

Abstract
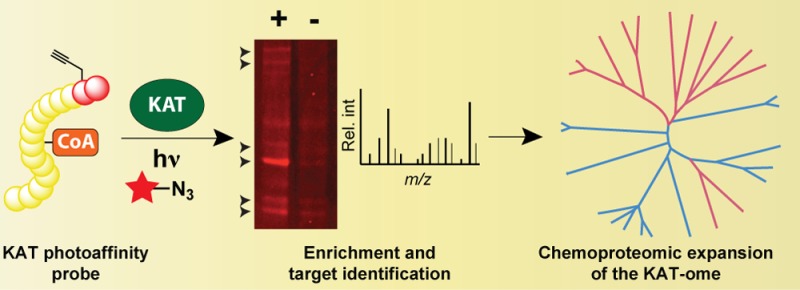
Lysine acetyltransferases (KATs) play a critical role in the regulation of gene expression, metabolism, and other key cellular functions. One shortcoming of traditional KAT assays is their inability to study KAT activity in complex settings, a limitation that hinders efforts at KAT discovery, characterization, and inhibitor development. To address this challenge, here we describe a suite of cofactor-based affinity probes capable of profiling KAT activity in biological contexts. Conversion of KAT bisubstrate inhibitors to clickable photoaffinity probes enables the selective covalent labeling of three phylogenetically distinct families of KAT enzymes. Cofactor-based affinity probes report on KAT activity in cell lysates, where KATs exist as multiprotein complexes. Chemical affinity purification and unbiased LC–MS/MS profiling highlights an expanded landscape of orphan lysine acetyltransferases present in the human genome and provides insight into the global selectivity and sensitivity of CoA-based proteomic probes that will guide future applications. Chemoproteomic profiling provides a powerful method to study the molecular interactions of KATs in native contexts and will aid investigations into the role of KATs in cell state and disease.
Introduction
Lysine acetylation plays a critical role in the regulation of transcription, metabolism, and other central biological functions. Acetylation of lysine residues can impact protein and genome function through multiple mechanisms, including physical relaxation of histone–DNA interactions,1 recruitment of bromodomain-containing effector proteins,2 covalent active-site modification,3 and regulation of protein stability.4 Lysine acetylation is a dynamic PTM that represents an equilibrium between the activity of two opposing enzyme classes: lysine acetyltransferase (KAT) enzymes, which impose the mark, and lysine deacetylases (KDACs), which remove it.5 While KDACs have been extensively investigated as epigenetic drug targets, several analyses indicate KATs can also drive cellular transformation and cancer progression in a tissue-specific manner. For example, fusion of the KAT enzyme MOZ to TIF2 is the primary genetic lesion associated with a subset of leukemias and imbues differentiated red blood cells with cancer stem cell-like properties.6 Nonmutant KAT activities can also support oncogenic gene expression programs, functioning as essential coactivators for transcription factors such as c-Myc and E2A-PBX gene fusions in cancer.7
Proteome-wide studies have revealed lysine acetylation is a prevalent PTM, rivaling phosphorylation in terms of substrate diversity with ∼4700 human acetylation sites identified to date.8−10 However, in contrast to the hundreds of known protein kinases, a recent phylogenetic analysis identified only 18 KATs in the human genome.11 The majority of these canonical KATs fall into four families: GCN5/PCAF, P300/CBP, MYST, and NCOA, with the rest consisting of transcription factor-related and orphan (sequence disparate) KAT activities (Supplementary Figure S1). KAT family members demonstrate significant intrafamily but little interfamily sequence homology, hindering bioinformatics approaches to KAT discovery and classification. The functional characterization of KATs is also limited by their regulation by protein partners and PTMs, factors that are difficult to recapitulate in vitro.12,13 Furthermore, while individual KATs have been shown to be susceptible to inhibition by small molecules and cellular acetyl-CoA/CoA ratio, methods for comparing the selectivity of these perturbations among multiple KATs in parallel do not exist.14,15 Thus, our ability to discover and characterize acetylation-mediated signaling would be greatly advanced by the development of new methods for the global analysis of KAT activity in cellular contexts.
Chemoproteomic profiling provides a powerful alternative to traditional biochemical assays for measuring enzyme activity in complex biological settings. In this approach, also commonly referred to as activity-based protein profiling (ABPP), active-site probes for an enzyme class of interest are modified with chemical handles enabling detection or affinity enrichment. Covalent labeling or enrichment of an enzyme by the affinity probe is then used as a proxy for enzyme activity.16 Chemoproteomic probes for KDAC enzymes have been used to discover novel KDAC complexes17 and characterize inhibitor selectivity in cell lysates.18 Similar approaches have also been pursued to study KAT enzymes, utilizing electrophile-containing analogues of the CoA cofactor.19,20 However, these probes have not been widely applied to profile KAT activity, owing to the fact that most KATs do not utilize mechanisms involving active-site nucleophiles.21
Here we report a general strategy for chemoproteomic profiling of KAT activity (Figure 1a). Bisubstrate inhibitors targeting three phylogenetically distinct KAT families were converted to clickable photoaffinity probes to enable KAT labeling and detection. Cofactor-based affinity probes quantitatively report on KAT-inhibitor interactions, are applied to identify a previously unknown acyltransferase activity possessed by the canonical KAT enzyme Gcn5, and report on KAT activity in cell lysates. Affinity purification and unbiased LC–MS/MS profiling of probe targets led to the identification of two noncanonical KAT enzymes, highlighting the existence of several orphan lysine acetyltransferases present in the human genome. In addition to providing insight into the global selectivity and sensitivity of CoA-based chemical proteomic probes that will guide future applications, these studies demonstrate the ability of chemical tools for profiling KAT activity to provide new insights into KATs and their molecular interactions in complex biological contexts.
Figure 1.
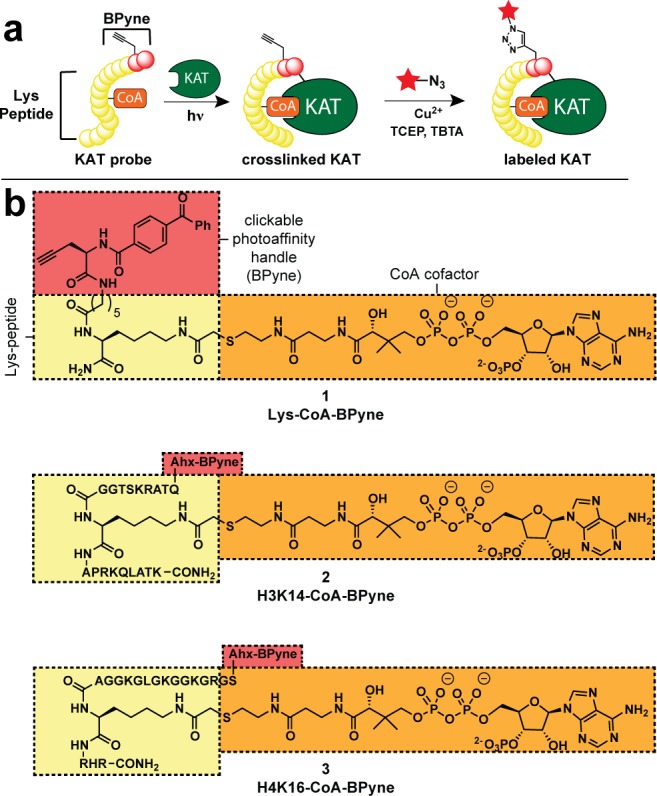
Cofactor-based affinity probes for the analysis of KAT activity. (a) Clickable photoaffinity labeling scheme. (b) Structures of KAT probes 1–3. Ahx = 6-aminohexanoic acid.
Results and Discussion
Synthesis and Evaluation of Chemoproteomic Probes for KAT Enzymes
We envisioned a general strategy for chemical profiling of KAT activity based on combining molecular recognition elements from KAT bisubstrate inhibitors with a clickable photoaffinity tag for covalent cross-linking and detection (Figure 1a). Pioneered by Cole and co-workers, KAT bisubstrate inhibitors link CoA with the ε-amino group of a lysine-containing peptide to form high affinity interactions with both the substrate and cofactor binding sites of KAT enzymes.22 These molecules inhibit KAT activity with nanomolar potencies, with selectivity for specific KATs encoded by the sequence of the bisubstrate peptide. On the basis of literature precedent, we hypothesized that modification of the N-termini of KAT bisubstrate inhibitors might be tolerated without a large loss in inhibitory potency. This provides a potential site for incorporation of the clickable photoaffinity element benzophenone-l-propargylglycine (BPyne; Figure 1), necessary for covalent labeling and detection.23,24 To test the scope of this approach, we synthesized a suite of KAT probes based on bisubstrate scaffolds that have been shown to target three major families of KATs: P300/CBP (Lys-CoA-BPyne; 1), GCN5/PCAF (H3K14-CoA-BPyne; 2), and MYST (H4K16-CoA-BPyne; 3) (Figure 1; Supplementary Scheme S1).22,25 KAT probes 1–3 were constructed from BPyne-peptide-bromoacetamide precursors, synthesized on Rink amide resin utilizing an orthogonal Lys-Dde protecting group and postcleavage HPLC purification. Nucleophilic displacement of BPyne-peptidyl-bromoacetamides, with commerical CoA, followed by final HPLC purification provided probes 1–3 on scales (1–100 μmol) sufficient for biological evaluation (Supplementary Scheme S1).
To test the affect of our structural modifications on molecular recognition of KAT enzymes, we assayed probes 1–3 against recombinant p300, pCAF, and Mof (MYST1) and compared their inhibitory activity to that of non-BPyne-containing “parent” inhibitor scaffolds (4–6; Supplementary Figure S2). Assayed at a single concentration (1 μM), probes 1–3 demonstrate inhibitory potencies and selectivities that closely mimic those of parent inhibitor scaffolds 4–6 (Supplementary Figures S3 and S4). Dose-response analysis of Lys-CoA-BPyne (1) demonstrated an IC50 of 26.7 nM toward p300 (95% confidence interval [CI95] = 11.75–60.64), within error of the inhibition by parent compound 4 (IC50 = 34.5 nM, CI95 = 17.5–67.8 nM; Supplementary Figure S5). Similar results are seen upon comparison of H3K14-CoA-BPyne 2 and parent bisubstrate 5. Together, these results suggest the BPyne subunit has minimal effects on KAT active-site recognition.
Selective In Vitro Labeling of KAT Enzymes
Next, we evaluated the utility of 1–3 as KAT labeling reagents in vitro. KAT probes 1–3 were incubated with purified recombinant KATs, photoirradiated at 365 nm, and subjected to Cu-catalyzed [3 + 2] cycloaddition (“click chemistry”) with a fluorescent azide tag.26 SDS-PAGE analysis of labeling reactions demonstrated dose-dependent fluorescent labeling of KATs at probe concentrations between 1 and 10 μM. Notably, each probe showed sensitive detection of the KAT enzyme family it was designed to target, i.e., KAT probe 1 reacted effectively with p300 (P300/CBP), probe 2 with pCAF (GCN5/PCAF),22 and probe 3 with Mof (MYST) (Figure 2a).25 Fluorescent labeling required probe, photo-cross-linking, and Cu2+; omission of any single component abolished labeling (Supplementary Figure S6). KAT probes 2 and 3, which are ∼3 kDa, caused a significant gel shift upon photoirradiation that was visible upon Coomassie staining (Figure 2a, Supplementary Figure S6). This property allowed us to quantify and optimize our photo-cross-linking conditions using gel densitometry. In our optimized photo-cross-linking protocol, we found that ∼33–40% of pCAF was covalently labeled by 2 after 60 min of irradiation at 365 nm on ice. These conditions are consistent with the literature,27 and cross-linking yields were not found to increase with extended photoirradiation times.
Figure 2.
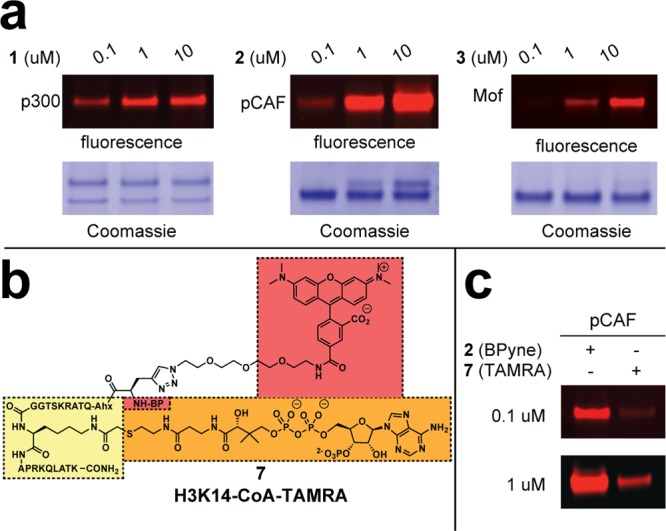
(a) Concentration dependence of probe labeling for preferred KAT partners. (b) Structure of fluorescent KAT photoaffinity probe 7. (c) Relative labeling of pCAF by clickable (left) and fluorescent (right) photoaffinity probes at low (0.1 μM) and high (1 μM) probe concentrations.
The alkyne handle of probes 1–3 was chosen for its minimal footprint and versatility toward conjugation of diverse azide reporters. However, it is not strictly necessary for ex vivo affinity profiling applications. In order to evaluate the utility of our bioorthogonal detection strategy, we compared the labeling of pCAF by clickable probe 2 with a fluorescent KAT photoaffinity probe, TAMRA-H3K14-CoA 7 (Figure 2b). Both probes facilitate fluorescent detection of pCAF at high probe concentrations (1 μM), suggesting fluorescent bisubstrates may be useful profiling agents for KAT enzymes. However, clickable probe 2 exhibits visibly greater labeling than fluorescent probe 7 at low probe concentrations (0.1 μM; Figure 2b). This suggests click chemistry detection strategies improve probe sensitivity, possibly by abrogating negative interactions of the fluorescent TAMRA reporter on probe-protein recognition that reduce photo-cross-linking.28
Moving toward more complex settings, we assessed the specificity and selectivity of each KAT probe (1–3) using a cocktail composed of p300, pCAF, and the MYST family acetyltransferase Mof. Specific probe labeling events were defined as those susceptible to competition by 50–100 equiv of parent KAT bisubstrate inhibitors 4–6 (Supplementary Figure S7), while selectivity refers to the subset of KAT enzymes labeled by each probe. Interestingly, each probe showed specific labeling of a unique subset of KAT enzymes at 1 μM, even in the presence of other KAT superfamily members. For example, Lys-CoA-BPyne 1 showed strong labeling of p300 and pCAF but did not significantly label the MYST family member Mof (Figure 3). The p300 signal was specifically competed in the presence of excess parent inhibitor, while a small portion of the pCAF signal remained, suggesting a combination of specific and nonspecific interactions between the 1-pCAF pair. H3K14-CoA-BPyne 2 demonstrated a clear preference for specific labeling of pCAF and weaker, but detectable, labeling of Mof (Figure 3/Supplementary Figure S8). H4K16-CoA-BPyne 3 most strongly labeled the MYST family member Mof and exhibited fainter labeling of p300 and pCAF (Figure 3). In addition to its parent bisubstrate, the Mof-3 interaction was competed by excess Lys-CoA (4), which had no similar effect on the interaction of pCAF with 2 (Supplementary Figure S8). For all three probes, selectivities were found to be reduced at higher probe concentrations (10 μM), where 1–3 exhibit considerable cross-reactivity (Supplementary Figure S8). Given its combination of broad-spectrum reactivity and straightforward synthesis of probe and competitor, these findings suggest Lys-CoA-BPyne 1 may be the most well-suited of the probes for applications requiring a general chemoproteomic reporter of KAT activity. In contrast, when applied at suitably low concentrations, peptidyl-KAT probes 2 and 3 may be better suited for applications that require more selective labeling of specific KAT families or as components of KAT chemoproteomic probe cocktails designed to achieve broad superfamily coverage.28
Figure 3.

Selectivity of KAT labeling by probes 1–3 (1 μM) assayed in a mixture of proteins from the P300/CBP, GCN5/PCAF, and MYST family. Specific labeling events show sensitivity to competition by parent bisubstrate inhibitors (4–6; 200 equiv).
Chemoproteomic Probes Report on KAT–Small Molecule and KAT–Cofactor Interactions
Having demonstrated the ability of our probes to label three classes of KATs, we next sought to investigate their ability to report on changes in KAT activity resulting from exposure to diverse molecular stimuli. First we investigated their ability to report on the affinity and selectivity of small molecule inhibitors.18,29 Co-incubation of pCAF with the known KAT inhibitor garcinol decreased labeling by KAT probe 2 in a dose-dependent manner (Figure 4). Quantification of fluorescent pCAF labeling by gel densitometry yielded an IC50 of 4.5 ± 1.2 μM for garcinol, consistent with the literature IC50 value of 5 μM.30 Similarly, labeling of p300 by Lys-CoA BPyne 1 was sensitive to inhibition by the small molecule inhibitor C646 (Supplementary Figure S9).31
Figure 4.
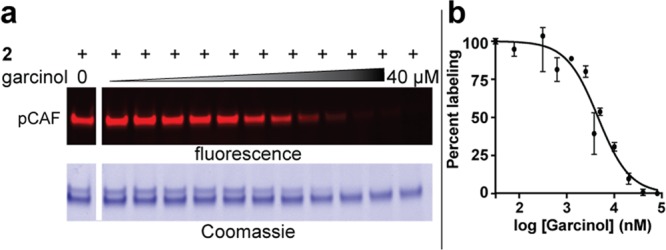
Competitive profiling of KAT active-site occupancy. (a) Fluorescent and Coomassie gels from pCAF-garcinol competition experiment. Conditions: 2 (1 μM), garcinol (0, 0.08, 0.16, 0.33, 0.63, 1.26, 2.5, 3.75, 5, 10, 20, 40 μM). (b) Dose-response analysis of competitive labeling generated via gel densitometry analysis of fluorescent labeling by KAT probe 2.
In addition to small molecule inhibition, recent studies have suggested KAT activity may be sensitive to changes in cellular acyl-CoA pools, providing a potential mechanism to link changes in the metabolic state of the cell to differential histone acylations and epigenetic control of gene expression.15,32 To test whether chemoproteomic probes could provide insight into these mechanisms, we investigated the ability of four different acyl-CoAs (acetyl, propionyl, butyryl, and crotonyl-CoA) to compete with 2 for the active site occupancy of Gcn5, a KAT whose activity has been proposed to be metabolically regulated (Figure 5).15,33 We found that high concentrations of acetyl-CoA efficiently competed labeling by probe 2, consistent with its role as a universal KAT cofactor. Propionyl-CoA also antagonized labeling, while butyryl-CoA was a partial antagonist, and crotonyl-CoA did not impede labeling (Figure 5b). While the ability of Gcn5 to utilize propionyl- and butyrl-CoA as cofactors has not been previously explored, our results are consistent with a previous biochemical analysis of human pCAF (which has a highly homologous KAT domain). These studies indicated a kinetic preference for acyl group donors of acetyl-CoA (kcat/Km ≈ 535 s–1 M–1) > propionyl-CoA (kcat/Km ≈ 92 s–1 M–1) ≫ butyrl-CoA and that malonyl-CoA was not utilized as a substrate.34 Indeed, we confirmed Gcn5 utilized propionyl- and butyrl-CoA as substrates via LC–MS/MS analysis (Supplementary Figure S10).
Figure 5.
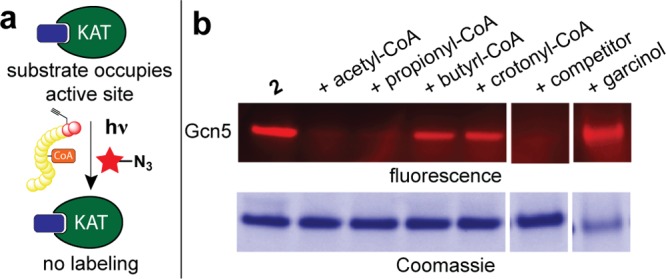
(a) Scheme for competitive substrate profiling. (b) Fluorescent and Coomassie gels from Gcn5-acyl-CoA competition experiment. Conditions: 2 (10 μM), acyl-CoAs (1000 μM), garcinol (40 μM).
These data suggest competitive affinity profiling provides a useful approach to rapidly gain new insights into KAT inhibitor and substrate selectivity.
Chemical Affinity Profiling of KAT Activity in Cell Lysates
The activity of KAT enzymes such as Gcn5 and pCAF is ideally studied in cellular settings. Recent commentary has suggested the failure of conventional high-throughput screening campaigns to yield selective KAT inhibitors may be due to the inability of these screens to interrogate the ability of small molecules to disrupt native KATs, which can exist as multiprotein complexes.11 Therefore, methods to monitor KAT activity directly from cell extracts are potentially valuable for next-generation inhibitor discovery efforts. Overexpression extracts of epitope-labeled ATAC (Ada Two-A containing) complex constitute an advanced model system applied for the study of GCN5 family KATs in their endogenous setting.35 We thus asked whether we could use chemoproteomic probe 2 to directly detect KAT activity in these systems, without the need for prefractionation, affinity purification, or antibodies. Accordingly, pCAF overexpression extracts from HEK-293 cells were treated with 2, followed by photo-cross-linking and click chemistry. Weak but detectable labeling of a protein corresponding to the molecular weight of pCAF was observed by fluorescence (Figure 6a). Notably, labeling was sensitive to competition by H3K14-CoA, and immunoblotting confirmed comigration with pCAF. To further verify labeling, we subjected proteins labeled by 2 in pCAF overexpression extracts to click chemistry with biotin azide, followed by affinity purification, tryptic digest, and LC–MS/MS protein identification. Notably, pCAF peptides were specifically identified in overexpression extracts treated with 2 (Figure 6b), but not in extracts pretreated with excess competitor 5 or lysates derived from HEK-293 cells transfected with an empty vector control (Figure 6a, bottom, Supplementary Table S1). In addition to overexpression extracts, clickable photoaffinity probes 1 and 2 are capable of detecting p300 and pCAF, respectively, when spiked into HeLa cell proteomes (Supplementary Figure S11). We used this characteristic to assess the sensitivity of probe 2 and estimate a lower limit at which GCN5/PCAF family enzymes may be detected. This is especially relevant since our inability to enrich pCAF from empty vector transfected HEK-293 lysates indicates expression level may be a limiting factor for KAT identification. We found that probe 2 could detect as low as 2.5 pmols of pCAF in a standard gel-based experiment against a proteomic background (Figure 6b). These findings calibrate the ability of chemoproteomic probes to monitor KAT activity in model systems and cell lysates and may be useful for the development of chemoproteomic approaches to screen for selective inhibitors of KATs and KAT-containing multiprotein complexes.36,37
Figure 6.
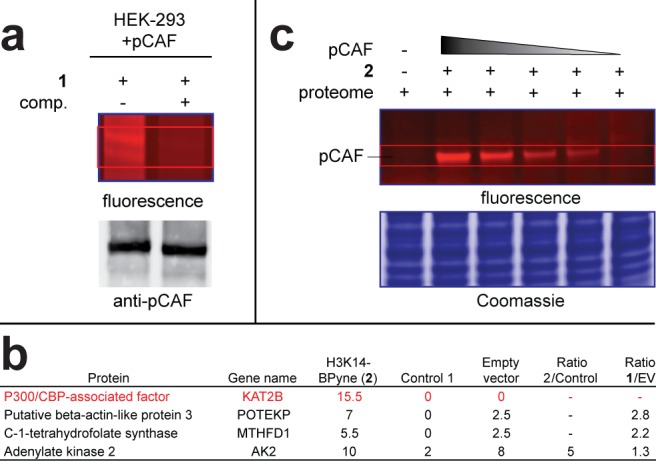
Labeling of KATs in cell lysates. (a) Labeling of pCAF in HEK-293 overexpression extracts. Comp. = competitor. (b) Proteins identified in LC–MS/MS experiments as targets of H3K14-CoA-BPyne 2 in pCAF transfected HEK-293 extracts. Control = competitor treated lane, EV = extract derived from HEK-293 cells transfected with empty vector. (c) Limit of detection of recombinant pCAF spiked into HeLa cell proteome. Conditions: 2 (1 μM), proteome (7 μg), recombinant pCAF (0, 6.25, 5, 3.75, 2.5, 1.25 pmol).
Chemoproteomic Profiling of KAT Activity: Probe Reactivity and Orphan KATs
Having demonstrated the utility of chemoproteomic probes for the targeted study of KAT activity in cellular contexts, we last sought to explore their utility for the discovery of potentially novel KAT activities directly from cancer cell proteomes. Labeling of whole cell extracts by 1–3 demonstrated a distinct pattern of specific protein labeling events for each probe, with KAT probe 1 targeting the largest number of proteins (Figure 7). To identify the proteomic targets of broad-spectrum KAT probe 1, HeLa cells were lysed, photo-cross-linked in the presence of 1, and subjected to click chemistry with biotin-azide followed by streptavidin enrichment, tryptic digest, and LC–MS/MS analysis. We identified 16 proteins that were abundant (>5 spectral counts) and showed >5-fold preferential enrichment in the absence of competitor (Figure 7b, Supplementary Table S2). Peptides matching canonical KAT p300 and CBP were not observed in our MS/MS data set, likely due to their low abundance in whole cell lysates (confirmed by Western blot; Supplementary Figure S12). Of the 16 proteins, 2 were acetyltransferases (green), 5 were CoA-binding proteins related to primary metabolism (red), with the remaining hits composed of highly abundant proteins and members of the proteasome regulatory complex. To validate LC–MS/MS identifications, we performed probe labeling experiments of two overexpressed and purified targets of 1: ATP-citrate lyase (Acly), the highest abundance pulldown target of 1, and N-α-acetyltransferase 50 (Naa50), an acetyltransferase. Both enzymes exhibited competitor-sensitive labeling, indicative of specific molecular recognition by KAT probe 1 (Figure 7c). Conserved domain analysis indicated that each protein specifically labeled by 1 either contains or is closely associated with a protein containing a binding site for an adenine nucleotide-containing cofactor (CoA, ATP, or NAD(P); Figure 7c). The off-target engagement of adenosine-binding proteins by KAT probe 1 parallels the widespread reactivity that has been observed for ATP- and GTP-containing chemical proteomic probes in biological settings.38−40 This is likely to be a general limitation of chemical proteomic probes incorporating adenine cofactor-based chemical scaffolds and highlights opportunities for design improvements as well as the requirement for orthogonal validation strategies to support chemical proteomic KAT discovery efforts.
Figure 7.

Cofactor-based affinity profiling of endogenous KAT activity in a cancer cell proteome. (a) Labeling of HeLa cell proteomes by KAT probes 1–3 (10 μM). Specific labeling events show sensitivity to competition by parent bisubstrates (100 equiv). (b) Proteins identified in LC–MS/MS experiments as targets of Lys-CoA-BPyne 1. Values represent the average spectral counts of two biological replicates. Green, acetyltransferases; Red, CoA-binding proteins. (c) Affinity labeling of recombinant protein verifies Acly and Naa50 as targets of 1.
In our data set, two acetyltransferases were enriched: Nat10 and Naa50. Nat10 is a noncanonical KAT (referred to here as orphan KATs) with a GNAT-related fold that displays histone and microtubule acetyltransferase activity in cells. Biologically, Nat10 has been observed to play a role in the regulation of telomerase function and nuclear shape and was recently identified as a druggable target for the treatment of Hutchinson–Gilford progeria syndrome.41,42 Naa50 (Nat13) is the catalytic component of the NatE acetyltransferase complex that is required for proper sister chromatid adhesion and chromatin condensation in vivo.43,44 The identification of Naa50 as a target of Lys-CoA-BPyne 1 initially struck us as paradoxical, as this enzyme belongs to the N-terminal acetyltransferase (NAT) family and has been shown to favor protein acetylation of N-terminal Met residues.45,46 However, literature investigation revealed that, of the 7 NAT catalytic subunits encoded in the human genome, Naa50 is the only member to have biochemically characterized ε-lysine acetyltransferase activity, providing a molecular rationale for its targeting by 1.47 The selective identification of Naa50 by 1 suggests that chemoproteomic profiling may have applications in identifying new KAT activities present in acetyltransferase families that are distinct in sequence from canonical KATs.
The proteomic identification of two orphan KAT activities by affinity probe 1 led us to re-evaluate the lysine acetyltransferase literature and consider whether other KAT activities were also missing from the list of 18 canonical human KATs (Supplementary Figure S1). This analysis identified 14 proteins, including Nat10 and Naa50, not in the list of 18 canonical KATs for which evidence of lysine acetyltransferase activity has been observed. Sequence alignment and similarity analyses were used to construct an expanded phylogenetic tree of acetyltransferase proteins, divided into canonical (P300/CBP, GCN5/PCAF, MYST, NCOA) and orphan KATs (Figure 8). Notably, many KATs from the initial list of 18 (ATAT1, TF3C4, ELP3; Supplementary Figure S1) cluster more closely with orphan KATs, indicative of greater sequence similarity. Together, these proteins encompass the most comprehensive list of human KATs assembled to date. However, we take care to point out that the experimental evidence supporting the activity of all 32 KATs, canonical and orphan, ranges widely. In particular, this list contains two proteins (Oga/Ncoat, an orphan KAT, and Src1, a canonical KAT) for which conflicting observations of KAT activity have been made.48,49 These discrepancies support the need for universal methodologies capable of directly assaying KAT activity in endogenous cells, toward which our current study provides an initial step. In the meantime, our chemoproteomics-inspired census of KAT activity provides fertile ground for functional investigation of orphan KAT enzymes using traditional structural and biochemical approaches, studies that may facilitate a more complete understanding of lysine acetylation in living systems.
Figure 8.
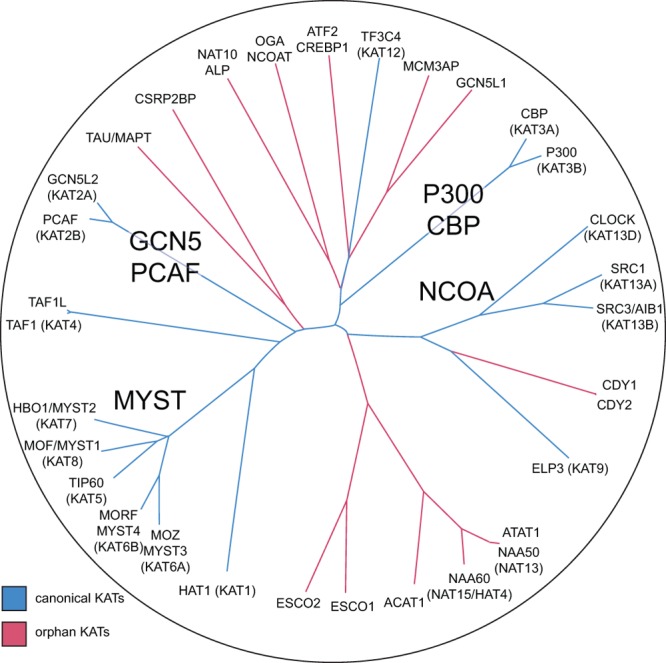
Expanded phylogenetic tree of KAT enzymes, including canonical KATs and orphan KAT activities observed in this study or annotated in the literature. KATs are denoted by gene name and relevant pseudonyms. Uniprot accession numbers and literature references for KAT activity are provided in the Supporting Information (Table S3).
Conclusion
In summary, here we have described a suite of probes for cofactor-based affinity profiling of KAT activity. We have defined key structural features necessary for the covalent labeling and detection of three families of KAT enzymes and demonstrated the utility of these probes to monitor KAT activity in settings ranging from purified enzymes to KAT overexpression extracts to native proteomes. Chemoproteomic probes of KAT activity provided new insights into KAT inhibitor and cofactor selectivity and highlighted the existence of several noncanonical orphan KAT activities that may contribute to cellular acetylation signaling pathways. In addition to these advances, it is also important to call attention to the limitations of our initial study. For example, while we demonstrated the ability of KAT affinity probes to report on pCAF activity in HEK-293 overexpression extracts, we were unable to detect canonical KAT enzymes such as CBP and Gcn5 directly from HeLa proteomes. This may be due to the low abundance of these KATs in whole cell lysates (Supplementary Figure S13) or the low cross-linking yields of our clickable photoaffinity probes, which covalently label only ∼33–40% of recombinant pCAF in vitro. This challenge may be addressed in future studies through scale-up, nuclear prefractionation, or utilization of multidimensional protein identification technology (MuDPIT) to increase LC–MS/MS detection sensitivity.50 Alternatively, chemical proteomic studies of kinase and KDAC activity have shown that noncovalent affinity probe resins can enable enrichment of specific enzyme classes without the need for photo-cross-linking,18,51 providing another potential route for the analysis of low abundance KATs. Furthermore, while the activity of KAT complexes have been shown to be preserved in cell extracts, these activities would be ideally studied in living cells and probes 1–3 are not cell-permeable. Cell-penetrating peptides have been used to promote uptake of KAT bisubstrate inhibitors, and similar approaches may facilitate live cell profiling of KAT activity.23 These improvements will be important to expand the scope of chemical proteomic analyses of KAT activity. Regardless, the ability of our current suite of chemoproteomic probes to highlight an expanded landscape of catalytic lysine acetylation provides an example of the power of this approach as currently constituted and sets the stage for the development of chemoproteomic strategies to identify KAT inhibitors and the functional characterization of canonical and orphan KAT activities in cellular settings. Such studies are currently underway, and will be reported in due course.
Experimental Details
Biochemistry and Cell Biology
Recombinant pCAF, catalytic domain (aa 492–658) was obtained from Cayman Chemical. Recombinant p300, catalytic domain (aa 1284–1673) and recombinant Gcn5, catalytic domain (497–663), were obtained from Enzo. The plasmid encoding recombinant MOF, catalytic domain (aa 147–449) was obtained from Addgene. His-tagged recombinant Mof was expressed in E. coli BL21 and purified via immobilized nickel affinity chromatography using standard conditions. Acly was obtained from US Biological. Naa50 was obtained from Origene. Garcinol and C646 were obtained from Cayman Chemical. Streptavidin-agarose was purchased from Pierce. SDS-PAGE was performed using Bis-Tris NuPAGE gels (4–12%) and MES running buffer in Xcell SureLock MiniCells (Invitrogen) according to the manufacturer’s instructions. SDS-PAGE fluorescence was visualized using an ImageQuant Las4010 Digitial Imaging System (GE Healthcare). Total protein content on SDS-PAGE gels was visualized by Blue-silver coomassie stain, made according to the published procedure.52 Separation-based assays for KAT activity were performed on a LabChip EZ Reader instrument (PerkinElmer) kindly provided by Dr. Jay Schneekloth. Fluorescence assays for the KAT enzyme Mof were analyzed on a Biotek Synergy 2 (Biotek).
KAT Inhibition Assays
Recombinant KAT activity was measured by electrophoretic mobility shift assay (EMSA) as previously reported.53 This assay measures the separation of FITC-labeled KAT substrate peptides (Histone H3 5-23 QTARKSTGGKAPRKQLATK-Ahx-FITC; Histone H4 1-19 SGRGKGGKGLGKGGAKRHR-Ahx-FITC) from their acetylated products following incubation with recombinant KAT and acetyl-CoA. A model separation is shown in Supplementary Figure S14. P300 and pCAF assays were performed in 30 μL of reaction buffer (50 mM HEPES, pH 7.5, 50 mM NaCl, 2 mM EDTA, 0.05% Tween 20, 10 μg/mL BSA) with KAT (p300 [50 nM] or pCAF [10 nM]) and FITC-peptide (FITC-H4 for P300; FITC-H3 for pCAF; 1 μM). Acetylation of FITC-H3/H4 peptide by p300 and pCAF was confirmed by LC–MS. Reactions were plated in 384-well plates, allowed to equilibrate at room temperature for 10 min, and initiated by addition of acetyl-CoA (final concentration = 5 μM). Plates were then transferred to a Lab-Chip EZ-Reader at ambient temperature and analyzed by microfluidic electrophoresis. Optimized separation conditions were downstream voltage of −400 V, upstream voltage of −2900 V, and a pressure of −2.0 psi for FITC-H3 and downstream voltage of −500 V, upstream voltage of −1500 V and a pressure of −2.0 psi for FITC-H4. Percent conversion is calculated by ratiometric measurement of substrate/product peak heights. Percent activity represents the percent conversion of KAT reactions treated with inhibitors 1–6 relative to untreated control KAT reactions, measured in triplicate, and corrected for nonenzymatic acetylation. Mof showed low activity toward FITC-H3/H4 peptide substrates and was monitored by fluorogenic KAT assay using an unlabeled H4 substrate peptide as previously reported.54 Dose-response analysis of p300 and pCAF inhibition by KAT probes 1 and 2 and parent inhibitors 4 and 5 were performed in triplicate and analyzed by nonlinear least-squares regression fit to Y = 100/(1 + 10∧(Log IC50 – X)*H), where H = Hill slope (variable). IC50 values represent the concentration that inhibits 50% of KAT activity. All calculations were performed using Prism 6 (GraphPad) software.
Fluorescent Labeling of KAT Enzymes for SDS-PAGE Analysis
Purified KAT enzymes (0.5–5 μg) or whole cell proteomes (20 μg) were incubated with KAT probes 1–3 (1 μM probe for recombinant labelings; 10 μM probe for proteomic labelings) in PBS (pH 7.0) for 1 h. Control experiments to correct for nonspecific cross-linking were treated with 1–3 in the presence of 100 equiv of competitors 4–6. Following equilibration, samples were photo-cross-linked on ice for 1 h using a 365 nm UV light in a FB-UVXL-1000 UV cross-linker. Probe labeling was detected by Cu(I)-catalyzed [3 + 2] cycloaddition (“click chemistry”). Click reactions were initiated by sequential addition of TAMRA-azide 8 (100 μM; 5 mM stock solution in DMSO, structure given in Supplementary Figure S15), TCEP (1 mM; 100 mM stock in H2O), tris(benzyltriazolylmethyl)amine ligand (TBTA; 100 μM; 1.7 mM stock in DMSO/tert-butanol 1:4), and CuSO4 (1 mM; 50 mM stock in H2O). Samples were vortexed and incubated at room temperature for 1 h. Cycloaddition reactions were quenched by addition of 5x SDS-loading buffer (strongly reducing) and subjected to SDS-PAGE (22 μL per well). Excess probe fluorescence was removed by destaining in a solution of 50% MeOH/40% H2O/10% AcOH overnight. Gels were then washed with water and fluorescently visualized using a ImageQuant Las4010 (GE Healthcare) with green LED excitation (λmax 520–550 nm) and a 575DF20 filter. For KAT probes 2 and 3, a characteristic intense low molecular weight fluorescence signal ∼3 kDa was observed (corresponding to the fluorescently labeled KAT probe [2, 3]), indicative of a high-yielding click chemistry reaction.
Cell Culture and Isolation of Whole-Cell Lysates
HeLa S3 cells (ATCC; Manassas VA) were cultured at 37 °C under 5% CO2 atmosphere in a culture medium of DMEM supplemented with 10% FBS and glutamine. HEK-293 cells were obtained from the NCI Tumor Cell Repository. For isolation of whole cell proteomes, HeLa cells were grown to 80–90% confluency, washed 3× with ice-cold PBS, scraped, and pelleted by centrifugation (1400g × 3 min, 4 °C). After removal of PBS cell pellets were stored at −76 °C or immediately processed. Cell pellets were resuspended in 1–2 mL of ice-cold PBS (10–20 × 106 cells/mL) and lysed by sonication (QSonica XL2000 100 W sonicator, 3 × 10 s pulse, 50% power, 60 s between pulses). Lysates were pelleted by centrifugation (14,000g × 30 min, 4 °C) and quantified on a Qubit 2.0 Fluorometer using a Qubit Protein Assay Kit. Proteomes were diluted to 2 mg/mL and stored in 1 mg aliquots at −76 °C until further processing.
Western Blotting
SDS-PAGE gels were transferred to nitrocellulose membranes (Novex, Life Technologies) by electroblotting at 30 V for 1 h using a XCell II Blot Module (Novex). Membranes were blocked using StartingBlock (PBS) Blocking Buffer (Thermo Scientific) for 20 min and then incubated overnight at 4 °C in a solution containing the primary antibody of interest (anti-Gcn5 [3305], anti-CBP [3378], Cell Signaling, 1:1000 dilution) in the above blocking buffer with 0.05% Tween 20. The membranes were next washed with TBST buffer and incubated with a secondary HRP-conjugated antibody (anti-rabbit IgG, HRP-linked [7074], Cell Signaling, 1:1000 dilution) for 1.5 h at room temperature. The membranes were again washed with TBST, treated with chemiluminescence reagents (Western Blot Detection System, Cell Signaling) for 1 min, and imaged for chemiluminescent signal using an ImageQuant Las4010 Digitial Imaging System (GE Healthcare).
Enrichment of KAT Enzymes for Proteomic Analysis
Whole cell proteomes were adjusted to a final protein concentration of 1 mg/mL and incubated with the indicated probe (1 or 2; 10 μM) for 1 h. HEK-293 enrichments utilized 0.5 mg of proteome as starting material, while HeLa enrichments utilized 1 mg of proteome. Control samples to correct for nonspecific cross-linking were preincubated with each probe’s cognate competitor (4 or 5; 100 equiv). Following equilibration, samples were split into 5 × 200 μL aliquots and photo-cross-linked on ice for 1 h using a 365 nm UV light in a FB-UVXL-1000 UV cross-linker. Cross-linked samples were then recombined and subjected to Cu(I)-catalyzed [3 + 2] cycloaddition with TAMRA biotin-azide 9 (Supplementary Figure S15) as previously described. Final concentrations for click reactions were as follows: HeLa proteome (1 mg/mL in PBS), probe 1 (10 μM), TAMRA biotin-azide (40 μM), TCEP (1 mM), TBTA (100 μM), tert-butanol (4.8%), and CuSO4 (1 mM). Samples were vortexed and incubated at room temperature for 1 h. Ice-cold 4:1 MeOH/CHCl3 (2.5 mL) was then added directly to the reaction mixture and mixed vigorously by vortexing. The biphasic solution was centrifuged (4000g × 20 min, 4 °C), and protein precipitated at the interface as a solid disk. Liquid layers were carefully discarded, and the resulting precipitate was resuspended in ice-cold 1:1 MeOH/CHCl3 (1 mL), sonicated on ice to resuspend, and repelleted by centrifugation (14,000g × 10 min, 4 °C). This wash step was repeated with ice-cold MeOH (1 mL). The resulting cell pellet was air-dried to remove excess methanol and redissolved in 1.2% SDS (1 mL) using iterative cycles of heating (95 °C) and sonication. Redissolved protein was allowed to cool to room temperature and added to 5 mL of PBS to give a final SDS concentration of 0.2%. Samples were then treated with 100 μL of streptavidin-agarose resin (prewashed 3× with 1 mL of PBS) and rotated for 1 h at room temperature. Streptavidin-agarose bound samples were then washed sequentially with 0.2% SDS in PBS (3 × 10 mL) and PBS (3 × 10 mL). Samples were then prepared for on-bead digest by reduction with 10 mM tris(2-carboxyethyl)phosphine (TCEP) and alkylation with 12 mM iodoacetamide. Samples were diluted to 2 M urea with 50 mM Tris-Cl pH 8.0 (400 μL total volume), followed by addition of trypsin and 2 mM CaCl2. Digests were allowed to proceed overnight at 37 °C. After extraction, tryptic peptide samples were acidified to a final concentration of 5% formic acid and frozen at −80 °C for LC–MS/MS analysis.
Liquid Chromatography–Mass Spectrometry and Data Analysis
Tryptic peptides enriched by probe 1 were loaded onto a reverse phase capillary column and analyzed by LC separation in combination with tandem MS. Peptides were eluted using a gradient of 5–42% over 40 min with the flow rate through the column set at 0.20 μL/min. Data was collected in a dual-pressure linear ion trap mass spectrometer (ThermoFisher LTQ VelosPro) set in a data-dependent acquisition mode. The 15 most intense molecular ions in the MS scan were sequentially and dynamically selected for subsequent collision-induced dissociation (CID) using a normalized collision energy of 35%. Tandem mass spectra were searched against UniProt H. sapiens protein database (01-13 release) using SEQUEST (ThermoFisher). Search parameters were fixed as follows: (i) enzyme specificity: trypsin; (ii) variable modification: methionine oxidation and cysteine carbamidomethylation; (iii) precursor mass tolerance ±1.40 amu; and (iv) fragment ion mass tolerance ±0.5 amu. Only those tryptic peptides with up to two missed cleavage sites meeting a specific SEQUEST scoring criteria (Delta Correlation (ΔCn) ≥ 0.08 and charge state dependent cross correlation (Xcorr) ≥ 1.9 for [M + H]1+, ≥ 2.2 for [M + 2H]2+, and ≥3.1 for [M + 3H]3+) were considered as legitimate identifications. Spectral count values depicted in Figure 7 represent an average of two biological replicates. Raw spectral counts for biological duplicates of probe-enriched and control experiments are provided in Supplementary Table S2.
Phylogenetic Analysis
Amino acid sequences for canonical and orphan lysine acetyltransferases were obtained from Uniprot. Accession numbers are provided in Table S3 (Supporting Information). A pairwise alignment was generated using Clustal Omega,55 and a phylogenic tree was constructed using the neighbor-joining method. All phylogenetic trees were displayed in hyperbolic space using Hypertree, with branches of the tree designated by different colors and labeled by name where appropriate.56
Acknowledgments
The authors thank Dr. Ming Zhou (Laboratory of Proteomics and Analytical Technology) for LC–MS/MS analyses, Dr. Hans Luecke (NIDDK) for the pCAF overexpression plasmid, Dr. Brian Lewis (NCI) for helpful discussions, and Dr. Michael Giano of the Schneider lab for assistance with peptide synthesis. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (ZIA BC011488-01).
Supporting Information Available
Synthetic materials and methods, characterization data, and supplementary figures and schemes. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Pogo B. G.; Allfrey V. G.; Mirsky A. E. Proc. Natl. Acad. Sci. U.S.A. 1966, 55, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A.; Chitsaz F.; Abbasi A.; Misteli T.; Ozato K. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B.; Bunkenborg J.; Verdin R. O.; Andersen J. S.; Verdin E. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R.; Tao R.; Gao X.; Li T.; Zhou X.; Guan K. L.; Xiong Y.; Lei Q. Y. Mol. Cell 2013, 51, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. J. Nucleic Acids Res. 2004, 32, 959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntly B. J.; Shigematsu H.; Deguchi K.; Lee B. H.; Mizuno S.; Duclos N.; Rowan R.; Amaral S.; Curley D.; Williams I. R.; Akashi K.; Gilliland D. G. Cancer Cell 2004, 6, 587. [DOI] [PubMed] [Google Scholar]
- Dawson M. A.; Kouzarides T. Cell 2012, 150, 12. [DOI] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. Science 2009, 325, 834. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Xu W.; Jiang W.; Yu W.; Lin Y.; Zhang T.; Yao J.; Zhou L.; Zeng Y.; Li H.; Li Y.; Shi J.; An W.; Hancock S. M.; He F.; Qin L.; Chin J.; Yang P.; Chen X.; Lei Q.; Xiong Y.; Guan K. L. Science 2010, 327, 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Cao J.; Gao X.; Zhou Y.; Wen L.; Yang X.; Yao X.; Ren J.; Xue Y. Nucleic Acids Res. 2011, 39, D1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Nat. Rev. Drug Discovery 2012, 11, 384. [DOI] [PubMed] [Google Scholar]
- Lee K. K.; Workman J. L. Nat. Rev. Mol. Cell Biol. 2007, 8, 284. [DOI] [PubMed] [Google Scholar]
- Thompson P. R.; Wang D.; Wang L.; Fulco M.; Pediconi N.; Zhang D.; An W.; Ge Q.; Roeder R. G.; Wong J.; Levrero M.; Sartorelli V.; Cotter R. J.; Cole P. A. Nat. Struct. Mol. Biol. 2004, 11, 308. [DOI] [PubMed] [Google Scholar]
- Cai L.; Sutter B. M.; Li B.; Tu B. P. Mol. Cell 2011, 42, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen K. E.; Hatzivassiliou G.; Sachdeva U. M.; Bui T. V.; Cross J. R.; Thompson C. B. Science 2009, 324, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M. J.; Cravatt B. F. Chem. Rev. 2006, 106, 3279. [DOI] [PubMed] [Google Scholar]
- Salisbury C. M.; Cravatt B. F. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Hopf C.; Savitski M. M.; Dittmann A.; Grandi P.; Michon A. M.; Schlegl J.; Abraham Y.; Becher I.; Bergamini G.; Boesche M.; Delling M.; Dumpelfeld B.; Eberhard D.; Huthmacher C.; Mathieson T.; Poeckel D.; Reader V.; Strunk K.; Sweetman G.; Kruse U.; Neubauer G.; Ramsden N. G.; Drewes G. Nat. Biotechnol. 2011, 29, 255. [DOI] [PubMed] [Google Scholar]
- Yu M.; de Carvalho L. P.; Sun G.; Blanchard J. S. J. Am. Chem. Soc. 2006, 128, 15356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang Y.; Thompson P. R.; Wang L.; Jiang L.; Kelleher N. L.; Cole P. A. Angew. Chem., Int. Ed. 2007, 46, 7621. [DOI] [PubMed] [Google Scholar]
- Berndsen C. E.; Albaugh B. N.; Tan S.; Denu J. M. Biochemistry 2007, 46, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau O. D.; Kundu T. K.; Soccio R. E.; Ait-Si-Ali S.; Khalil E. M.; Vassilev A.; Wolffe A. P.; Nakatani Y.; Roeder R. G.; Cole P. A. Mol. Cell 2000, 5, 589. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Balasubramanyam K.; Cebrat M.; Buck D.; Guidez F.; Zelent A.; Alani R. M.; Cole P. A. J. Am. Chem. Soc. 2005, 127, 17182. [DOI] [PubMed] [Google Scholar]
- Xie N.; Elangwe E. N.; Asher S.; Zheng Y. G. Bioconjugate Chem. 2009, 20, 360. [DOI] [PubMed] [Google Scholar]
- Wu J.; Xie N.; Wu Z.; Zhang Y.; Zheng Y. G. Bioorg. Med. Chem. 2009, 17, 1381. [DOI] [PubMed] [Google Scholar]
- Speers A. E.; Cravatt B. F. Chem. Biol. 2004, 11, 535. [DOI] [PubMed] [Google Scholar]
- Dorman G.; Prestwich G. D. Biochemistry 1994, 33, 5661. [DOI] [PubMed] [Google Scholar]
- Sieber S. A.; Niessen S.; Hoover H. S.; Cravatt B. F. Nat. Chem. Biol. 2006, 2, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D.; Hardouin C.; Boger D. L.; Cravatt B. F. Nat. Biotechnol. 2003, 21, 687. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K.; Altaf M.; Varier R. A.; Swaminathan V.; Ravindran A.; Sadhale P. P.; Kundu T. K. J. Biol. Chem. 2004, 279, 33716. [DOI] [PubMed] [Google Scholar]
- Bowers E. M.; Yan G.; Mukherjee C.; Orry A.; Wang L.; Holbert M. A.; Crump N. T.; Hazzalin C. A.; Liszczak G.; Yuan H.; Larocca C.; Saldanha S. A.; Abagyan R.; Sun Y.; Meyers D. J.; Marmorstein R.; Mahadevan L. C.; Alani R. M.; Cole P. A. Chem. Biol. 2010, 17, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.; Su X.; He B. ACS Chem. Biol. 2012, 7, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaugh B. N.; Arnold K. M.; Denu J. M. ChemBioChem 2011, 12, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemhuis H.; Packman L. C.; Nightingale K. P.; Hollfelder F. ChemBioChem 2008, 9, 499. [DOI] [PubMed] [Google Scholar]
- Martinez E.; Palhan V. B.; Tjernberg A.; Lymar E. S.; Gamper A. M.; Kundu T. K.; Chait B. T.; Roeder R. G. Mol. Cell. Biol. 2001, 21, 6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson D. A.; Franke A. S.; Weitzel D. H.; Speer B. L.; Hughes P. F.; Hagerty L.; Fortner C. N.; Veal J. M.; Barta T. E.; Zieba B. J.; Somlyo A. V.; Sutherland C.; Deng J. T.; Walsh M. P.; MacDonald J. A.; Haystead T. A. ACS Chem. Biol. 2013, 8, 2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin D. A.; Brown S. J.; Rosen H.; Cravatt B. F. Nat. Biotechnol. 2009, 27, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli M. P.; Szardenings A. K.; Liyanage M.; Nomanbhoy T. K.; Wu M.; Weissig H.; Aban A.; Chun D.; Tanner S.; Kozarich J. W. Biochemistry 2007, 46, 350. [DOI] [PubMed] [Google Scholar]
- Sadler N. C.; Angel T. E.; Lewis M. P.; Pederson L. M.; Chauvigne-Hines L. M.; Wiedner S. D.; Zink E. M.; Smith R. D.; Wright A. T. PLoS One 2012, 7, e47996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George Cisar E. A.; Nguyen N.; Rosen H. J. Am. Chem. Soc. 2013, 135, 4676. [DOI] [PubMed] [Google Scholar]
- Fu D.; Collins K. Mol. Cell 2007, 28, 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrieu D.; Britton S.; Demir M.; Rodriguez R.; Jackson S. P. Science 2014, 344, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimenta-Marques A.; Tostoes R.; Marty T.; Barbosa V.; Lehmann R.; Martinho R. G. Dev. Biol. 2008, 323, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F.; Chu C. W.; Kong X.; Yokomori K.; Zou H. J. Cell Biol. 2007, 177, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F.; Chu C. W.; Kong X.; Yokomori K.; Zou H. J. Cell Biol. 2007, 177, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foyn H.; Jones J. E.; Lewallen D.; Narawane R.; Varhaug J. E.; Thompson P. R.; Arnesen T. ACS Chem. Biol. 2013, 8, 1121. [DOI] [PubMed] [Google Scholar]
- Evjenth R.; Hole K.; Karlsen O. A.; Ziegler M.; Arnesen T.; Lillehaug J. R. J. Biol. Chem. 2009, 284, 31122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butkinaree C.; Cheung W. D.; Park S.; Park K.; Barber M.; Hart G. W. J. Biol. Chem. 2008, 283, 23557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard H. M.; Harries J. C.; Hussain S.; Bevan C.; Heery D. M. Mol. Cell. Biol. 2001, 21, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessani N.; Niessen S.; Wei B. Q.; Nicolau M.; Humphrey M.; Ji Y.; Han W.; Noh D. Y.; Yates J. R. 3rd; Jeffrey S. S.; Cravatt B. F. Nat. Methods 2005, 2, 691. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. Nat. Biotechnol. 2007, 25, 1035. [DOI] [PubMed] [Google Scholar]
- Candiano G.; Bruschi M.; Musante L.; Santucci L.; Ghiggeri G. M.; Carnemolla B.; Orecchia P.; Zardi L.; Righetti P. G. Electrophoresis 2004, 25, 1327. [DOI] [PubMed] [Google Scholar]
- Fanslau C.; Pedicord D.; Nagulapalli S.; Gray H.; Pang S.; Jayaraman L.; Lippy J.; Blat Y. Anal. Biochem. 2010, 402, 65. [DOI] [PubMed] [Google Scholar]
- Trievel R. C.; Li F. Y.; Marmorstein R. Anal. Biochem. 2000, 287, 319. [DOI] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Soding J.; Thompson J. D.; Higgins D. G. Mol. Syst. Biol. 2011, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham J.; Sudarsanam S. Bioinformatics 2000, 16, 660. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.