

Abstract

We describe a strategy for rendering peptides resistant to proteolysis by formulating them as high-density brush polymers. The utility of this approach is demonstrated by polymerizing well-established cell-penetrating peptides (CPPs) and showing that the resulting polymers are not only resistant to proteolysis but also maintain their ability to enter cells. The scope of this design concept is explored by studying the proteolytic resistance of brush polymers composed of peptides that are substrates for either thrombin or a metalloprotease. Finally, we demonstrate that the proteolytic susceptibility of peptide brush polymers can be tuned by adjusting the density of the polymer brush and offer in silico models to rationalize this finding. We contend that this strategy offers a plausible method of preparing peptides for in vivo use, where rapid digestion by proteases has traditionally restricted their utility.
Introduction
The biocompatibility and ease of programming of peptides have long inspired their development as therapeutics,1−4 signaling agents,5 and sensors.6 However, significant problems limit their use in vivo, including short durations of activity resulting from rapid digestion by endogenous proteases and efficient renal clearance due to their generally low molecular weights.1−4 Proteolytic digestion of circulating peptides can be rapid, occurring with half-lives of less than a few minutes, owing to the abundance of active proteases in both serum and tissues.4 The greatest threats to peptide integrity are found in the lumen of the small intestine, which contains gram quantities of proteases secreted by the pancreas (i.e., α-chymotrypsin, trypsin, and carboxypeptidases), as well as in the brush border membrane of epithelial cells, which houses some 15 peptidases that together cleave amide bonds in peptides and proteins with little specificity.4,7 In practice, unmodified therapeutic peptides are typically directly injected at the site of interest to minimize proteolytic degradation, and many are used only as last-resort, salvage treatments in patients with multidrug resistant afflictions.4 Harnessing the inherent specificity, affinity, and low immunogenicity of peptides in therapeutic and diagnostic applications will require the development of simple, widely applicable, and easy-to-access methods that protect active peptides from proteolysis, but do not hinder their function.
Traditional strategies for limiting enzymatic degradation involve chemical modification of the peptide, including the incorporation of unnatural amino acids (e.g., d-amino acids),8−13 terminal capping via acetylation of the N-terminus or amidation of the C-terminus,14 introduction of backbone modifications such as N-methylation,15−17 use of stabilizing linkers,18−21 cyclization,22−24 and conjugation to polyethylene glycol (PEG).25−29 Hence, chemistries are chosen such that peptides are no longer recognized by, or become inaccessible to, the active site of a proteolytic enzyme. However, because these strategies modify the connectivity, or amino acid identity of the peptide, they can reduce its bioactivity, often necessitating multiple rounds of structure–function studies to restore the activity of the material.30 Strategies that do not require direct modification of the peptide chemical structure typically involve manipulation of their three-dimensional spatial arrangement via chemical conjugation of the peptide to a higher molecular weight structure. Architectures of this type include peptide–polymer conjugates31 or systems involving the display of multiple copies of the peptide on a small molecule scaffold.32−34 However, in practice syntheses of these materials often require multiple conjugation and purification steps, or the preparation of complicated scaffolds that are not generalizable or conveniently deployed.
We present a new methodology for protecting active peptides from proteolysis by packaging them into high-density brush polymers via ring opening metathesis polymerization (ROMP), using an easily prepared initiator. This strategy is inspired by observations we have made in our laboratory over several years, involving the behavior of brush polymers that result from the graft-through polymerization of norbornyl-peptide monomers via ROMP. Specifically, we have observed that polymerization can result in structures that resist proteolysis relative to their monomeric analogues.35,36 Given these observations, we hypothesized that polymerized peptides, while protected from proteolysis, would maintain their intended biological function and that this phenomenon might be a general feature of peptides arranged in this manner. If so, such an approach could provide a general, accessible route to the development of proteolytically resistant peptide displays capable of performing the functions inherent to the peptide, such as binding a receptor or ligand, initiating a signaling pathway, penetrating a cell, or inducing a therapeutic effect. We envision that such a strategy will provide a feasible route for the preparation and delivery of peptides in future therapeutic applications, where the peptide is active on the polymer or is released at a given place and time via a cleavable linkage.
To test the core concept, we prepared two canonical cell-penetrating peptides (CPPs), Tat (YGRKKRRQRRR)37−39 and Arg8 (RRRRRRRR),40−42 as modified norbornene monomers and polymerized them to generate brush polymers via ROMP. There is a long history of appending peptides with interesting functional properties, such as an ability to penetrate cells43−45 or a propensity for localizing inside a cellular nucleus,46−48 directly to a material of interest in order to bestow that same property on the new material. Indeed, CPPs have been used in this regard and have been shown to effectively penetrate cells when chemically conjugated to small molecule drugs, therapeutic peptides, and other structures.43−45 While effective for model studies, the downside to this strategy is that CPPs (and all other targeting peptides composed of naturally occurring amino acids) are generally still susceptible to proteolytic degradation, when conjugated as individual, linear peptide sequences.45 We reasoned that a CPP organized as a high-density brush polymer might be resistant to proteolysis but still able to efficiently penetrate and carry a cargo into cells. Here, we show that polymerized Tat and Arg8 are at least as efficient at penetrating cells as individual CPPs. Additionally, we generate ∼10–50 nm diameter spherical micellar assemblies of a CPP-containing brush polymer and demonstrate that this formulation also penetrates cells efficiently. Importantly, both polymer and particle formulations of the CPPs are resistant to proteolysis under conditions that freely degrade the peptides. Therefore, in a critical demonstration of this concept, the materials still penetrate cells after exposure to multiple proteases unlike the standard CPPs, which are inactivated by cleavage.
The generality of this approach was tested by polymerizing two additional peptide substrates for two different classes of enzymes: a matrix metalloprotease (MT1-MMP) and a serine protease (thrombin). These studies further confirm that polymerized peptides are more resistant to proteolytic digestion than their monomeric analogues. We conjectured that this resistance is derived from the high packing density of the peptides in the polymer brush and we tested this concept through both experimental and computational analyses of blend copolymers with varying densities of peptide. Computational studies validated experimental findings and supported the notion that copolymerization of the peptide monomer with a monomer containing a short oligoethylene glycol (OEG) sequence alleviates steric congestion, and potentially noncovalent interactions among the peptide strands. These phenomena otherwise prevent access to the active site of a protease, and are features that typically impart proteolytic resistance to globular protein structures. Together, these data suggest that bioactive peptides can be packaged as polymers to attenuate proteolytic degradation in a tunable fashion, and, since the identity of the amino acid sequence is unaltered, the inherent function of the peptide will, in many cases, be preserved.
Results and Discussion
Polymerization of Cell Penetrating Peptides
Validation of the proteolytic resistance and bioactivity of polymerized CPPs required preparation of well-defined brush polymers with low dispersity via a living polymerization method. High-density brush polymers of known cell-penetrating peptides, Tat and Arg8, together with appropriate control polymers, were prepared via living ROMP by a popular initiator, ((H2IMES)(pyr)2(Cl)2Ru=CHPh)49 (1, Figure 1). ROMP by this initiator was selected for preparation of these materials for a variety of reasons. First, the initiator exhibits fast initiation and slower propagation kinetics, which typically afford polymers with exceptionally narrow molecular weight distributions. Second, it is highly functional group tolerant, enabling the incorporation of a wide range of chemical functionality via polymerization of groups pendant to a norbornene moiety, including fluorophores,50 drugs,51−53 sugars,54 oligonucleotides,55 and peptides.35,56−59 We note that very few polymerization techniques have been shown to incorporate peptides directly by graft-through polymerization from a peptide-containing monomer. Reports on graft-through polymerization of peptides by reversible addition–fragmentation chain transfer (RAFT)60−63 or free-radical polymerizations64−66 describe only blend polymers with less than 50% incorporation of peptides. Additionally, the polymers produced by these methods generally have broader molecular weight distributions than those typically afforded by ROMP.35 Furthermore, a high degree of functionality and complexity can be readily generated on a single polymer via ROMP by preparing multiblock copolymers of appropriately functionalized norbornene monomers or via the use of chain transfer agents to end-label a polymer through a single cross metathesis event upon complete consumption of monomers.
Figure 1.

Synthetic routes for the polymerization of cell penetrating polymers and controls. Routes to the preparation of (A) homopolymers and (B) block copolymers. Note that the guanidinium moiety and Arg8 peptide are polymerized with protecting groups and deprotected after polymerization by treatment of the polymers with a TFA solution. For each polymer, m and n are the degrees of polymerization (DPs) given in Table 1. See the Experimental Section and Supporting Information for synthetic details.
Peptides used to generate ROMP monomers were prepared by solid phase peptide synthesis (SPPS) using standard fluorenylmethyloxycarbonyl (FMOC) chemistry. Peptide monomers were prepared by coupling a carboxylic acid-modified norbornene to the N-terminus of the desired peptide sequence on resin. We have shown previously that peptide monomers with side chain protecting groups and a five-carbon linker between the peptide and the norbornene unit generally polymerize at faster rates than those with shorter linkers or those that possess no protecting groups.35 Therefore, we prepared all peptide monomers with this linker and used an Arg8 monomer with side chains protected. However, efforts to prepare and polymerize the protected Tat peptide were thwarted by poor solubility of the protected material in solvents compatible with the ROMP initiator. Therefore, the Tat peptide monomer was prepared without protecting groups (see Figure S1 in the Supporting Information for chemical structures of monomers). All peptides that were incorporated into polymers were also separately prepared (without norbornyl-groups) as fluorescein-labeled peptides via conjugation of 5/6-carboxyfluorescein to the ε-amino group of an N-terminal lysine, for use as controls to evaluate the cellular uptake efficiency of the peptides alone versus polymerized materials. (See Figure S2–S3 and Table S1 for characterization data of the peptide controls.)
Each peptide-based monomer was polymerized, and the resulting polymers were end-labeled with fluorescein to enable tracking of the uptake of the material and to serve as a model cargo (Figure 1A). In addition to polymers containing the canonical Tat and Arg8 CPPs, several control polymers were prepared, including a polymer of an uncharged, nonpeptide unit of oligoethylene glycol (OEG) and another consisting of a peptide side chain that did not contain any charged residues (GSGSG). For comparison, a polymer composed of monomers bearing a single guanidinium moiety67 was prepared as a graft-through analogue of polymers prepared via the graft-to technique employed in other studies.68,69 We note that these graft-to guanidinium-containing polymers are the only other cell penetrating polymers prepared by ROMP techniques that have been reported to date. The graft-to guanidinium polymer prepared in this work displayed poor solubility as a homopolymer and was therefore prepared as a block copolymer with a water-solubilizing OEG monomer (Figure 1B, where R1 = OEG and R2 = guanidinium). After polymerization, the polymers were characterized by size-exclusion chromatography with multiangle light scattering (SEC-MALS) to ascertain degree of polymerization (DP) and molecular weight distribution (dispersity or Mw/Mn) (Table 1 and chromatograms in Figure S4). Good agreement between the obtained DP and the theoretical DP based on the initial monomer-to-initiator ratio ([M]0/[I]0) was observed. Further, all dispersities were less than 1.11, indicating the expected narrow molecular weight distributions.
Table 1. Characterization of Cell Penetrating Polymer and Controlsa.
| block m |
block n |
|||||
|---|---|---|---|---|---|---|
| polymer side chain identity (R1, R2 groups) | Mnb | Mw/Mnc | DP (m)d | Mnb | Mw/Mnc | DP (n)d |
| OEG | 3 600 | 1.026 | 10 (10) | – | – | – |
| GSGSG | 5 700 | 1.049 | 10 (12) | – | – | – |
| Tat | 8 600 | n/a | 5 (6) | – | – | – |
| Arg8 | 36 000 | 1.08 | 8 (6) | – | – | – |
| OEG-b-Guanidinium | 8 300 | 1.016 | 23 (15) | 12 000 | 1.07 | 8 (12) |
| Phenyl-b-GSGSGe | 14 000 | 1.021 | 54 (70) | 18 000 | 1.021 | 8 (12) |
| Phenyl-b-Tate | 13 000 | 1.013 | 52 (70) | 22 000 | 1.11 | 5 (6) |
Block m and n refer to the first and second block to be polymerized as shown in Figure 1. Each polymer is named according to the identity of the monomer polymerized as drawn in Figure 1. Block copolymers are listed with block m first and block n second.
Number-average molecular weight from light scattering.
The dispersity of each block.
Experimentally determined degree of polymerization for block m and block n as denoted in Figure 1B, with theoretical values based on the amount of material used, in parentheses.
Amphiphilic polymers were formulated into nanoparticles, denoted in the text as the GSGSG Particle and Tat Particle. All data were obtained by SEC-MALS, except for those describing the Tat polymer, which did not elute on the SEC column and was instead characterized in a cuvette using batch-mode static light scattering. Without the SEC component, no information on the molecular weight distribution of this polymer was obtained. However, the amphiphilic polymer that contains Tat (Phenyl-b-Tat) eluted well on SEC and yielded close to the predicted DP in low dispersity. Repeated polymerizations of peptide-containing homopolymers yield consistent numerical values. The SEC-MALS chromatograms for each polymer are provided in Figure S4.
The Tat peptide-containing homopolymer, lacking side-chain protecting groups, performed poorly on SEC-MALS, presumably due to unfavorable interactions of the peptide with the size exclusion column. Therefore, no information on the molecular weight distribution of these polymers was obtained, but a molecular weight determination was achieved by measuring the bulk light scattering of the solution in a cuvette (without the size exclusion column); and complete consumption of the Tat monomer after polymerization was verified by 1H NMR (Figure S5).
Given the complexity of the Tat and Arg8 peptide-containing polymers (i.e., multiple charged and nucleophilic side chains), we investigated whether ROMP of these materials proceeds in a living fashion, in order to ensure that well-defined and well-ordered structures, devoid of cross-metathesis or premature termination, could regularly be accessed by this strategy. Confirming the living nature of the polymerization, a plot of Mn (obtained by SEC-MALS) vs [M]0/[I]0 for the Arg8 monomer yields a linear fit for [M]0/[I]0 less than 9 (Figure 2A and Table 2). At larger [M]0/[I]0 ratios, propagation ceased, presumably due to steric hindrance encountered from assembling multiple copies of the long, side-chain protected peptide sequence, whose molecular weight as a monomer is 3.5 kDa. A similar plot was obtained from data gathered for polymerization of the Tat polymer, collected by static light scattering (SLS) in a cuvette (Figure 2B and Table 2). Therefore, we conclude that both CPP monomers are polymerized in a living fashion to a DP of <9, despite the complexity and functionality of their side chains, making this an exceptionally convenient strategy for predictably generating polymeric architectures from peptide monomers.
Figure 2.

Plots correlating the number-average molecular weight (Mn) with the initial monomer–to-catalyst ratio ([M0/I0] for the polymerization of the Arg8 monomer (A) and the Tat monomer (B). Linear fits are indicative of a living polymerization. For both monomers, propagation ceased after the polymerization of ∼9 monomers.
Table 2. Characterization of the Polymerization of the Tat and Arg8 Monomers at Multiple Initial Monomer-to-Catalyst Ratiosa.
| Arg8 polymerization | |||
|---|---|---|---|
| [M]0:[I]0b | Mnc | DPd | Mw/Mne |
| 5 | 11 000 | 3 | 1.05 |
| 10 | 20 000 | 6 | 1.05 |
| 15 | 30 000 | 9 | 1.03 |
| 20 | 27 000 | 8 | 1.05 |
| 40 | 31 000 | 9 | 1.07 |
| 60 | 23 000 | 6 | 1.08 |
| Tat polymerization | |||
|---|---|---|---|
| [M]0:[I]0b | Mnc | DPd | Mw/Mne |
| 5 | 6 400 | 4 | n/a |
| 10 | 9 600 | 5 | n/a |
| 15 | 15 000 | 8 | n/a |
| 20 | 14 000 | 8 | n/a |
| 40 | 16 000 | 9 | n/a |
| 60 | 15 000 | 8 | n/a |
The polymers listed are all homopolymers as shown in Figure 1a.
The initial monomer-to-catalyst ratio used.
The number-average molecular weight obtained.
The degree of polymerization obtained.
The dispersity of polymers. All data for the Arg8 monomer polymerization were collected by SEC-MALS. Data for the Tat monomer polymerization were obtained in a cuvette via static light scattering. The data reported represent values from a single batch of polymerizations performed on the same day.
In addition to exploring the activity of a single polymer chain, we also aimed to examine the proteolytic resistance and bioactivity of large assemblies of peptide-containing polymers. We envisioned that nanoscale assemblies of multiple peptide–polymers would be large enough to avoid renal clearance thresholds in future applications that might otherwise prevent long circulation times of peptides or lower molecular weight polymers, but it was unclear whether these large assemblies would resist proteolysis, or enter cells. To generate nanoparticles, we prepared amphiphilic polymers of two peptides: Tat and a GSGSG control peptide (Figure 1B. Note: Arg8-based nanoparticles could not be generated with consistently spherical morphologies matching GSGSG controls or Tat systems and hence were excluded from this study). The design of these amphiphiles is such that the phenyl-modified norbornene monomers operate as hydrophobic moieties to drive self-assembly of the amphiphiles into micellar nanoparticles containing many copies of polymer. To prepare these amphiphiles, a hydrophobic monomer was polymerized to completion prior to addition of the peptide monomer to the living polymer (Figure 1B, where R1 is phenyl and R2 is GSGSG or Tat). Self-assembly of these amphiphilic polymers into a nanoscale structure was then accomplished by slow dialysis of the material from an organic cosolvent, in which the amphiphile is completely dissolved (DMF), into a selective solvent, in which only the peptide brush is soluble (aqueous phosphate-buffered saline, PBS). The amphiphilic polymers of Tat and GSGSG were found, by DLS and TEM, to form spherical micelles of ∼10−50 nm diameter (Figure 3).
Figure 3.

Characterization of Tat and GSGSG particles. TEM images and DLS data for the Tat particle (A,C) and the GSGSG particle (B,D). Scale bars are 200 nm.
Assessing Cellular Uptake in HeLa Cells by Flow Cytometry and Live-Cell Confocal Microscopy
Fluorescence-based in vitro assays were performed in HeLa cells to compare the cellular uptake of the peptide controls, polymers, and nanoparticles. The goal was to determine whether polymerization of the CPPs had an impact on their ability to facilitate cellular entry or on their mechanisms of cellular uptake. In these studies, flow cytometry was used to quantify the amount of cellular uptake, and live-cell confocal microscopy was used to verify internalization and examine the localization of the internalized material.
In flow cytometry experiments, relative to the vehicle control (PBS), all CPP-containing peptide polymers and particles gave robust fluorescent counts, approximately 2-fold higher than those of the peptides alone (Figure 4 and individual histograms in Figures S6–S8). These data verify that CPPs maintain or have enhanced function when incorporated into brush polymers or larger polymeric assemblies. We also note that the Tat and Arg8 polymers gave responses similar to those of the guanidinium polymer, which is an analogue of the only other cell-penetrating ROMP polymer reported to date.
Figure 4.

Quantitative comparison of cellular uptake of peptides, polymers and particles at 2.5 μM after 30 min incubation with HeLa cells by flow cytometry. On the y-axis, normalized mean fluorescence refers to the mean fluorescence counts detected for the material divided by the mean fluorescence counts exhibited by the vehicle control (PBS). Representative histograms showing the fluorescence counts for each material are given in Figures S6–S8.
To probe whether the cellular uptake of the polymerized materials was due to the peptide amino acid sequence and not the polymer backbone itself, or the result of the arrangement of any peptide into a brush polymer, we investigated the uptake of polymeric materials containing an OEG brush and a GSGSG brush, both of which do not enter cells as their monomer units. The control materials showed negligible fluorescence signals (less than a 2-fold increase in fluorescence relative to vehicle), similar to the small molecule fluorescein tag itself. Therefore, these data indicate that the amino acid sequences of Tat and Arg8 drive the internalization of the polymers.
It was also important to confirm that the fluorescence observed in the initial studies resided within the cytoplasm, rather than on the cell’s external surface. To this end, we chose to perform live-cell confocal microscopy, because fixation of cells by formaldehyde, methanol or other agents, can cause artifacts due to the release of fluorescently labeled materials entrapped in endosomes.70,71 In particular, we performed Z-stack analyses at 1 μm step sizes on live cells treated with each peptide-based material. Across multiple Z-slices for cells treated with all Tat-, Arg8- and guanidinium- containing materials, at the same concentration used in flow cytometry experiments, a combination of punctate and diffuse fluorescence was observed (Figure 5, for individual Z-slices, see Figures S9–S11), indicative of compartmentalized and cytosolic localization, respectively. By contrast, no fluorescence was seen for any of the negative controls (GSGSG and OEG polymers), which do not contain cationic moieties and do not penetrate cells in any detectable manner.
Figure 5.
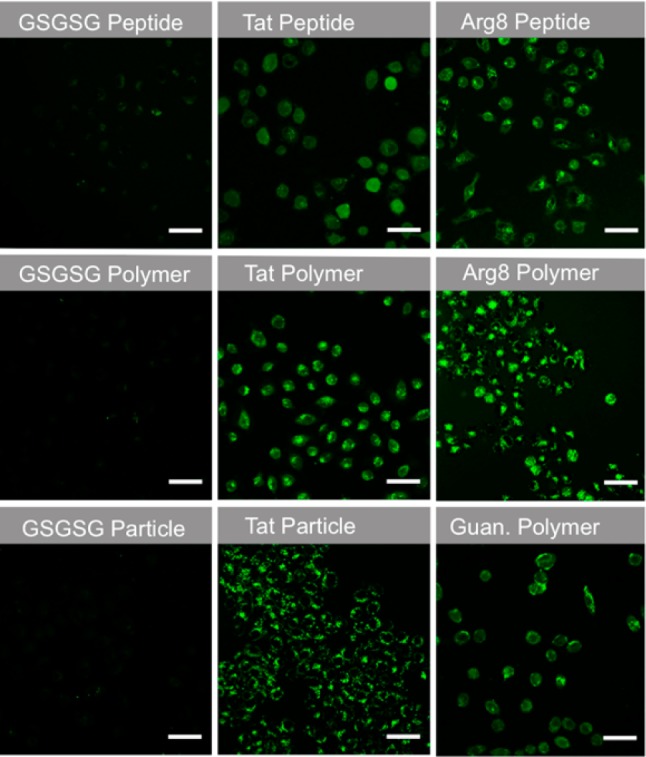
Live-cell confocal images of peptides, polymers and nanoparticles labeled with fluorescein. Images are the average maximum intensity from six consecutive 1 μm slices. Scale bars are 50 μm. For the Tat variants, the six individual Z-slices that were used for the averaged images shown in this figure are provided in Figures S9–S11.
We also verified that polymerization did not render the peptides toxic to cells. The viability of cells treated with the Tat and GSGSG peptide, polymer and nanoparticles was assayed via the CellTiter-Blue assay. When compared to the vehicle control, HeLa cells treated with all formulations of the materials at 5 μM, twice the concentration used in the uptake studies described above, remained >92% viable for 48 h (Figure S12).
Exploring the Mechanism of Cellular Uptake
There is much debate in the literature over the mechanism of entry of CPPs. However, it is generally agreed that the cellular uptake of these materials requires association with anionic species at the cell membrane (i.e., sulfated proteoglycans or phospholipid polar headgroups) followed by internalization via endocytosis or membrane disruption.72,73 To investigate whether the monomeric, polymeric, and nanoparticle formulations of the CPPs follow similar internalization routes, we subjected cells to thermal inhibition and common pharmacological compounds that disrupt different aspects of membrane trafficking and endocytosis.
First, membrane trafficking was arrested74,75 by reducing the incubation temperature to 4 °C. This resulted in a dramatic decrease in the fluorescent signals for the Tat, Arg8, and guanidinium polymers and nanoparticles by flow cytometry, but had no influence on the values from the GSGSG controls (Figure 6A). Similar effects were seen with an inhibitor of dynamin-dependent endocytosis (dynasore)76 and also with methyl-β-cyclodextrin (MβCD),77−79 an agent known to remove membrane cholesterol, and thereby alters the fluidity of the membrane. Each condition resulted in no change in the fluorescence values obtained for the GSGSG controls, which is consistent with the notion that these uncharged materials do not internalize. The polymers containing guanidinium, Tat and Arg8 side chains all showed uptake, as is expected for these modes of inhibition. Note that, in all cases, pharmacological and thermal inhibition exhibit only a small effect on the flow cytometry readings of the Tat and Arg8 peptides. This behavior for CPP-type peptides has been observed by others. Specifically, Dowdy and co-workers have suggested that the apparent temperature-independence of peptide uptake is due to false-positive readings resulting from membrane-bound, noninternalized peptides.80 To circumvent this problem, the cells could be washed with heparin, a polyanionic saccharide, which competes with the cell membrane for binding of the polycationic CPPs. In the Dowdy work, heparin washes significantly reduced the aberrant signals seen in flow cytometry recordings during incubation at 4 °C, but did not completely abolish them, as observed in our studies (Figure 6A, Tat and Arg8 peptide data). Together, these results suggest that cell penetration of the peptides (individual CPPs) is due, in part, to membrane disruption or endocytotic processes and that these mechanisms of entry are maintained or enhanced upon polymerization.
Figure 6.
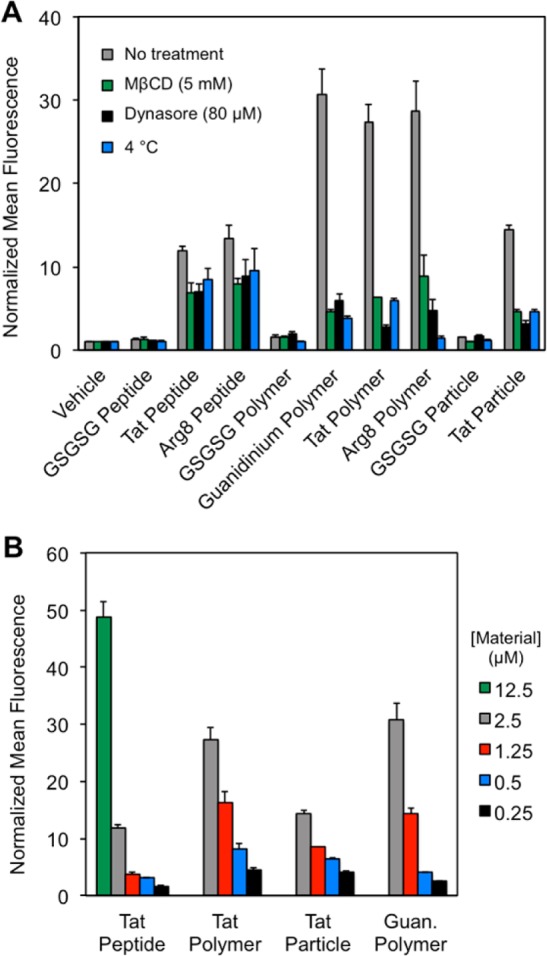
Experiments probing the mechanism of cellular entry. (A) Pharmacological and thermal probes of cellular uptake mechanisms. HeLa cells were pretreated with MβCD (9.5 mM) or dynasore (80 μM) for 30 min prior to incubation with the material of interest or preincubated at 4 °C. (B) Concentration dependence of the cellular uptake of key materials. All reported flow cytometry data are described as a fold-shift relative to the vehicle control. All experiments described here were performed in DMEM with 10% FBS. Heparin washes were performed as described in the Experimental Section.
To verify that the wide range of components found in fetal bovine serum (FBS) do not play a role in facilitating or inhibiting cellular entry of the materials, experiments were also performed in FBS-free media. No significant difference in mean fluorescence from any material was observed by flow cytometry in the presence or absence of FBS, suggesting that FBS components, such as growth factors, lipids, hormones, etc., do not influence uptake of these materials (Figure S13).
In order to examine how the concentration of the Tat peptide-containing materials impacted their cellular uptake, we performed flow cytometry experiments at several concentrations of material. It is important to note that concentration in these experiments was with respect to the fluorophore, where there is one fluorophore per peptide or polymer, but many copies of fluorophore per particle. (For concentration determination, see Figure S14.) In general, the peptide is less competent in cell penetration than the polymer or particle formulations, with cellular uptake of the peptide nearly abolished at 1.25 μM. This is in contrast to the polymer and nanoparticles that were still taken up by cells at concentrations as low as 0.5 μM (Figure 6B).
Studies on linear peptides have shown that 8–16 guanidiniums are optimal for cell penetration, with activity dramatically decreasing when over 16 guanidiniums are used.40,41,81 Likewise, polynorbornyl polymers bearing guanidinium moieties showed decreased internalization when 25 guanidiniums were incorporated, compared to when 10 were employed.68 Therefore, it is somewhat surprising that we see such efficient penetration for the Tat side chain 5-mer homopolymer and nanoparticle, which contains at least 30 guanindinium units (Figure 6B). In fact, the polymer penetrates cells as efficiently (within a factor of 2 fluorescence counts) as the relevant peptide analogue, even in scenarios in which 5-fold fewer fluorophores are present to achieve the same effective concentration of peptide (such as 2.5 μM Tat polymer and 12.5 μM Tat peptide). These data suggest that the arrangement of the brush polymer may aid in the cellular uptake mechanism, which could require assembly of multiple CPPs for proper transport across the membrane. Indeed, oligomerization of CPPs into “carpet” bundles and direct transportation of these bundles across the membrane has been proposed for many years as the so-called “carpet mechanism”.72,73 Alternatively, efficient cellular entry could be due to tangled pendant peptide chains presenting a lower effective number of charged residues to the cell membrane.
Proteolysis Studies of Cell Penetrating Peptides, Polymers and Nanoparticles
We next assessed whether these cell-penetrating materials were resistant to proteolysis, as hypothesized. We focused on the proteolytic cleavage of materials containing the Tat peptide, given that it has a more diverse amino acid sequence than the Arg8 peptide and would therefore have more unique cleavage sites.
Tat-containing materials, at the same concentration used in flow cytometery and confocal microscopy studies described above (2.5 μM), were challenged for 20 min with various proteases at high enzyme concentration (∼1 μM) prior to determining the extent of proteolytic cleavage and residual bioactivity. Such activity was assayed by three separate methods: reverse-phase high-performance liquid chromatography (RP-HPLC), flow cytometry, and confocal microscopy. In these assays, RP-HPLC was used to determine the degree to which proteolytic treatment degrades the integrity of the peptide as a monomer or as part of a polymeric formulation. The bioactivity of enzymatically digested materials was then assessed in cellular assays by both flow cytometry and confocal microscopy. To determine whether the location of the peptide cleavage site(s) affects the sensitivity of the peptide to enzymatic cleavage, several different proteases were tested: trypsin (7 predicted cleavage sites), chymotrypsin (2 predicted cleavage sites), and the protease cocktail Pronase, which has the potential to digest the peptide backbone at every amino acid position.
We employed RP-HPLC to assess the percent of intact material following enzymatic digestion (Figure 7A). Standard curves were generated that compared peak areas of the uncleaved Tat peptide, polymer, or nanoparticle at an appropriate concentration range, such that the concentration of material remaining after incubation with enzyme could be estimated (Figure S15). Note, this is only an estimate of polymer or particle concentration, as the absorbance measurements of the intact polymer will be affected by the norbornyl polymer backbone, the phenyl coblock, and the fluorescein end-label, which will still be present after proteolytic digestion. In these assays, no differences in the peak area or retention time of the polymer or particle were observed after treatment with any of the proteases tested and the RP-HPLC chromatograms are identical with and without enzyme treatment (i.e. no new peaks formed in the chromatogram) (Figure S16), suggesting that the Tat polymers and particles are resistant to proteolysis. By contrast, complete consumption of the Tat peptide was detected, along with the appearance of new peaks in the RP-HPLC chromatograms. Correspondingly, the mean fluorescence counts of the polymer or particle measured by flow cytometry are largely unaffected by protease treatment (presumably since the peptide chains have not been digested). However, proteolytic digestion of the Tat peptide diminished the intensity of the fluorescence signal to less than 10% of the value obtained prior to enzymatic digestion (Figure 7B). The same trends were observed by both RP-HPLC and flow cytometry when the peptide concentration was kept uniform (12.5 μM peptide, 2.5 μM polymer) to normalize the number of potential cleavage events. This is an especially notable difference given that the peptides had 5 times the number of fluorescein (1 per peptide) equivalents per peptide than the polymer (1 per 5 peptides). Furthermore, a time-course plot of RP-HPLC and flow cytometry data reveal that the Tat polymer is stable to chymotrypsin treatment over 14 h and the material retains the ability to enter cells after incubation with the enzyme (Figure S17).
Figure 7.

Assessment of the effects of protease treatment on the integrity and bioactivity of the Tat peptide, polymer, and particle by three methods. (A) RP-HPLC data showing the quantity of remaining material postenzymatic treatment. Standard curves comparing peak area to the concentration of the intact peptide, polymer, or particle were prepared to determine percent cleavage (Figure S15). (B) Flow cytometry data of the materials after proteolytic digestion. Data is reported as the percentage of fluorescence seen after enzyme treatment relative to the value seen without treatment. (C) Confocal microscopy images comparing cells incubated with materials that have been pretreated with chymotrypsin alongside cells incubated with materials that have not received this pretreatment. Images are the maximum average intensity from six consecutive 1 μm slices. Scale bars are 50 μm. In all cases Tat-containing materials at the indicated concentrations were treated with 1 μM of protease for 20 min at 37 °C.
Finally, confocal microscopy was used to verify trends observed by RP-HPLC and flow cytometry. In these experiments, the Tat peptide, Tat polymer, and Tat nanoparticle were pretreated with chymotrypsin (under identical conditions as used in the flow cytometry and RP-HPLC assays) prior to incubation with HeLa cells. These cells were then imaged by live-cell confocal microscopy alongside cells incubated with the same materials that had not been subjected to the enzyme pretreatment. A dramatic comparison emerges in which cells treated with protease-digested Tat peptides show minimal fluorescence relative to those treated with undigested peptides (Figure 7C). In stark contrast, the Tat polymer and particles give identical fluorescence images with or without enzyme treatment.
Demonstrating the Generality of the Approach with Two Additional Protease Substrates
To test whether our strategy could be extended to different proteases and to peptide sequences other than the highly charged Tat and Arg8 sequences, we polymerized two additional peptide substrates. Importantly, the two peptides each have a more extensive sampling of amino acid side chains compared to the CPP sequences and are optimized substrates for degradation by two different enzymes: a serine protease and a metalloprotease.
We first examined the generality of our approach by preparing a peptide substrate for thrombin, a coagulation factor protease. A monomer bearing the thrombin substrate sequence (GALVPRGS) was readily prepared via SPPS with a short, water-solubilizing peptide sequence (GERDG) at the C-terminus (Figure S18), and was polymerized by ROMP to several degrees of polymerization (characterization data for polymers are given in Figure S19 and Table S2). The monomer peptide and homopolymers were treated with thrombin, and the resulting product mixture was analyzed by RP-HPLC (Figure S20). These analyses indicate that the monomeric peptide was readily degraded by thrombin, as evidenced by the disappearance of the monomer peak and corresponding appearance of product peaks, however, homopolymers at several degrees of polymerization were resistant to cleavage relative to the monomer, albeit not completely shut off from proteolysis, confirming the generality of the approach (Figure 8A,B).
Figure 8.

Chemical structures and cleavage kinetics of monomeric and polymeric peptide substrates for thrombin and the matrix metalloprotease (MT1-MMP). (A) Structures of a set of homopolymers containing a thrombin peptide substrate. (B) Cleavage kinetics of thrombin-sensitive monomers and homopolymers at DP = 10, 20, and 30. (C) Structure of the fluorogenic substrate homopolymer. (D) Cleavage kinetics of the fluorogenic homopolymer relative to monomer, by multiple proteases in addition to the protease for which the substrate is optimized, MT1-MMP.
We next examined an optimized peptide substrate sequence for a cancer-associated membrane-bound matrix metalloprotease (MMP).82 Here, we omitted the N-terminal Cys residue from the optimized peptide sequence, CRPAHLRDSG, because free thiols (lacking protecting groups) are difficult to polymerize by ROMP, due to coordination to the initiators.83,84 Since this peptide was not expected to function in an orthogonal bioactivity assay, as for the CPP studies above, we prepared the sequence as a fluorogenic substrate, to readily and rapidly quantify cleavage events at low concentrations of material to obtain detailed kinetic information. For more information on the preparation of monomers, polymers and additional assays and kinetics details, see the Supporting Information and also Figures S21–S29 and Tables S3–S4.
In kinetic assays, the fluorogenic monomer was readily cleaved by an assortment of proteases (Figure 8C,D), but not by MMP-9 for which it is not a substrate (Figure S28). In contrast, the homopolymer (DP = 20) exhibits very little cleavage upon treatment with multiple proteases, as seen in the 24-h time course plots (Figure 8B). Initial enzymatic reaction rates (V0), obtained by monitoring each reaction for the first 40 min (less than 25% cleavage seen for all materials), indicate that the monomer is cleaved 17- to 95-fold faster than the homopolymer at comparable peptide concentrations (Table 3). Michaelis–Menten plots were obtained for the cleavage of the monomer by MT1-MMP, yielding a specificity constant (kcat/Km) of 0.52 ± μM–1min–1 and a Km of 11 μM (Figure 9A). A Michaelis–Menten plot of the homopolymer time-course data reveals that saturation kinetics have not been reached at ∼7 times the calculated Km of the monomer, as suggested by the near linear fit to the data obtained (Figure 9B). Note that the fluorescence observed in assays of the homopolymer approached the lower limit of detection, resulting in large standard errors in the data. Moreover, solubility limits of the homopolymer prevented a full Michaelis–Menten plot from being generated. Nevertheless, these data clearly indicate that the protease exhibits lower affinity for and activity on the peptide substrate when it is incorporated into a brush polymer.
Table 3. Initial Velocities (V0) for the Proteolysis of Fluorogenic Monomer and Homopolymer by Assorted Proteasesa.
| protease | monomer V0 (μMmin–1) | homopolymer V0 (μMmin–1) |
|---|---|---|
| MT1-MMP | 0.11 ± 0.01 | 0.0066 ± 0.002 |
| Thermolysin | 0.27 ± 0.03 | 0.0055 ± 0.001 |
| Trypsin | 0.18 ± 0.01 | 0.002 ± 0.001 |
| Pronase | 0.2 ± 0.01 | 0.0021 ± 0.001 |
| MMP-9 | 0.005 ± 0.001 | – |
The terms monomer and homopolymer correspond to the fluorogenic homopolymer and polymers. The fluorogenic peptide is optimized so that it is not a substrate for MMP-9. No fluorescence was detected during treatment of the homopolymer with MMP-9.
Figure 9.

Michaelis–Menten plots. Proteolysis of (A) the fluorogenic monomer where kcat = 5.7 ± 0.4 min–1, Km = 11 ± 2 μM, and kcat/Km = 0.52 ± μM–1min–1 and (B) the fluorogenic homopolymer by MT1-MMP, where the estimated Km is >70 μM.
Tuning the Proteolytic Susceptibility of Peptide-Containing Brush Polymers
We were curious about the origin of proteolytic resistance and also recognized that, in certain circumstances, it might be disadvantageous to render a peptide entirely refractory to proteolysis, for example, in the case of a peptide sensor for a protease,57,85 or when designing a device that targets tissue or releases a drug in response to proteolytic digestion.86,87 Therefore, we sought to tune the proteolytic susceptibility of the fluorogenic substrate for MT1-MMP described in the previous section. We envisioned that the proteolytic resistance of homopolymers might result from packing or other stabilizing peptide–peptide interactions, leading to steric protection against enzymatic cleavage. This picture led to the hypothesis that proteolytic susceptibility would be restored by spacing the peptides out along the polymer backbone. Spacing was accomplished by preparation of random blend copolymers that incorporated a monomer “spacer” or “diluent” at varying blend ratios (Figure 10). The spacer we chose was a water-soluble OEG monomer, which is inert to proteolytic enzymes. Random blend copolymers (total DP = 20) were prepared at substrate to OEG ratios of 1:19, 5:15, 10:10, and 15:5 (Figure 10A). A detailed description of the preparation and characterization of these blend polymers is given in the Supporting Information (Figure S30 and Table S5). A general trend emerged in which proteolytic activity of MT1-MMP was greatest when more spacers were incorporated. Indeed, the 1:19 blend polymer proved to be as susceptible to proteolytic degradation as the substrate monomer (Figure 10B). These data suggest that the protection from proteolysis observed in our systems does not result simply from the attachment of the peptide to a high molecular weight polymer, but rather from its arrangement into a high-density peptide brush.
Figure 10.
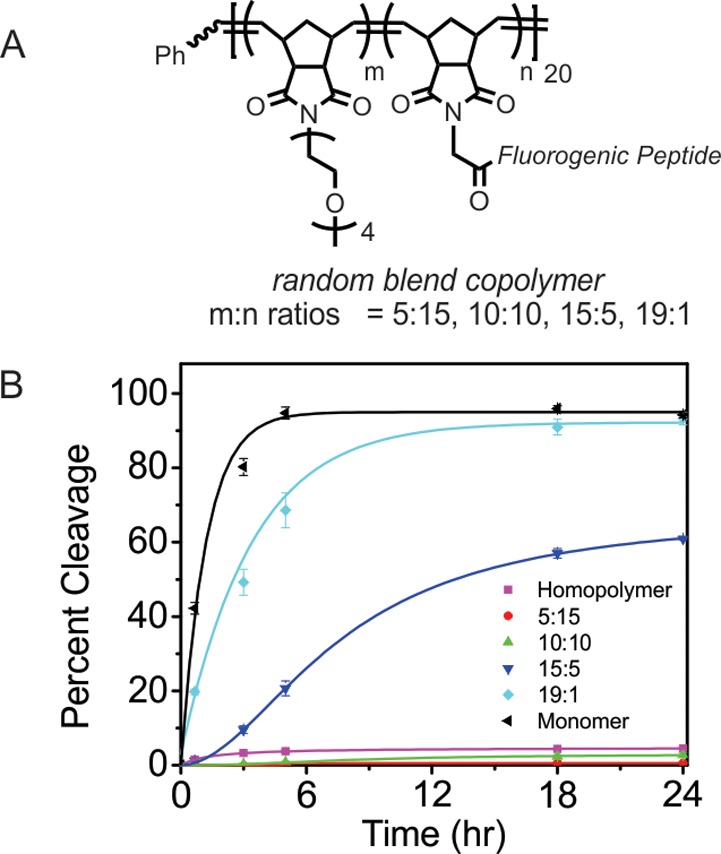
Chemical structure of the random blend copolymers and cleavage kinetics of the monomer, homopolymer, and a series of random blend copolymers of the fluorogenic peptide substrate with OEG diluent. (A) Structure of a series of random blend copolymers of the fluorogenic peptide substrate monomer and an OEG monomer. (B) Comparison of the cleavage kinetics of the fluorogenic random blend copolymers, (ratio is m:n as shown in A, overall DP = 20) homopolymer (DP = 20) and monomer.
Molecular Dynamics Simulations
To further examine the proteolytic susceptibility trends observed with the random blend copolymers of the fluorogenic substrate and OEG moiety, a series of molecular dynamics simulations were performed, examining analogous blend copolymers with discrete structures and no dispersity (i.e., a single molecular entity was modeled for each structural analogue). For computational simplicity, all polymers were constructed in silico to have a DP of 10, instead of 20, in four key arrangements meant to best simulate idealized scenarios: a homopolymer of ten repeated fluorogenic substrates (Figure 11A); two blend copolymers with an OEG:peptide ratio of 9:1, one having the peptide at one end of the polymer (Figure 11B), and the other having the peptide at position five (Figure 11C); and one with an intermediate peptide:OEG ratio of 5:5 (Figure 11D).
Figure 11.

Representative conformations and surface-accessibility data for in silico models of the fluorogenic substrate/OEG copolymers. Chemical structure and a representative conformation of the discrete, monodisperse, simulated polymers: homopolymer (A); the 9:1 ratio blend copolymer with the peptide at the end of the polymer (B); the 9:1 ratio blend copolymer with the peptide in the middle (i.e., position 5) (C); and the intermediate 5:5 ratio blend copolymer (D). (E) Plots of the probe-accessible surface area of the four structures, averaged over the last 40 ns of each heat–cool cycle. Blue bars represent the surface area accessible per peptide to a spherical probe with a radius of 3.14 nm (size on the order of a typical protease) and the gray bars represent the same measurement using a probe radius of 0.14 nm (approximately the size of a water molecule).
Simulations were performed on each structure, starting with the molecule in an artificial, extended conformation with a straight norbornyl backbone, extended peptides, and OEG brushes arrayed at right angles to the polymer backbone. In the simulations, each structure was equilibrated for an initial 20 ns at 300 K, after which each simulation was split and continued in two ways: one simulation for each molecule was continued for 100 ns at 300 K and the other was further randomized by a single heating (500 K) and cooling (300 K) cycle before continuing at 300 K for remainder of the 100 ns simulation.
In every simulation, the initial extended structure of each molecule collapsed quickly into a more compact conformational ensemble. Representative conformations of each construct were obtained by applying a root-mean-square deviation (RMSD)-based clustering algorithm to the last 40 ns of the respective heat–cool simulations. In these structures, the homopolymer and 5:5 copolymer have collapsed into an elongated globule, with their peptide chains tangled around the polymer backbone. In contrast, the single peptide chain in the 9:1 copolymers, are visible as relatively isolated components at the surface of the constructs. A detailed discussion of differences in radius of gyration of each structure and also on conformational fluctuations in the structures as quantified by RMSDs is given in the Supporting Information and Figures S31–S33 and Table S6.
We examined the role of hydrogen bonding in facilitating the compression or tangling of the peptide-containing structures by computing the numbers of intrasolute hydrogen-bonds during the last 40 ns of each heat–cool trajectory (Table S6). The homopolymer averaged 0.5 amino acid-amino acid peptide bonds per residue (88 interactions over 17 residues per monomer), which is about half the ratio typically seen for a folded protein.88 The three copolymers averaged somewhat fewer (0.4) hydrogen bonds per residue, with very few hydrogen bonds to the OEG moieties (less than four in all cases). Overall, these hydrogen bond counts are consistent with a view that, although the polymers have collapsed, they are not as well structured as typical globular proteins and the inclusion of OEG units leads to a decrease in hydrogen bonding. Thus, the addition of OEG units effectively “dilutes” the density of the peptide-brush by reducing the overall degree of hydrogen bonding in the polymer structure. Moreover, the OEG moieties block what would otherwise be stabilizing interactions among the peptides brushes without compensating with new OEG-peptide interactions.
Finally, the accessibility of the peptide components of the various constructs to large and small molecules was examined by computing their time-averaged probe-accessible surface area (SA), using a large (3.14 nm) probe sphere whose size is similar to that of a protein, and a small (0.14 nm) water-sized probe sphere (Figure 11E). A general trend was seen with the large probe, where larger surface accessibility was seen as the peptide content decreased. The greatest accessibility is observed for the 9:1 copolymer with the peptide at position 5. This is the closest representation to the experimental 19:1 random blend copolymer (based on the polymerization method, Figure S30 and Table S5), which is nearly as susceptible to proteolytic degradation as the peptide monomer in vitro. In contrast, the probe-accessible surface areas obtained with the water-sized probe are relatively uniform across all constructs. These results suggest that the peptides in all five constructs maintain similar accessibility to small molecules, like water, but that the tighter packing of the more peptide-rich constructs reduces accessibility to protein-size molecules, such as the proteases examined experimentally. Moreover, these data suggest that peptides whose function depends on interaction with a small molecule or a receptor with a relatively accessible binding site will exhibit ample binding to the peptide substrates in any of the constructs considered, including the homopolymer. The implication of this finding is that the bioactivity of many peptides will be maintained after polymerization of the sequence into a high-density brush. For peptides whose function depends on interaction with proteins or macromolecules with tight or cramped binding pockets (such as a protease), polymerization into a high-density brush polymer may impede function, as is consistent with the data presented in this work.
In sum, the simulation results suggest that all of the constructs studied here tend to collapse into fairly compact globular conformations, and that a higher peptide content leads to formation of more stabilizing intramolecular hydrogen bonds and reduced accessibility of the peptides to proteins in solution. This picture is qualitatively consistent with the experimental observation that constructs with high peptide content are better protected from enzymatic degradation. Although the simulations are subject to error due to their limited duration and uncertainties in the force field, they yield a usefully detailed representation of the systems under study and offer a plausible explanation for the key experimental results.
Electrostatic repulsion is another potential contributor to the proteolytic protection observed in these systems. Wooley and co-workers recently reported that charge-matched nanoparticles and proteases show a decrease in proteolytic activity, presumably due to repulsion at large charge densities.89 Given the range of proteases used in the present study, including protease cocktails, it is difficult to correlate the proteolytic susceptibility of the polymers with the pairing of the overall polymer charge with the isoelectric point of each enzyme. However, tuning charge−charge repulsion along with steric interactions could provide an additional route for optimization of related systems in future work.
Conclusions
Herein we present a new, easily deployed methodology for formulating peptides into well-defined brush polymers, which preserve bioactivity but protect the peptides from proteolysis. Our strategy involves the direct (graft-through) polymerization of peptide-containing norbornene monomers via ROMP. This obviates the need for the extra purification steps required for more traditional synthetic routes to peptide-containing polymers, which involve chemical conjugation of the peptide to a preformed polymer, as would be required in a graft-to synthesis scheme. The wide range of peptide substrates and proteases for which the present strategy succeeds suggests that it could be applicable to many peptide based therapeutic agents or biosensors that must retain function despite exposure to harsh proteolytic milieu. This approach offers an attractive alternative to existing methods of protecting peptides from proteolysis because it is simple, does not alter the amino acid sequence of the peptide, and enables easy functionalization with other useful moieties or cargo.
It is possible that polymerization of a peptide into a high-density brush could not only render the peptide resistant to proteolysis, but also affect its function. The computational data suggests that peptides whose functions rely upon interaction with a large molecule or with a tight or cramped active site of an enzyme would encounter significant steric hindrance that could inhibit function. However, peptides that bind small molecules or accessible receptors on a cell should retain bioactivity. Certainly, the accessibility of polymeric peptide brushes in terms of receptor binding is supported by studies of polynorbornyl polymers containing RGD targeting peptides, which bind integrin receptors more efficiently than their peptide analogues.58 As such, while we have provided a strategy for adjusting the density of the polymer brush, one could imagine applications or environments in which even more flexibility or design control is needed. In these scenarios, the robust capabilities of this ROMP strategy are ideal. For example, the incorporation of a longer linker between the norbornyl polymer backbone and the peptide could provide a means for separating the peptide from the backbone, thereby making the peptides more flexible and sterically accessible. This could include linkers that are cleavable under appropriate biological conditions or by exogenous sources. Indeed, appropriate pH-52 or UV-sensitive linkers90 have been described for ROMP polymers.
In addition to proteolysis, there are two other problems that often limit the bioavailability and clinical efficacy of peptide-based therapeutics: rapid renal clearance and inefficiencies in cellular uptake. Regarding the first, it should be noted that the exclusion limit for glomerular filtration includes molecules or assemblies whose molecular weight exceeds 50 kDa.3 It is thus relevant that the size of peptide constructs generated by ROMP can be controlled by preparation of amphiphilic polymers that self-assemble into various structures and sizes, based on their hydrophilic-to-hydrophobic ratios, as exemplified by the ∼10–50 nm spherical micelles generated here with the Tat peptide. Such particles are not expected to be subject to rapid renal elimination, and work in our laboratory is exploring the requirements for in vivo retention of ROMP-derived materials in general. Regarding the second problem, we have shown here that the ROMP copolymer strategy affords a robust ability to synthesize polymers and polymer assemblies that cross cell membranes efficiently, and we anticipate that these could transport a variety of cargo into cells. For example, therapeutic peptides could be easily incorporated, as multiple copies in a block copolymer; or as a single copy in a polymer, configured as an end-label. Indeed, dual labeled polymers with a polymerized CPP and a releasable therapeutic moiety could provide a unique approach to addressing transmembrane delivery of materials into cells.
In summary, we have presented a new method for packaging peptides, which renders them resistant to proteolysis in a tunable fashion but does not alter their amino acid sequence, and therefore preserves their biological activities. We envision that this strategy can be employed broadly to render therapeutic peptides or peptide-based sensors resistant to proteolysis, thus enhancing their bioavailability and clinical efficacy.
Experimental Section
Materials
Amino acids used in SPPS were purchased from Aapptec and NovaBiochem. All other materials were obtained from Sigma-Aldrich and used without further purification unless otherwise noted. Initiator 1 ((H2IMES)(pyr)2-(Cl)2Ru=CHPh) was prepared as described previously.49 Analytical scale RP-HPLC was performed with a Jupiter Proteo90A Phenomenex column (150 × 4.60 mm) using a Hitachi-Elite LaChrom L2130 pump with a UV–vis detector (Hitachi-Elite LaChrome L-2420) monitoring at 214 nm. Peptides were purified with a Jupiter Proteo90A Phenomenex column (2050 × 25.0 mm) on a Waters DeltaPrep 300 System. For all RP-HPLC assays, gradient solvent systems were used in which Buffer A was 0.1% TFA in water and Buffer B was 0.1% TFA in acetonitrile. Polymer dispersities and molecular weights were determine by size-exclusion chromatography (Phenomenex Phenogel 5 u 10, 1–75 K, 300 × 7.80 mm in series with a Phenomenex Phenogel 5 u 10, 10–1000 K, 300 × 7.80 mm with 0.05 M LiBr in DMF as the running buffer at a flow rate of 0.75 mL/min) using a Shimadzu pump equipped with a multiangle light scattering detector (DAWN-HELIO, Wyatt Technology) and a refractive index detector (HITACHI L2490 or a Wyatt Optilab T-rEX detector) normalized to a 30 K MW polystyrene standard. For SEC-MALS chromatograms in which a multimodal distribution is observed by light scattering but not in the RI chromatogram, we analyzed only the peak width that has an associated RI component. DLS measurements were performed on a DynaPro NanoStar (Wyatt Tech). TEM images were obtained by depositing samples on carbon-formavar-coated copper grids (Ted Paella, Inc.), which were then stained with 1% w/w uranyl acetate and then imaged on a Techanai G2 Sphera operating at an accelerating voltage of 200 kV. All concentrations of fluorescent materials were obtained by measuring UV absorbance of the fluorophore on a ThermoScientific Nanodrop 2000c and the data was fit to the standard curves described in Supporting Information. Fluorescent data was recorded on a fluorescence plate reader, PerkinElmer HTS 7000 Plus Bio Assay Reader (excitation: 340 nm; emission: 465 nm), or on a Photon Technology International fluorescence reader. 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded on a Varian Mercury Plus spectrometer. Chemical shifts are reported in ppm relative to the DMF-d7 or CDCl3 residual peaks.
Guanidinium Monomer Synthesis
A 10 mL round-bottom flask equipped with a stir bar was charged with an amine-terminated norbornene (2-(2-aminoethyl)-3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3-(2H)-dione) (70 mg, 0.24 mmol, 1 equiv), which was prepared as described previously50 and dissolved in 4.5 mL of dry DMF under N2 (g). To this was added N,N-bis(boc)-1-guanylpyrazole (105 mg, 0.34 mmol, 1 equiv) and diisopropylethylamine (120 μL, 0.68 mmol, 2 equiv). The reaction mixture was stirred at room temperature for 12 h. The solution was concentrated to dryness and resuspended in 25 mL of CH2Cl2, then washed with water (×3) and then brine. The CH2Cl2 layer was collected, dried over Na2SO4 (s) and concentrated to dryness. The material was then purified by flash column chromatography on silica gel (33% EtOAC in hexanes) to yield a white powder in 92% yield (140 mg, 0.31 mmol) Rf 0.37 (33% EtOAc in hexanes): 1H NMR (400 MHz, CDCl3, 298 K) δ 11.43 ppm (1H, b), 8.45 (1H, b), 6.27 (2H, t, J = 1.5 Hz), 3.71 (2H, dd, J = 7.0, 4.4), 3.64 (2H, m), 3.25 (2H, d, J = 1.5 Hz), 2.7 (2H, m), 1.51 (1H, d, J = 1.1 Hz), 1.48 (9H, s), 1.47 (9H, s), 1.25 (1H, d, J = 1.8 Hz); 13C NMR (100 MHz, CDCl3, 298 K) δ 178.1, 157.0, 156.6, 153.0, 137.9, 137.7, 83.3, 79.5, 48.0, 45.0, 43.1, 40.0, 38.0, 28.3, 28.1; High-resolution MS analysis (ESI-TOFMS) m/z calculated 449.2395, found 449.2394.
Peptide Synthesis
Peptides were synthesized using standard Solid Phase Peptide Synthesis (SPPS) procedures on an AAPPTec Focus XC automated synthesizer. The Arg8 peptide was synthesized with the Pbf protecting group left on the side chains by the use of highly acid-sensitive Sieber Amide resin. All other peptides were prepared protecting group-free on Rink Amide MBHA resin. A typical SPPS procedure involved FMOC deprotection with 20% methylpiperidine in DMF (one 5 min deprotection followed by one 15 min deprotection), and 45 min amide couplings using 3.75 equiv of the FMOC-protected, and side chain-protected amino acid, 4 equiv of HBTU and 8 equiv of DIPEA. Peptide couplings that were incomplete by Kaiser test were drained and then subjected to fresh reagents. Monomers were prepared by amide coupling to N-(hexanoic acid)-cis-5-norbornene-exo-dicarboximide (prepared via a published protocol59) or to N-(glycine)-cis-5-norbornene-exo-dicarboximide56 for the fluorogenic substrates at the N-terminus of the peptide. Fluorescein-labeled peptides were assembled by addition of Boc-Lys(FMOC)-OH to the N-terminus of the peptide, followed by removal of the FMOC protecting group and amide coupling to 5/6-carboxy fluorescein. Following completion of the synthesis, peptides were cleaved from the resin. The side-chain protected Arg8 peptide was cleaved from the Sieber amide resin by five 2 min rinses with 2% TFA in DCM. All other peptides were cleaved and deprotected by treatment with TFA/H2O/TIPS in a 9.5:2.5:2.5 ratio for 2 h. The peptides were then precipitated in cold ether and purified by RP-HPLC. The identity of each peptide was confirmed by ESI-MS or MALDI-MS and purities were verified by observation of a single peak in analytical RP-HPLC chromatograms (Figures S2, S20A, and S22 and Tables S1 and S3).
Polymerizations
Polymerizations were carried out in a glovebox under N2 (g). A typical protocol used to generate a polymer with DP = 10 involved mixing the monomer (0.0125 mmol, 10 equiv, 25 mM) with the catalyst (0.00125 mmol, 1 equiv, 2.5 mM) in dry DMF (0.5 mL). Homopolymerizations that have not been reported previously in the literature were preformed in DMF-d7 and followed by 1H NMR to confirm complete consumption of the monomer and to determine the time period required to reach completion. Polymers for cell penetration studies were end-labeled with a copy of fluorescein using a chain transfer agent (1.5 equiv) for 2 h as described previously,50 followed by termination with ethyl vinyl ether (10 equiv) for 1 h at room temperature. Block copolymers were prepared by polymerizing the first monomer (either phenyl or PEG) to completion and then adding the second monomer (a peptide or the guanidinium group), followed by end labeling with the fluorescein chain transfer agent and finally termination with ethyl vinyl ether. Fluorescein-labeled polymers were treated with NH4OH (aq) for 20 min to remove the pivolate protecting group, as described previously.50 The resulting polymers were directly characterized by SEC-MALS.
The side-chain protected Arg8 polymer was precipitated with cold ether and collected by centrifugation. The resulting powder was dissolved in 2 mL of a mixture of TFA/H2O/TIPS (95:2.5:2.5) and stirred for 4 h at room temperature. The product was precipitated with cold ether, collected by centrifugation and dried. In preparation for in vitro studies, all polymers were washed (×3) with cold ether (to remove the Ru catalyst) and then dissolved in PBS and dialyzed in an effort to remove any residual monomer or catalyst. The Tat and GSGSG particles were generated by dissolving the amphiphilic polymers in DMF, and then diluting with an equivalent volume of PBS over 1 h and finally dialyzing this solution into PBS over 48 h with 3 buffer changes using dialysis cups of MWCO 3500 (Thermo Scientific, cat. #69552).
RP-HPLC Analysis of CPP Proteolysis
The extent of proteolytic degradation of the Tat peptide, polymer and particle by trypsin (Gibco Life Tech., cat. #15090–046), α-chymotrypsin (Fisher Scientific, cat. #ICN1522722) and Pronase (Roche, cat. #10165921001) was assessed by comparison of chromatograms in RP-HPLC. In these experiments, the material at the indicated concentration was incubated with each protease (at 1 μM) for 20 min, and then the enzymes were heat denatured at 65 °C for 15 min, and the solution was immediately injected onto an analytical RP-HPLC. Given that treatment with each protease gives multiple fragments of the Tat sequence, a standard curve for each starting material was prepared to assess the percentage of intact material remaining after proteolytic digestion (Figure S15). Note that the standard curves for the polymer and particle will be biased due to the fact that after cleavage, the polymer backbone and fluorophore should remain intact, and will comprise part of the measured peak area. Nevertheless, no new peaks were seen in the chromatograms of the polymer or particle post enzyme treatment (Figure S16), suggesting that these materials are not susceptible to cleavage by the proteases.
Cell Culture
HeLa cells were purchased from ATCC (CCL-2). Cells were cultured at 37 °C under 5% CO2 in phenol-red- containing Dulbecco’s Modified Eagle Medium (DMEM; Gibco Life Tech., cat. #11960–044) supplemented with 10% fetal bovine serum (Omega Scientific, cat. #FB02) and with 1× concentrations of nonessential amino acids (Gibco Life Tech., cat. #11140–050) sodium pyruvate (Gibco Life Tech., cat. #11360–070), l-glutamine (Gibco Life Tech., cat. #35050–061), and the antibiotics penicillin/streptomycin (Corning Cellgro, cat. #30–002-C1). Cells were grown in T75 culture flasks and subcultured at ∼75–80% confluency (every ∼3–4 days).
Flow Cytometry
HeLa cells were plated at a density of 90 000 cells per well of a 24-well plate 18 h prior to treatment. Materials dissolved in Dulbecco’s Phosphate Buffered Saline (DPBS without Ca2+ or Mg2+; Corning Cellgro, cat. #21–031-CM) at 10× the desired concentration were added to the wells, and the plates were incubated for 30 min at 37 °C. The medium was then removed, and the cells were washed twice with DPBS and then incubated three times for 5 min with heparin (0.5 mg/mL in DPBS; Affymetrix, cat. #16920), and finally rinsed again with DPBS. The cells were then trypsinized (0.25% trypsin; Gibco Life Tech., cat. #15090–046) for 10 min, cold medium was added, and the cells were transferred to Eppendorfs, centrifuged to pellets and then resuspended in a minimal amount of cold DPBS. Fluorescence activated cell sorting data (10 000 events on three separate cultures) was acquired on an Accuri C6 flow cytometer set to default “3 blue 1 red” configuration with standard optics and slow fluidics (14 μL/min). For proteolysis studies, the indicated concentration of Tat peptide, homopolymer or particle was pretreated with 1 μM of trypsin, chymotrypsin or Pronase for 20 min in DPBS, after which the protease was heat denatured for 15 min at 65 °C. The cells were then incubated, prepared and analyzed by flow cytometry as described above. For mechanistic studies, cells were preincubated with the indicated compound for 30 min at 37 °C prior to addition of the cell-penetrating material. The following concentrations were used: 80 μM dynasore (Enzo Life Sciences, cat. #270–502-M005) and 9.5 mM MβCD (Fischer Scientific, cat. #AC377110050). For studies at reduced temperature, cells were incubated at 4 °C for 30 min prior to and during the incubation with the compound of interest. All subsequent washes and manipulations were also done with ice-cooled media and other materials. Data is reported as the normalized mean fluorescence, which is the mean fluorescence yielded by the material/the mean fluorescence from the vehicle control.
Live-Cell Confocal Microscopy
HeLa cells were seeded on glass-bottom 24-well plates at a cell density of 90 000 cells per well 18 h prior to treatment. The medium was removed and then replaced with medium lacking phenol red (Gibco Life Tech., cat# 31053–028) to minimize background fluorescence. Materials dissolved in DPBS (at 10× the desired concentration) were added to the wells and the plates were incubated for 30 min at 37 °C. The washing procedure used in the flow cytometry experiments (2 × DPBS, 3 × heparin for 5 min, 1 × DPBS) was followed here. Following removal of the final DPBS rinsate, fresh media (phenol red- free) was added to each well. Live cells were imaged on an Olympus FV1000 confocal microscope. For proteolysis studies, the indicated concentration of Tat peptide, homopolymer and particle were pretreated with 1 μM of trypsin, chymotrypsin or Pronase for 20 min in DPBS, after which the protease was heat denatured for 15 min at 65 °C. The cells were then incubated, prepared and analyzed by confocal microscopy as described above.
Cell Viability Assays
The CellTiter-Blue assay (Promega, cat. #G8081) measures the reduction of resazurin to resorufin via fluorescence. HeLa cells were plated at a density of 4000 cells per well of a 96-well plate 18 h prior to treatment. Materials dissolved in DPBS at 5 μM were added to the wells along with a 10% DMSO positive control. Cells were incubated for 48 h at 37 °C. The medium was removed and 80 μL of fresh media lacking phenol red was added. To this was added 20 μL of the CellTiter-Blue reagent and the cells were then incubated for 2 h prior to measuring fluorescence in a plate reader using 560 nm excitation and 590 nm emission. The fluorescence measurements were corrected for background fluorescence from the CellTiter-Blue reagent by subtracting the fluorescence reading of wells treated with the reagent in the absence of cells. Fluorescence values were then referenced as a percentage of the value obtained for the PBS vehicle control.
Polymerization of Thrombin Substrate
Polymerizations were carried out in a glovebox under a N2 (g) atmosphere. To generate the polymers containing the thrombin peptide sequence, the monomer (0.007 mmol, 10 equiv, 23 mM for DP = 10; 0.013 mmol, 20 equiv, 45 mM for DP = 20; 0.021 mmol, 30 equiv, 70 mM for DP = 30) was mixed with the catalyst (0.0007 mmol, 1 equiv, 2.3 mM) in DMF-d7 (0.3 mL) and monitored by 1H NMR to confirm complete consumption of the monomer and to determine the time period required to reach completion. Upon completion, the polymers were quenched with ethyl vinyl ether for 10 min, and then precipitated with cold ether and dried under a vacuum. The resulting polymers were directly characterized by SEC-MALS.
RP-HPLC Analysis of Thrombin Proteolysis
The extent of proteolytic degradation of the thrombin peptide polymers by thrombin (Sigma, cat. #T6884–100UN) was assessed by comparison of chromatograms in reverse-phase HPLC. In these experiments, the monomer and polymers were dissolved in PBS buffer (2.2 mM with respect to peptide). Thrombin (10 units) was added to each sample and an HPLC trace was immediately obtained followed by subsequent HPLC injections every 45 min. A standard curve of the authentic C-terminal fragment was generated to convert the peak area to percent cleavage.
Fluorogenic Peptide Studies
The fluorogenic peptide NorG-E(EDANS)RPAHLRDSGK(dabcyl)GSGSG was prepared by SPPS as described above where the EDANS was added as a modified Glu (FMOC-Glu(EDANS)-OH; AAPTec, cat. #AFE150) while dabcyl (Anaspec, cat #81800) was conjugated to the ε-amino group of a lysine. This monomer was polymerized into a homopolymer with DP = 20, determined by bulk light scattering. Note that the fluorogenic peptide sequence did not run as a polymer on the SEC column needed for SEC-MALS. Blend copolymers with a PEG monomer were prepared by first assessing the rate of polymerization of the two monomers. At the concentration of monomer studied, the PEG monomer was quick to polymerize (complete within 15 min), while the fluorogenic substrate polymerized at a rate of 1.78 monomers per hr (Figure S30). To ensure reasonable interdigitation of the two monomers in the random blend copolymer, the PEG monomer was added via syringe pump at appropriate rates to prepare peptide: PEG polymers at a ratio of 1:19, 5:15, 10:10, 15:5 and 19:1 as described in Table S5. Cleavage of the homopolymer monomer and blend copolymers (40 μM) by the noted protease in (at 25 nM) in reaction buffer (50 mM Tris (pH 7.4), 1 mM ZnCl2, 150 mM NaCl, 5 mM CaCl2) was monitored by measurement of fluorescence in a plate reader or fluorimeter. The proteases, MT1-MMP (catalytic domain; Calbiochem, cat. #476935), MMP-9 (catalytic domain; Enzo Life Sciences cat. #BML-AW360–0010), thermolysin (Promega, cat. #V4001), trypsin (Gibco Life Tech., cat. #15090–046) and Pronase (Roche, cat. #10165921001) were purchased from commercial sources. Standard curves and assay details are described in the Supporting Information.
Computational Methods
Details of the polymer constructs that were simulated are as follows. In all cases, the polymer backbone was composed of 10 norbornene units, flexibly linked by olefin bonds, with a 1:1 mix of cis and trans units. In the homopolymer, each norbornene residue is linked to the N-terminus of the fluorogenic peptide NorGE(EDANS)RPAHLRDSGK(DABCYL)GSGSG) (Figure 11A), where the EDANS and DABCYL fluorophores are linked to E and K residues, respectively. The C-terminus of each peptide was amide capped. In the 5:5 blend copolymer, five of these peptide-dye chains were linked to norbornenes 1, 2, 5, 6, and 10 (counting from the end of the phenyl ring); and five OEG chains, each with four ethylene glycol units, were linked to the remaining norbornenes (Figure 11D). In the 9:1 blend copolymers, all positions are occupied by OEGs except that the tenth or fifth norbornene (Figure 11B or 11C) is occupied by the fluorogenic peptide.
All-atom molecular dynamics simulations were performed to study the conformations of the simulated polymers, using the explicit water model TIP3P91 and the Gromacs 4.6 software package.92 All bonded and Lennard–Jones terms of the polymer backbone and dye moieties were assigned by the General Amber Force Field (GAFF)93 and partial atomic charges were assigned using AM1-BCC.94,95 Parameters from the Amber ff99SB-ILDN force field96 were assigned to the peptide components. All simulations started from extended polymer backbone and peptide configuration and were performed using periodic boundary conditions. Each polymer construct was solvated in a cubic simulation box with edge lengths set to the longest dimension of the molecule plus 2 nm. This led to box sizes with edge lengths of 10–15 nm. The systems were first energy minimized with the steepest-descent algorithm, and then equilibrated for 10 ns under constant volume and temperature conditions and then another 10 ns under constant temperature and pressure conditions. The Particle–Mesh–Ewald (PME) method97 was used for electrostatic interactions, and the cutoff distance of the Lennard–Jones (LJ) interactions was 10 Å. In some simulations, a heat–cool cycle was used immediately after the equilibration phase to boost the systems out of local energy minima and search for additional stable conformational states. Here, the temperature was increased from 300 to 500 K linearly over 2 ns; the simulation was run for 1 ns at 500 K; and the temperature was then reduced back to 300 K linearly over 2 ns, and kept at 300 K for 95 ns of production dynamics. For comparison, regular MD simulations at constant 300 K were also performed for 100 ns following the same equilibration phase. We also simulated the two 9:1 blend copolymers without the dye components (see Supporting Information), to verify that the dye molecules do not influence our major conclusions. For these simulated polymers, one Cl− counterion was added in order to neutralize the +1 charge.
Acknowledgments
We acknowledge generous support for this research from the AFOSR through PECASE (FA9550-11-1-0105) to N.C.G. and from a BRI grant (FA99550-12-1-0414) in support of collaborative work between the groups of M.K.G and N.C.G. In addition, we acknowledge support from the NIH through a Director’s New Innovator Award (1DP2OD008724), through the NIBIB (1R01EB011633) and for a Transformative Award (NHLBI R01HL117326). Additional support comes from ARO (W911NF-14-1-0169) including a DURIP for instrumentation (W911NF-13-1-0321). A.P.B. was generously supported by the American Cancer Society – North Texans Creating Tomorrow’s Miracles Postdoctoral Fellowship. Cassandra Callmann is thanked for assistance with TEM and is supported by the CRIN program, UCSD. We acknowledge the UCSD Light Microscopy Facility (P30 Grant NS047101) and the UCSD Cryo-Electron Microscopy facility (NIH R37GM-033050, and the Agouron Institute support to T. Baker).
Supporting Information Available
Additional experimental procedures, data and figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s):M.K.G. has an equity interest in, and is a cofounder and scientific advisor of, VeraChem LLC.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Craik D. J.; Fairlie D. P.; Liras S.; Price D. Chem. Biol. Drug Des. 2013, 81, 136. [DOI] [PubMed] [Google Scholar]
- Kaspar A. A.; Reichert J. M. Drug Discovery Today 2013, 18, 807. [DOI] [PubMed] [Google Scholar]
- McGregor D. P. Curr. Opin. Pharmacol. 2008, 8, 616. [DOI] [PubMed] [Google Scholar]
- Vlieghe P.; Lisowski V.; Martinez J.; Khrestchatisky M. Drug Discovery Today 2010, 15, 40. [DOI] [PubMed] [Google Scholar]
- Barbieri F.; Bajetto A.; Pattarozzi A.; Gatti M.; Würth R.; Thellung S.; Corsaro A.; Villa V.; Nizzari M.; Florio T. Int. J. Pept. 2013, 2013, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos E.; Vazquez O.; Mascarenas J. L.; Vazquez M. E. Chem. Soc. Rev. 2009, 38, 3348. [DOI] [PubMed] [Google Scholar]
- Woodley J. F. Crit. Rev. Ther. Drug Carrier Syst. 1994, 11, 61. [PubMed] [Google Scholar]
- Hamamoto K.; Kida Y.; Zhang Y.; Shimizu T.; Kuwano K. Microbiol. Immunol. 2002, 46, 741. [DOI] [PubMed] [Google Scholar]
- Miller S. M.; Simon R. J.; Ng S.; Zuckermann R. N.; Kerr J. M.; Moos W. H. Bioorg. Med. Chem. Lett. 1994, 4, 2657. [Google Scholar]
- Powell M. F.; Stewart T.; Otvos L. Jr.; Urge L.; Gaeta F. C.; Sette A.; Arrhenius T.; Thomson D.; Soda K.; Colon S. M. Pharm. Res. 1993, 10, 1268. [DOI] [PubMed] [Google Scholar]
- Steer D. L.; Lew R. A.; Perlmutter P.; Smith A.; Aguilar M. I. Curr. Med. Chem. 2002, 9, 811. [DOI] [PubMed] [Google Scholar]
- Weinstock M. T.; Francis J. N.; Redman J. S.; Kay M. S. Pept. Sci. 2012, 98, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H.; Kodama H.; Osada S.; Kato F.; Jelokhani-Niaraki M.; Kondo M. Biosci., Biotechnol., Biochem. 2003, 67, 2269. [DOI] [PubMed] [Google Scholar]
- Brinckerhoff L. H.; Kalashnikov V. V.; Thompson L. W.; Yamshchikov G. V.; Pierce R. A.; Galavotti H. S.; Engelhard V. H.; Slingluff C. L. Jr. Int. J. Cancer 1999, 83, 326. [DOI] [PubMed] [Google Scholar]
- Biron E.; Chatterjee J.; Ovadia O.; Langenegger D.; Brueggen J.; Hoyer D.; Schmid H. A.; Jelinek R.; Gilon C.; Hoffman A.; Kessler H. Angew. Chem., Int. Ed. 2008, 47, 2595. [DOI] [PubMed] [Google Scholar]
- Chatterjee J.; Gilon C.; Hoffman A.; Kessler H. Acc. Chem. Res. 2008, 41, 1331. [DOI] [PubMed] [Google Scholar]
- Gordon D. J.; Sciarretta K. L.; Meredith S. C. Biochemistry 2001, 40, 8237. [DOI] [PubMed] [Google Scholar]
- Blackwell H. E.; Grubbs R. H. Angew. Chem., Int. Ed. 1998, 37, 3281. [DOI] [PubMed] [Google Scholar]
- Kim Y. W.; Grossmann T. N.; Verdine G. L. Nat. Protoc. 2011, 6, 761. [DOI] [PubMed] [Google Scholar]
- Schafmeister C. E.; Po J.; Verdine G. L. J. Am. Chem. Soc. 2000, 122, 5891. [Google Scholar]
- Verdine G. L.; Hilinski G. J. Methods Enzymol. 2012, 503, 3. [DOI] [PubMed] [Google Scholar]
- Osapay G.; Prokai L.; Kim H. S.; Medzihradszky K. F.; Coy D. H.; Liapakis G.; Reisine T.; Melacini G.; Zhu Q.; Wang S. H.; Mattern R. H.; Goodman M. J. Med. Chem. 1997, 40, 2241. [DOI] [PubMed] [Google Scholar]
- Rozek A.; Powers J. P.; Friedrich C. L.; Hancock R. E. Biochemistry 2003, 42, 14130. [DOI] [PubMed] [Google Scholar]
- Su C. M.; Jensen L. R.; Heimer E. P.; Felix A. M.; Pan Y. C.; Mowles T. F. Horm. Metab. Res. 1991, 23, 15. [DOI] [PubMed] [Google Scholar]
- Lee H.; Jang I. H.; Ryu S. H.; Park T. G. Pharm. Res. 2003, 20, 818. [DOI] [PubMed] [Google Scholar]
- Li W.; Wang Y.; Zhu X.; Li M.; Su Z. J. Biotechnol. 2002, 92, 251. [DOI] [PubMed] [Google Scholar]
- Nischan N.; Chakrabarti A.; Serwa R. A.; Bovee-Geurts P. H. M.; Brock R.; Hackenberger C. P. R. Angew. Chem., Int. Ed. 2013, 52, 11920. [DOI] [PubMed] [Google Scholar]
- Ramon J.; Saez V.; Baez R.; Aldana R.; Hardy E. Pharm. Res. 2005, 22, 1374. [DOI] [PubMed] [Google Scholar]
- Monfardini C.; Schiavon O.; Caliceti P.; Morpurgo M.; Harris J. M.; Veronese F. M. Bioconjugate Chem. 1995, 6, 62. [DOI] [PubMed] [Google Scholar]
- Ruijtenbeek R.; Kruijtzer J. A. W.; van de Wiel W.; Fischer M. J. E.; Flück M.; Redegeld F. A. M.; Liskamp R. M. J.; Nijkamp F. P. ChemBioChem 2001, 2, 171. [DOI] [PubMed] [Google Scholar]
- Ruttekolk I. R.; Chakrabarti A.; Richter M.; Duchardt F.; Glauner H.; Verdurmen W. P. R.; Rademann J.; Brock R. Mol. Pharmacol. 2011, 79, 692. [DOI] [PubMed] [Google Scholar]
- Falciani C.; Lozzi L.; Pini A.; Corti F.; Fabbrini M.; Bernini A.; Lelli B.; Niccolai N.; Bracci L. Chem. Biol. Drug Des. 2007, 69, 216. [DOI] [PubMed] [Google Scholar]
- Pini A.; Falciani C.; Bracci L. Curr. Protein Pept. Sci. 2008, 9, 468. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Imura T.; Leman L. J.; Curtiss L. K.; Maryanoff B. E.; Ghadiri M. R. J. Am. Chem. Soc. 2013, 135, 13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammeyer J. K.; Blum A. P.; Adamiak L.; Hahn M. E.; Gianneschi N. C. Polym. Chem. 2013, 4, 3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku T.-H.; Chien M.-P.; Thompson M. P.; Sinkovits R. S.; Olson N. H.; Baker T. S.; Gianneschi N. C. J. Am. Chem. Soc. 2011, 133, 8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A. D.; Pabo C. O. Cell 1988, 55, 1189. [DOI] [PubMed] [Google Scholar]
- Green M.; Loewenstein P. M. Cell 1988, 55, 1179. [DOI] [PubMed] [Google Scholar]
- Puglisi J. D.; Chen L.; Blanchard S.; Frankel A. D. Science 1995, 270, 1200. [DOI] [PubMed] [Google Scholar]
- Futaki S.; Suzuki T.; Ohashi W.; Yagami T.; Tanaka S.; Ueda K.; Sugiura Y. J. Biol. Chem. 2001, 276, 5836. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Mitchell D. J.; Pattabiraman K.; Pelkey E. T.; Steinman L.; Rothbard J. B. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. J.; Kim D. T.; Steinman L.; Fathman C. G.; Rothbard J. B. J. Pept. Res. 2000, 56, 318. [DOI] [PubMed] [Google Scholar]
- Heitz F.; Morris M. C.; Divita G. Br. J. Pharmacol. 2009, 157, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren E.; Torchilin V. P. Trends Mol. Med. 2012, 18, 385. [DOI] [PubMed] [Google Scholar]
- Wang F.; Wang Y.; Zhang X.; Zhang W.; Guo S.; Jin F. J. Controlled Release 2014, 174, 126. [DOI] [PubMed] [Google Scholar]
- Cartier R.; Reszka R. Gene Ther. 2002, 9, 157. [DOI] [PubMed] [Google Scholar]
- Escriou V.; Carriere M.; Scherman D.; Wils P. Adv. Drug Delivery Rev. 2003, 55, 295. [DOI] [PubMed] [Google Scholar]
- Lange A.; Mills R. E.; Lange C. J.; Stewart M.; Devine S. E.; Corbett A. H. J. Biol. Chem. 2007, 282, 5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford M. S.; Love J. A.; Grubbs R. H. Organometallics 2001, 20, 5314. [Google Scholar]
- Thompson M. P.; Randolph L. M.; James C. R.; Davalos A. N.; Hahn M. E.; Gianneschi N. C. Polym. Chem. 2014, 5, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin P. A.; Smith D.; Nguyen S. T. Chem. Commun. 2005, 3793. [DOI] [PubMed] [Google Scholar]
- Rao N. V.; Mane S. R.; Kishore A.; Das Sarma J.; Shunmugam R. Biomacromolecules 2012, 13, 221. [DOI] [PubMed] [Google Scholar]
- Johnson J. A.; Lu Y. Y.; Burts A. O.; Xia Y.; Durrell A. C.; Tirrell D. A.; Grubbs R. H. Macromolecules 2010, 43, 10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M. C.; Mortell K. H.; Hegeman A. D.; Kiessling L. L. J. Mol. Catal. 1997, 116, 209. [Google Scholar]
- James C. R.; Rush A. M.; Insley T.; Vuković L.; Adamiak L.; Král P.; Gianneschi N. C. J. Am. Chem. Soc. 2014, 136, 11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad R. M.; Grubbs R. H. Angew. Chem., Int. Ed. 2009, 48, 8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M. E.; Randolph L. M.; Adamiak L.; Thompson M. P.; Gianneschi N. C. Chem. Commun. 2013, 49, 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard H. D.; Okada S. Y.; Grubbs R. H. J. Am. Chem. Soc. 2001, 123, 1275. [DOI] [PubMed] [Google Scholar]
- Patel P. R.; Kiser R. C.; Lu Y. Y.; Fong E.; Ho W. C.; Tirrell D. A.; Grubbs R. H. Biomacromolecules 2012, 13, 2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D. S.; Johnson R. N.; Pun S. H. J. Controlled Release 2012, 157, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. N.; Burke R. S.; Convertine A. J.; Hoffman A. S.; Stayton P. S.; Pun S. H. Biomacromolecules 2010, 11, 3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellinger J. G.; Pahang J. A.; Shi J.; Pun S. H. ACS Macro Lett. 2013, 2, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Schellinger J. G.; Pun S. H. J. Biol. Eng. 2013, 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke R. S.; Pun S. H. Bioconjugate Chem. 2010, 21, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata H.; Sanda F.; Endo T. Macromolecules 1997, 30, 2902. [Google Scholar]
- O’Brien-Simpson N. M.; Ede N. J.; Brown L. E.; Swan J.; Jackson D. C. J. Am. Chem. Soc. 1997, 119, 1183. [Google Scholar]
- Som A.; Tezgel A. O.; Gabriel G. J.; Tew G. N. Angew. Chem., Int. Ed. 2011, 50, 6147. [DOI] [PubMed] [Google Scholar]
- Kolonko E. M.; Kiessling L. L. J. Am. Chem. Soc. 2008, 130, 5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolonko E. M.; Pontrello J. K.; Mangold S. L.; Kiessling L. L. J. Am. Chem. Soc. 2009, 131, 7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky J. M.; Leslie S. J.; Arora P. S.; Beerman T. A.; Dervan P. B. Bioorg. Med. Chem. 2002, 10, 3313. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Melikov K.; Vives E.; Ramos C.; Verbeure B.; Gait M. J.; Chernomordik L. V.; Lebleu B. J. Biol. Chem. 2003, 278, 585. [DOI] [PubMed] [Google Scholar]
- Patel L. N.; Zaro J. L.; Shen W. C. Pharm. Res. 2007, 24, 1977. [DOI] [PubMed] [Google Scholar]
- Trabulo S.; Cardoso A. L.; Mano M.; De Lima M. C. P. Pharmaceuticals 2010, 3, 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover J. A.; Willingham M. C.; Pastan I. Cell 1984, 39, 283. [DOI] [PubMed] [Google Scholar]
- Vonderheit A.; Helenius A. PLoS Biol. 2005, 3, e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty G. J.; McMahon H. T. Annu. Rev. Biochem. 2009, 78, 857. [DOI] [PubMed] [Google Scholar]
- Lopez C. A.; de Vries A. H.; Marrink S. J. PLoS Comp. Biol. 2011, 7, e1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal S. K.; Skretting G.; Garred O.; Vilhardt F.; van Deurs B.; Sandvig K. Mol. Biol. Cell 1999, 10, 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidovetzki R.; Levitan I. Biochim. Biophys. Acta 2007, 1768, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan I. M.; Wadia J. S.; Dowdy S. F. J. Controlled Release 2005, 102, 247. [DOI] [PubMed] [Google Scholar]
- Nakase I.; Niwa M.; Takeuchi T.; Sonomura K.; Kawabata N.; Koike Y.; Takehashi M.; Tanaka S.; Ueda K.; Simpson J. C.; Jones A. T.; Sugiura Y.; Futaki S. Mol. Ther. 2004, 10, 1011. [DOI] [PubMed] [Google Scholar]
- Ouyang M.; Huang H.; Shaner N. C.; Remacle A. G.; Shiryaev S. A.; Strongin A. Y.; Tsien R. Y.; Wang Y. Cancer Res. 2010, 70, 2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl M.; Ding S.; Lee C. W.; Grubbs R. H. Org. Lett. 1999, 1, 953. [DOI] [PubMed] [Google Scholar]
- Trnka T. M.; Grubbs R. H. Acc. Chem. Res. 2000, 34, 18. [DOI] [PubMed] [Google Scholar]
- Chien M.-P.; Thompson M. P.; Lin E. C.; Gianneschi N. C. Chem. Sci. 2012, 3, 2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien M.-P.; Carlini A. S.; Hu D.; Barback C. V.; Rush A. M.; Hall D. J.; Orr G.; Gianneschi N. C. J. Am. Chem. Soc. 2013, 135, 18710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien M.-P.; Thompson M. P.; Barback C. V.; Ku T.-H.; Hall D. J.; Gianneschi N. C. Adv. Mater. 2013, 25, 3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickle D. F.; Presta L. G.; Dill K. A.; Rose G. D. J. Mol. Biol. 1992, 226, 1143. [DOI] [PubMed] [Google Scholar]
- Samarajeewa S.; Zentay R. P.; Jhurry N. D.; Li A.; Seetho K.; Zou J.; Wooley K. L. Chem. Commun. 2014, 50, 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. A.; Lu Y. Y.; Burts A. O.; Lim Y.-H.; Finn M. G.; Koberstein J. T.; Turro N. J.; Tirrell D. A.; Grubbs R. H. J. Am. Chem. Soc. 2010, 133, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price D. J.; Brooks C. L. 3rd J. Chem. Phys. 2004, 121, 10096. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; Hess B.; Lindahl E. Bioinformatics 2013, 29, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. J. Comput. Chem. 2004, 25, 1157. [DOI] [PubMed] [Google Scholar]
- Jakalian A.; Bush B. L.; Jack D. B.; Bayly C. I. J. Comput. Chem. 2000, 21, 132. [Google Scholar]
- Jakalian A.; Jack D. B.; Bayly C. I. J. Comput. Chem. 2002, 23, 1623. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Proteins 2010, 78, 1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. J. Chem. Phys. 1995, 103, 8577. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.