, and will subsequently catalyze C2 methylation if the site is unmethylated. A key feature of the unusual mechanisms of catalysis proposed for these enzymes is the attack of a methylene radical, derived from a methylcysteine residue, onto the carbon center undergoing methylation to generate a paramagnetic protein–nucleic acid cross-linked species. This species has been thoroughly characterized during Cfr-dependent C8 methylation, but does not accumulate to detectible levels in RlmN-dependent C2 methylation. Herein, we show that inactive C118S/A variants of RlmN accumulate a substrate-derived paramagnetic species. Characterization of this species by electron paramagnetic resonance spectroscopy in concert with strategic isotopic labeling shows that the radical is delocalized throughout the adenine ring of A2503, although predominant spin density is on N1 and N3. Moreover, 13C hyperfine interactions between the radical and the methylene carbon of the formerly [methyl-13C]Cys355 residue show that the radical species exists in a covalent cross-link between the protein and the nucleic acid substrate. X-ray structures of RlmN C118A show that, in the presence of SAM, the substitution does not alter the active site structure compared to that of the wild-type enzyme. Together, these findings have new mechanistic implications for the role(s) of C118 and its counterpart in Cfr (C105) in catalysis, and suggest involvement of the residue in resolution of the cross-linked species via a radical mediated process.
Introduction
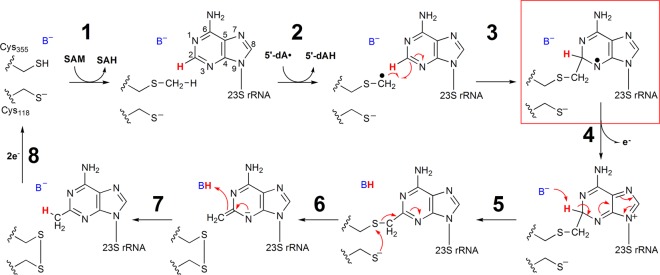
RlmN and Cfr are two evolutionarily related proteins that catalyze methylation of adenosine 2503 (A2503) of 23S bacterial rRNA.1−5 RlmN catalyzes only C2 methylation of this nucleotide,6 while Cfr catalyzes C2 methylation only after it catalyzes C8 methylation.1 A2503 is ultimately located in the peptidyltransferase center of the bacterial ribosome near the entrance to the exit channel for the nascent polypeptide;7−10 however, studies have shown that these two reactions take place before 23S rRNA is incorporated into the intact ribosome.11 Methylation of C2 of A2503 by RlmN is nearly ubiquitous in eubacteria and is believed to enhance translational fidelity.12 Recent studies have shown that RlmN is also responsible for methylation of adenosine 37 of several Escherichia coli (Ec) tRNAs.13 Although the carbon center of the appended methyl group derives from the activated methyl moiety of S-adenosyl-l-methionine (SAM), RlmN and Cfr are not classical methyltransferases and do not catalyze the transfer of an intact methyl group from a methyl donor to the final product.14,15 Instead, these two enzymes belong to the radical SAM (RS) superfamily of proteins, which use a 5′-deoxyadenosyl 5′-radical (5′-dA·) generated from a reductive cleavage of SAM to initiate radical-based transformations.16−18 As in all structurally characterized RS enzymes, in RlmN, SAM ligates to a unique iron ion of a requisite [4Fe–4S] cluster cofactor via its amino and carboxylate functionalities.19−22 Although this binding mode facilitates reductive cleavage of the molecule to the 5′-dA· in most RS enzymes, it also facilitates SN2-based transfer of the methyl group from SAM to a conserved cysteinyl residue (C355 in RlmN) in the first chemical step of the RlmN and Cfr reactions.21,23 Upon release of the product, S-adenosylhomocysteine (SAH), and rebinding of another molecule of SAM, this second SAM molecule undergoes reductive fragmentation to generate the 5′-dA·, which abstracts a hydrogen atom from the methylcysteine (mCys) residue.14 The resulting methylene radical adds to C2 (C8 in Cfr) of the aromatic ring in a Minisci-like reaction to afford a paramagnetic cross-linked protein–nucleic acid species (Scheme 1).24,25 Upon loss of an electron and a proton, the cross-link is proposed to be resolved via disulfide-bond formation with the participation of a second, strictly conserved, Cys residue (C118 in RlmN) (Scheme 1).14 Consistent with this step of the reaction, C → A or C → S variants of C118 are unable to catalyze complete methylation of the substrate and form stable adducts with rRNA both in vivo and in vitro.2,14,26
Scheme 1. Mechanism for RlmN Proposed by Grove et al.14.
Recently, experimental evidence for the paramagnetic protein–nucleic acid cross-linked species was provided in the Cfr reaction during C8 methylation of A2503 using a 155-nucleotide long RNA substrate analogue (155-mer). When Cfr and the 155-mer were mixed with SAM and a required low-potential reductant, a substrate-derived paramagnetic species was observed. Although the unpaired electron resided predominantly on N7 of A2503, it was also delocalized throughout the adenine ring of the nucleotide. Further isotopic labeling studies showed that the species existed in a covalent cross-link with the terminal methylene carbon of a formerly C338 mCys residue, confirming the identity of the paramagnetic intermediate originally proposed by Grove et al.14,25
The mechanism shown in Scheme 1 proposes that loss of an electron from the substrate radical intermediate (step 4) precedes proton abstraction from C2 (C8 Cfr), and that the role of C118 is to participate in the resolution of the protein–nucleic acid cross-linked species by forming a disulfide bond with C355 (C338 in Cfr). Herein, we show that the cognate paramagnetic species observed during C8 methylation of A2503 by wild-type (wt) Cfr does not accumulate to detectible levels during C2 methylation by wt RlmN but does accumulate in an RlmN C118S or C118A variant. This observation suggests a possible refinement of the mechanism shown in Scheme 1, wherein a proton abstraction prior to electron transfer would allow for resolution of the protein–nucleic acid cross-link via a radical fragmentation mechanism involving C118.
Materials and Methods
Materials
Vent polymerase and Antarctic phosphatase were purchased from New England Biolabs (Ipswich, MA). Nuclease P1 from Penicillium citrinum was purchased from Sigma-Aldrich (St. Louis, MO). All oligonucleotide primers were obtained from Integrated DNA Technologies (Coralville, IA) and used as received. Talon metal affinity resin was purchased from Clontech (Mountain View, CA). PD-10 prepoured gel-filtration columns were purchased from GE Biosciences (Piscataway, NJ). S-Adenosylmethionine and S-adenosyl-[methyl-13C]methionine were synthesized enzymatically and purified as described previously.27 [2-2H]Adenosine triphosphate (97% enrichment) was purchased from Cambridge Isotopes (Andover, MA). The 155-mer and [2-2H]A-155-mer RNA substrates (encompassing positions 2454–2608 of E. coli 23S rRNA) were prepared using runoff transcription as previously described.25Ec flavodoxin (Fld) and flavodoxin reductase (Flx) were purified as previously described.28
Preparation of RlmN and Cfr Variants
The C118S and E105A variants of RlmN and the C105A variant of Cfr were generated using the Stratagene QuikChange II kit (Agilent Technologies) with primers listed in Table S1. One amendment to the procedure is that Vent polymerase was substituted for Pfu polymerase. Codon changes were confirmed by DNA sequencing at the Pennsylvania State University Genomics Core Facility. RlmN C118S and E105A, and Cfr C105A, were overproduced and purified in their apo (protein lacking its [4Fe–4S] cluster) forms and subsequently reconstituted with iron and sulfide to restore their [4Fe–4S] clusters as previously described.23,25 These three proteins are referred to as apo RlmNC118S→RCN, apo RlmNE105A→RCN, and apo CfrC105A→RCN, respectively. RlmN C118A was overproduced and purified in its holo form as previously described.14,23
Assays for Methyl Transfer by Apo RlmNC118S→RCN
Methylation of C355 on apo RlmNC118S→RCN was monitored by SAH production as previously described.23 Briefly, a 200 μL reaction containing 100 mM Tris-HCl, pH 8.5, 10 mM MgCl2, and 105 μM apo RlmNC118S→RCN was incubated in the absence of SAM for 5 min at 25 °C. A 10 μL aliquot was then removed and mixed with 10 μL of a solution containing 100 μM l-tryptophan, used as an internal standard (IS), and 100 mM H2SO4. The reaction was initiated by addition of SAM to a final concentration of 2 mM, and at appropriate times, 10 μL aliquots were removed and added to 10 μL of a quenching solution consisting of 100 μM l-tryptophan and 100 mM H2SO4. SAH concentrations were determined by liquid chromatography with detection by mass spectrometry (LC–MS) using a standard curve of known concentrations and applying a correction factor of 2 to account for the 2-fold dilution of the original assay mixture.23
Turnover Assays for wt RlmN or RlmNE105A→RCN
Assays contained either 10 μM apo RlmNE105A→RCN or wt RlmN, 50 mM Tris–HCl, pH 8.4, 10 mM MgCl2, 100 μM RNA, and 2 mM SAM in a total volume of 100 μL. Reactions were initiated by the addition of 2 mM dithionite, and aliquots were removed at designated times and quenched by addition to a solution of 50 mM H2SO4 and 100 μM l-tryptophan. Each sample was then mixed with 20 μL of 2× P1 nuclease buffer (250 mM sodium acetate, pH 6.0, 45 mM NaCl and 4 mM ZnCl2), 0.5 U of P1 nuclease, and 10 U of Antarctic phosphatase, and digestion of the RNA was carried out at 37 °C for 12 h. The samples were subjected to centrifugation to remove precipitate, and the supernatants were analyzed as previously described.23,25 The m2A standard was synthesized as previously described.11
Preparation of RlmNC118S→RCN and CfrC105A→RCN Electron Paramagnetic Resonance (EPR) Samples
A 500 μL reaction mixture containing 100 mM Tris-HCl, pH 8.5, 2 mM MgCl2, 300 μM 155-mer (or [2-2H]A-155-mer), and 529 μM apo RlmNC118S→RCN was incubated at room temperature for 10 min. The reaction mixture was then divided into two 250 μL aliquots, which were incubated an additional 5 min at 37 °C with 1 mM SAM or 1 mM [methyl-13C]SAM. Upon initiation of the reactions by addition of 10 mM (final concentration) dithionite, they were transferred to EPR tubes, incubated for ∼30 s at 37 °C, and then frozen by inserting the EPR tubes in cryogenic liquid isopentane (approximately −140 °C). To show the time-dependent formation of the radical species, 150 μM RlmNC118S→RCN was first mixed with 50 mM Tris-HCl, pH 8.5, 2 mM MgCl2, 200 μM 155-mer, and 1 mM SAM in a total volume of 1 mL. The reaction was then initiated by the addition of 5 mM dithionite (final concentration), and at designated times, 200 μL aliquots were removed and added to EPR tubes, which were quickly frozen in cryogenic liquid isopentane. EPR measurements were carried out as described below. Preparation of time-dependent samples of CfrC105A→RCN with the 155-mer was carried out as described above, with the exception that 200 μM CfrC105A→RCN and 300 μM 155-mer were used.
EPR Measurements
All EPR measurements were carried out on a Bruker Elexsys E580 X-band spectrometer equipped with a SuperX-FT microwave bridge and using a Bruker ER 4122 SHQE SuperX high-sensitivity cavity in combination with an ER 4112-HV Oxford Instruments variable temperature helium flow cryostat. All measurements were performed using a 40.96 ms conversion time, a 20.48 ms time constant, and 1024 points. Spectra were averaged over 500–2000 scans.
Density Functional Theory
All calculations were performed using Gaussian 03 Rev. E. within the spin-unrestricted Density Functional Theory (DFT) level.29 The geometries of all models were optimized utilizing the BP86 functional30,31 without any restrictions. In the optimizations, Ahlrichs triple-ζ valence basis set (TZV)32 with one set of polarization functions was used for all atoms (TZVP). The single point calculations with subsequent extraction of EPR parameters were performed using the B3LYP functional33,34 with TZVP basis sets on all atoms. Both geometry optimization and single point calculations were performed using Gaussian’s implementation of continuum solvation model COSMO (conductor-like screening model)35 in the PCM (polarizable continuum models) framework termed as C-PCM with ε = 4.0.36
Crystallographic Characterization of RlmN C118A
Crystals of RlmN C118A (10 mg/mL in 10 mM HEPES, pH 7.5) were obtained in a Coy anaerobic chamber using the hanging-drop vapor-diffusion method with 7.5% (w/v) PEG 6000, 0.1 M HEPES, pH 7.5, 5% (v/v) 2-methyl-2,4-pentanediol as the precipitant and 0.25 M LiCl as the well solution. Crystals appeared within 1 week at room temperature and were mounted on rayon loops for data collection. Samples were flash-frozen by direct plunge into liquid nitrogen after cryoprotection in precipitating solution supplemented with 30% (v/v) PEG 400. To determine the structure of C118A RlmN with SAM, crystals were soaked in a 5 mM solution of SAM in mother liquor for 30 min at room temperature and harvested as described above. All crystallographic data sets were collected at the Life Sciences Collaborative Access Team beamlines at the Advanced Photon Source and processed using the HKL2000 software package.37 The structure was solved by molecular replacement with PHASER using the structure of wt Ec RlmN (PDB accession code 3RF9) as the search model. Refinement and model building were performed with REFMAC538 and Coot.39 In each structure, two RlmN molecules were found in the asymmetric unit. In the C118A RlmN+SAM structure, the final model consists of residues 17–348 in chain A, residues 17–375 in chain B, two [4Fe-4S] clusters, and two molecules of SAM. In the C118A RlmN structure, the final model consists of residues 17–352 and residues 361–374 in chains A and B (residues 353–360 are disordered in both chains), two [4Fe–4S] clusters, and 231 water molecules. Ramachandran plots generated with Molprobity40 indicate 100% of residues in allowed and additionally allowed regions. Structural superpositions were performed using the secondary-structure matching method.41 Electron density maps were calculated with FFT.42 Figures were generated with the PyMOL Molecular Graphics System (Schrödinger, LLC). Table S3 reports all data collection and refinement statistics.
Results and Discussion
The Central Mechanistic Hypothesis of This Study
The key proposed mechanistic feature of the reactions catalyzed by RlmN and Cfr is the formation of a paramagnetic protein–nucleic acid cross-link intermediate between the methylene carbon of an mCys residue and the target carbon center (C2 or C8) of the nucleotide substrate (Scheme 1). This species was recently observed and thoroughly characterized during Cfr-catalyzed C8 methylation of a 155-mer RNA substrate analogue.25 In similar experiments with RlmN, conducted in this study, this species was not detected, suggesting that its rate of decay is faster than its rate of formation. In the mechanism depicted in Scheme 1, loss of an electron from the paramagnetic species is proposed to precede proton abstraction; however, the exact sequence of steps is unknown and other possibilities exist. For example, direct proton abstraction from the target carbon center before loss of an electron would allow for resolution of the protein–nucleic acid cross-linked species via a radical mechanism, as shown in Scheme 2 for RlmN-catalyzed C2 methylation. The role of the second active-site cysteine in the mechanism, among other possibilities, could be (i) to abstract the target proton (Scheme 2B); (ii) to stabilize a resulting thiyl radical, perhaps via formation of a disulfide-radical or disulfide-radical anion (Scheme 2A); (iii) or to do both (Scheme 2C). This alternative mechanism would predict that decay of the paramagnetic species depends on prior proton abstraction and perhaps the assistance of the second active-site cysteine in stabilizing the nascent thiyl radical. In such a scenario, RlmN variants containing substitutions at the second active site cysteine might be expected to exhibit an EPR-detectable paramagnetic signal under turnover conditions.
Scheme 2. Possible Roles for C118 in RlmN.
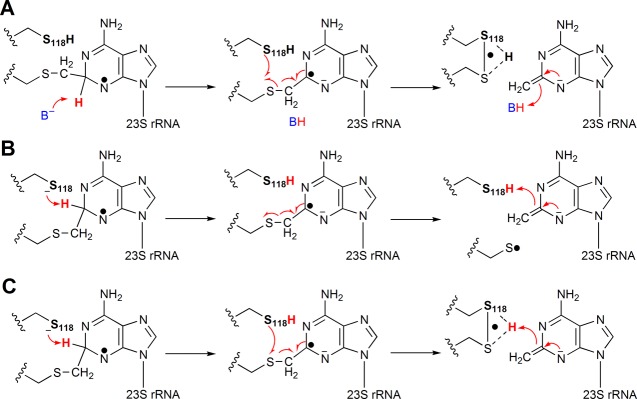
(A) Cys118 participates in a disulfide radical species upon deprotonation of the cross-linked intermediate at C2 and subsequent fragmentation. (B) Cys118 acts as the base that abstracts the C2 proton, initiating fragmentation of the cross-linked species to generate an oxidizing thiyl radical. (C) Cys118 acts as the base that abstracts the C2 proton and participates in the disulfide radical species formed upon fragmentation of the cross-linked intermediate.
Previous structural studies on RlmN suggested that E105 might play a dual role in catalysis, functioning as a general base to remove a proton from C355 to facilitate SN2-based methyl transfer, and as the general base that removes the C2 proton from the adenine nucleotide of the cross-linked intermediate (Scheme 1).21 This determination resulted, in part, from mechanistic studies that suggested that the base that removes the C2 proton is monoprotic, because the C2 hydrogen migrates to the methyl group of the product with no exchange with solvent hydrons.14,43 If E105 does, in fact, function in the latter capacity, it would be expected that an E105A variant would exhibit a drastically reduced rate of m2A formation due to an inability or decreased ability to resolve the cross-linked intermediate. Figure S1 depicts the rate of m2A formation for wt RlmN and the E105A variant. As can be observed, both enzymes catalyze multiple turnovers with similar initial rates. Therefore, it seems unlikely that E105 acts in this capacity, raising the possibility that C118 might function in this role, given that it is the only other conserved monoprotic amino acid in the vicinity of what is believed to be the active site.21
Purification and Characterization of Apo RlmNC118S→RCN
As we have shown previously, alanyl or seryl substitutions at C118 of RlmN lead to purification of a protein displaying a UV/vis spectrum with a maximum absorption feature around 263 nm rather than the typical absorption at 280 nm displayed by the wt protein or other Cys → Ala variants.14 This blue-shift in wavelength was shown to derive from rRNA that was covalently bound to the protein.14 More recent mass spectrometric studies showed unambiguously that the covalent linkage in this variant involves an adduct between the C355 mCys residue and A2503.26 Therefore, any attempt to study turnover with this variant would be difficult, if not impossible, given that it is isolated in an inactive state due to alkylation of a key residue. Our earlier work, however, showed that the iron–sulfur (Fe/S) cluster in RlmN and Cfr is required for generation of the mCys residue, the first chemical step in the proposed catalytic mechanism.23 Thus, strategies to produce RlmN and Cfr in the absence of their Fe/S clusters allow for purification of the proteins in their unmethylated states.
The EcrlmN gene encoding the C118S substitution was expressed in the presence of o-phenanthroline, a chelating agent that binds iron, and the corresponding apo protein was purified and subsequently reconstituted under anaerobic conditions. The as-isolated (AI) protein contained 0.18 iron and 0.23 sulfide ions, respectively, whereas the reconstituted (RCN) protein (apo RlmNC118S→RCN) contained 2.7 iron and 3.7 sulfide ions (Table S2). The UV/vis spectra of the AI (black trace) and RCN (red trace) proteins (Figure S2A) are also consistent with the presence of a [4Fe–4S] cluster in the RCN protein and its relative absence in the AI protein. Additionally, the maximum absorption of the displayed spectrum is at 278 nm rather than ∼263 nm, as is observed for this RlmN variant when isolated under conditions in which free iron is not chelated before induction of expression. In Figure S2B, a reaction (22 °C) of 100 μM apo RlmNC118S→RCN with SAM is displayed. As is observed with wt RlmN or wt Cfr that is overproduced and isolated under these conditions, treatment of the RCN RlmN C118S variant with SAM results in rapid production of SAH (83 μM), which is indicative of methyl transfer to Cys355.23,25 Previous Mössbauer studies on RlmN showed that the amount of SAH formed is directly proportional to the stoichiometry of [4Fe–4S] clusters on the protein.23 Therefore, these studies indicate that ∼80% of RlmNC118S→RCN is appropriately reconstituted.
Observation and Characterization of a Radical Species Produced by Apo RlmNC118S→RCN
As indicated above, wt RlmN does not accumulate a detectable substrate-derived paramagnetic species during turnover. However, when continuous wave EPR measurements were carried out at 70 K on a sample of RlmNC118S→RCN treated with dithionite, SAM, and the unlabeled (i.e., natural abundance) 155-mer, a spectrum characteristic of an unpaired electron strongly coupled to a single hydrogen nucleus (Figure 1, not labeled) was readily detected. Simulation of the spectrum of the radical generated by RlmNC118S→RCN indicates a fairly isotropic 1H hyperfine (HF) coupling constant (Aiso) of 65 MHz (see Table 1, A1), and the broadness of the spectral lines required inclusion of additional anisotropic HF coupling from 14N nuclei. Spin quantification of the signal using flavodoxin semiquinone (37.2 μM) as a standard resulted in a total spin concentration of ∼30 μM for the RlmNC118S→RCN paramagnetic species. This concentration corresponds to ∼10% of that of the 155-mer (∼300 μM), which is limiting in the reaction.
Figure 1.
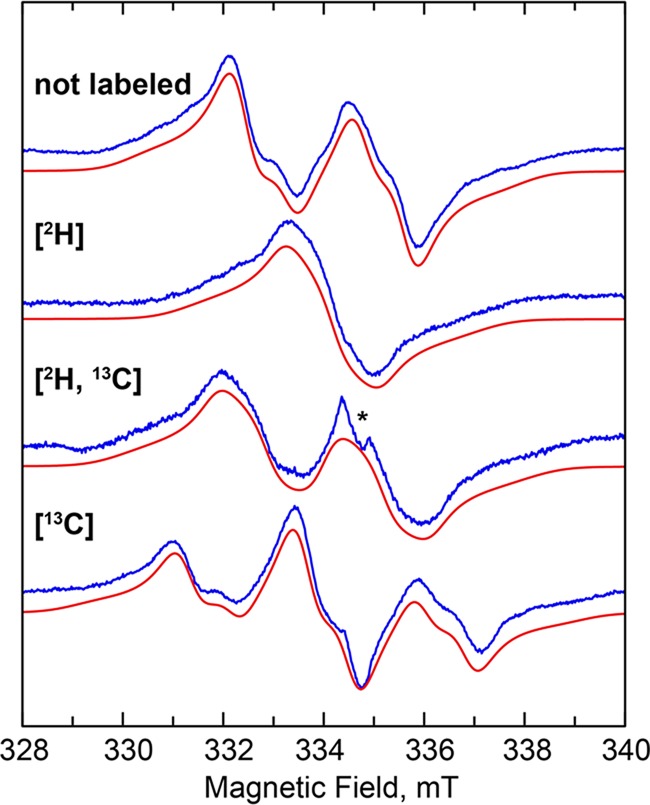
CW EPR spectra of the cross-linked substrate radical: using isotopically unenriched substrates (not labeled), the 155-mer substrate with 2H at C2 ([2H]), apo RlmNC118S→RCN containing a 13C-methyl mCys residue ([13C]), and both the labeled 155-mer and labeled protein ([2H, 13C]). Experimental data (blue) and corresponding simulations (red), taking into account the hyperfine coupling constants shown in Table 1. Experimental conditions: temperature, 70 K; modulation amplitude, 5 G; microwave power, 0.12 mW; microwave frequency, 9.38 GHz.
Table 1. Hyperfine Coupling Constants Extracted from Simulation of the CW EPR Spectra Shown in Figure 2.
| HF coupling constants (MHz) |
|||||
|---|---|---|---|---|---|
| nucleus | A1 | A2 | A3 | Aiso | assignment |
| A1, 1H | 55 | 64.7 | 74.6 | 64.8 | C2–H |
| A2, 1H | 0 | 26 | 5 | 10 | C8–H |
| A3, 14N | 55 | 0 | 0 | 18 | N3 |
| A4, 14N | 19 | 0 | 0 | 6.3 | N1 |
| A5, 13C | 65 | 65 | 65 | 65 | met-Cys |
A subsequent kinetic analysis of formation of this radical (Figure 2, blue diamonds) shows that it forms with a rate constant of ∼1.38 min–1 but does not decay (see Figure S3A for spectra). By contrast, the published paramagnetic intermediate observed in the wt Cfr reaction (Figure 2, dashed line) forms with a rate constant of 1.44 min–1 and decays with a rate constant of 0.96 min–1.25 If C118 in RlmN is required for decay of the paramagnetic intermediate shown in Scheme 1, then it might be expected that C105, the cognate residue in Cfr, would function in a similar capacity in the Cfr-catalyzed reaction. Also shown in Figure 2 (red circles) is the time-dependent formation of a radical observed under turnover conditions with Cfr C105A (see Figure S3B for spectra). The radical forms with a rate constant of ∼0.61 min, but similarly to RlmN C118S, does not decay, as does the radical intermediate in the wt protein.
Figure 2.
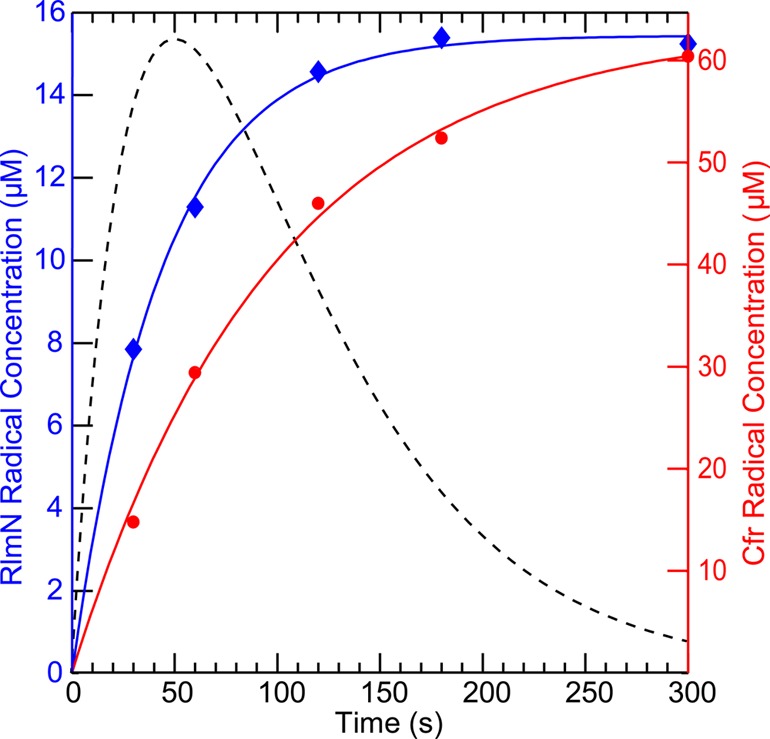
Time-dependent formation of the radical species observed during turnover of RlmNC118S→RCN (blue diamonds) and CfrC105A→RCN (red circles). Time dependence of radical formation was fitted using an A → B kinetic model, affording a rate constant of k = 1.4 min–1 for RlmNC118S→RCN (solid blue line) and k = 0.61 min–1 for CfrC105A→RCN (solid red line). For comparison, the time dependence of the radical formed by wt Cfr during C8 methylation is shown as a dashed black line (concentrations were normalized to the maximum concentration of the RlmN-generated radical). Concentrations were estimated using the EPR signal of flavodoxin semiquinone (37.2 μM) as a standard. Corresponding EPR spectra are presented in the Supporting Information in Figure S2.
To establish that the RlmN-associated paramagnetic signal was derived from a radical species on an adenosine nucleotide, a similar EPR sample was prepared with a 155-mer RNA substrate that was generated via runoff transcription using ATP labeled with deuterium solely at C2 ([2-2H]A-155-mer). This substrate significantly simplified the EPR spectrum, indicating that the largest 1H HF interaction is experienced by the proton on C2 (Figure 1, [2H]). However, despite deuterium incorporation at C2, the sample still exhibited a complex spectral envelope, suggesting the presence of more than one strongly coupled 1H nucleus. By analogy with the m8A substrate radical generated by Cfr under turnover conditions,25 we attempted to simulate the spectrum by including one additional 1H HF coupling and a highly anisotropic 14N HF coupling. Although the fit of the spectra improved dramatically, the shape of the high- and low-field portions of the spectra could not be reproduced completely without overbroadening of the central sharp features. On this basis we included an additional 14N HF coupling, which allowed the complete simulation of the line shape of all the EPR spectra. Table 1 shows the parameters used to simulate the EPR spectra in Figure 1.
To verify that the substrate radical is covalently bound to the mCys residue of RlmN, we prepared RlmNC118S→RCN containing a 13C-labeled mCys residue by overproducing it in its apo form and reacting it with [methyl-13C]SAM after reconstitution, and then used this protein to generate EPR samples under turnover conditions. As shown in Figure 1 ([13C]), the resulting spectrum showed an additional splitting that can be attributed to an isotropic 13C HF coupling of about 64 MHz, resulting in a pseudo 1:2:1 patterned EPR signal. When the [2-2H]A-155-mer RNA substrate was used with the 13C-labeled mCys protein, the spectrum was dramatically simplified, leaving a doublet-like spectrum, owing to the strong 13C coupling (Figure 1, [2H, 13C]).
On the basis of our spectroscopic analysis, we can unambiguously assign the strongest coupled proton to that at C2. The isotropic character of its HF coupling is indicative of sp3 hybridization at this position. The second largest 1H HF coupling is rather anisotropic, which is a typical characteristic of a ring proton.44 Therefore, we are inclined to assign this 1H coupling to the proton on C8. The two 14N couplings have large uncertainties in their HF coupling constants because they were deduced from the broadening of the shoulders in the EPR spectra rather than from actual splittings. Nevertheless, as it is rather apparent from the magnitude of the 1H–C2 and 13C-mCys HF coupling constants, most of the spin density is located on and in the vicinity of C2 of the adenine ring. Thus, we can assign the obtained A3 and A4 HF coupling constants to the N1 and N3 nuclei.
DFT calculations were performed to corroborate the experimental analysis. Similarly to that observed in the paramagnetic intermediate generated during C8 methylation by wt Cfr,25 the spin density of the paramagnetic species produced by RlmNC118S→RCN was found to be delocalized over the adenine ring (see Figure 3). On the basis of the calculated Mulliken spin populations, the larger 14N HF coupling constant (A3) can be assigned to N3 and the smaller one (A4) to N1. Therefore, although the spin density is delocalized, we can formally assign the radical to the N3 position based on the largest contribution to the spin population (Figure 3).
Figure 3.
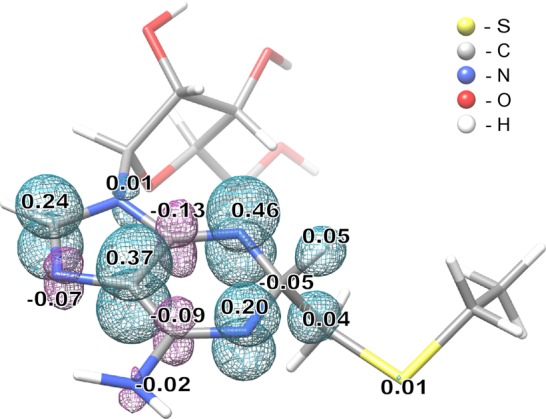
DFT model of the cross-linked substrate radical. Model obtained using an unrestricted B3LYP formalism and a TZVP basis set on all atoms, and including Conductor-like Screening Model (COSMO) as a solvation model. Numbers indicate Mulliken spin populations (only atoms with spin populations above 0.01 are indicated). Cyan and magenta mesh surfaces represent positive and negative spin density distribution, respectively.
Similarly to our observations of the paramagnetic species produced during Cfr-catalyzed C8 methylation, our analysis of the radical produced by RlmNC118S→RCN also shows that the major observed 1H HF interaction is isotropic, which we assign to the proton on the target carbon that undergoes a change in hybridization from sp2 to sp3. One notable difference between the paramagnetic species produced by each of the enzymes is the nature of the second strongest 1H HF coupling, which is assigned to the only other proton directly attached to the adenine ring. In the radical produced by RlmN, this proton would be at C8, while in the radical produced by Cfr, this proton would be at C2. The primarily isotropic nature of the HF coupling of the C2 proton in the radical produced by Cfr departs from what is observed typically for aromatic ring protons; they normally exhibit anisotropic HF coupling.25 This observation suggests that in the C8 radical species, the C2 carbon must be somewhat distorted from planarity. By contrast, in the electronic structure of the radical species observed herein in RlmN-dependent C2 methylation, the C2 hydrogen exhibits strong isotropic HF coupling, while the C8 hydrogen, as expected, exhibits relatively weak anisotropic HF coupling, in line with that typically observed for aromatic ring protons. We note that Cfr can catalyze C2 methylation subsequent to C8 methylation, while RlmN only catalyzes C2 methylation. Therefore, the distorted geometry of the C2–H bond observed in the Cfr-derived radical might be a consequence of Cfr’s dual role.
Crystallographic Characterization of C118A RlmN
To ensure that radical formation in RlmN C118S is due to the absence of a cysteine at position 118 rather than any large-scale structural changes in the active site arising from the substitution, X-ray crystal structures of RlmN C118A were solved in the presence and absence of SAM (Table S3, PDB accession codes 4PL1 and 4PL2, see Figure S4 for representative electron density maps). We used a C118A variant instead of the C118S variant of RlmN for crystallization; however, the EPR spectrum of the radical species is similar in both proteins (Figure S5), indicating that conclusions drawn from the structures of RlmN C118A will translate well to the C118S variant. The X-ray structure of RlmN C118A with SAM (2.60 Å resolution) confirms that the structure of the active site is virtually identical to that of the wt protein (0.3 Å rmsd for 359 Cα atoms) (Figure 4A,C), demonstrating that the variant is correctly configured for catalysis. These observations lend strong support to the proposal that the C118 variant yields an observable radical species in RlmN by blocking its natural decay pathway (Scheme 2).
Figure 4.

A comparison of the active sites in wt and C118A RlmN. X-ray structures of C118A RlmN were determined in the presence (A) and absence (B) of SAM and compared to the analogous wt RlmN structures with (C) and without (D) SAM (PDB accession codes 3RFA and 3RF9, respectively). Selected residues and SAM are shown in stick format and the [4Fe–4S]2+ cluster is shown as a space-filling model. Arrows in panels B and D illustrate conformational changes associated with absence of the SAM cosubstrate.
The X-ray structure of RlmN C118A without SAM (2.20 Å resolution) reveals a dramatic conformational change in a conserved MG(M/I)GE motif near the [4Fe–4S]2+ cluster (Figure 4A,B) when compared to wt RlmN with and without SAM (Figure 4C,D).21 In wt RlmN, this methionine-rich region resides under the Fe/S cluster in a beta strand to loop transition adjacent to C118. In the RlmN C118A structure without SAM, however, the first two residues in the MGMGE sequence shift 4 Å toward the iron–sulfur cluster, partially occupying the SAM binding pocket and making a close approach (∼3.5 Å) to the unique iron site. The MGMGE motif displays a striking capacity for flexibility in this structure, and a subtler version of this conformational change (although occurring in the opposite direction) is observed in the analogous wt RlmN structures (Figure 4C,D). In wt RlmN, the absence of the SAM cosubstrate triggers a rotomer change in C118 and a peptide flip in M176 of the MGMGE motif. As a result, M176 relaxes back into a canonical beta-sheet hydrogen bonding pattern and shifts further away from the iron–sulfur cluster. The conformational changes in the MGMGE sequence in both structures appear to be driven by altered interactions between the carbonyl backbone of M176 and the side chain of C118, suggesting that the structure of the MG(M/I)GE region is highly responsive to the surrounding environment in RlmN and tightly linked to the position of residue 118.
Conclusion
In our original working hypotheses for RlmN- and Cfr-dependent methylation of A2503 in rRNA, electron transfer from the paramagnetic protein–nucleic acid cross-linked intermediate was proposed to occur before proton abstraction, and resolution of the cross-linked species was proposed to occur solely by a polar process, involving disulfide-bond formation between two active-site cysteines. The proposed role for C355 of RlmN (C338 of Cfr) as the lynchpin in catalysis is bolstered by (i) high-resolution mass spectrometry and X-ray crystallography,21 wherein a methyl group was shown to be attached to the sulfur atom of C355 of AI RlmN; (ii) deuterium transfer experiments,14 wherein the 5′-dA· was shown to abstract a H· from the mCys residue, and not to abstract a H· from C2 or C8 of the nucleotide substrate; and (iii) EPR studies of Cfr,25 in which the methylene carbon of a formerly [methyl-13C]mCys338 residue was found to exhibit substantial HF coupling with the delocalized spin on the adenine ring of the substrate, indicating that it was covalently attached to the nucleotide base. By contrast, the exact role of the second active-site cysteine residue (C118 in RlmN and C105 in Cfr) is less clear. Our studies,14 as well as studies by McCusker et al.,26 suggest that this residue plays a role in resolution of the cross-linked species; its substitution in RlmN with Ser or Ala residues leads to generation of a stable RlmN–nucleic acid cross-link species, which is unable to advance to the designated product.14,26 Our observation of a paramagnetic protein–nucleic acid cross-linked species during the reaction of RlmN C118S/A but not the wt protein suggests that loss of the electron from this species is gated by proton abstraction, and that C118 of RlmN participates somehow in this process (Scheme 2). Furthermore, proton abstraction before electron loss would allow for resolution of the cross-linked species via a radical mechanism rather than a polar mechanism, similar to the mechanism by which the cross-linked species is formed. Moreover, a radical fragmentation mechanism for resolution of the cross-linked species is consistent with the observations of McCusker et al., in which the addition of external thiols to an isolated RlmN C118S protein–nucleic acid cross-linked species did not lead to resolution of the linkage. In one possible scenario, C118 could be the base that abstracts the C2 proton; its substitution with an incompatible amino acid would therefore inhibit decay of the radical species, allowing it to be detected (Scheme 2A). A second possibility is that another active-site base abstracts the C2 proton of the paramagnetic protein–nucleic acid cross-linked species to yield a radical anion, and the resulting species undergoes radical fragmentation to a C355/C118 disulfide radical (anion) (Scheme 2B). Delivery of an electron from the Fe/S cluster or possibly directly from flavodoxin regenerates the two active-site thiols and readies the enzyme for another round of methyl transfer to generate the active-site mCys residue. A third possibility is that C118 acts both as the base and the radical disulfide (anion) partner (Scheme 2C).
All steps leading up to the cross-linked radical intermediate are based on strong experimental evidence. By contrast, the mechanism by which the cross-linked radical intermediate decays to product is not well understood. It is well-known that carbon-radical addition to heteroaromatic species is accompanied by protonation of the target, as in the Minisci reaction,24,45 and it is likely that RlmN and Cfr activate their substrates in this manner. Our spectroscopic and DFT studies of the radical species observed in Cfr turnover indicate that the N7 nitrogen is not protonated in this intermediate; however, hydrogen bonding to N1 and/or N3 of the substrate would be expected to polarize the adenine base sufficiently for radical addition. Deprotonation at C8 or C2 (RlmN) of the cross-linked intermediate appears to occur before its fragmentation; however, it is well documented that the acidity of carbon acids in cation-radical species is greatly enhanced over their closed-shell counterparts, such as in the toluene cation-radical, which has a calculated pKa value of −11.46 Moreover, formation of neutral radicals also significantly depresses pKa values of adjacent or allylic carbon acids, as has been shown for an enoxy radical intermediate during catalysis by 2-hydroxy-4-methylpentanoyl-CoA dehydratase, in which the acidity of the carbon acid in the radical species is depressed over 25 orders of magnitude compared to its closed-shell counterpart.47
Whether radical fragmentation of the resulting radical anion species affords a thiyl radical or a disulfide-radical anion is not clear; both species have been observed in enzymatic catalysis—most notably in the reaction catalyzed by ribonucleotide reductase.48 However, thiyl radicals are strong oxidants, whereas disulfide radical anions have both reducing and oxidizing properties depending on their protonation states or the environment in which they reside.49 At present, we favor a scenario in which the second active-site cysteine functions as a base and the cross-linked species is resolved via a radical fragmentation mechanism to generate a thiyl radical that is subsequently reduced by one electron.
Acknowledgments
This work was supported by NIH Grants GM101957 (S.J.B.) and GM101390 (M.T.G), Tobacco Settlement Funds (TSF13/14 SAP 4100062216) to S.J.B., and a Pathway to Independence award to A.K.B. Use of the Advance Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory was supported by the U.S. DOE contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Supporting Information Available
Tables S1–S3 and Figures S1–S5. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ A.S., T.L.G., and M.I.R. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Giessing A. M. B.; Jensen S. S.; Rasmussen A.; Hansen L. H.; Gondela A.; Long K. S.; Vester B.; Kirpekar F. RNA 2009, 15, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminska K. H.; Purta E.; Hansen L. H.; Bujnicki J. M.; Vester B.; Long K. S. Nucl. Acids. Res. 2010, 38, 1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrenberg C.; Schwarz S.; Jacobsen N. E.; Hansen L. H.; Vester B. Mol. Microbiol. 2005, 57, 1064. [DOI] [PubMed] [Google Scholar]
- Long K. S.; Poehlsgaard J.; Kehrenberg C.; Schwarz S.; Vester B. Antimicrob. Agents Chemother. 2006, 50, 2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz S.; Werckenthin C.; Kehrenberg C. Antimicrob. Agents Chemother. 2000, 44, 2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh S.-M.; Xiong L.; Bae T.; Mankin A. S. RNA 2008, 14, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N.; Freborn B.; Nissen P.; Penczek P.; Grassucci R. A.; Sweet R.; Frank J.; Moore P. B.; Steitz T. A. Cell 1998, 93, 1105. [DOI] [PubMed] [Google Scholar]
- Harms J.; Schluenzen F.; Zarivach R.; Bashan A.; Gat S.; Agmon I.; Bartels H.; Francheschi F.; Yonath A. Cell 2001, 107, 679. [DOI] [PubMed] [Google Scholar]
- Schuwirth B. S.; Borovinskaya M. A.; Hau C. W.; Zhang W.; Vila-Sanjurjo A.; Hoton J. M.; Cate J. H. D. Science 2005, 310, 827. [DOI] [PubMed] [Google Scholar]
- Selmer M.; Dunham C. M.; Murphy F. V. IV; Weislbaumer A.; Petry S.; Kelley A. C.; Weir J. R.; Ramakrishnan V. Science 2006, 313, 1935. [DOI] [PubMed] [Google Scholar]
- Yan F.; LaMarre J. M.; Röhrich R.; Wiesner J.; Jomaa H.; Mankin A. S.; Galoníc Fujimori D. J. Am. Chem. Soc. 2010, 132, 3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Laslop N.; Ramu H.; Klepacki D.; Mankin A. S. EMBO J. 2010, 29, 3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benítez-Páez A.; Villarroya M.; Armengod M.-E. RNA 2012, 18, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove T. L.; Benner J. S.; Radle M. I.; Ahlum J. H.; Landgraf B. J.; Krebs C.; Booker S. J. Science 2011, 332, 604. [DOI] [PubMed] [Google Scholar]
- Yan F.; Fujimori D. G. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker S. J.; Grove T. L. F1000 Biol. Rep. 2010, 2, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey P. A.; Booker S. J. Adv. Protein Chem. 2001, 58, 1. [DOI] [PubMed] [Google Scholar]
- Sofia H. J.; Chen G.; Hetzler B. G.; Reyes-Spindola J. F.; Miller N. E. Nucleic Acids Res. 2001, 29, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs C.; Broderick W. E.; Henshaw T. F.; Broderick J. B.; Huynh B. H. J. Am. Chem. Soc. 2002, 124, 912. [DOI] [PubMed] [Google Scholar]
- Walsby C. J.; Hong W.; Broderick W. E.; Cheek J.; Ortillo D.; Broderick J. B.; Hoffman B. M. J. Am. Chem. Soc. 2002, 124, 3143. [DOI] [PubMed] [Google Scholar]
- Boal A. K.; Grove T. L.; McLaughlin M. I.; Yennawar N. H.; Booker S. J.; Rosenzweig A. C. Science 2011, 332, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vey J. L.; Drennan C. L. Chem. Rev. 2011, 111, 2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove T. L.; Radle M. I.; Krebs C.; Booker S. J. J. Am. Chem. Soc. 2011, 133, 19586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minisci F. Synthesis 1973, 1973, 1. [Google Scholar]
- Grove T. L.; Livada J.; Schwalm E. L.; Green M. T.; Booker S. J.; Silakov A. Nat. Chem. Biol. 2013, 9, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker K. P.; Medzihradszky K. F.; Shiver A. L.; Nichols R. J.; Yan F.; Maltby D. A.; Gross C. A.; Galonić Fujimori D. J. Am. Chem. Soc. 2012, 134, 18074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwig D. F.; Booker S. J. Biochemistry 2004, 43, 13496. [DOI] [PubMed] [Google Scholar]
- Lanz N. D.; Grove T. L.; Gogonea C. B.; Lee K. H.; Krebs C.; Booker S. J. Methods Enzymol. 2012, 516, 125. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Montgomery J. J. A.; Vreven T.; Kudin K. N.; Burant J. C.; Millam J. M.; Iyengar S. S.; Tomasi J.; Barone V.; Mennucci B.; Cossi M. S. G.; Rega N.; Petersson G. A.; Nakatsuji H.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Klene M.; Li X.; Knox J. E.; Hratchian H. P.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Ayala P. Y.; Morokuma K.; Voth G. A.; Salvador P.; Dannenberg J. J.; Zakrzewski V. G.; Dapprich S.; Daniels A. D.; Strain M. C.; Farkas O.; Malick D. K.; Rabuck A. D.; Raghavachari K.; Foresman J. B.; Ortiz J. V.; Cui Q.; Baboul A. G.; Clifford S.; Cioslowski J.; Stefanov B. B.; Liu G.; Liashenko A.; Piskorz P.; Komaromi I.; Martin R. L.; Fox D. J.; Keith T.; Al-Laham M. A.; Peng C. Y.; Nanayakkara A.; Challacombe M.; Gill P. M. W.; Johnson B.; Chen W.; Wong M. W.; Gonzalez C.; Pople J. A.; Gaussian 03, Revision E.01; Gaussian, Inc.: Wallingford, CT, 2004.
- Becke A. D. Phys. Rev. A 1988, 38, 3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Wang Y. Phys. Rev. B 1988, 38, 12228. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. J. Chem. Phys. 1994, 100, 5829. [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]
- Lee C. T.; Yang W. T.; Parr R. G. Phys. Rev. B 1988, 37, 785. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schuurmann G. J. Chem. Soc., Perkin Trans. 2 1993, 799. [Google Scholar]
- Barone V.; Cossi M. J. Phys. Chem. A 1998, 102, 1995. [Google Scholar]
- Otwinowski Z.; Minor W. Methods Enzymol. 1997, 276, 307. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Acta Crystallogr. 1997, D53, 240. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Acta Crystallogr. 2004, D60, 2126. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. Acta Crystallogr. 2010, D66, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Acta Crystallogr. 2004, D60, 2256. [DOI] [PubMed] [Google Scholar]
- Ten Eyck L. F. Methods Enzymol. 1985, 115, 324. [DOI] [PubMed] [Google Scholar]
- Yan F.; Fujimori D. G. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil J. A.; Bolton J. R.; Wertz J. E.. Electron Paramagnetic Resonance. Elementary Theory and Practical Applications; John Wiley & Sons, Inc.: New York, 1994. [Google Scholar]
- Minisci F.; Citterio A.; Perchinunno M.; Bertini F. Gazz. Chim. Ital. 1975, 105, 1083. [Google Scholar]
- Bordwell F. G.; Bausch M. J. J. Am. Chem. Soc. 1986, 108, 2473. [DOI] [PubMed] [Google Scholar]
- Smith D. M.; Buckel W.; Zipse H. Angew. Chem., Int. Ed, 2003, 42, 1867. [DOI] [PubMed] [Google Scholar]
- Lawrence C. C.; Bennati M.; Obias H. V.; Bar G.; Griffin R. G.; Stubbe J. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surdhar P. S.; Armstrong D. A. J. Phys. Chem. 1987, 91, 6532. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.