


Abstract
cgDNA is a package for the prediction of sequence-dependent configuration-space free energies for B-form DNA at the coarse-grain level of rigid bases. For a fragment of any given length and sequence, cgDNA calculates the configuration of the associated free energy minimizer, i.e. the relative positions and orientations of each base, along with a stiffness matrix, which together govern differences in free energies. The model predicts non-local (i.e. beyond base-pair step) sequence dependence of the free energy minimizer. Configurations can be input or output in either the Curves+ definition of the usual helical DNA structural variables, or as a PDB file of coordinates of base atoms. We illustrate the cgDNA package by comparing predictions of free energy minimizers from (a) the cgDNA model, (b) time-averaged atomistic molecular dynamics (or MD) simulations, and (c) NMR or X-ray experimental observation, for (i) the Dickerson–Drew dodecamer and (ii) three oligomers containing A-tracts. The cgDNA predictions are rather close to those of the MD simulations, but many orders of magnitude faster to compute. Both the cgDNA and MD predictions are in reasonable agreement with the available experimental data. Our conclusion is that cgDNA can serve as a highly efficient tool for studying structural variations in B-form DNA over a wide range of sequences.
INTRODUCTION
Sequence-dependent DNA mechanical properties are believed to be essential in various biological processes, such as DNA looping (1), nucleosome positioning (2), and gene regulation (3). Many of the important DNA-protein interactions occurring in both eukaryotic and prokaryotic cells involve a combination of chemistry and geometry, and depend upon the probability of DNA adopting specific configurations as predicted by a free energy that is of an essentially mechanical nature (4). Consequently, understanding how the mechanics of DNA is influenced by its sequence is an important problem in modern structural biochemistry; there is a need for efficient models to quantify and explore sequence dependence at different length scales and over a wide range of sequence variation. Of particular interest are the biologically relevant intermediate, or mesoscopic, length scales of a few tens to several hundreds of base pairs. A comprehensive study of sequence dependence at such length scales remains difficult by all-atom molecular dynamics (or MD) simulations, due to the intensity of the required numerical simulations, despite the increasing achievements in the field (5). On the other hand, sequence-dependent effects are, by definition, below the resolution of standard, uniform, coarse-grain models, such as the worm-like chain, or homogeneous elastic rod descriptions. Accordingly, the focus of our interest is sequence-dependent, coarse-grain models of DNA, which is a rather active field; as an update to pertinent citations appearing since our own previous contribution to the area (6), we mention (7–9, reviews), (10, an improved parameterization of a 3 atoms-per-nucleotide coarse-grain model), and (11, a webserver to access various databases and models of nucleic acid flexibility).
In this article, we introduce the cgDNA software package, which is a suite of Matlab (or Octave) scripts that implement the nearest-neighbour, rigid base, mesoscopic scale, sequence-dependent, coarse-grain model of B-form DNA in solvent under standard environmental conditions that was introduced in (6). In this model each base on each strand of a DNA molecule is considered as a rigid entity that interacts directly with each of its five nearest neighbours (two on the same strand and three on the opposite strand, see Supplementary Material Section S1 or (6)). Concomitantly, the free energy is approximated as a shifted quadratic function of the usual double-helical DNA internal coordinates (tilt, roll, twist, propeller, stagger, etc. (12)) along the fragment, which determine the relative 3D rotational and translational displacements between all neighbouring bases both along and across the two backbone strands. The shift in the quadratic function represents the configuration of minimal accessible free energy, i.e. the ground state, of the DNA molecule with the prescribed sequence. Equivalently, the ground state is the ensemble average shape within a Boltzmann distribution generated from the shifted quadratic free energy.
The cgDNA prediction of the free energy function for DNA fragments of an arbitrary sequence necessarily involves a parameter set for the underlying model. At the time of writing there is only one such parameter set available, which, as described in prior work (6,13), was estimated from a large-scale database (14) of atomic-resolution MD time-series simulations in explicit solvent under standard conditions. Further cgDNA parameter sets are currently under development to reflect both physical changes in conditions associated with the solvent (e.g. temperature, ion concentration and species), and modelling changes due to differences in training set MD simulation protocols and duration, and in the parameter estimation techniques used to extract the coarse-grain parameters from the resulting time series. In cgDNA, the shifted quadratic, free energy of an entire DNA molecule (of a prescribed sequence of arbitrary length) is obtained by summing (over all base pairs and all junctions) sequence-dependent, shifted quadratic, free energies associated with intra-base-pair, base-base interactions, and inter-base-pair, or cross-junction, base–base interactions. Accordingly, after accounting for the symmetry of reading a DNA sequence along the 5′ → 3′ direction of either backbone, a cgDNA parameter set comprises the coefficients in two (one for each of the distinct base pairs) intra-base-pair shifted quadratic functions (each of dimension six), along with the coefficients of 10 (one for each of the distinct dinucleotide sequence steps) inter-base-pair, or junction, shifted quadratic functions (each of dimension 18).
The cgDNA model is, we believe, unique among existing coarse-grain models of DNA because it encompasses predictions of ground states with a non-local sequence dependence beyond the dinucleotide sequence composition context, and this despite its parameter set having only dinucleotide sequence dependence, and the only physical interactions being between nearest-neighbour bases. This surprising property is explained mathematically by the fact that within the model the computation of the ground state, or accessible free energy minimizer, involves the inversion of the predicted cgDNA stiffness matrix, which is banded, but not block diagonal, so that its inversion is a non-local operation. The equivalent explanation of the non-local sequence dependence of the ground state in physical terms is the phenomenon of frustration—each base cannot simultaneously minimize the pair-wise free energy in all of its five nearest-neighbour interactions. Frustration in a rigid base model of DNA means that the ground state can be pre-stressed, and the consequences of this pre-stress or frustration can propagate along the double chain of bases to affect the configuration of the ground state globally, with the effects being significant in at least the tetranucleotide sequence context. The concept of frustration is a standard one in the materials science literature. With specific reference to biomolecules it has also been described in the context of protein folding (15,16), protein-DNA interactions (17,18) and both protein- (19) and DNA-based nanostructures (20), but we are unaware of the importance of frustration to the coarse-graining of the DNA double helix being discussed before the treatment in (6). For DNA, frustration can arise in double-chain coarse-grain models involving nearest-neighbour interactions of rigid bases; in contrast frustration does not naturally arise in the more standard, for example (21,22), single-chain coarse-grain models involving nearest-neighbour interactions of rigid base pairs. Because nearest-neighbour rigid base-pair models do not contain a mechanism for frustration, they cannot predict the non-local sequence dependence of the ground state, which is known to arise (14,23–25), without having a very large parameter set that explicitly encodes all tetranucleotide or beyond sequence contexts (6).
The basic user input to cgDNA is the sequence of a DNA fragment, then (for a given underlying choice of parameter set) cgDNA first returns the associated ground-state configuration and overall stiffness matrix of that particular sequence fragment. The free energy difference between any two conformations of the given sequence fragment can subsequently be evaluated. The computations internal to cgDNA are carried out in a non-dimensional and numerically well-scaled version of the Curves+ (12) version of the DNA internal helical parameters that are fully motivated and described in (6). But to facilitate the comparison of predictions from different sources, the cgDNA conformations can be input or output in the (dimensional, unscaled) Curves+ (12) helical coordinates, or as PDB format coordinates (26) of each non-hydrogen atom in each base. The PDB format files provide access to other coordinate systems, for example the 3DNA (27) version of the DNA internal helical parameters.
The coarse-grain model underlying cgDNA has already (6,13) been shown to rather accurately (and perhaps unsurprisingly) reconstruct the ground states and stiffnesses as observed in the direct MD simulations of the 12–18 bp oligomers that were used as the training set for the model parameter set estimation. In particular the nearest-neighbour, rigid base, level of coarse graining was demonstrated to capture rather accurately the strong sequence dependence of both the ground state and the stiffness matrix that was observed in the MD simulations. The cgDNA model was also shown to successfully predict non-local changes in the ground state consequent upon single point mutations in the oligomer sequence, and these predictions were shown to be accurate when compared to direct MD simulations that were not used in the parameter training set.
We here further illustrate the predictive capabilities of the cgDNA coarse-grain modelling package in the context of four DNA oligomers, specifically the widely studied Dickerson–Drew dodecamer and three oligomers containing A-tracts (which are believed to have exceptionally bent ground states), where for each oligomer both independent MD simulations and experimental (crystal structure or nuclear magnetic resonance (NMR)) data are available for comparison of ground-state conformations. Due both to space constraints, and the lack of suitable model-independent experimental data on sequence-dependent conformational free energy differences for naked B-form DNA, we here restrict our attention to comparisons between different predictions of ground-state conformations. The selection of sequences to be compared follows (28,29). Our conclusion is that cgDNA predictions of sequence-dependent oligomer ground states are of comparable accuracy to those of MD simulations, which are both in reasonable agreement with the experimental observations. However, for each sequence the cgDNA predictions are several orders of magnitude faster to compute than MD simulations. Consequently, the cgDNA approach opens the possibility of scanning variations in shape of ground states over a large range of sequences. In this sense cgDNA can be regarded as an alternative, more detailed, approach to that of (30) which is optimized for scanning shape variation in very long sequences (on genomic scales), but which does not consider the full and detailed structure of the DNA molecule.
MATERIALS AND METHODS
Model
The cgDNA model is a coarse-grain description of B-form DNA (with only standard Watson–Crick base pairings) in which each base is modelled as a rigid object. Each base has a frame embedded in it according to the conventions detailed in Curves+ (12). The basic user input to cgDNA is a base composition sequence 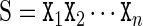 , listed in the 5′ to 3′ direction along one strand, where
, listed in the 5′ to 3′ direction along one strand, where 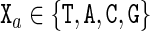 . The base pairs associated with this sequence are denoted
. The base pairs associated with this sequence are denoted 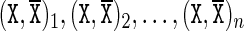 , where
, where 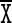 is defined as the Watson–Crick complement of
is defined as the Watson–Crick complement of 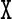 ,
,  , etc. As is more fully described in the Supplementary Material (Section S1), the relative rotation and translation between the base frames embedded in the two bases
, etc. As is more fully described in the Supplementary Material (Section S1), the relative rotation and translation between the base frames embedded in the two bases 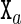 and
and 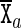 in a base pair are described by an intra-base-pair coordinate vector ya with six entries comprising three rotation coordinates (Buckle-Propeller-Opening) and three translation coordinates (Shear-Stretch-Stagger). A base-pair frame is defined as an appropriate average of the two base frames in the base pair, and the relative rotation and translation between adjacent base-pair frames is described by an inter-base-pair coordinate vector
in a base pair are described by an intra-base-pair coordinate vector ya with six entries comprising three rotation coordinates (Buckle-Propeller-Opening) and three translation coordinates (Shear-Stretch-Stagger). A base-pair frame is defined as an appropriate average of the two base frames in the base pair, and the relative rotation and translation between adjacent base-pair frames is described by an inter-base-pair coordinate vector 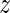 a with the six entries comprising three rotation coordinates (Tilt-Roll-Twist) and three translation coordinates (Shift-Slide-Rise). Any conformation of the DNA fragment is therefore determined by the coordinate vector w = (y1,
a with the six entries comprising three rotation coordinates (Tilt-Roll-Twist) and three translation coordinates (Shift-Slide-Rise). Any conformation of the DNA fragment is therefore determined by the coordinate vector w = (y1, 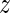 1, y2,
1, y2, 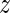 2, …,
2, …, 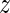 n − 1, yn) of length 12n − 6, where the six degrees of freedom of overall translation and rotation in space have been eliminated.
n − 1, yn) of length 12n − 6, where the six degrees of freedom of overall translation and rotation in space have been eliminated.
The cgDNA model of a DNA molecule with sequence S is a free energy of the form
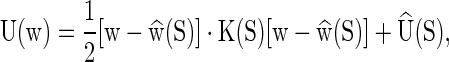 |
(1) |
where w is the oligomer configuration vector of size (12n − 6) as described above, and the sequence-dependent coefficients are a symmetric, positive-definite oligomer stiffness matrix K(S) of size (12n − 6) × (12n − 6), a vector of coordinates 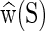 that defines the ground (or minimum accessible free energy) state of the fragment, and the energy
that defines the ground (or minimum accessible free energy) state of the fragment, and the energy 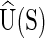 of the ground state. As the free energy (1) is quadratic the associated Boltzmann equilibrium distribution of the configuration coordinates w can (as discussed in the Supplementary Material S1) be assumed to be well-approximated by a Gaussian (or multivariate normal) with mean
of the ground state. As the free energy (1) is quadratic the associated Boltzmann equilibrium distribution of the configuration coordinates w can (as discussed in the Supplementary Material S1) be assumed to be well-approximated by a Gaussian (or multivariate normal) with mean  and covariance proportional to K−1(S).
and covariance proportional to K−1(S).
Given the sequence S of an oligomer as an input, the cgDNA model constructs the stiffness matrix K(S) and ground-state shape 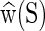 from a comparatively small parameter set. As further detailed in the Supplementary Material (Section S1), within the cgDNA model the stiffness matrix K(S) vanishes outside a stencil of overlapping 18 × 18 blocks, which reflects the restriction within the model to nearest-neighbour base coupling terms in the free energy. Moreover, for an arbitrary sequence, the stiffness matrix K(S) can simply be assembled by overlaying overlapping small submatrices. Each submatrix is itself selected from a small number of possibilities in the model parameter set according to either the composition of the associated base pair
from a comparatively small parameter set. As further detailed in the Supplementary Material (Section S1), within the cgDNA model the stiffness matrix K(S) vanishes outside a stencil of overlapping 18 × 18 blocks, which reflects the restriction within the model to nearest-neighbour base coupling terms in the free energy. Moreover, for an arbitrary sequence, the stiffness matrix K(S) can simply be assembled by overlaying overlapping small submatrices. Each submatrix is itself selected from a small number of possibilities in the model parameter set according to either the composition of the associated base pair 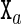 or of the associated dinucleotide step
or of the associated dinucleotide step 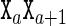 . This means that the entries in the stiffness matrix K(S) have only a local dependence on the sequence S; because of the overlap pattern, there is trinucleotide sequence dependence of the 6 × 6 diagonal intra–intra blocks, while all other entries have dinucleotide sequence dependence.
. This means that the entries in the stiffness matrix K(S) have only a local dependence on the sequence S; because of the overlap pattern, there is trinucleotide sequence dependence of the 6 × 6 diagonal intra–intra blocks, while all other entries have dinucleotide sequence dependence.
While the stiffness matrix K(S) is narrowly banded, it is not block diagonal, which means that its inverse is dense, albeit with blocks whose entries decay quickly away from the diagonal. Moreover, the entries in the inverse have a non-local dependence on the entries in the original matrix, and so a non-local dependence on the sequence S. Consequently, in the Boltzmann equilibrium distribution predicted by the cgDNA model there are correlations between the configurations of any two distant bases, albeit that these correlations fall off quickly with separation along the oligomer. Moreover, the values of these correlations have a non-local dependence on the sequence, due to the non-local nature of the operation of inverting the matrix K(S).
Within the cgDNA model there is a similar simple construction procedure for the ground-state vector  for any sequence S. However, as further detailed in the Supplementary Material (Section S1), this construction involves the inverse of the stiffness matrix K(S), and it is this feature which means that the cgDNA model encompasses non-local sequence dependence of the ground-state vector
for any sequence S. However, as further detailed in the Supplementary Material (Section S1), this construction involves the inverse of the stiffness matrix K(S), and it is this feature which means that the cgDNA model encompasses non-local sequence dependence of the ground-state vector 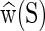 .
.
The current version of cgDNA is not parametrized to give physical significance to differences in the value of the ground-state energy 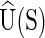 for two different sequences S1 and S2; in essence there is currently an associated arbitrary choice of constant in the ground-state energy of each different sequence. However, for a given sequence S, there is no impediment to computing either the associated normalized Boltzmann equilibrium distribution, or the differences in free energies of two different configurations w1 and w2, because the value of
for two different sequences S1 and S2; in essence there is currently an associated arbitrary choice of constant in the ground-state energy of each different sequence. However, for a given sequence S, there is no impediment to computing either the associated normalized Boltzmann equilibrium distribution, or the differences in free energies of two different configurations w1 and w2, because the value of 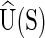 does not appear in either expression.
does not appear in either expression.
Software
The cgDNA software package is a suite of Matlab (http://www.mathworks.com) programs for implementing the cgDNA rigid base model; the package is freely available for download at http://lcvmwww.epfl.ch/cgDNA. The cgDNA software can also be run within the open source code Octave (http://www.gnu.org/software/octave/). The package depends upon a cgDNA parameter set, and currently cgDNAparamset1 is available as part of the download. As described in detail in (6), this parameter set was estimated using a large training set of MD trajectories of B-DNA oligomers of lengths 18 and 12 (an extended version of the ABC set of oligomers (14)). The simulations were performed in explicit solvent with physiological concentrations of (potassium chloride) salt using the latest AMBER force field, according to a well-established protocol (14). Other parameter sets are currently under development to reflect both different solvent conditions, and differing MD protocols and parameter estimation techniques, and will be added to the web page as they become available. The parameter set is provided as a Matlab data file with a relatively simple, documented format, so that the user may in principle also provide their own parameter values. The current version of cgDNA is by construction Gaussian, i.e. multivariate normal, so that in particular all scalar marginals of its associated Boltzmann distribution are univariate normal. This need not be the case for histograms generated by the MD training set, and notable deviations from normality have been observed and commented on for certain sequence fragments (14). Nevertheless, at the level of resolution of a sequence-dependent, rigid base model, such as cgDNA, the deviations from Gaussian appear minor compared to the sequence-dependent variations exhibited in the model. This issue was previously discussed in (6), and in particular the helical parameter histograms for all of the MD data used to train the cgDNA model parameter set can be found in that articles Supplementary Web Page http://lcvmwww.epfl.ch/cgDNA/uvw.
For a given parameter set, the basic user input is a string 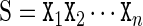 with
with 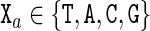 representing the 5′ to 3′ sequence along the reference strand. For the software, the sequence can be as short as a single dinucleotide step, to make a two base-pair oligomer or dimer; the effective upper bound on input sequence length depends on the available local computer memory. The package has been tested on sequences of 5K base pairs (which generates a 60K × 60K symmetric, sparse, stiffness matrix). Nevertheless, the practical target sequence length is fragments with between ten to a few hundreds of base pairs. The lower limit is dictated by a decrease in confidence in the quadratic cgDNA model for bases close to the end of a fragment, which in turn is related to fraying that is observed in MD simulations, see, for example, the comparison between 12mer and 14mer reconstructions shown in Figure 3. For most end sequences, and for the solvent conditions detailed in (6), a conservative procedure seems to be to disregard, or at least be cautious of, predictions for bases that are three base pairs or closer to the end of a fragment. With this rule of thumb a 10mer only has 4 base pairs that are sufficiently far from the ends. In contrast, the practical upper length limit is dictated by the observation that the sequence-dependent mechanical properties of naked DNA, particularly in the absence of any self-avoidance interactions, do not seem to be physically pertinent for fragments of many hundreds of base pairs in length.
representing the 5′ to 3′ sequence along the reference strand. For the software, the sequence can be as short as a single dinucleotide step, to make a two base-pair oligomer or dimer; the effective upper bound on input sequence length depends on the available local computer memory. The package has been tested on sequences of 5K base pairs (which generates a 60K × 60K symmetric, sparse, stiffness matrix). Nevertheless, the practical target sequence length is fragments with between ten to a few hundreds of base pairs. The lower limit is dictated by a decrease in confidence in the quadratic cgDNA model for bases close to the end of a fragment, which in turn is related to fraying that is observed in MD simulations, see, for example, the comparison between 12mer and 14mer reconstructions shown in Figure 3. For most end sequences, and for the solvent conditions detailed in (6), a conservative procedure seems to be to disregard, or at least be cautious of, predictions for bases that are three base pairs or closer to the end of a fragment. With this rule of thumb a 10mer only has 4 base pairs that are sufficiently far from the ends. In contrast, the practical upper length limit is dictated by the observation that the sequence-dependent mechanical properties of naked DNA, particularly in the absence of any self-avoidance interactions, do not seem to be physically pertinent for fragments of many hundreds of base pairs in length.
Figure 3.
Selected ground-state coordinate values for the sequences A4T4 and A4T4_mod. See also caption of Figure 1. Here, a non-local sequence context dependence is visible in some coordinates in the cgDNA, MD and NMR results (see main text). NMR results are for the sequence A4T4 whereas MD results are for the sequence A4T4_mod. cgDNA predictions are given for both A4T4 (green) and A4T4_mod (black); the difference in predictions of 12mer and 14mer ground states gives an indication of end effects within the cgDNA model.
Given a parameter set and input sequence S, the cgDNA package immediately computes the free energy minimizer (or ground state) 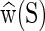 and associated stiffness matrix K(S). These oligomer-based coefficients can then be visualized in a variety of ways. And the user may also compute free energy differences between any two configurations of the given DNA fragment. The internal computations within cgDNA are performed using a non-dimensional and numerically well scaled form of the Curves+ coordinates as detailed in (6,13). However, in order to make possible a wide range of comparison of data, cgDNA includes procedures for both reading and writing DNA fragment configurations in either of the (dimensional, unscaled) Curves+ (12) helical coordinates or as PDB format coordinates (26) of each non-hydrogen atom in each base. Through the intermediary of PDB coordinates other variables, for example, the 3DNA (27) versions of the DNA internal helical parameters, corresponding to any configuration can also be either read in or out.
and associated stiffness matrix K(S). These oligomer-based coefficients can then be visualized in a variety of ways. And the user may also compute free energy differences between any two configurations of the given DNA fragment. The internal computations within cgDNA are performed using a non-dimensional and numerically well scaled form of the Curves+ coordinates as detailed in (6,13). However, in order to make possible a wide range of comparison of data, cgDNA includes procedures for both reading and writing DNA fragment configurations in either of the (dimensional, unscaled) Curves+ (12) helical coordinates or as PDB format coordinates (26) of each non-hydrogen atom in each base. Through the intermediary of PDB coordinates other variables, for example, the 3DNA (27) versions of the DNA internal helical parameters, corresponding to any configuration can also be either read in or out.
In contrast, the stiffness matrix K(S) is currently only available within cgDNA when expressed with respect to the internal Curves+ coordinates. The basic reason for this limitation is that the free energy U(w) does not remain quadratic under a general nonlinear change of coordinates, so that even the meaning of a stiffness matrix expressed in different coordinates is not entirely straightforward. Although this issue can be addressed in a number of ways, we here opt for simplicity and provide results in only one set of coordinates, namely the cgDNA internal ones.
Further details of the cgDNA software can be found in the Supplementary Material (Section S2) and in the documented program files.
Data for comparison
The predictive capabilities of the cgDNA modelling package are assessed here by making comparisons between different approximations to ground-state configurations: direct cgDNA predictions, experimental observation of shape, and time-averaged structures along all-atom MD simulations in explicit solvent. For this purpose we consider the DNA molecules with the sequences shown in Table 1, where the availability of the differing types of prediction of the associated ground states are also indicated. The naming conventions largely follow (28,29). Since the pair of sequences A4T4 and A4T4_mod differ only at the ends, they are grouped together throughout, as is the pair of sequences T4A4 and T4A4_mod.
Table 1. Sequences and data used in the assessment of the cgDNA package. Check marks (and where appropriate citations) indicate the availability and external source of each type of prediction of the shape of the free energy minimizer of each fragment. See main text for structure accession numbers and detailed description of the MD simulations.
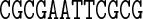
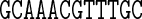
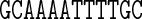
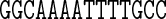
The data used in our comparisons were obtained from a variety of sources. The experimental data consists of NMR structures of the sequences A4T4 and T4A4 (PDB codes 1rvh and 1rvi, (34)) and the Dickerson–Drew dodecamer sequence DD (PDB code 1NAJ, (31)), along with X-ray crystallography structures of the sequence DD (PDB code 1FQ2, (32)) and the sequence A3CGT3 (PDB code 1HQ7, (33)). For each of the published NMR structures, we computed cgDNA shape parameters for their ‘best representative conformer’ as indicated in each PDB file. The alternative of computing the average over all published structures gave rather similar results.
The MD structural estimates for the sequences DD, A4T4_mod and T4A4_mod were taken from (28) and (29); specifically we used time averages from the KCl_Dg simulation (duration 2.4 μs) from Supplementary Table S2 of (28), and from the A4T4_mod and T4A4_mod simulations (both of duration 150 ns) in Supplementary Table S2 of (29). Finally, for the sequence A3CGT3, we ourselves performed a 200 ns all-atom MD simulation in explicit solvent using the protocol described in the previous section. All the simulations used for comparison employed the same protocol as in the cgDNA MD training set, but for longer durations (14). In particular, the DD trajectory is currently still among the longest duration MD simulations of DNA that have been published.
The above mentioned data was originally and variously expressed in either PDB, 3DNA, cgDNA scaled, or Curves+ coordinates. The necessary conversions were then made using cgDNA procedures, and the comparison of ground states given below are all expressed in (dimensional, unscaled) Curves+ helical parameters.
RESULTS
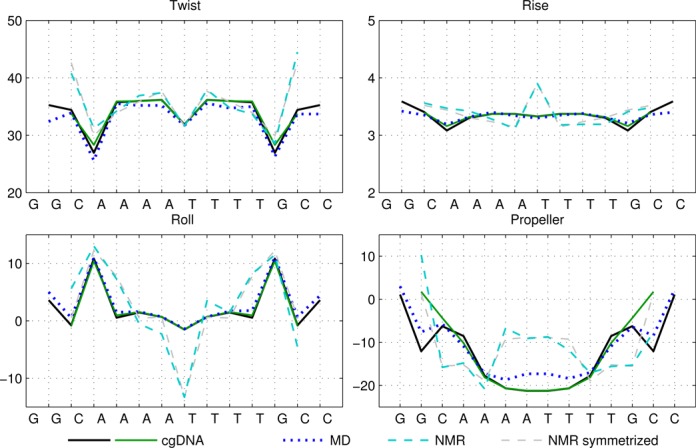
Figures 1–4 show the ground-state values of the Twist, Roll, Rise and Propeller helical coordinates at each position along the molecule for each of the sequences in Table 1, as determined in a number of different ways. Analogous plots for all the helical coordinates are provided in Supplementary Figures S6–S9. In each figure, the sequence is indicated on the abscissa, with intra-base-pair coordinates at each base pair, and inter-base-pair coordinates at each junction, or base-pair step. It can be observed that for each molecule, the ground state predicted by cgDNA is rather close to that predicted directly from MD simulation throughout the interior and end regions of the fragments. In particular, the difference between cgDNA and MD predictions are small compared to the variations along each fragment, and between fragments. (In Figure 1, MD estimates at the ends were not available for the sequence DD.)
Figure 1.
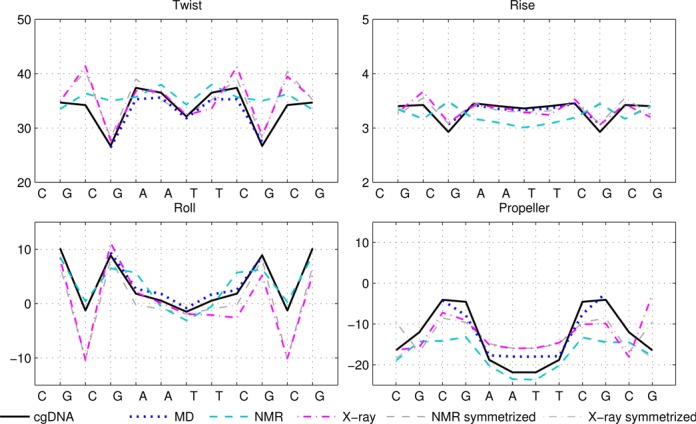
Selected ground-state coordinate values for the sequence DD (Dickerson–Drew dodecamer). The cgDNA predictions are in good agreement with the MD, and reasonable agreement with NMR and X-ray results. MD results for end base pairs are not available for this sequence. In Figures 1–4, rotations (Twist, Roll and Propeller) are measured in degrees and Rise is in Å. Sequence position is indicated on the horizontal axis and coordinate values are interpolated by piecewise linear curves.
Figure 4.
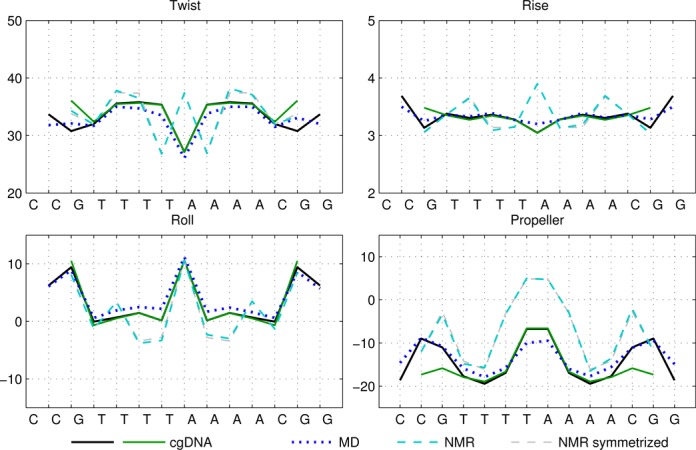
Selected ground-state coordinate values for the sequences T4A4 and T4A4_mod. See also captions of Figures 1 and 3.
It can also be seen that both the cgDNA and MD predictions are in reasonably good agreement with the available experimental data. As detailed in Table 1, all of the sequences that are considered are palindromes, for which the physical properties of the two DNA strands are indistinguishable, so that the ground-state configuration (or ensemble average solution structure) must have a corresponding symmetry. In Curves+ conventions the symmetry in the two possible choices of reading strand is expressed in the condition that the ground-state coordinate values should either be even or odd functions of position (depending on the coordinate type) about the centre of the molecule (34). However, as already remarked in (27), the experimental (especially crystallographic) data are not perfectly consistent with this property. For example, it is evident that the Twist and Roll estimates from the X-ray structures shown in Figures 1 and 2 are not perfectly symmetric. This absence of symmetry could be attributed to a variety of sources, for example packing effects in the case of X-ray crystallography. Nevertheless, in order to make the best possible comparison with the cgDNA prediction of the ground state, which should be symmetric, we are free to read from either of the two possible strands in the available experimental observations. Consequently, for each NMR or X-ray estimate in Figures 1–4, we also include a symmetrized value, which was computed as the arithmetic average of the two coordinate values obtained from each of the two possible choices of reference strand. These symmetrized NMR and X-ray estimates are consistent with the palindromic nature of the molecule and are in better agreement with the cgDNA predictions and MD averages. More generally, the cgDNA and MD results are in reasonable agreement with the NMR and X-ray data throughout, but with some notable exceptions, for example Roll and Propeller in Figure 3, and Propeller in Figure 4.
Figure 2.
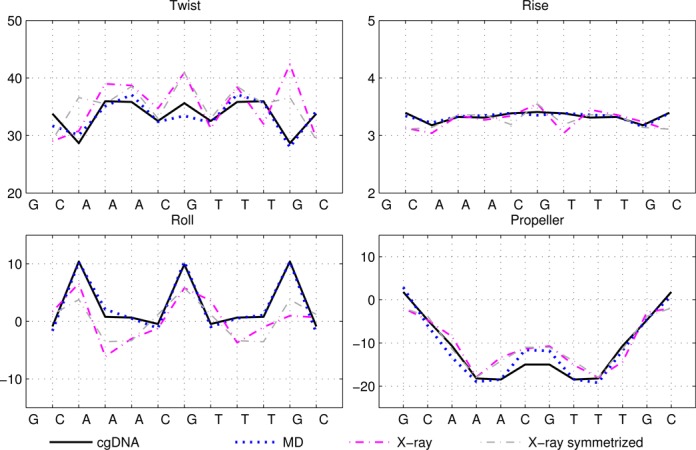
Selected ground-state coordinate values for the sequence A3CGT3. See also caption of Figure 1.
A unique feature of the rigid base model underlying the cgDNA package is its ability to capture non-local sequence context effects for the ground-state coordinates. The fact that some coordinates exhibit measurable context effects has been previously noted (6,13–14,23–25,30), and is visible here in our example A-tract sequences. For instance, as can be seen in Figure 4, the value of Roll for the central AA dinucleotide in the tetranucleotide 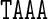 differs from the value of Roll for the central AA dinucleotide in the tetranucleotide
differs from the value of Roll for the central AA dinucleotide in the tetranucleotide 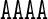 ; the context dependence is visible in the cgDNA and MD results as well as in the NMR data, although there is some discrepancy between the values. Similarly, in Figure 3, the value of Propeller for the central A nucleotide in the pentanucleotide
; the context dependence is visible in the cgDNA and MD results as well as in the NMR data, although there is some discrepancy between the values. Similarly, in Figure 3, the value of Propeller for the central A nucleotide in the pentanucleotide 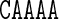 differs from the value of Propeller for the central A nucleotide in the pentanucleotide
differs from the value of Propeller for the central A nucleotide in the pentanucleotide 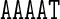 . A similar observation holds for the value of Propeller in the central A nucleotide of the pentanucleotides
. A similar observation holds for the value of Propeller in the central A nucleotide of the pentanucleotides 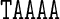 and
and 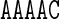 in Figure 4. For the cases considered here, the context effects are visibly stronger for the experimental data than for the MD or cgDNA results. Further examples illustrating context effects, including the impact of a single point mutation, can be found in (6,13).
in Figure 4. For the cases considered here, the context effects are visibly stronger for the experimental data than for the MD or cgDNA results. Further examples illustrating context effects, including the impact of a single point mutation, can be found in (6,13).
DISCUSSION
We have described cgDNA, a new conformation prediction package for B-form DNA in standard environmental conditions. It is based on a sequence-dependent rigid base model of DNA and employs a relatively small, mononucleotide- and dinucleotide-dependent parameter set. For a molecule of any given length and sequence, cgDNA provides a direct, explicit prediction of the ground-state conformation of the molecule, along with the ground-state stiffness (or inverse covariance) matrix. These two sequence-dependent quantities are precisely what are required to evaluate DNA free-energy differences between any two conformations of a given oligomer. For this (or any other) purpose, cgDNA shapes can be both input and output in either the Curves+ definitions of the DNA structural coordinates, or as a PDB file of atomic coordinates, so that any desired additional analysis can be made.
The current cgDNA parameter set has been trained on MD simulations of rather short fragments 12–18 bp. However, the cgDNA ground-state shape and stiffness matrix can be reconstructed for oligomers up to at least a few thousand base pairs in length. Such a ground state and stiffness matrix are the required inputs to implement a Monte Carlo sampling procedure, which, as will be reported elsewhere, allows comparison of cgDNA model predictions of quantities such as DNA persistence length with longer length scale experimental data.
cgDNA can closely replicate the sequence-dependent conformations of bases in B-form DNA in solvent as predicted by fully atomistic MD simulation, and this for sequences that were not in the original cgDNA parameter training set. But cgDNA is computationally several orders of magnitude faster. As the cgDNA parameters are trained on MD simulations, it is natural that the level of agreement between the cgDNA predictions and the experimental data can be no better than that between the MD simulation predictions and experiment. However, in all cases that we have examined it is also the case that the cgDNA predictions are no worse than the time-averaged MD predictions, and both are reasonably close to the experimental data when compared, for example, to deviations from palindromic symmetry that are exhibited in the experimental observations. Nevertheless, the differences between the cgDNA and MD predictions are in all cases considerably smaller than the differences with experiment. Certainly, both MD potentials and consequently cgDNA parameter sets could be improved, but the comparatively large discrepancies with experiment may well also be related to the difficulties and particularities of the available experimental techniques in predicting DNA free-energy minimizers in solution.
As with any coarse-grain model, the computational efficiency of cgDNA comes at the price of a decrease in resolution, so that (in addition to training cgDNA parameter sets, e.g. for differing solvent conditions) MD simulations remain essential to examine the details of any non-Gaussian (e.g. melting or fraying) events, or to obtain any atomistically detailed information. Nevertheless, and while acknowledging that improved model parameter sets would certainly be desirable, our conclusion is that for the purpose of predicting sequence-dependent B-form DNA ground states at the resolution of helical parameters cgDNA can already effectively serve as a substitute for MD simulation. Once a cgDNA parameter set has been established, the additional computational effort to compute the ground state of each new sequence with the cgDNA software is trivial compared to that of an additional MD simulation, so that cgDNA can be used both for rapidly scanning over a much larger diversity of oligomer sequences, and for much longer sequences—if desired, ground states of DNA fragments of several thousand base pairs can easily be computed. cgDNA provides an efficient tool for studying sequence-dependent structural variations in B-form DNA, both within and between molecules, and is sufficiently fast to allow long, or many, sequences to be scanned easily.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
It is a pleasure for the authors to be able to thank J.S. Mitchell for valuable suggestions and discussions, J. Glowacki for programming help, R. Lavery for comments on an early draft, and the anonymous referees for their constructive criticisms.
FUNDING
Swiss National Science Foundation [200021-126666 and 200020-143613 to J.H.M.]. Funding for open access charge: Swiss National Science Foundation [200020-143613 to J.H.M.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Schleif T. DNA looping. Annu. Rev. Biochem. 1992;61:199–223. doi: 10.1146/annurev.bi.61.070192.001215. [DOI] [PubMed] [Google Scholar]
- 2.Segal E., Fondufe-Mittendorf Y., Chen L., Thastrom A., Field Y., Moore I., Wang J., Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michor F., Liphardt J., Ferrari M., Widom J. What does physics have to do with cancer? Nat. Rev. Cancer. 2011;11:657–670. doi: 10.1038/nrc3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calladine C., Drew H., Luisi B.F., Travers A.A. Understanding DNA. 3rd edn. Waltham, Massachusetts: Elsevier Academic Press; 2004. [Google Scholar]
- 5.Cheatham T.E., Case D.A. Twenty-five years of nucleic acid simulations. Biopolymers. 2013;99:969–977. doi: 10.1002/bip.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez O., Petkevičiūtė D., Maddocks J. A sequence-dependent rigid-base model of DNA. J. Chem. Phys. 2013;138:055102. doi: 10.1063/1.4789411. [DOI] [PubMed] [Google Scholar]
- 7.Potoyan D.A., Savelyev A., Papoian G.A. Recent successes in coarse-grained modeling of DNA. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2013;3:69–83. [Google Scholar]
- 8.Doye J.P.K., Ouldridge T.E., Louis A.A., Romano F., Sulc P., Matek C., Snodin B.E., Rovigatti L., Schreck J.S., Harrison R.M., Smith W.P. Coarse-graining DNA for simulations of DNA nanotechnology. Phys. Chem. Chem. Phys. 2013;15:20395–20414. doi: 10.1039/c3cp53545b. [DOI] [PubMed] [Google Scholar]
- 9.Noid W.G. Perspective: coarse-grained models for biomolecular systems. J. Chem. Phys. 2013;139:090901. doi: 10.1063/1.4818908. [DOI] [PubMed] [Google Scholar]
- 10.Hinckley D.M., Freeman G.S., Whitmer J.K., de Pablo J.J. An experimentally-informed coarse-grained 3-site-per-nucleotide model of DNA: Structure, thermodynamics, and dynamics of hybridization. J. Chem. Phys. 2013;139:144903. doi: 10.1063/1.4822042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hospital A., Faustino I., Collepardo-Guevara R., González C., Gelpí J.L., Orozco M. NAFlex: a web server for the study of nucleic acid flexibility. Nucleic Acids Res. 2013;41:W47–W55. doi: 10.1093/nar/gkt378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavery R., Moakher M., Maddocks J., Petkeviciute D., Zakrzewska K. Conformational analysis of nucleic acids revisited: curves+ Nucleic Acids Res. 2009;37:5917–5929. doi: 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petkevičiūtė D. Ph.D. thesis #5520. EPFL; 2012. A DNA coarse-grain rigid base model and parameter estimation from molecular dynamics simulations. [Google Scholar]
- 14.Lavery R., Zakrzewska K., Beveridge D., Bishop T., Case D., Cheatham T. III, Dixit S., Jayaram B., Lankas F., Laughton C., et al. A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA. Nucleic Acids Res. 2010;38:299–313. doi: 10.1093/nar/gkp834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Onuchic J.N., Luthey-Schulten Z., Wolynes P.G. Theory of protein folding: the energy landscape perspective. Annu. Rev. Phys. Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 16.Truong H.H., Kim B.L., Schafer N.P., Wolynes P.G. Funneling and frustration in the energy landscapes of some designed and simplified proteins. J. Chem. Phys. 2013;139:121908. doi: 10.1063/1.4813504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levy Y., Onuchic J.N., Wolynes P.G. Fly-casting in protein-DNA binding: frustration between protein folding and electrostatics facilitates target recognition. J. Am. Chem. Soc. 2007;129:738–739. doi: 10.1021/ja065531n. [DOI] [PubMed] [Google Scholar]
- 18.Marcovitz A., Levy Y. Frustration in protein-DNA binding influences conformational switching and target search kinetics. Proc. Natl. Acad. Sci. U.S.A. 2011;108:17957–17962. doi: 10.1073/pnas.1109594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forman C.J., Fejer N.S., Chakrabarti D., Barker P.D., Wales D.J. Local frustration determines molecular and macroscopic helix structures. J. Phys. Chem. B. 2013;117:7918–7928. doi: 10.1021/jp4040503. [DOI] [PubMed] [Google Scholar]
- 20.Liedl T., Högberg B., Tytell J., Ingber D.E., Shih W.M. Self-assembly of three-dimensional prestressed tensegrity structures from DNA. Nat. Nanotechnol. 2010;5:520–524. doi: 10.1038/nnano.2010.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olson W., Gorin A., Lu X.-J., Hock L., Zhurkin V. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. U.S.A. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lankaš F., Šponer J., Langowski J., Cheatham T. III. DNA basepair step deformability inferred from molecular dynamics simulations. Biophys. J. 2003;85:2872–2883. doi: 10.1016/S0006-3495(03)74710-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yanagi K., Privé G.G., Dickerson R.E. Analysis of local helix geometry in three B-DNA decamers and eight dodecamers. J. Mol. Biol. 1991;217:201–214. doi: 10.1016/0022-2836(91)90620-l. [DOI] [PubMed] [Google Scholar]
- 24.Packer M., Dauncey M., Hunter C. Sequence-dependent DNA structure: tetranucleotide conformational maps. J. Mol. Biol. 2000;295:85–103. doi: 10.1006/jmbi.1999.3237. [DOI] [PubMed] [Google Scholar]
- 25.Araúzo-Bravo M.J., Fujii S., Kono H., Ahmad S., Sarai A. Sequence-dependent conformational energy of DNA derived from molecular dynamics simulations: toward understanding the indirect readout mechanism in protein-DNA recognition. J. Am. Chem. Soc. 2005;127:16074–16089. doi: 10.1021/ja053241l. [DOI] [PubMed] [Google Scholar]
- 26.Berman H.M., Battistuz T., Bhat T.N., Bluhm W.F., Bourne P.E., Burkhardt K., Feng Z., Gilliland G.L., Iype L., Jain S., et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 27.Lu X.-J., Olson W. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dršata T., Pérez A., Orozco M., Morozov A., Šponer J., Lankaš F. Structure, stiffness and substates of the Dickerson-Drew dodecamer. J. Chem. Theory Comput. 2013;9:707–721. doi: 10.1021/ct300671y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lankaš F., Špačková N., Moakher M., Enkhbayar P., Šponer J. A measure of bending in nucleic acids structures applied to A-tract DNA. Nucleic Acids Res. 2010;38:3414–3422. doi: 10.1093/nar/gkq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou T., Yang L., Lu Y., Dror I., Machado A., Ghane T., Felice R.D., Rohs R. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013;41:W56–W62. doi: 10.1093/nar/gkt437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Z., Delaglio F., Tjandra N., Zhurkin V., Bax A. Overall structure and sugar dynamics of a DNA dodecamer from homo- and heteronuclear dipolar couplings and 31P chemical shift anisotropy. J. Biomol. NMR. 2003;26:297–315. doi: 10.1023/a:1024047103398. [DOI] [PubMed] [Google Scholar]
- 32.Sines C., McFail-Isom L., Howerton S., VanDerveer D., Williams L. Cations mediate B-DNA conformational heterogeneity. J. Am. Chem. Soc. 2000;122:11048–11056. [Google Scholar]
- 33.Locasale J., Napoli A., Chen S., Berman H., Lawson C. Signatures of protein-DNA recognition in free DNA binding sites. J. Mol. Biol. 2009;386:1054–1065. doi: 10.1016/j.jmb.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stefl R., Wu H., Ravindranathan S., Sklenár V., Feigon J. DNA A-tract bending in three dimensions: solving the dA4T4 vs. dT4A4 conundrum. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1177–1182. doi: 10.1073/pnas.0308143100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.