

Abstract
Werner syndrome (WS) is a human chromosomal instability disorder associated with cancer predisposition and caused by mutations in the WRN gene. WRN helicase activity is crucial in limiting breakage at common fragile sites (CFS), which are the preferential targets of genome instability in precancerous lesions. However, the precise function of WRN in response to mild replication stress, like that commonly used to induce breaks at CFS, is still missing. Here, we establish that WRN plays a role in mediating CHK1 activation under moderate replication stress. We provide evidence that phosphorylation of CHK1 relies on the ATR-mediated phosphorylation of WRN, but not on WRN helicase activity. Analysis of replication fork dynamics shows that loss of WRN checkpoint mediator function as well as of WRN helicase activity hamper replication fork progression, and lead to new origin activation to allow recovery from replication slowing upon replication stress. Furthermore, bypass of WRN checkpoint mediator function through overexpression of a phospho-mimic form of CHK1 restores fork progression and chromosome stability to the wild-type levels. Together, these findings are the first demonstration that WRN regulates the ATR-checkpoint activation upon mild replication stress, preventing chromosome fragility.
INTRODUCTION
The Werner syndrome protein, WRN, is a member of the RecQ family of DNA helicases mutated in the cancer-prone disease Werner syndrome (WS). WS cells show a marked delay in S-phase progression and are extremely sensitive to agents perturbing DNA replication (1–3). Based on WRN enzymatic activities and substrate preferences in vitro, it is thought that WRN may participate in multiple DNA metabolic pathways in vivo, such as replication, recombination and repair (4–6). WRN has been implicated in the correct and fruitful recovery from replication fork arrest (1,7–8). Given a coordinate action of WRN and DNA polymerase delta in the replication of DNA substrates containing G4 tetraplex structures (9), the crucial requirement of the WRN helicase activity in maintaining common fragile site (CFS) stability and a role of WRN in alleviating stalled forks arrested at secondary structures formed at CFS FRA16D (10,11), WRN could work in the removal of the obstacles encountered by the replisome during replication to facilitate fork progression.
Several studies suggested a possible cross-talk between WRN and ATR (12–14). ATR deficiency is associated with CFS expression (15), and defects of other checkpoint proteins, including CHK1, lead to instability at CFS (16). Of interest, we previously demonstrated that WRN regulates CFS stability by acting in a pathway associated with ATR (10). Despite WS cells exhibit an ATR-like instability at CFS (10), and that WRN has been found phosphorylated by ATR under replication stress (12), there is no evidence of a functional requirement of WRN in the establishment of the replication checkpoint upon moderate replication perturbation such as the one that causes breaks at CFS.
In this study, we disclose a previous unidentified role for WRN in mediating the ATR-checkpoint activation under mild replication stress. Moreover, our findings provide a novel mechanistic framework to the WRN-dependent stabilization of CFS.
MATERIALS AND METHODS
Cell cultures
HEK293T cells were obtained from American Type Culture Collection (VA, USA). HEK293T cells proficient and deficient for WRN were generated by stably expressing, respectively, a scrambled shRNA (shCTRL) or shRNA against WRN (shWRN) (OriGene). Cells were cultured in the presence of hygromycin (200 μg/ml; EMD Chemicals Inc.) to maintain selective pressure for shRNA expression. AG11395 (WRN-deficient) fibroblasts retrovirally-transduced with full-length cDNA encoding wild-type WRN (WSWRN), missense-mutant form of WRN with inactive helicase (WRN-K577M) or expressing a Flag-tagged full-length WRN plasmid carrying Ala substitutions at all the six S/TQ sites (WSWRN6A) were generated as previously described (10,12).
All the cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% Fetal bovine serum (FBS) (Boehringer Mannheim) and incubated at 37°C in a humidified 5% CO2 atmosphere.
Plasmids and transfection
Plasmids expressing the Flag-tagged WRNwt or the Flag-tagged WRN6A were generated as earlier reported (12). The phospho-mimic (Flag-CHK1317/345D) mutant form of CHK1, a kind gift from Professor K.K. Khanna (Queensland Institute of Medical Research, Australia), was constructed as previously described (17). To express the plasmids, cells were transfected using the Neon™ Transfection System Kit (Invitrogen), according to the manufacturer's instructions.
Immunoprecipitation, cell fractionation and western blot analysis
Immunoprecipitation and chromatin fractionation experiments were performed as previously described (12,18). Quantification was performed on scanned images of blots using ImageJ software, and values shown on the graphs represent a percentage compared with the matched untreated control normalized against the protein content evaluated through Lamin B1 immunoblotting.
Cell lysates were prepared as previously reported (18). Antibodies used for western blot (WB) were commercially obtained for Flag-Tag (Sigma-Aldrich), phospho-S/TQ, phospho-CHK1-Ser345 (Cell Signaling Technologies), WRN, CHK1, Cyclin A, RAD9 (Santa Cruz Biotechnology, Inc.), Lamin B1 (Abcam), GAPDH (EMD Millipore), RPA32 (Calbiochem), TopBP1 (Bethyl Laboratories). Horseradish peroxidase-conjugated goat species-specific secondary antibodies (Santa Cruz Biotechnology, Inc.) were used.
Chromosomal aberration and fluorescence in situ hybridization analyses
Fragile sites were induced treating cells with aphidicolin (Aph; Sigma-Aldrich). Harvesting of the cells, metaphase chromosome preparations and fluorescence in situ hybridization (FISH) analysis was performed and analyzed as previously reported (10).
DNA fiber analysis
Cells were pulse-labeled with 25 μM chlorodeoxyuridine (CldU) and then labeled with 250 μM iododeoxyuridine (IdU) at the times specified, with or without treatment as reported in the experimental schemes. DNA fibers were prepared and spread out as previously described (12). For immunodetection of labeled tracks, the following primary antibodies were used: rat anti-CldU/BrdU (Abcam) and mouse anti-IdU/BrdU (Becton Dickinson). Images were acquired randomly from fields with untangled fibers using Eclipse 80i Nikon Fluorescence Microscope, equipped with a VideoConfocal (ViCo) system. The length of labeled tracks were measured using the Image-Pro-Plus 6.0 software, and values were converted into kilobase using the conversion factor 1 μm = 2.59 kb as reported (19). A minimum of 100 individual fibers were analyzed for each experiment and the mean of at least three independent experiments presented.
Statistical analysis
All the data are presented as means of at least three independent experiments. Statistical comparisons of WS or WRN-mutant cells to their relevant control were analyzed by Student's t-test. P < 0.0001 was considered significant.
RESULTS
WRN deficiency results in defective ATR-dependent checkpoint activation under mild replication stress
To assess the role for WRN in ATR pathway activation in response to mild replication stress, we examined the phosphorylation status of the main target of ATR, CHK1. To compare isogenic cell lines, HEK293T cells stably expressing scrambled (WRN-wt) or WRN-targeting shRNA (WRN-kd) were generated. WRN-kd cells showed about 80% depletion of WRN protein under the experimental conditions used in this study (Figure 1A). Treatment with low dose of Aph induced a time-dependent phosphorylation of CHK1 in WRN-wt cells, already noticeable after 1 h and peaking at 24 h (Figure 1A), suggesting that also a modest replication perturbation can trigger a quick checkpoint response. In contrast, CHK1 phosphorylation was absent, or very weak, in WRN-kd cells, and it was detectable only at the late time-points even if not at the wild-type levels (Figure 1A). However, unlike the nanomolar dose of Aph, treatment with 1 mM HU, which leads to a robust genome‐wide replication arrest, induced comparable CHK1 phosphorylation levels in both WRN-wt and WRN-kd cells (Figure 1B and unpublished data). Although CHK1 phosphorylation was hampered in WRN-deficient cells, similar amounts of Cyclin A were detected after treatments in both cell lines, suggesting that defective CHK1 phosphorylation was not attributable to a smaller proportion of S-phase population in WRN-kd cells (Figure 1B). To prove that the defective phenotype is maintained in cell lines from human patients, we investigated CHK1 activation in an isogenic pair of uncorrected or WRN wild-type-corrected (WSWRN) SV40-transformed WS fibroblasts (10). As shown in Figure 1C, Aph treatment induced CHK1 phosphorylation in WSWRN cells in a manner similar to that seen in WRN-wt cells, whereas in WS cells it resulted in no or minimal activation of CHK1. Nonetheless, treatment of cells with high doses of Aph or HU, which cause a complete replication arrest, led to comparable CHK1 phosphorylation in both cell lines (Supplementary Figure S1). Interestingly, a defective phosphorylation of CHK1 after low dose of Aph was consistently observed in WS-derived hTERT-immortalized primary fibroblasts (Supplementary Figure S2), suggesting that the phenotype is unlikely due to the cell transformation, but rather it may relate with the absence of WRN.
Figure 1.
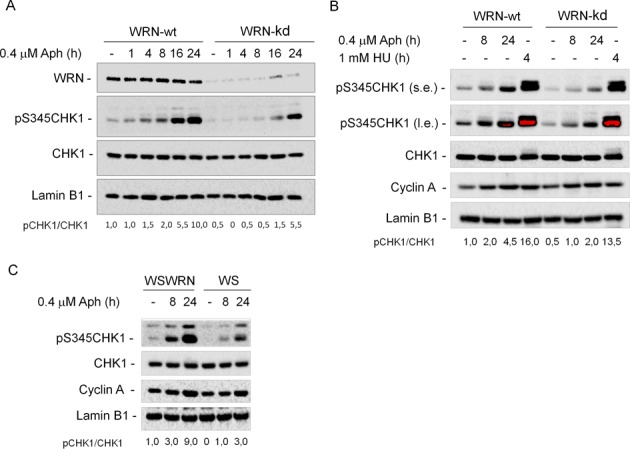
WRN is required for CHK1 activation following mild replication stress. (A) WB detection of CHK1 phosphorylation in total extracts of WRN-wt and WRN-kd cells untreated (-) or treated with Aph, as indicated. In WRN-kd cells, downregulation of the WRN protein was verified using a specific anti-WRN antibody. The presence of activated, i.e. phosphorylated, CHK1 was assessed using S345 phospho-specific antibody (pS345). Total amount of CHK1 was determined with an anti-CHK1 antibody. Equal loading was confirmed probing with an anti-Lamin B1 antibody. (B) WRN-wt and WRN-kd cells were treated with Aph or HU and processed as described in (A). (s.e., short-exposure; l.e., long-exposure). Cyclin A was used to quantify S-phase cells. (C) WRN syndrome (WS) cells and WS cells complemented with the wild-type WRN (WSWRN) were treated with 0.4 μM Aph for the indicated time. Cell lysates were analyzed by WB for the presence of phosphorylated CHK1 as described in (A). Cyclin A was used to quantify S-phase cells. The ratio of phosphorylated protein to total protein normalized to the untreated wild type is reported below each lane. All cell lines were tested at least three times and representative experiments are shown.
ATR-checkpoint response in WRN-deficient cells was also investigated by flow-cytometric analysis. As expected, Aph slowed down cell cycle progression of WSWRN cells, and delayed S-G2 phase transition (Supplementary Figure S3A). In contrast, WS cells exhibited a higher proportion of G2/M phase cells accumulated starting from 8 h of treatment (Supplementary Figure S3A), with a more pronounced accumulation of cells in the M phase, as evaluated by immunostaining for the mitosis-specific marker phospho-histone H3 (Supplementary Figure S3B).
Overall, these findings imply that WRN plays a critical function in response to mild replication stress, and support a possible role for WRN as specific mediator of CHK1 activation.
Often, checkpoint mediators are themselves targets of apical kinases, and WRN is indeed phosphorylated by ATR after both hydroxyurea (HU) and camptothecin (CPT) treatments (12,14). To reinforce the existence of a functional relationship between WRN and the ATR-dependent checkpoint after mild replication stress, we asked whether WRN was targeted by ATR kinase under Aph treatment. HEK293T cells were treated with low dose of Aph and cell lysates were subjected to WRN IP. Using phospho-S/TQ antibodies, which specifically recognize phosphorylated ATM/ATR substrates, phosphorylation of WRN was barely detectable in untreated conditions, but strongly increased after treatments (Figure 2A), suggesting that moderate stress is able to induce WRN modification. In an independent experiment, we transfected HEK293T cells with plasmids expressing the Flag-tagged full-length wild-type WRN (wt) or a mutant Flag‐WRN6A, which makes WRN unphosphorylable by ATR after replication stress (12). Following Aph treatment, cell extracts were prepared and subjected to Immunoprecipitaion (IP) using anti-Flag tag antibodies. The expression of wild-type WRN resulted in the expected phosphorylation at S/TQ sites, whereas mutant WRN6A protein abrogated pS/TQ immunoreactivity (Figure 2B), suggesting that, even after low dose of Aph, WRN is mainly phosphorylated at C-terminus residues.
Figure 2.

ATR-dependent WRN phosphorylation upon Aph treatment. (A) WB detection of WRN phosphorylation in HEK293T cells treated or not with 0.4 μM Aph for the indicated time-points. Cell extracts were immunoprecipitated using anti-WRN antibody followed by immunoblotting with an anti-S/TQ antibody. Total WRN was used to assess the amount of WRN immunoprecipitated. The ratio of phosphorylated protein to total protein is reported below each lane. (B) Analysis of WRN status in HEK293T cells transfected with plasmids expressing the Flag-tagged full-length wild-type WRN (wt) or a full-length carrying Ala substitutions at all the six S/TQ sites (6A) and treated or not with Aph as indicated. Cell extracts were prepared 48 h post-transfection and used to immunoprecipitate ectopic WRN with anti-Flag tag antibody, followed by immunoblotting with an anti-S/TQ antibody. Total WRN was used to assess the amount of wild-type or mutant form of WRN immunoprecipitated. All experiments are representative images of at least two replicates.
Collectively, these results demonstrate that mild replication stress, like that inducing breaks at CFS, may trigger CHK1 phosphorylation that requires the presence of WRN.
WRN phosphorylation by ATR, but not WRN helicase activity, is required for checkpoint activation upon mild replication stress
To verify whether WRN phosphorylation by ATR is a prerequisite for checkpoint activation after replication stress induced by Aph, we studied the ability of WS cells stably expressing the mutant form of WRN unphosphorylable by ATR (WSWRN6A) (12) to phosphorylate CHK1. WSWRN, WS and WSWRN6A cells were used and CHK1 phosphorylation evaluated. As expected, WSWRN cells were proficient in activation of CHK1 after treatment (Figure 3A). In contrast, WSWRN6A cells showed defective CHK1 phosphorylation as WS cells. Therefore, these data suggest that phosphorylation of WRN may play a role in the ATR-checkpoint activation in response to moderate replication stress.
Figure 3.
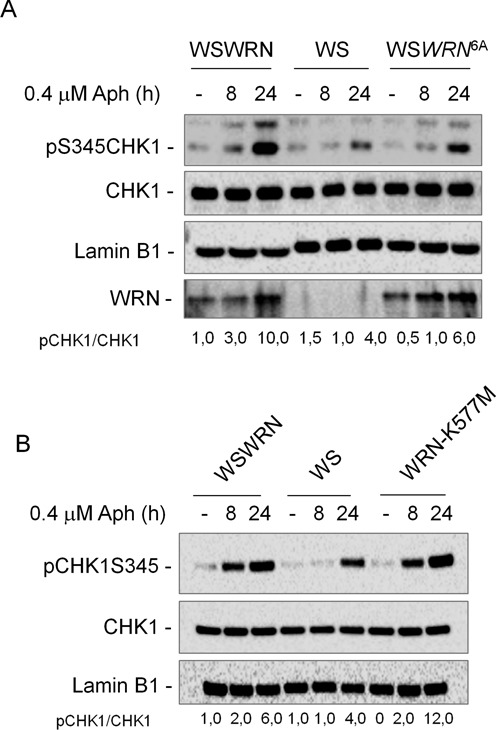
ATR-dependent WRN phosphorylation, but not defective WRN helicase activity, fails to activate CHK1 upon moderate replication stress. (A) WRN-deficient (WS) cells and WS cells complemented with wild-type (WSWRN) or expressing an unphosphorylable mutant form of WRN (WSWRN6A) were treated or not with Aph for the indicated times. CHK1 phosphorylation was analyzed by WB using phospho-specific antibodies (pS345). Total amount of CHK1 was determined with an anti-CHK1 antibody. Equal loading was confirmed probing with an anti-Lamin B1 antibody. Total WRN was used to assess the amount of wild-type or mutant form of WRN. (B) WS cells and WS cells complemented with wild-type (WSWRN) or expressing a mutant form of WRN affecting helicase (WRN-K577M) were treated or not with Aph for the indicated times. CHK1 phosphorylation was analyzed by WB using phospho-specific antibodies (pS345). Total amount of CHK1 was determined with an anti-CHK1 antibody. Lamin B1 was used as loading control. The ratio of phosphorylated protein to total protein and then normalized to the untreated wild type is listed below each lane. All experiments are representative images of at least three replicates.
WRN helicase activity, which is crucial in maintaining CFS stability (10), could facilitate formation of the single-strand DNA (ssDNA) leading to checkpoint activation (20). Thus, to verify whether suppression of helicase function hinders CHK1 phosphorylation, WS fibroblasts and WS cells stably expressing the wild-type WRN (WSWRN) or its helicase-dead form (WRN-K577M) were treated with Aph and cell lysates subjected to WB. The data confirmed that, in WSWRN cells, CHK1 was properly phosphorylated after Aph treatment, and comparable levels of CHK1 phosphorylation were observed in WRN-K577M cells, indicating that expression of the helicase-dead WRN protein is sufficient to recover from the defective phenotype (to restore checkpoint activity) (Figure 3B). Consistently, flow-cytometric experiments indicated that loss of the WRN helicase activity had no effect on the delay of cell cycle progression induced by Aph (Supplementary Figure S4).
We therefore concluded that the WRN helicase activity is not implicated in the activation of the ATR-dependent pathway in response to low levels of replication stress, whereas the ATR-dependent phosphorylation of WRN results essential for CHK1 activation.
Analysis of the ATR-signaling pathway in WRN-deficient and mutant cells after low levels of replication perturbation
Activation of the ATR pathway depends on RPA binding to ssDNA (20). To determine whether impaired RPA-coated ssDNA accumulation contributes to defective checkpoint activation observed in the absence of WRN, we measured the amount of chromatin-bound RPA32, a component of the RPA heterotrimer, in Aph-treated WSWRN and WS cells. Biochemical analysis of fractionated cell extracts showed that RPA32 was only slightly more chromatin-associated in WS cells, either exposed to Aph or left untreated, respect to WSWRN cells (Figure 4A). Indeed, even though WS cells showed more chromatin-bound RPA32, they also seemed to contain more RPA32 in total extracts (Figure 4A, bottom). In agreement with this result, a time course analysis of non-extractable RPA32 foci by immunofluorescence revealed that, in the absence of WRN, a higher number of RPA32-positive nuclei accumulated under unperturbed conditions, while only a slight increase at 8 h Aph treatment was detected, even if the RPA32 fluorescence appeared to be more intense than in the control cells (Supplementary Figure S5A).
Figure 4.
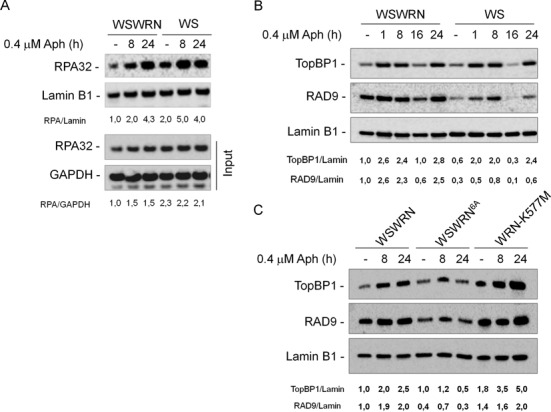
Analysis of chromatin loading of ATR-pathway sensors and mediators in cells upon mild replication stress. (A) WB analysis for chromatin loading of RPA32 in wild-type (WSWRN) and WS cells untreated or treated with Aph for various times as indicated. WB analysis of chromatin recruitment of RPA32 in WSWRN and WS cells. Total amount of RPA32 was determined with an anti-RPA32 antibody. Lamin B1 was used as loading control. WB of whole cell extracts (Input) is reported.The ratio of the RPA32/Lamin B1 signal (chromatin) or of the RPA32/gliceraldeide-3-fosfato deidrogenasi (GAPDH) signal (input) has been normalized to that of wild-type and it is reported below each lane. (B) WSWRN and WS cells were treated with Aph for various times or left untreated as indicated. Total amount of TopBP1 and RAD9 were determined with an anti-TopBP1 or anti-RAD9 antibody, respectively. Lamin B1 was used as loading control. (C) WSWRN cells and WS cells expressing an ATR-unphosphorylable form of WRN (WSWRN6A) or mutant form of WRN helicase (WRN-K577M) were treated or not with Aph for the indicated times. Total amount of TopBP1 and RAD9 and equal loading of total proteins were determined as in (B). The ratio of the TopBP1/Lamin B1 signal or of the RAD9/Lamin B1 has been normalized to that of wild-type and then reported below each lane. All experiments are representative images of at least three replicates.
Next, to support the evidence that loss of WRN does not affect the generation of ssDNA after exposure to low dose of Aph, the extent of ssDNA formation was quantified through native BrdU staining. The analysis revealed that the percentage of ssDNA-positive nuclei was roughly similar in both WSWRN and WS cells (Supplementary Figure S5B), but the number of foci per cell was higher in WS cells (Supplementary Figure S5C).
Together, these results indicate that, in WS cells, CHK1 activation is not defective because of the inability to form the RPA-coated ssDNA after moderate replication perturbation.
It is known that the TopBP1 mediator protein is required for efficient CHK1 phosphorylation, and that it is recruited to stalled forks by the chromatin-bound RAD1-RAD9-HUS1 (9.1.1) complex (21,22). In addition, WRN and the 9.1.1 complex physically interact (23). To test the possibility that WRN could stabilize 9.1.1/TopBP1 association following mild replication stress, we analyzed chromatin loading of TopBP1 and RAD9 by WB after cellular fractionation in WSWRN and WS cells treated or not with Aph at the indicated times. As shown in Figure 4B, in WSWRN cells, the amount of chromatin-bound TopBP1 started to increase at 1 h after treatment, and remained high at 8 and 24 h, even if it decreased at 16 h in agreement with other reports (24). Similarly, RAD9 was loaded on the chromatin at all times of Aph exposure except at 16 h (Figure 4B). In contrast, in WS cells low levels of RAD9 loaded to chromatin were detectable under untreated and low-dose Aph-treated conditions (Figure 4B). Interestingly, the expression of the unphosphorylable form of WRN, but not, as expected, loss of WRN helicase activity, compromised chromatin association of checkpoint mediators, and a clear reduction of TopBP1 and RAD9 recruitment was found in WSWRN6A cells at 24 h of Aph (Figure 4C).
Therefore, defective CHK1 activation upon replication perturbation induced by Aph, in WS and WSWRN6A cells, appears related to the levels of chromatin-bound checkpoint mediators.
Replication fork dynamics are altered in WRN-deficient and mutant cells
Having demonstrated that correct CHK1 activation relies on both the presence of WRN and its phosphorylation by ATR, and since CHK1 has been implicated in maintaining fork integrity and proper cell cycle progression under conditions of replicative stress (25,26), we examined how the absence of WRN or expression of mutant forms could influence replication dynamics. We first monitored replication in WSWRN, WS, WSWRN6A and WRN-K577M cells under unperturbed conditions as indicated in the scheme (Figure 5A). Under these conditions, WSWRN showed an average fork progression rate of 1.1 kb/min (Figure 5B). By contrast, WRN-deficient cells or cells expressing the mutant forms of WRN displayed a significant reduction of fork speed (about 2-fold) as compared to control cells (Figure 5B). A logical expectation from this result might be that replication stress exacerbated the effect; however, all the cell lines reached identical values of fork velocity upon Aph treatment (Figure 5C). This implies that fork progression is mainly affected by WRN deficiency or expression of WRN-mutated forms, and that Aph is able to further delay forks only in control cells.
Figure 5.
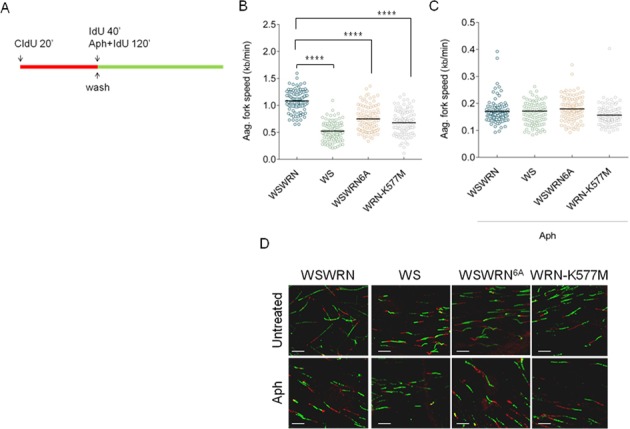
Replication fork progression is impaired in WRN-deficient and mutant cells. (A) Experimental scheme of dual labeling of DNA fibers. (B) Analysis of replication fork velocity in WSWRN (wild-type), WRN-deficient (WS) and mutant cells (WSWRN6A and WRN-K577M) under unperturbed conditions or (C) exposed to 0.4 μM Aph as indicated in (A). The length of the green tracks was measured. Median values are represented as horizontal black lines. (D) Representative images of actual DNA fibers from WSWRN, WRN-deficient and mutant cells. Scale bars, 10 μm. **** = Statistically significant P < 0.0001 (Student's t-test).
It has been previously proposed that cells lacking CHK1 activity show reduced rate of replication fork progression and increased origin firing (27). Thus, we verified whether the inability of Aph to reduce further fork speed in WRN-deficient and WRN-mutant cells could be the end result of new origins activated to compensate for replication problems. We used a modified double labeling protocol that enables us to study fork recovery from Aph (Figure 6A). Under such conditions, all the cell lines showed an analogous ability to replicate after removal of Aph, evaluated as length of green tracks (Figure 6B and C). Interestingly, using roscovitine, a CDK inhibitor that prevents origin firing, a reduced length of green tracks in WS, WSWRN6A and WRN-K577M cells was detected, but no effect was found in WSWRN cells (Figure 6B). Such a finding could indicate that DNA is partly replicated from ongoing forks and partly from a downstream fork fired during roscovitine treatment. Consistent with more new origin firing in WS, WSWRN6A and WRN-K577M cells, roscovitine also decreased the number of isolated green tracks detected during the recovery from Aph (Figure 6D).
Figure 6.
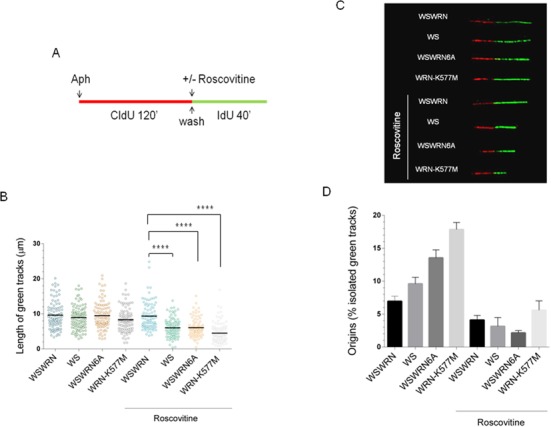
Replication fork restart is affected by roscovitine in WRN-deficient and mutant cells. (A) Experimental scheme of dual labeling of DNA fibers in presence or absence of roscovitine. (B) Analysis of replication fork recovery in WSWRN (wild-type), WRN-deficient (WS) and mutant cells (WSWRN6A and WRN-K577M) treated as indicated in (A). Aph (0.4 μM) and roscovitine (100 μM) were added for the indicated time. Median values are represented as horizontal black lines. (C) Representative images of replication tracks from WSWRN, WRN-deficient and mutant cells with or without roscovitine treatment. (D) Percentage of new origin firing in cells treated as in (B). Data are reported as mean from three independent experiments. Error bars represent standard error. **** = Statistically significant P < 0.0001 (Student's t-test).
Therefore, we conclude that the increased replication elongation observed in WRN-deficient or WRN-mutant cells is due to increased origin firing of neighboring dormant origins, and that this back-up mechanism obscures a more severe reduction of fork progression occurring in these cells.
Expression of phospho-mimic CHK1 mutant recovers cells from sensitivity to Aph treatment and restores fork elongation
Since low levels of replication stress induce CFS expression, we asked whether defective checkpoint activation found in WS and WSWRN6A cells could be related to CFS instability. We first investigated the sensitivity of WSWRN6A to Aph. A dose-dependent enhancement of chromosomal aberrations, similarly to that previously seen in WS cells (10), was observed in WSWRN6A, which showed approximately 2-fold more aberrations than control cells at the higher dose (Figure 7A). These results suggest that WSWRN6A cells are sensitive to Aph treatment, and that loss of ATR-mediated WRN phosphorylation is responsible for chromosome instability.
Figure 7.
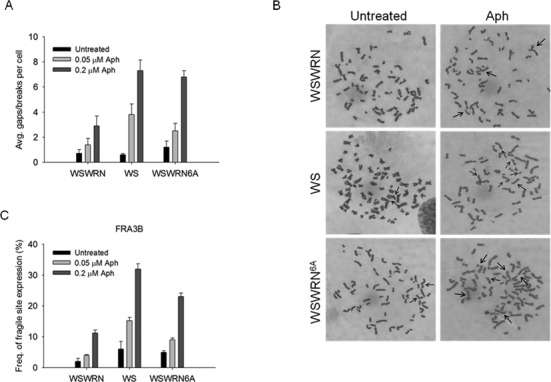
Enhanced CFS induction in cells expressing an ATR-unphosphorylable form of WRN. (A) Average overall chromosome gaps and breaks in WS cells (WS), WS cells expressing an ATR-unphosphorylable form of WRN (WSWRN6A) and in WS cells in which wild-type WRN was reintroduced (WSWRN). Cells were treated with two different doses of Aph for 24 h before harvesting. Data are presented as means of three independent experiments. Error bars represent standard error. (B) Representative Giemsa-stained metaphases of wild-type (WSWRN), WS and WSWRN6A fibroblasts untreated or treated with 0.2 μM Aph. Arrows indicate chromosomal aberrations. (C) Frequency of gaps and breaks at CFS by FISH analysis. Cells were treated as described in (A). FISH was performed using BACs probes corresponding to the FRA3B region. Frequency of fragile site induction is presented as the percentage of chromosome 3 homolog with gaps and breaks at FRA3B. Data are presented as means of three independent experiments. Error bars represent standard error on the mean.
Next, we examined if increased chromosomal damage induced by Aph in WSWRN6A took place at CFS. As shown in Figure 7C, instability at CFS FRA3B was induced in a dose-dependent manner in WSWRN6A cells, with a number of gaps and breaks higher than in control cells, and at levels comparable to those of WS cells. Interestingly, enhanced FRA3B induction was detected even in the absence of Aph in both WSWRN6A and WS cells (Figure 7C). Similar results were observed testing other CFS (Supplementary Figure S6).
These findings demonstrate that phosphorylation of WRN by ATR is essential not only for correct CHK1 phosphorylation but also, and most notably, for CFS stability. Thus, we wondered whether forced activation of CHK1 could overcome sensitivity to Aph in WS and WSWRN6A cells, that is in cells where CHK1 is not phosphorylated. Cells were transfected with a construct expressing a mutant form of CHK1 in which Ser317 and Ser345 were changed into Asp (CHK1317/345D), mimicking the phosphorylated status of the protein (17). WB analysis confirmed that the Flag-tagged phospho-mimic CHK1 mutant (Flag-CHK1317/345D) was expressed at comparable levels in both cell lines (Figure 8A). After transfection, cells were treated with Aph and metaphase chromosomes were collected and scored for total gaps and breaks. As expected, Aph treatment resulted in enhanced chromosomal damage in WS and WSWRN6A cells (Figure 8B). Interestingly, expression of the CHK1317/345D protein into both WS and WSWRN6A cells resulted in the rescue of the chromosomal fragility induced by low dose of Aph and also determined a slight reduction of the spontaneous number of aberrations per cell in WSWRN6A cells (Figure 8B). These results indicate that DNA breakage observed in both WS and WSWRN6A cells upon mild replication perturbation actually correlates with incorrect CHK1 activation.
Figure 8.
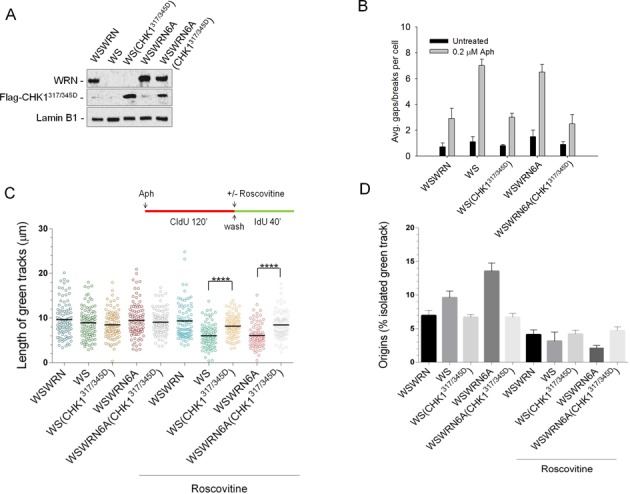
Expression of phospho-mimic CHK1 rescues sensitivity to Aph and replication fork slowing in WS and WSWRN6A cells. (A) WS and WSWRN6A cells were transfected with Flag-tagged CHK1 S317D/S345D mutant (CHK1317/345D). Lysates were collected 48 h thereafter and expression levels of Flag-CHK1317/345D were determined by immunoblotting with anti-Flag antibody. WSWRN cells were used as negative control. Total WRN was used to assess the amount of wild-type or mutant form of WRN. Lamin B1 was used as loading control. (B) Effect of the expression of the Flag-CHK1317/345D plasmid on chromosomal damage. After transfection, cells were treated or not with Aph for 24 h. The graph shows the average overall chromosome gaps and breaks. Data are presented as means of three independent experiments. Error bars represent standard error on the mean. (C) Evaluation of replication fork recovery in WS and WSWRN6A cells transfected with Flag-CHK1317/345D. Cells were treated as indicated in the experimental scheme. Aph (0.4 μM) and roscovitine (100 μM) were added for the indicated time. Median values are represented as horizontal black lines. (D) Percentage of new origin firing in cells treated as in (C). Data are reported as mean from three independent experiments. Error bars represent standard error. **** = Statistically significant P < 0.0001 (Student's t-test).
Next, we asked whether the introduction of the phospho-mimic CHK1 mutant could allow cells to recover from fork stalling, possible without activating new origins. WS and WSWRN6A cells were transfected or not with the Flag-CHK1317/345D plasmid, and then treated with Aph and roscovitine prior to analyze their replication recovery (Figure 8C). Analysis of the length of replicating tracks on DNA fibers showed that the presence of phospho-mimic CHK1 mutant enabled both cell lines to resume replication in a wild-type fashion, overcoming the requirement of new origin firing, as demonstrated by the similar track lengths in cells treated or not with roscovitine (Figure 8C), and the analysis of isolated green tracks, showing that less dormant origins were activated (Figure 8D).
Altogether, these data strongly support the hypothesis that, in WS and WSWRN6A cells, the inability to trigger CHK1 activation leads to replication fork demise and origin firing in the attempt to complete replication.
DISCUSSION
We previously determined that WRN in association with the ATR pathway prevents CFS breakage, but with largely unknown mechanisms (10). Here, we provide evidence for a crucial role of the WRN protein in mediating CHK1 phosphorylation specifically following low level of replicative stress, such as that causing CFS expression.
Loss of CHK1 has been related to CFS expression, and CHK1 undergoes phosphorylation upon mild replication perturbation (16). Although CHK1 has been widely found activated in response to low doses of Aph (16,28–29), a recent work suggested that CHK1-dependent protection of CFS might be unrelated to its phosphorylation-dependent activation (24). Consistently with earlier reports (16,28–29), we detect CHK1 phosphorylation after Aph treatment, albeit at reduced levels if compared to strong replication perturbation as that induced by HU. Since CHK1 phosphorylation was demonstrated from independent studies using different cell lines, it is unlikely that the genetic background of cells could influence this specific phenotype. However, the percentage of S-phase cells might markedly affect the levels of CHK1 phosphorylation, especially in the presence of partial inhibition of DNA synthesis, perhaps accounting for these discrepancies between the data. Noteworthy, in the study where CHK1 phosphorylation has not been detected after low Aph doses, chromatin loading of ATR and its mediators, RAD9 and TopBP1, has been demonstrated (24). Our findings confirm that mild replication stress determines chromatin accumulation of RAD9 and TopBP1 similarly to what observed in response to robust replication perturbation induced genome-wide.
Interestingly, RAD9 and TopBP1 recruitment to chromatin involves the presence of WRN under unperturbed conditions as well as upon mild replication stress, suggesting that the reported association of WRN with 9.1.1, whose loss increases CFS expression (23), is important for CHK1 activation. Therefore, our results indicate that the presence of the WRN protein may be required for CHK1 activation, suggesting that WRN acts as a checkpoint mediator protein, specifically when replication is partially inhibited. A similar function of a RecQ helicase has been demonstrated in yeast, where Sgs1 plays a non-catalytic role in correct activation of Rad53 after replication stress (30), bridging RPA and Rad53 together (31). Consistent with a possible role as mediator protein, WRN is phosphorylated at S/TQ sites by ATR also after Aph-induced replication stress, and this modification of WRN appears to be crucial for promoting efficient CHK1 phosphorylation upon mild replication perturbation, as well as for stabilization of CFS.
In WS cells expressing the ATR-unphosphorylable mutant form of WRN, we detect a decrease of RAD9 and TopBP1 levels respect to the other cell lines, especially at the later time. This is not surprising since the phospho-WRN mutant is a double mutant that cannot be targeted by ATR and ATM (12), and we cannot exclude that loading on chromatin of mediators at later times could rely on the ATM-dependent phosphorylation of WRN. Altogether, it is plausible that, upon moderate replication perturbation, WRN may act as a mediator of CHK1 activation, and this function can be regulated by the ATR-dependent phosphorylation. Conversely, WRN helicase mutant does not affect CHK1 activation upon mild replication stress and, consistently, accumulates ATR mediators properly.
In the absence of ATR or CHK1 fork destabilization can occur (26,32–34). The inability of WRN-deficient cells or cells expressing the ATR-unphosphorylable form of WRN to properly activate CHK1 could similarly lead to fork undermining. Indeed, loss of WRN or expression of the ATR-unphosphorylable mutant form affects replication fork progression under unperturbed cell growth, consistently with previous studies (35,36). Also expression of the helicase-dead form of WRN strongly reduces fork speed in untreated cells, suggesting that reduced elongation rates and chromosome instability are connected in cells with absent or dysfunctional WRN protein. In the same cells, mild replication stress further delays fork speed, but in a lesser extent than in control cells. Interestingly, WRN-deficient cells and cells expressing the helicase-dead or unphosphorylable WRN display an apparently normal ability to recover DNA replication after mild replication perturbation; however, their apparently normal elongation rates are strikingly reduced upon origin firing inhibition by roscovitine. This phenotype possibly indicates that loss of WRN function results in a severe elongation defect or fork stalling, triggering additional local origin firing to complete replication. Consistently, WS cells show more ssDNA formation under unperturbed replication or after Aph. Such increased generation of ssDNA could derive from persistent fork stalling and from regions left unreplicated behind such forks or from additional origin firing. Moreover, the presence of more origin firing in WRN-deficient cells and cells expressing the helicase-dead or the ATR-unphosphorylable WRN mutant is not a mere consequence of deregulated CHK1 activation, which has been reported to induce unscheduled origin firing (25), as it is even more evident in cells expressing the helicase-dead WRN mutant, which shows wild-type CHK1 phosphorylation. Similarly, since cells expressing the helicase-dead form of WRN are able to support CHK1 activation but also show replication defects, failing to recover forks from Aph-induced replication perturbation, it is plausible that this reduction of fork speed is linked to a cooperation of CHK1 and WRN. Cells lacking CHK1 reduce fork elongation by half respect to the control under unperturbed conditions (27,37), exactly the same extent of reduction associated to loss of WRN function. Expression of a phospho-mimic CHK1 mutant in WS cells or cells expressing the ATR-unphosphorylable form of WRN recovers cells from sensitivity to Aph and restores fork elongation to wild-type level, suggesting that experimentally-induced reactivation of CHK1, bypassing the WRN mediator function, is sufficient to recover perturbed forks and chromosome instability. Thus, stabilization of perturbed forks by CHK1 can probably overcome loss of WRN, most likely through hyperactivation of compensatory pathways. This result is consistent with our earlier data showing that loss of WRN determines ssDNA gap accumulation and increased number of RAD51 foci after Aph treatment (38). Indeed, RAD51 is involved in the maintenance of CFS stability (39) and in the repair of gaps left behind the stalled fork, and most importantly is activated by CHK1 (40,41). One possibility to explain the effect of enhanced recovery in WS cells expressing a constitutively-active CHK1 may be related to a more efficient RAD51-dependent post-replication gap repair to stabilize unreplicated regions. On the other hand, in cells expressing the ATR-unphosphorylable WRN, the introduction of the constitutively-active CHK1 might be sufficient to prevent fork destabilization, thus allowing fork recovery through the helicase activity of WRN, which is likely fully functional in these cells.
Altogether, our data allow to propose a model, which explains how WRN can function to guarantee CFS stability, through a checkpoint-dependent and independent way. The ATR-dependent WRN phosphorylation is required to stimulate CHK1 activation that is instrumental for stabilization of stalled forks, whereas the helicase activity of WRN might be necessary to support replication restart, possibly by the resolution of DNA secondary structures, to promote CFS stability. Thus, if WRN protein is lost or cannot be phosphorylated by ATR, replication checkpoint cannot be activated leading to destabilization of replication forks, and subsequently, to enhanced CFS expression. Similarly, the inability to rescue stalled forks, due to loss of WRN helicase activity, results in enhanced CFS instability (10).
Therefore, our findings suggest a novel role of WRN as checkpoint mediator in response to moderate replication stress and give strong mechanistic support to the notion that defective fork repair/recovery undermines integrity of chromosomes at CFS. Moreover, they may contribute to shed light on the origin of chromosome instability in WS and more in general to clarify how genome instability accumulates in preneoplastic lesions, thus promoting cancer development.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
This work is dedicated to the memory of Gianfranco Franchitto. The authors express their grateful thanks to Professor K.K. Khanna for sharing research material (Queensland Institute of Medical Research, Australia).
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Associazione Italiana per la Ricerca sul Cancro [IG #11871 to A.F., IG #13398 to P.P.]. Funding for open access charge: Associazione Italiana per la Ricerca sul Cancro [IG #11871 to A.F.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Pichierri P, Franchitto A, Mosesso P, Palitti F. Werner's syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol. Biol. Cell. 2001;12:2412–2421. doi: 10.1091/mbc.12.8.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poot M, Gollahon K A, Rabinovitch P S. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Human genetics. 1999;104:10–14. doi: 10.1007/s004390050903. [DOI] [PubMed] [Google Scholar]
- 3.Poot M, Yom J S, Whang S H, Kato J T, Gollahon K A, Rabinovitch P S. Werner syndrome cells are sensitive to DNA cross-linking drugs. FASEB journal. 2001;15:1224–1226. doi: 10.1096/fj.00-0611fje. [DOI] [PubMed] [Google Scholar]
- 4.Brosh R.M., Jr DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer. 2013;13:542–558. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rossi M.L., Ghosh A.K., Bohr V.A. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst.) 2010;9:331–344. doi: 10.1016/j.dnarep.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suhasini A.N., Brosh R.M. Jr. DNA helicases associated with genetic instability, cancer, and aging. Adv. Exp. Med. Biol. 2013;767:123–144. doi: 10.1007/978-1-4614-5037-5_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baynton K., Otterlei M., Bjoras M., von Kobbe C., Bohr V.A., Seeberg E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J. Biol. Chem. 2003;278:36476–36486. doi: 10.1074/jbc.M303885200. [DOI] [PubMed] [Google Scholar]
- 8.Sakamoto S., Nishikawa K., Heo S.J., Goto M., Furuichi Y., Shimamoto A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells. 2001;6:421–430. doi: 10.1046/j.1365-2443.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 9.Kamath-Loeb A.S., Loeb L.A., Johansson E., Burgers P.M., Fry M. Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J. Biol. Chem. 2001;276:16439–16446. doi: 10.1074/jbc.M100253200. [DOI] [PubMed] [Google Scholar]
- 10.Pirzio L.M., Pichierri P., Bignami M., Franchitto A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J. Cell Biol. 2008;180:305–314. doi: 10.1083/jcb.200705126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah S.N., Opresko P.L., Meng X., Lee M.Y., Eckert K.A. DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 2010;38:1149–1162. doi: 10.1093/nar/gkp1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ammazzalorso F., Pirzio L.M., Bignami M., Franchitto A., Pichierri P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010;29:3156–3169. doi: 10.1038/emboj.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otterlei M., Bruheim P., Ahn B., Bussen W., Karmakar P., Baynton K., Bohr V.A. Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J. Cell Sci. 2006;119:5137–5146. doi: 10.1242/jcs.03291. [DOI] [PubMed] [Google Scholar]
- 14.Pichierri P., Rosselli F., Franchitto A. Werner's syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene. 2003;22:1491–1500. doi: 10.1038/sj.onc.1206169. [DOI] [PubMed] [Google Scholar]
- 15.Casper A.M., Nghiem P., Arlt M.F., Glover T.W. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 16.Durkin S.G., Arlt M.F., Howlett N.G., Glover T.W. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene. 2006;25:4381–4388. doi: 10.1038/sj.onc.1209466. [DOI] [PubMed] [Google Scholar]
- 17.Gatei M., Sloper K., Sorensen C., Syljuasen R., Falck J., Hobson K., Savage K., Lukas J., Zhou B.B., Bartek J., et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J. Biol. Chem. 2003;278:14806–14811. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- 18.Franchitto A., Pirzio L.M., Prosperi E., Sapora O., Bignami M., Pichierri P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J. Cell Biol. 2008;183:241–252. doi: 10.1083/jcb.200803173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson D.A., Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou L., Elledge S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 21.Delacroix S., Wagner J.M., Kobayashi M., Yamamoto K., Karnitz L.M. The Rad9-Hus1-Rad1 (9–1–1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumagai A., Lee J., Yoo H.Y., Dunphy W.G. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 23.Pichierri P., Nicolai S., Cignolo L., Bignami M., Franchitto A. The RAD9-RAD1-HUS1 (9.1.1) complex interacts with WRN and is crucial to regulate its response to replication fork stalling. Oncogene. 2012;31:2809–2823. doi: 10.1038/onc.2011.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koundrioukoff S., Carignon S., Techer H., Letessier A., Brison O., Debatisse M. Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet. 2013;9:e1003643. doi: 10.1371/journal.pgen.1003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feijoo C., Hall-Jackson C., Wu R., Jenkins D., Leitch J., Gilbert D.M., Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zachos G., Rainey M.D., Gillespie D.A. Chk1-dependent S-M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol. Cell. Biol. 2005;25:563–574. doi: 10.1128/MCB.25.2.563-574.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petermann E., Woodcock M., Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl Acad. Sci. U.S.A. 2010;107:16090–16095. doi: 10.1073/pnas.1005031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Q., Guntuku S., Cui X.S., Matsuoka S., Cortez D., Tamai K., Luo G., Carattini-Rivera S., DeMayo F., Bradley A., et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 29.Ozeri-Galai E., Schwartz M., Rahat A., Kerem B. Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene. 2008;27:2109–2117. doi: 10.1038/sj.onc.1210849. [DOI] [PubMed] [Google Scholar]
- 30.Bjergbaek L., Cobb J.A., Tsai-Pflugfelder M., Gasser S.M. Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. EMBO J. 2005;24:405–417. doi: 10.1038/sj.emboj.7600511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hegnauer A.M., Hustedt N., Shimada K., Pike B.L., Vogel M., Amsler P., Rubin S.M., van Leeuwen F., Guenole A., van Attikum H., et al. An N-terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. EMBO J. 2012;31:3768–3783. doi: 10.1038/emboj.2012.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maya-Mendoza A., Petermann E., Gillespie D.A., Caldecott K.W., Jackson D.A. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 2007;26:2719–2731. doi: 10.1038/sj.emboj.7601714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopes M., Cotta-Ramusino C., Pellicioli A., Liberi G., Plevani P., Muzi-Falconi M., Newlon C.S., Foiani M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- 34.Tercero J.A., Diffley J.F. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Lopez A.M., Jackson D.A., Iborra F., Cox L.S. Asymmetry of DNA replication fork progression in Werner's syndrome. Aging Cell. 2002;1:30–39. doi: 10.1046/j.1474-9728.2002.00002.x. [DOI] [PubMed] [Google Scholar]
- 36.Sidorova J.M., Kehrli K., Mao F., Monnat R., Jr Distinct functions of human RECQ helicases WRN and BLM in replication fork recovery and progression after hydroxyurea-induced stalling. DNA Repair (Amst.) 2013;12:128–139. doi: 10.1016/j.dnarep.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petermann E., Caldecott K.W. Evidence that the ATR/Chk1 pathway maintains normal replication fork progression during unperturbed S phase. Cell Cycle. 2006;5:2203–2209. doi: 10.4161/cc.5.19.3256. [DOI] [PubMed] [Google Scholar]
- 38.Murfuni I., De Santis A., Federico M., Bignami M., Pichierri P., Franchitto A. Perturbed replication induced genome wide or at common fragile sites is differently managed in the absence of WRN. Carcinogenesis. 2012;33:1655–1663. doi: 10.1093/carcin/bgs206. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz Michal, Zlotorynski Eitan, Goldberg Michal, Ozeri Efrat, Rahat Ayelet, le Sage Carlos, P C Chen Benjamin, J Chen David, Agami Reuven, Kerem Batsheva. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Genes Dev. 2005;19:2715–2726. doi: 10.1101/gad.340905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashimoto Y., Ray Chaudhuri A., Lopes M., Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sorensen C.S., Hansen L.T., Dziegielewski J., Syljuasen R.G., Lundin C., Bartek J., Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.