


Abstract
DNA damage response and repair proteins are centrally involved in genome maintenance pathways. Yet, little is known about their functional role under non-DNA damage-inducing conditions. Here we show that Rad9 checkpoint protein, known to mediate the damage signal from upstream to downstream essential kinases, interacts with Aft1 transcription factor in the budding yeast. Aft1 regulates iron homeostasis and is also involved in genome integrity having additional iron-independent functions. Using genome-wide expression and chromatin immunoprecipitation approaches, we found Rad9 to be recruited to 16% of the yeast genes, often related to cellular growth and metabolism, while affecting the transcription of ∼2% of the coding genome in the absence of exogenously induced DNA damage. Importantly, Rad9 is recruited to fragile genomic regions (transcriptionally active, GC rich, centromeres, meiotic recombination hotspots and retrotransposons) non-randomly and in an Aft1-dependent manner. Further analyses revealed substantial genome-wide parallels between Rad9 binding patterns to the genome and major activating histone marks, such as H3K36me, H3K79me and H3K4me. Thus, our findings suggest that Rad9 functions together with Aft1 on DNA damage-prone chromatin to facilitate genome surveillance, thereby ensuring rapid and effective response to possible DNA damage events.
INTRODUCTION
Genetic material must be maintained throughout life so that it remains functionally intact and is faithfully transmitted to progeny. To meet this challenge, cells have evolved a set of complementary DNA damage response (DDR) pathways and dedicated protein machineries that arrest cell-cycle progression, thus providing a time window for repair. The strong cancer predisposition observed in certain inherited human disorders as well as the increasing number of ageing-related syndromes with defects in DNA repair emphasize the biological impact of genome care taking mechanisms in cellular life (1).
Rad9 protein represents one of the most well-studied members of the DDR pathway in the model eukaryotic organism Saccharomyces cerevisiae (2). It is a 148 kD multidomain protein containing two BRCA1 C-Terminal (BRCT) domains which are required for its oligomerization and the recognition of phosphorylated histones (γH2A) upon DNA damage (3–7). Similar to the mammalian p53BP1, Rad9 protein contains a conserved Tudor domain that recognizes H3K79 methylated histones after double-strand break (DSB) formation (8). ScRad9 acts at G1/S, intra-S and G2/M checkpoints (2,9–11); the protein is phosphorylated during normal cell-cycle progression but it becomes hyperphosphorylated by Mec1 and Tel1 kinases in response to DNA damage (12,13). In this form, ScRad9 acts as a scaffold that brings Rad53 effector kinase in close proximity to Mec1 and Tel1 kinases, and facilitates its auto-phosphorylation, a critical step for DDR signaling (14,15).
The involvement of basal transcription factors in distinct DNA repair pathways (16,17) as well as the preferential repair of the transcribed DNA strand of genes (18) and of promoter surrounding sequences in mammals (19) have established a functional link between DNA repair and transcription. Upon ultraviolet (UV)-induced DNA damage, ScRad9 is required for the repair of transcribed and non-transcribed DNA strands of active genes but, importantly, not for the repair of transcriptionally inactive sequences (20). However, under non-DNA damage-inducing conditions, Rad9 binds chromatin, though its functional role at non-damaged DNA sites remains unknown (4,15). Mutations of the Tudor domain almost completely abolish Rad9 chromatin association during G1 phase in undamaged cells suggesting that this domain recognizes distinct histone methylation marks to enable association with bulk chromatin (4). Under untreated conditions, the Rad9 Tudor domain was also shown to associate with chromatin through its interaction with H3K79me and its BRCT-mediated dimerization (21). Upon DNA damage, activated Rad9 also interacts with γ-H2A or with Dpb11 replication initiation protein, during M phase, at DNA damage sites. Besides UV-induced DNA lesions, Rad9 was recently shown to be required along with Mec1, Rad51 and Rad54 for DNA DSB repair by the homologous recombination pathway (22,23).
Here, we employed a series of biochemical, genetics and functional genomics approaches to dissect the functional contribution of Rad9 in undamaged cells. We showed that Rad9 physically and genetically interacts with the transcription factor Aft1 and is recruited to fragile chromatin sites in the absence of exogenously inducing conditions in an Aft1-dependent manner. Aft1 is the main transcription factor that regulates iron homeostasis in yeast (24) but it appears to be a multitask protein affecting a diverse range of cellular processes, including cell wall stability, DNA damage, protein transport and mitochondrial function (25–27). Fine mapping of Rad9 protein localization to chromatin under physiological conditions revealed substantial genome-wide parallels between Rad9-bound chromatin regions and histone marks linked to transcriptional activation, such as H3K36me3, H3K79me3 and H3K4me. Taken together, these findings suggest that Rad9 and Aft1 function together on DNA damage-prone sites to enhance genome surveillance. This way, a rapid and effective response to possible DNA damage events is ensured.
MATERIALS AND METHODS
Strains and growth conditions
The background strain used in this study is the BJ5457 that lacks proteases (MATa ura3–52 trp1 lys2–801 his3[Delta]200 pep4::HIS3 prb1[Delta]1.6R can1 Gal+). Epitope tagging of the proteins was performed as previously described with the insertion of 9 or 13Myc oligos using the proper plasmids: pYM1 kanMX6 (3HA), pYM6 k1TRP1 (9Myc) (28) or pFA6a-13Myc-TRP1 (29) to insert the tag with the respective marker. The primers used for the epitope tagging and gene deletions are listed in Supplementary Table S1 along with the constructed strains. aft1Δ is S288C-derived (30). Cells were grown in Synthetic Complete (SC) or Yeast Peptone Dextrose (YPD) media as described in (31). SC medium contains 0.67% nitrogen base (including 1.23 μM FeCl3 and 0.25 μM CuSO4), the 20 aminoacids, uracil, adenine and 2% glucose. The copper-dependent transcription was induced by the addition of BCS chelator of Cu+1 and Cu+2 (bathocuproine disulfonic acid-Na2 salt, Serva) in a final concentration of 100 μM. The metal-dependent transcription was induced by the addition in a final concentration of 100 μM of the chelator of Cu+1/Cu+2/Fe+1/Fe+2 BPS (bathophenanthroline disulfonic acid-Na2 salt, Serva). For the galactose induction experiment, cells were grown in YP Raffinose and galactose was added in a final concentration of 2% for 75 min.
6-Azauracil (6-AU) growth assay
Cells were grown in rich medium (YPD) to an OD600 ∼1. Seven serial dilutions (1/10) were spotted on YPD plates which also contained or not (control) 300 μg/ml 6-AU (Sigma A1757). Plates were incubated for 2 days at 30°C.
BioLector growth assays
Exponentially growing cells of the different strains were collected and 1.5 ml micro-cultures were set (at least in triplicate) in a 48-well flowerplate starting from an OD600 = 0.25. Micro-cultures were left growing in BioLector machine (m2p labs), shaking and under ideal humidity conditions until they reached stationary phase. OD600 (biomass) was monitored every 20 min and recorded electronically. The growth assay was performed twice. Results from the OD measurements of 3–6 independent micro-cultures of each strain were averaged point-by-point and normalized over the first value of each series before plotting the growth curves.
Constructs
The pDB20-flag-URA3 plasmid was used for the insertion and overexpression of Rad9. The pYX142-LEU2 plasmid was used for the insertion of Rad9–9Myc (NcoI-SlaI) and 9Myc (SmaI-SlaI). They were used for the overexpression of the proteins tested in co-immunoprecipitation (co-IP) and chromatin immunoprecipitation (ChIP) experiments. Plasmids for bacterial expression of 6×His-N-Aft1, 6×His-C-Aft1 and GST-Nhp6a, used in the in vitro protein interaction assay, were previously described (32), whereas plasmids for GST-N-Rad9 and GST-BRCT-Rad9 bacterial expression, used in the same assay, were constructed by insertion of a 1.5 kb (+1/+1513) fragment corresponding to N-Rad9 and a 0.95 kb (+2986/+3930) fragment corresponding to C-Rad9, respectively, between the BamHI and EcoRI sites of pGEX-2T vector.
ChIP
ChIP assays were performed as previously described (33) with some adaptations (Supplementary Protocol S1). Briefly, cells were grown until a final OD550 ∼0.8 (BCS was applied for 3 h and BPS for 6 h when needed), cross-linked with formaldehyde for 20 min at room temperature, broken by vortexing for 40 min at 4°C with glassbeads, sheared by sonication in fragments peaking at ∼400 bp. IP was performed using the sc-789 c-myc rabbit polyclonal immunoglobulin G (Santa Cruz) antibody (2 μg/IP). The extract of 6 × 107 cells was used per IP or INPUT sample. The ratio of IP/INPUT sample used in real-time quantitative polymerase chain reaction (qPCR) was 1/25; the dye used was SYBR green. The enrichment values were normalized to those of INPUT samples (non-immune) and are presented as fold change over the enrichment value obtained by amplification of control regions (background enrichment of the tested proteins) as mentioned in the figures. The primers used are listed in Supplementary Table S2.
Co-IP and GST-pull down assays
Rad9-Aft1 interaction was checked as follows: (i) Yeast cells were grown in SC medium with the proper auxotrophies to an OD550 ∼0.8, collected, broken by vortexing and properly washed (Supplementary Protocol S2). Note that 2 μg of the proper antibody were added and after an overnight incubation at 4°C, G sepharose beads were added followed by 2.5 h incubation at 4°C. rad9Δ strain which expressed endogenous Aft1–9Myc was transformed with high copy pDB20-flag plasmid and pDB20-Rad9-flag expressing flag-tagged Rad9 in order to get rad9Δ Aft1–9Myc pDB20 Rad9-flag strain. rad9Δ Aft1–9Myc pDB20 flag was used as control. Anti-flag was used as a bait (F3165 Sigma-Aldrich) followed by probing with anti-Myc (sc-789 Santa Cruz Biotechnology) in western blot assay. (ii) Protein extracts were prepared from wild type (wt) yeast cells grown in YPD/BPS-BCS, endogenously expressing Rad9–9Myc or Rad9–9Myc along with Aft1–3HA. Portions (1/40) of each extract were analyzed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting using anti-HA (sc-805 Santa Cruz Biotechnology) or anti-Myc to detect Aft1–3HA and Rad9–9Myc, respectively. Remaining extracts were incubated with EZview Red anti-HA affinity gel (Sigma) and then flow-through as well as pulled-down proteins were analyzed by SDS-PAGE and immunoblotting using anti-Myc to detect Rad9–9Myc. (iii) GST, GST-Nhp6a (positive control), GST-N-Rad9 and GST-BRCT-Rad9 peptides were expressed in Escherichia coli ER2566 cells and bound on glutathione agarose beads. 6xHis-N-Aft1 and 6xHis-C-Aft1 peptides were produced in E. coli ER2566 cells and purified by Ni-NTA agarose beads. Each eluted Aft1 derivative was incubated in vitro with each glutathione bound peptide. Beads were washed and retained peptides were eluted in gel loading buffer and analysed by SDS-PAGE and immunoblotting using anti-His antibody (Penta-His mouse, 34660 Qiagen). The electrophoretic pattern of the GST-tagged (total amounts) as well as the 6xHis-tagged (input amounts) proteins used in the assay was checked by coomassie blue staining.
Reverse transcriptase-qPCR (RT-qPCR) analyses
RNA was extracted using the hot acid phenol method. RT was performed as described (34) and transcript enrichment was calculated by qPCR. Normalization of the expression levels was done over a constitutively expressed gene (CMD1).
ChIP on chip analysis
The array used was S. cerevisise Tiling 1.0R Array manufactured by Affymetrix with probes tiled at a 5 bp resolution. The protocol proposed by Affymetrix was followed, adjusted and optimized to the needs of yeast (Supplementary Protocol S3). Cells were grown to a final concentration of OD550 = 0.8 in SC (and added BCS/BPS for 3 and 6 h incubation, respectively) or YP raffinose followed by addition of galactose (2%) for 75 min. Soluble chromatin solution from ∼7 × 107 cells was used per IP sample. INPUT chromatin (non-immune) from each experiment was used to normalize our results. CEL files obtained after scanning were loaded onto TAS v1.1 software to calculate the signal and P-values for each interrogated genomic probe position in a two sample comparison analysis. Visualization of the intensity values was done by using the Integrated Genome Browser, from which we obtained the results in two different P-value cut-offs (10−3 and 5 × 10−3). Further statistical analysis of the results was performed with the use of R statistical software (http://www.r-project.org/). All Tiling Array raw data are submitted to the EMBL-EBI ArrayExpress database (ArrayExpress accession E-MEXP-3838, E-MTAB-2200 and E-MTAB-2201 conforming to the MIAME guidelines).
Functional and average gene analyses
Functional analysis to determine the overrepresentation of Gene Ontology (GO) terms was performed with the use of the BiNGO plugin in the Cytoscape platform (35,36). Average gene analysis was performed as previously described (37,38). For the average gene analysis, each of the 5769 genes of S. cerevisiae [Saccharomyces Genome Database (SGD) version, sacCer1] was divided in 100 equally sized bins and the average signal value was calculated for each bin. In this way, every gene was shrunk into 100 points regardless of its total length with the first point corresponding to the Transcription Start Site (TSS) and the last one to the Transcription Termination Site (TTS). Subsequently, an area of ±500 bp from the TSS/TTS, respectively, was divided into 50 bins of 10 bp for every gene. In this way, each gene was represented as a profile consisting of 200 points (50 upstream, 100 genic, 50 downstream). In the occasions where another gene was located within less than 500 bp upstream or downstream, the area ended on the spot where the neighbouring gene was met. The plots obtained represent the average value of the 200 bins for the 5769 genes. GC-content of the sequences where the protein was enriched was calculated with EMBOSS GeeCee tool of Galaxy (http://main.g2.bx.psu.edu/), in comparison to a random sample of sequences of equal number and size. GC-content value distributions were compared with a two-sided t-test. Details on data analysis can be found in Supplementary Protocol S4.
Genome-wide expression analysis
The array used was GeneChip Yeast Genome 2.0 Array manufactured by Affymetrix including ∼5744 probe sets for 5841 of the 5845 genes present in S. cerevisiae. rad9Δ and wt cells were grown in SC (with the addition of the chelators BCS and BPS), RNA was isolated with hot acid phenol method (34) and the one-cycle cDNA synthesis protocol proposed by Affymetrix was followed for target preparation. Hybridization, washing and scanning was performed as proposed (Supplementary Protocol S5). Intensity values were calculated by Affymetrix Expression Console Software as well as by other commercial software, such as FlexArray using Robust Multi-array Average (RMA) to normalize our data (39). The average of the raw intensity values was calculated for two biological replicates and a two sample equal variance-homoscedastic t-test analysis was performed in order to obtain targets with a P-value ≤ 0.05 and a fold change of ±1.2 (rad9Δ versus wt). Functional analysis of the results was performed using the BiNGO plugin in the Cytoscape platform as described in Supplementary Protocol S4. All Expression Array raw data are submitted to the EMBL-EBI ArrayExpress database (ArrayExpress accession E-MEXP-3838, conforming to the MIAME guidelines).
RESULTS
Rad9 interacts with Aft1 and localizes to Aft1-regulated genes in an Aft1-dependent manner
We identified Aft1 as Rad9-interacting partner and verified this physical interaction by in vivo and in vitro assays. For a co-IP assay in yeast extracts, Rad9-flag tagged protein was expressed by a pDB20 high copy plasmid and Aft1–9Myc protein was tagged in the genome. Cells were grown in SC medium under conditions for iron regulon induction (by adding the iron ion chelator BPS, causing iron starvation) in which Aft1 is localized to the nucleus (24) (Figure 1A). The in vivo interaction was further confirmed by co-IP assay of Aft1–3HA and Rad9–9Myc proteins that were both tagged in the genome (Figure 1B). By performing GST-pull down assays we were able to identify the interacting domains of the two proteins. We found that Rad9 BRCT domain (at the C-terminus of Rad9) interacted with the N-terminal part of Aft1 which includes its DNA-binding domain (32) (Figure 1C).
Figure 1.
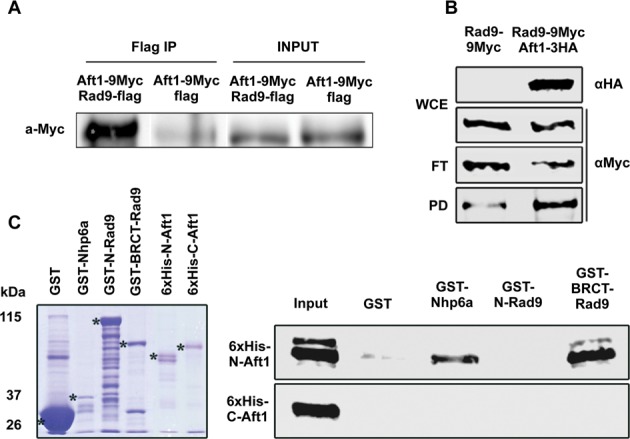
Rad9 associates with Aft1 in vivo with its BRCT domain interacting with N-Aft1 in vitro.(A) In rad9Δ background, Aft1 was tagged with 9Myc epitopes. Rad9 tagged with flag epitope was inserted in the high copy plasmid pDB20 and this construct was inserted to rad9Δ strain in order to get rad9Δ Aft1–9Myc pDB20-Rad9-flag strain. rad9Δ Aft1–9Myc pDB20-flag strain was used as control. In SDS-PAGE and immunoblotting analysis, anti-Flag was used as a bait and probing was done with anti-Myc. Input and IP samples are shown. The co-IP experiment was repeated twice. (B) Protein extracts were prepared from wt cells grown in YPD/BPS-BCS, endogenously expressing Rad9–9Myc or Rad9–9Myc along with Aft1–3HA. Portions (1/40) of each extract (WCE) were analyzed by SDS-PAGE and immunoblotting using anti-HA or anti-Myc to detect Aft1–3HA and Rad9–9Myc, respectively. Remaining extracts were incubated with EZview anti-HA agarose beads and then flow-through (FT) as well as pulled-down (PD) proteins were analyzed by SDS-PAGE and immunoblotting using anti-Myc to detect Rad9–9Myc. (C) Bacterially expressed GST (negative control), GST-Nhp6a (positive control) (32), GST-N-Rad9 or GST-BRCT-Rad9 proteins, bound on glutathione agarose beads, were incubated with bacterially expressed 6xHis-N-Aft1 or 6xHis-C-Aft1 derivatives eluted from Ni-NTA agarose beads. Glutathione beads were then washed and proteins bound on them were analysed by SDS-PAGE and immunoblotting using anti-His antibody. The input lane contains 20% of the total amount of each 6xHis-tagged protein incubated with the beads. Left panel: coomassie blue gel showing the electrophoretic pattern of the GST-tagged (total amounts) as well as the 6xHis-tagged (input amounts) proteins used in the assay.
Using ChIP experiments and qPCR, we further investigated the occupancy of the Rad9-Aft1 complex on previously identified Aft1 gene targets. We included in our analysis FTR1 and FRE1 genes known to be regulated by iron via Aft1 (40–42) and the copper-regulated CTR1 gene where we have found that Aft1 binds irrespective of a specific DNA consensus binding site (unpublished data) and positively affects its transcription. FRE7 copper-regulated gene was used as a negative control for Aft1 binding and regulation (43). Using conditions that favour the induction of both iron and copper regulons (BCS/BPS; chelators of copper and iron ions, respectively), we found that Rad9–9Myc was recruited to promoter and/or coding regions of the examined genes (Figure 2A–D). The recruitment of Rad9–9Myc to FTR1 and CTR1 genes was Aft1-dependent (Figure 2E). By contrast, Aft1–9Myc localization to the CTR1 or FTR1 gene promoters under the same experimental conditions did not require Rad9 (Figure 2F). The interaction of Rad9 with Aft1 was further analysed by examining the accumulation of CTR1 and FTR1 RNA under induction conditions in the presence and in the absence of RAD9 gene. Whereas Aft1 maintained its known activator role, we found Rad9 to substantially reduce the CTR1 and FTR1 RNA levels (Figure 2G).
Figure 2.
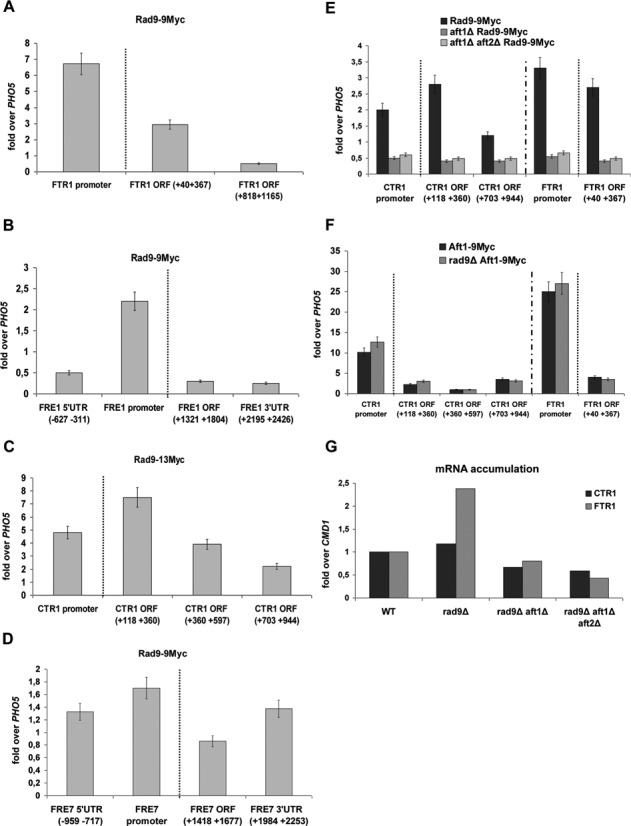
Rad9 is recruited to Aft1-regulated genes in an Aft1-dependent manner. By ChIP analysis the enrichment of Rad9–9Myc was studied in each amplicon (x-axis): on FTR1(A), on FRE1(B), on CTR1(C) and on FRE7 gene (D). The enrichment of Rad9–9Myc on CTR1 and FTR1 promoter and coding regions as obtained by ChIP analyses in Rad9–9Myc, aft1Δ Rad9–9Myc and aft1Δaft2Δ Rad9–9Myc strains is visualized in (E). Aft2 is an Aft1 paralogue protein, with partially redundant function (40,41). The Aft1–9Myc enrichment on CTR1 and FTR1 promoter and coding regions as obtained by ChIP analyses in Aft1–9Myc and rad9Δ Aft1–9Myc is visualized in (F). All strains were grown under induction conditions (see text). Normalization in all cases was performed firstly over INPUT chromatin and secondly by measuring the enrichment of Rad9–9Myc on PHO5 coding region, where Rad9–9Myc binding is minimal in such growth conditions (as tested by our group) and dividing Rad9–9Myc enrichment to this ‘non-specific’ enrichment. A dotted line in the ChIP experiments distinguishes the enrichment in the gene promoter from the enrichment in the coding regions. (G) RNA was isolated from strains (x-axis) grown under induction conditions and RT assay combined with qPCR was performed, aiming to assess changes in the CTR1 and FTR1 transcript levels. Normalization was done over change of CMD1 expression (not altered in these growth conditions), after firstly normalizing over the wt strain. The experiment was performed three times.
Taken together, our data show that Rad9 interacts with Aft1 transcription factor and that Aft1 is quantitatively required for Rad9 recruitment to promoter and coding regions of specific Aft1-regulated gene targets.
Rad9 and Aft1 exhibit a synthetic effect and may affect transcriptional elongation
Both Aft1 and Rad9 ChIP signals were enriched in all tested coding regions raising the possibility for their functional role in transcriptional elongation. To test this, we examined RAD9 and AFT1 null mutants for growth defects using a 6-AU assay; 6-AU reduces the rate of transcriptional elongation by inhibiting enzymes that are involved in purine and pyrimidine biosynthesis (44). We found that in 6-AU aft1Δ and—to a quite lesser extent—rad9Δ cells exhibited a ‘slow growth’ and ‘smaller colonies’ phenotype that closely resembled the one previously seen in snf2Δ mutant cells (45) (Figure 3A and B).
Figure 3.
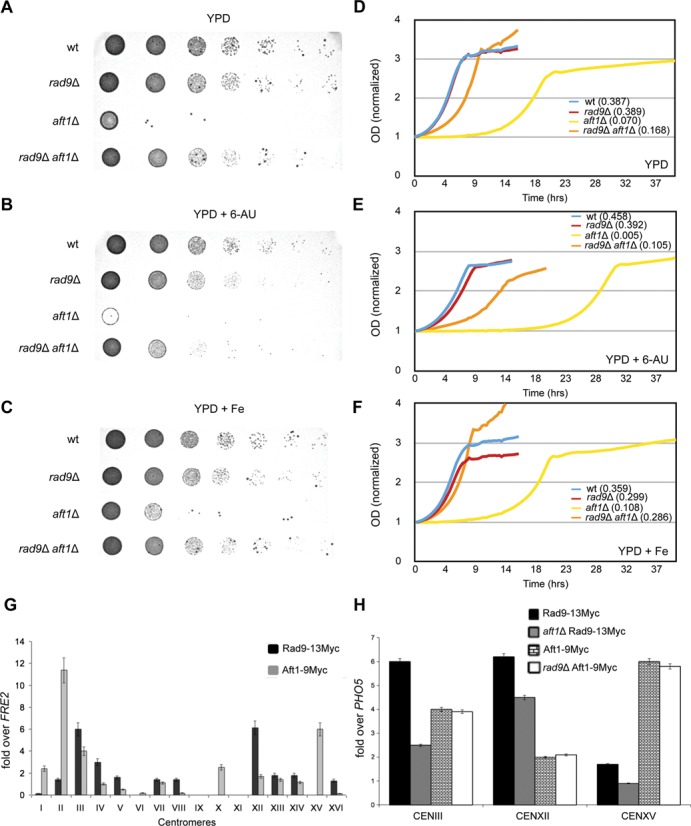
Rad9 and Aft1 exhibit synthetic effect and may affect transcriptional elongation. Cultures of the strains were grown in rich medium (YPD) to an OD600 = 1.0. Seven serial dilutions of the cells were spotted (A) on YPD plates, (B) on YPD plates which contained 300 μg/ml 6-AU and (C) on YPD plates which contained 50 μM FeCl3. Plates were incubated for 2 days at 30°C. Microscopy studies showed that the examined mutants have similar cell size in YPD rich medium (data not shown). The OD600 of exponentially growing cells (starting from an OD600 = 0.25) was measured every 20 min (D) inYPD, (E) in YPD plus 300 μg/ml 6-AU and (F) in YPD plus 50 μM FeCl3, until they reached stationary phase, using the BioLector technology. Values of each growth curve were normalized over the value of the first measurement in each case (wt, rad9Δ, aft1Δ, rad9Δaft1Δ). The relative growth rate of the strains in each condition was calculated and the values are shown in parentheses. (G) ChIP analysis that shows Rad9–13Myc and Aft1–9Myc enrichment to yeast centromeres. Normalization was performed firstly over INPUT chromatin and secondly by measuring the binding of Rad9–13Myc on FRE2 coding region (where Rad9 and Aft1 binding is minimal in the used growth conditions) and then dividing Rad9–13Myc or Aft1–9Myc enrichment by this ‘non-specific’ enrichment. Experiments were performed at least in duplicate. The same binding pattern was also obtained after normalizing over PHO5 coding region with similar results (data not shown). Due to high content in AT nucleotides in centromeric areas, some of the primers were not functional in real-time PCR (chr IX and XI). (H) Aft1-dependency of Rad9 localization to centromeres. The centromeric localization of Rad9–13Myc in wt and aft1Δ cells, as well as the centromeric localization of Aft1–9Myc in wt and rad9Δ cells was examined by ChIP in CENIII, CENXII and CENXV. All strains were grown under inducing conditions (SC BCS BPS). Normalization was performed firstly over INPUT chromatin and secondly by measuring the enrichment of Rad9–13Myc on PHO5, as described in the legend to Figure 2.
We also found that under standard growth conditions (YPD), RAD9 gene deletion significantly rescued the previously known aft1Δ growth defect (Figure 3A). This implied a synthetic function or effect of Rad9 and Aft1. Following the addition of FeCl3, aft1Δ growth defect as well as the double mutant synthetic phenotype were still apparent indicating a Rad9-Aft1 interaction independent of the classic role of Aft1 as a regulator of iron homeostasis (Figure 3C). Note that the rad9Δ aft1Δ synthetic phenotype was also apparent in 6-AU (Figure 3B).
The growth assay phenotypes were supported by the growth curves of the strains (wt, rad9Δ, aft1Δ, rad9Δaft1Δ) in YPD, 6-AU and FeCl3 (Figure 3D, E and F, respectively) for the construction of which we employed the BioLector technology. The growth curves correspond to the spot phenotypes and all the observed differences are significant (two-sided t-test P-value < 0.05), including the slow growth phenotype of both rad9Δ and aft1Δ strains in 6-AU.
In accordance to the above findings is the example of Rad9-Aft1 synthetic effect in centromeres. Centromeric areas are vital for preserving genome integrity through their role in chromosome segregation. Aft1 has been reported in only a few yeast centromeres (46), it is known to interact with proteins of kinetochores (26) and its deletion affects chromosomal segregation and cell growth (47). Furthermore, DNA damage checkpoint proteins were found in centromeres in mammals (including ScRad9's partial homologue Brca1) as reviewed (48). Consequently, we examined Rad9 recruitment on centromeres and for this we designed primers for all 16 yeast centromeric sequences and performed ChIP analysis. We were able to detect Rad9–13Myc and Aft1–9Myc enrichment to most of the yeast centromeres (Figure 3G). Moreover, the presence of Aft1 seemed to be necessary for Rad9 recruitment, whereas the opposite was not true, as shown by ChIP analyses on selected CEN loci (Figure 3H).
In conclusion, our data support a Rad9-Aft1 synthetic effect possibly exerted in centromeres and taking into account the sensitivity of rad9Δ and aft1Δ cells to 6-AU inhibitor we argue in favour of a potential involvement of the proteins to transcription elongation.
Rad9 affects the transcription of a small percentage of yeast genes
The above findings prompted us to examine whether Rad9 has a functional role in transcription beyond that of DNA repair. For this, we performed genome-wide expression analysis of rad9Δ cells grown in SC medium under the previously described induction conditions using the Affymetrix GeneChip Yeast Genome 2.0 Array. Our analysis revealed that ∼2% (131) of yeast genes were transcriptionally altered (two-sided t-test P-value 0.05) when compared to wt cells exhibiting an equal distribution of up- and down-regulated genes (61 and 70 genes, respectively, Supplementary Table S3 contains a complete list of up- and down-regulated genes). The gene expression data showed minimum variation across independent experiments with a positive correlation of expression patterns (Pearson's r = 0.388; P-value < 10−139, df = 5470). qPCR analysis confirmed the microarray gene expression profiles for a subset of genes (Supplementary Table S3).
Next, we grouped all genes according to their known or predicted biological function into GO categories (Molecular Function–MF, Biological Process–BP, Cellular Component–CC) using the BiNGO plugin of the Cytoscape software platform (35,36). We then tested which GO terms were significantly overrepresented among the significantly differentially expressed genes in rad9Δ cells and identified several biological processes including biogenesis and structure of ribosomes, metabolism, mitochondrial function, chromatin modification, transcriptional silencing and protein degradation (Table 1 and Supplementary Data sets S1–S3 contain the complete BP, MF and CC GO lists).
Table 1. Selected GO categories showing significant overrepresentation in the genes affected in rad9Δ cells grown in BCS/BPS.
| GO | Cellular Component | # of genes |
|---|---|---|
| 133 | Polarisome | 2 |
| 5677 | Chromatin silencing complex | 2 |
| 35097 | Histone methyltransferase complex | 2 |
| 5739 | Mitochondrion | 30 |
| 31966 | Mitochondrial membrane | 12 |
| 5746 | Mitochondrial respiratory chain | 3 |
| 22625 | large ribosomal subunit | 6 |
| 16020 | Membrane | 45 |
| 5937 | Mating projection | 7 |
| 42575 | DNA polymerase complex | 2 |
| 781 | Chromosome, telomeric region | 3 |
| 5856 | Cytoskeleton | 8 |
| GO | Molecular Function | # of genes |
| 16790 | Thiolester hydrolase activity | 4 |
| 42393 | Histone binding | 3 |
| 15077 | Monovalent inorganic cation transmembrane transporter activity | 4 |
| 4843 | Ubiquitin-specific protease activity | 2 |
| 8135 | Translation factor activity, nucleic acid binding | 3 |
| 3724 | RNA helicase activity | 2 |
| 3824 | Catalytic activity | 51 |
| GO | Biological Process | # of genes |
| 31935 | Regulation of chromatin silencing | 3 |
| 746 | Conjugation | 7 |
| 6303 | Double-strand break repair via non-homologous end joining | 3 |
| 6566 | Threonine metabolic process | 2 |
| 6081 | Cellular aldehyde metabolic process | 3 |
| 5991 | Trehalose metabolic process | 2 |
| 6081 | Various metabolic genes | 3 |
These findings show that Rad9 does not affect transcription in general; however, abrogation of functional Rad9 triggers a small number of gene expression changes that affect biological processes crucial for cellular survival and biogenesis.
Rad9 is localized to a significant subset of yeast genes only a small fraction of which is deregulated in RAD9 depletion
Having shown that Rad9 is recruited on specific gene coding regions, we then analysed the extent of its localization throughout the genome. In addition, we coupled the resulted genome-wide Rad9 binding profile with the genome-wide gene expression profile of rad9Δ presented above, in order to assess the level of interplay between Rad9 binding and gene deregulation upon RAD9 depletion. Using the Affymetrix GeneChip S. cerevisiae 1.0R Tiling Array, we examined Rad9–13Myc recruitment on chromatin of cells grown under induction conditions (BCS/BPS). [This array has 3.2 million perfect match/mismatch probe pairs tiled over the complete genome (except from centromeric loci) in a 5 bp resolution.] Rad9–13Myc enrichment peaks were defined with the use of standard Affymetrix software and regions of enrichment were called on the basis of a minimum t-test P-value threshold of 0.005. This analysis resulted in 1559 peaks overlapping with 935 genes and corresponding to 16.2% of the total genes in the genome which is about seven times more genes than the ones deregulated by Rad9 depletion (131 genes).
Rad9 binding was also detected in the small gene subset that was found to be deregulated in rad9Δ mutants (31 out of 131 genes, Fisher's exact test P-value < 0.01). Genes both bound by Rad9 and deregulated in rad9Δ are very likely to be under a direct control of Rad9 and were found to be enriched in metabolic functions (see Supplementary Table S4 for the gene list and Supplementary Data set S4 for the BP GO list). A mild preference of Rad9 binding to up-regulated genes was further intensified in the case of the rad9Δ deregulated genes (Figure 4C).
Figure 4.
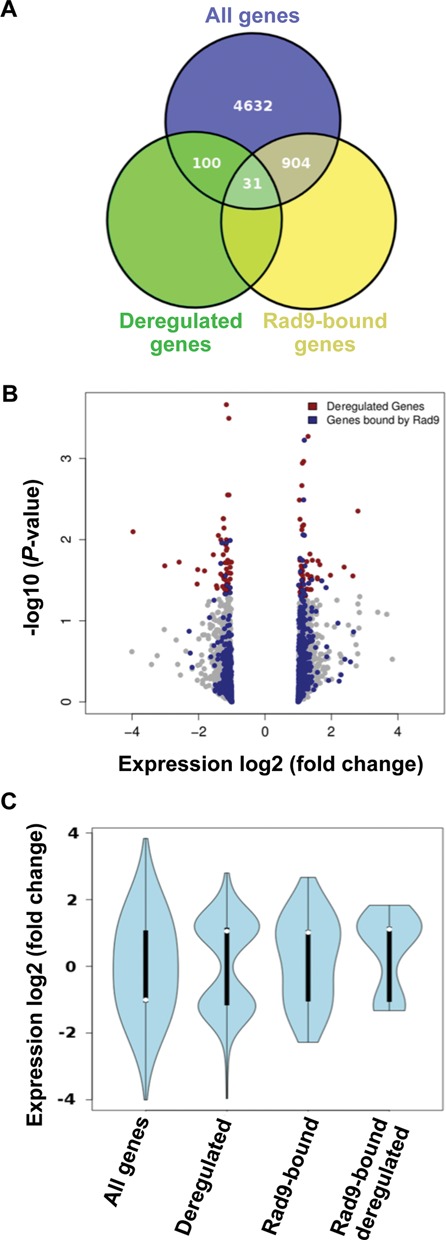
Expression analyses of rad9Δ cells and correlation to Rad9 localization. (A) Venn diagram representing total number of genes and overlaps with deregulated and Rad9-bound genes. Overlap between Rad9-bound and deregulated genes was significant on the basis of a Fischer's test (P-value < 0.01). (B) Volcano plot for a genome-wide expression profile of rad9Δ cells. All genes depicted in grey. Significantly deregulated genes (P < 0.05) depicted in red and Rad9 bound genes in blue. Note the ‘loading’ of the blue dots to the right side of the plot (upregulation). (C) ‘Violin-shaped’ boxplots summarizing the distribution of log2 (Fold-change) expression values for all genes, deregulated ones (131 genes with P < 0.05), Rad9 bound genes (935) and the interesection of Rad9 bound and deregulated genes (31 genes, see also Venn diagram). The shift towards upregulation is noted as a trend for the total Rad9-bound genes and becomes significant between the ‘Deregulated’ and ‘Rad9-bound deregulated’ gene sets (one-sided t-test P-value < 0.05).
Therefore, although it seems that Rad9 may have a direct role on the transcription levels of a few genes related to primary metabolic processes, it is clear that, under the examined non-DNA damage-inducing conditions, Rad9 localizes to a very significant fraction of gene coding chromatin.
Rad9 is recruited to highly active genes in an Aft1-dependent manner with a bias for coding regions
Under non-DNA damage-inducing conditions, Rad9–13Myc was recruited to ∼900–1200 genomic regions (depending on the threshold that we set: P = 0.001 or P = 0.005, respectively). Around 80% of these Rad9 binding regions were located within gene coding sequences (t-test P-value < 10−12) (Figure 5A-bar Rad9 and see Supplementary Figure S1 for a typical pattern of Rad9 localization on a 50 kb region of ChrXII), while ∼10% were strictly overlapping non-coding spacers and a remaining ∼10% was spanning both coding and non-coding regions of the same gene. Given that coding space in the yeast genome spans ∼70% of its total length, this signified a clearly non-random preference of Rad9 to occupy coding sequences (bars Rad9 and Genome). According to our GeneChip genomic data, Aft1–9Myc also displayed a slight preference to occupy coding sequences (bar Aft1 versus bar Genome). Regions occupied by both Rad9–13Myc and Aft1–9Myc were also enriched in coding sequences (bar Rad9+Aft1). The propensity of Rad9–13Myc to occupy coding sequences was almost unaffected by the absence of Aft1 protein (bar aft1Δ Rad9 versus bar Rad9), indicating that Aft1 has no qualitative effect on Rad9 overall genomic distribution. On the other hand, in the absence of Rad9, Aft1–9Myc peaks were distributed randomly across the entire yeast genome showing no preference for coding regions (bar rad9Δ Aft1 versus bar Genome).
Figure 5.
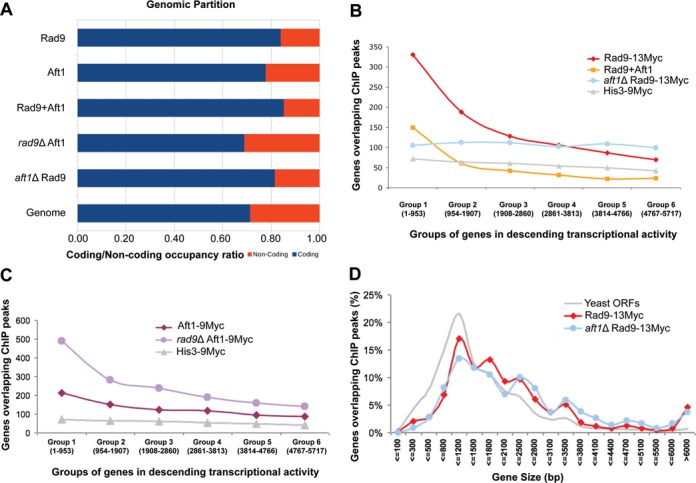
Genome-wide localization of Rad9 in the presence and absence of Aft1. (A) Partition of ChiP peaks into coding and non-coding chromatin for: Rad9–13Myc, Aft1–9Myc, common sites, rad9Δ Aft1–9Myc and aft1Δ Rad9–13Myc compared to the background partition for the complete yeast genome. A clear enrichment in coding sequences is especially notable for the common sites (Rad9–13Myc+Aft1–9Myc). All coding ratios except of rad9Δ Aft1–9Myc have a value of 0.79 or more compared to a genome average of 0.71. Bootstrap P-values of enrichment were calculated using 106 random permutations of the binding sites. All P-values were <10−6 except from rad9Δ Aft1–9Myc which showed a partition similar to the genome background. (B) Rad9–13Myc is localized to the most active yeast genes in an Aft1-dependent manner. Yeast genes were grouped in six equal groups (x-axis) in descending transcriptional activity in SC BCS BPS growth conditions (our microarray data). Rad9 targets (ChIP peaks significant to a P < 0.005 level) that fell within each group were calculated (y-axis) in wt or aft1Δ cells (red and blue lines, respectively). Distribution of common targets of Rad9 and Aft1 is also shown. His3–9Myc control protein is shown in grey. (C) The same analysis as in (B) was performed for Aft1–9Myc in wt or rad9Δ cells (dark and light purple lines, respectively) following curves with similar slopes. (D) Rad9–13Myc has a binding bias for long genes. S. cerevisiae genes were grouped in 20 bins depending on their size (x-axis). The percentage of the number of ChIP peaks from each experiment that fit each of the bins is plotted. The distribution of all yeast Open Reading Frames (ORFs) is plotted in grey line.
Given the non-random preference of Rad9 to occupy coding sequences we next analyzed the possible correlation of Rad9 binding with overall gene expression under the same conditions. We used data from a genome-wide expression analysis of the wt strain grown in the conditions described above and grouped yeast genes according to their transcriptional activity. Yeast genes were binned in six equal groups of 950 genes each with descending levels of expression, and the absolute number of genes overlapping a significant ChIP peak was calculated for each such group in the presence or absence of Aft1. We found that Rad9 has a strong binding bias to highly transcriptionally active genes and that this preference was to a great extent Aft1-dependent (Figure 5B). In fact, Rad9 and Aft1 common gene targets are even more enriched in the highly expressed genes (Figure 5B). Aft1–9Myc protein also showed a tendency to localize to highly active genes but this preference was almost independent of Rad9 (Figure 5C). A parallel analysis of the His3–9Myc cytoplasmic protein was used as negative control. The binding bias of Rad9–13Myc to highly active genes, was verified by shifting cells from glucose to galactose growth conditions. In these conditions specific genes responsible for the galactose metabolism are highly induced (49). We indeed observed a very strong binding of Rad9 to this group of genes (such as GAL1,GAL2,GAL3,GAL7,GAL10,GAL80) (see Supplementary Data set S5 for a presentation of Rad9 localization intensity to targets induced in galactose or glucose). Similarly, Rad9 was localized to the iron and copper regulon genes only when they were induced (e.g. only in the presence of iron and copper chelators), hence transcriptionally active (Supplementary Figure S2 shows the GO network of Rad9 targets under these conditions). To further evaluate Rad9 selectivity in its genomic localization, we also examined the genome-wide binding pattern of overexpressed Rad9 (plasmid-derived, ∼10 times more molecules than normal). We found Rad9 recruited to a comparable number of genes (which overlapped significantly with the targets of endogenous Rad9), indicating a selective binding to the genome (Supplementary Table S5 shows the GO groups overrepresented in the common targets of endogenous and overexpressed Rad9).
Further analysis of our data showed a positive correlation of Rad9–13Myc binding and gene length. Rad9–13Myc exhibited a bias for longer genes in both wt and aft1Δ cells (Figure 5D).
Additionally, we performed in silico qualitative analysis of the Rad9–13Myc targets in order to reveal overrepresented GO categories, in the presence or absence of Aft1. We first clustered the target genes into three groups, depending on the binding value of Rad9–13Myc, and then performed functional analysis for each group as described above. The results of this analysis are presented in detail in Supplementary Text S1 and a summary is shown in Tables 2 and 3 (P < 10−5). Rad9-binding, as expected, was enriched in the subset of highly expressed genes according to our expression data. These genes are related to major metabolic pathways (mainly of glucose and aminoacids), oxidation and reduction enzymatic activities and cell wall structure. Additionally, they are overrepresented in categories related to protein synthesis, cellular growth, translational elongation, regulation of translation and ribosomal function, and involved in catalytic activities, vitamin and biotin binding and post-transcriptional regulation of gene expression. In contrast, in aft1Δ cells, GO categories related to aminoacid metabolism, cell wall structure and cytosolic ribosomes were either absent or underrepresented. Instead, Rad9 was localized with high significance to groups of genes related to protein import into the nucleus and nuclear lumen structure. These differences are in agreement to our finding that Aft1 is required for Rad9 localization to highly active genes often related to cellular growth.
Table 2. Selected GO categories showing significant overrepresentation in Rad9–13Myc-enriched targets in wt cells grown in BCS/BPS (P < 10−5).
| GO | Cellular Component | # of genes |
|---|---|---|
| 9277 | Fungal-type cell wall | 21 |
| 5830 | Cytosolic ribosome | 20 |
| GO | Molecular Function | # of genes |
| 8182 | Translation elongation factor activity | 7 |
| 3824 | Catalytic activity | 127 |
| 16885 | Ligase activity, forming carbon-carbon bonds | 4 |
| 19842 | Vitamin binding | 12 |
| 16491 | Oxidoreductase activity | 29 |
| 16814 | Hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds, in cyclic amidines | 6 |
| 8943 | Glyceraldehyde-3-phosphate dehydrogenase activity | 3 |
| GO | Biological Process | # of genes |
| 6090 | Pyruvate metabolic process | 14 |
| 19752 | Carboxylic acid metabolic process | 46 |
| 19319 | Hexose biosynthetic process | 12 |
| 6006 | Glucose metabolic process | 16 |
| 6094 | Gluconeogenesis | 11 |
| 10608 | Post-transcriptional regulation of gene expression | 44 |
| 6445 | Regulation of translation | 43 |
| 55114 | Oxidation reduction | 29 |
| 8652 | Aminoacid biosynthetic process | 17 |
Table 3. Selected GO categories showing significant overrepresentation in Rad9–13Myc-enriched targets in aft1Δ cells grown in BCS/BPS (P < 10−5).
| GO | Cellular Component | # of genes |
|---|---|---|
| 31981 | Nuclear lumen | 114 |
| 5643 | Nuclear pore | 19 |
| GO | Molecular Function | # of genes |
| 5524 | ATP-binding | 67 |
| 16887 | ATPase activity | 32 |
| GO | Biological Process | # of genes |
| 6606 | Protein import into nucleus | 13 |
| 19222 | Regulation of metabolic process | 223 |
| 6913 | Nucleocytoplasmic transport | 40 |
| 6445 | Regulation of translation | 92 |
| 10608 | Post-transcriptional regulation of gene expression | 92 |
| 6403 | RNA localization | 32 |
In addition to the group of total peaks (all statistically important loci to which Rad9–13Myc was localized) (a), we conducted functional analysis for the following specific groups: peaks located entirely within coding regions (b), peaks strictly overlapping non-coding regions (c) and peaks spanning the coding and non-coding regions of the same gene (d), and looked for differences in the presence (i) or in the absence (ii) of Aft1, or regardless of Aft1 presence (iii) (Supplementary Text S2, Part 1 and for group (d) Supplementary Text S2, Part 2). The results of the above analyses (commented in detail in Supplementary Text S2) highlight the fact that Rad9 localization preferences for highly transcriptionally active groups of genes are Aft1-dependent. In addition, they are indicative of a Rad9 functional potential in multiple cellular pathways under non-DNA damage-inducing conditions, such as the ones related to primary metabolism and cell growth.
We have also performed the above functional analyses to the Aft1 targets in the presence or absence of Rad9. The results are presented in the tables of Supplementary Text S3. This comparison showed that transcriptionally active groups of genes (such as GO terms of ‘oxidoreductase activity’, ‘aminoacid metabolism’ and ‘glucose catabolism’) were among the common target groups of Rad9 and Aft1. Gene clusters related to iron transport/assimilation were, as expected, overrepresented in Aft1 targets. Rad9 absence did not significantly affect the Aft1 localization to highly active targets and generally had an overall subtle effect to Aft1 binding.
The Rad9 localization pattern corresponds to distribution of epigenetic marks
Rad9 has been proposed to be constitutively present to chromatin through the interaction of its Tudor domain with H3-K79me (21). Previous studies have analysed the genome-wide relative occupancy of mono-, di- and trimethylation of H3K4, trimethylation of H3K36 and H3K79, as well as acetylation of H3K9, H3K14 and H4 (37), so we compared these epigenetic mark patterns to our data. We first performed average gene analysis (37,38) to determine the mean genome-wide relative occupancy of Rad9–13Myc and confirmed a strong localization to gene coding regions with a bias to the 3′ portion of the ORF (Figure 6A). In aft1Δ cells, Rad9 relative occupancy patterns exhibited a marked shift towards the 3′ end of the gene peaking at the 3′-UTR (Figure 6B). Aft1–9Myc average relative occupancy was similar to that of Rad9–13Myc (Figure 6C), and was not significantly changed in rad9Δ cells (Figure 6D), which was consistent with our previous observations concerning how the two proteins may interact on chromatin.
Figure 6.
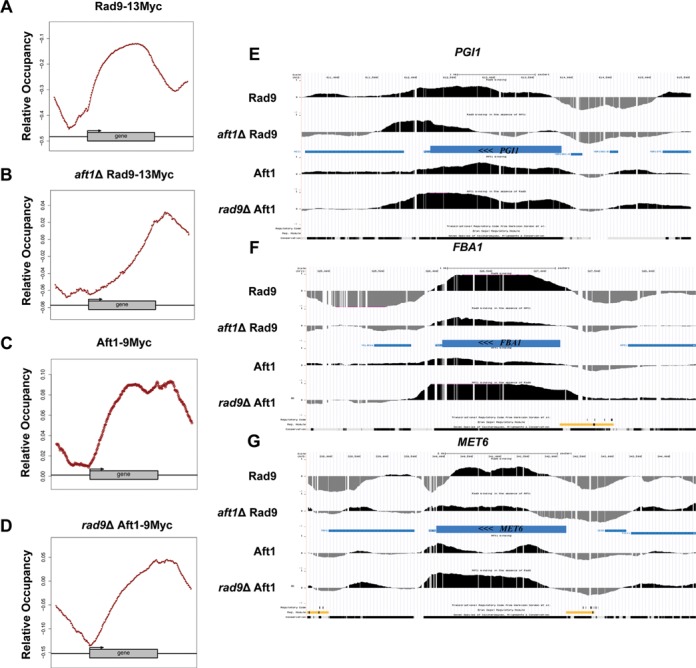
Average gene relative occupancy of Rad9 and Aft1 and localization patterns to methylated targets. Average gene analysis was performed to the genome-wide localization data as described in Materials and Methods. Relative occupancies of (A) Rad9–13Myc in SC BCS BPS, (B)aft1Δ Rad9–13Myc in SC BCS BPS, (C) Aft1–9Myc in SC BCS BPS and (D)rad9Δ Aft1–9Myc in SC BCS BPS are shown. (E–G) Rad9 and Aft1 ChIP localization signal on methylated targets. Three methylated genes according to Pokholok et al. study (36) were randomly selected. The average binding profiles of Rad9, aft1Δ Rad9, Aft1 and rad9Δ Aft1 on those genes are presented: (E) PGI1, (F) FBA1, (G) MET6. Binding patterns are representative of the genome-wide profiles. Note the direction of the transcription on the genes (right to left).
We next compared the average gene Rad9–13Myc localization patterns to the respective occupancy patterns of methylation and acetylation marks as obtained by Pokholok et al. (37). Supplementary Figure S3 illustrates a comparison of our data to those of Pokholok et al. study. Rad9 localization patterns were mostly similar to the H3K36me3 (a hallmark of transcriptional elongation) and H3K4me3 patterns that show a bias to the 3′ portion of genes and less similar to the H3K4me2 and H3K79me3 patterns that show a bias to the middle part of genes (Supplementary Figure S3). Conversely, all three types of acetylation (H3K9ac, H3K14ac, H4ac) which are correlated to promoter regions of genes, showed no similarities to the Rad9–13Myc relative occupancy patterns (Supplementary Figure S3).
In order to assess the correlation of Rad9 and Aft1 recruitment with the methylated state of chromatin, we selected three methylated genes randomly chosen in Pokholok et al. study (PGI1,FBA1,MET6) and examined the Rad9 and Aft1 distribution on them (in wt and in the absence of each other) (Figure 6E–G). Both Rad9 and Aft1 were localized to all three genes. In addition, consistently with the average gene localization patterns, Rad9 distribution in aft1Δ cells was shifted to the 3′ end of the genes.
Overall, Rad9–13Myc relative localization patterns exhibit similarities to specific methylation marks, some of which are related to active elongating transcription apparatus (37), consistent to the growth phenotype results for Rad9 and Aft1 proteins shown in Figure 3.
Rad9 is recruited to GC-rich regions and meiotic recombination hotspots in an Aft1-dependent manner
In order to expand our analysis of Rad9 and Aft1 localization in the genome we examined how it correlates to genomic regions which are highly active and/or prone to DNA damage. The relative occupancy of Rad9-targets, Aft1-targets and their intersection are presented in Figure 7A. For these gene categories and especially for the Rad9-Aft1 overlapping targets, we found a correlation between the localization pattern and the GC content of the respective target sequences. Increased transcriptional activity has been linked to GC-rich genomic regions in yeast (50) and mammals (51–53). In more detail, we compared the target sequences to an equal number of randomly selected sequence fragments with equal sequence lengths. The calculated median GC content of the sequences bound by Rad9, Aft1 or both was between ∼41% and 43%, while the corresponding value for the randomly selected regions was close to the yeast genome average of 38% (t-test P-value of 10−127, df = 2305) (Figure 7B). The correlation of Rad9-binding to the underlying sequence GC content was Aft1-dependent, since the GC-median percentage of Rad9-bound regions in aft1Δ cells was closer to random, as was the respective percentage of Aft1-bound regions in rad9Δ cells (Figure 7C). This shift to lower GC content is probably a reflection of the corresponding shifts of the binding patterns towards the 3′ end of the genes (Figure 7D), as both 5′ and 3′ UTR sequences are less GC-rich compared to the gene bodies (54).
Figure 7.
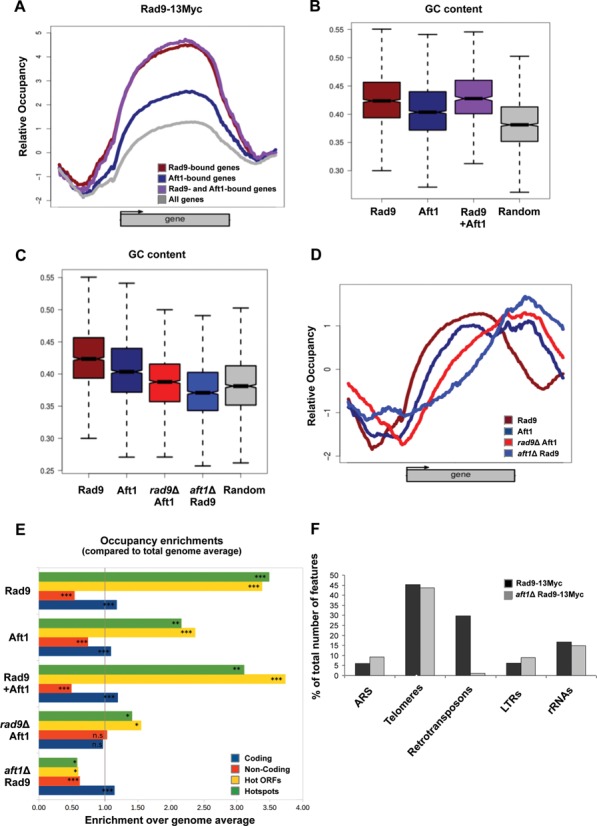
Rad9 localizes to GC-rich regions and meiotic recombination hotspots in an Aft1-dependent manner. (A) Rad9–13Myc ChIP signal mean patterns for genes bound by Rad9, Aft1 and by both proteins compared to the average for the total number of yeast genes. (B) GC content boxplots of genes bound by Rad9, Aft1 or both proteins in comparison to the complete set of genes in full correspondence with (A). Rad9-bound genes show the highest GC content. (C) GC content boxplots of binding sites (enriched loci) for Rad9 and Aft1, in the presence or absence of each other, compared to a set of randomly selected genomic positions. Rad9 sites also show the highest GC content, which is significantly lowered in the absence of Aft1. The same effect is observed for Aft1 in the absence of Rad9. (D) Mean binding patterns for Rad9 and Aft1 both in the presence and absence of each other (combined plot of Figure 6A–D). (E) Occupancy enrichment values for coding regions, non-coding spacers (see Figure 5A), hot ORFs and hotspots, expressed as the ratio of observed over expected occupancy based on the genome average. Significant (more than 2-fold) enrichment in Rad9, Aft1 and common binding sites was observed for both hot ORFs and hotspots while significant depletion was calculated for Rad9 sites in the absence of Aft1 and to a lesser extent for Aft1 sites in the absence of Rad9. Bootstrap values calculated for 106 random permutations. (***P < 10−6; **P < 10−4; *P < 10−3, n.s.=non-significant). (F) The five non-ORF feature categories that were most represented in Rad9–13Myc Tiling Arrays in the presence or absence of Aft1 (black and grey columns, respectively) are presented in the chart, as percentage of the total number of features in each category in the yeast genome.
We further studied average Rad9–13Myc relative occupancy patterns in relation to the transcriptional activity of the complete set of yeast genes (in wt and aft1Δ cells). More specifically, we divided all genes in five subgroups based either on k-means clustering approach or a stratification of their expression rates and then correlated the average gene occupancy of each subgroup with mean gene expression and GC content. The results of this analysis are presented in the figures of Supplementary Text S4 and support our conclusions so far: transcriptional activity is a major factor that defines Rad9 recruitment to chromatin under non-DNA damage-inducing conditions. The fact that this recruitment is Aft1-dependent highlights the importance of a functional link between Rad9 and Aft1 on a genome-wide scale.
Genomic regions rich in GC content in addition of being highly transcriptionally active are characterized by high probability of meiotic recombination events, namely, hotspots and hot ORFs of meiotic recombination (50,55–56). When we compared the binding pattern of Rad9 along chromosome III to the mapped loci of meiotic recombination hotspot regions, we observed a significant localization pattern similarity (Supplementary Figure S4). Indeed, a detailed analysis of occupancy enrichment showed that Rad9–13Myc was localized to meiotic recombination hotspot regions with high probability and this localization was Aft1-dependent (Figure 7E). A similar binding bias was observed for the Aft1–9Myc and also for the overlap of Aft1-Rad9 localization sites (Figure 7E). Given that the hotspot-enriched regions span less than 4% of the total genome, the aforementioned overlaps suggest highly significant non-random co-localizations.
In addition to transcriptionally highly active loci, Rad9–13Myc was also recruited to non-ORF elements as defined in the SGD (Supplementary Figure S5, for a snapshot of S. cerevisiae non-ORF features). Rad9 was present at ∼30% of retrotransposons, ∼6% of Long Terminal Repeats, almost half of the telomeres, ∼16% of rRNA genes and ∼6% of Autonomous Replicating Sequences (Figure 7F and see Supplementary Table S6 for the corresponding numbers of non-ORF features to which Rad9 is localized in the presence or absence of Aft1). Notably, the localization of Rad9 to retrotransposons, where both Rad9 and Aft1 are present, was Aft1-dependent (Figure 7F). This finding highlights the role of Aft1 in genome integrity since retrotransposons are loci prone to chromosome rearrangements and points to a combined function of the two proteins to genome integrity surveillance. Rad9 presence on telomeres was Aft1 independent, which is consistent with the fact that the two proteins are mostly localized to different telomeric regions. It is safe to assume that Rad9 is recruited to telomeres by additional mechanisms.
In conclusion, in addition to the GC-rich and transcriptionally highly active loci, Rad9 also localizes to vital for genome integrity regions, retrotransposons and centromeres in an Aft1-dependent manner.
DISCUSSION
Preservation of genetic information is of prime importance to all living systems. Rad9 is the S. cerevisiae transducer/adaptor protein known to mediate the DNA damage signal from the upstream to the downstream kinases in the DDR pathway [reviewed in (57,58)]. Previous biochemical studies have also reported the presence of Rad9 in the chromatin fraction of cells grown under non-DNA damage-inducing conditions (4,15,21) raising questions for additional functions of this protein. The dynamics of Rad9 recruitment on chromatin have not been studied before under a general functional perspective. Here we present a detailed study of the Rad9 role by performing both small and genome-wide expression and localization analyses. We reveal chromatin elements involved or associated with Rad9 recruitment under non-DNA damage-inducing conditions. Our results imply non-random, biased, constitutive Rad9 association with chromatin and suggest possible benefits for continuous maintenance of genome integrity.
Rad9 protein involvement in transcription
We identified the iron-repressed Aft1 transcription factor as a new Rad9-interacting protein in extracts of physiologically grown yeast cells. We also found that the two proteins interact via the Rad9 BRCT domain and N-terminal domain of Aft1. Importantly, genetic data by growth and ChIP assays showed a synthetic effect of the two proteins. These findings led us to examine whether Rad9 could have a role in transcription. Previous studies have shown that RAD9-dependent gene transactivation is required for DNA excision repair of active genes under DNA damage conditions (20). In this work, we examined Rad9 role in transcription under non-DNA damage induction. Genome-wide expression analyses of yeast cells showed that Rad9 does not affect transcription in general; the transcription levels of only ∼2% (131) of yeast genes were significantly altered in its absence (rad9Δ). Rad9 directly associated to one-fourth of these genes which are predominantly related to metabolic functions. The remaining three-fourths of the transcriptionally altered genes are probably affected by Rad9 indirectly.
Possible involvement in transcriptional elongation (with a rather redundant function) was suggested by a distinctly reduced growth phenotype observed in RAD9 gene-deleted strains tested by the 6-AU assay. This general observation is consistent with several other pieces of evidence: Rad9 localized to both promoter and protein coding regions of selected metal-regulated genes originally examined by manual ChIP assays, a finding confirmed by the genome-wide ChIP assays. Rad9 was enriched within the coding region of transcriptional units, with a preference towards the 3′ ends, as shown by average gene analysis of the genome-wide experiments. Finally, similar average occupancy patterns were observed between Rad9 and H3K36me3 which is considered an epigenetic hallmark of active transcript elongation (37), a modification catalyzed by Set2, a factor associated with the later stages of transcriptional elongation (59). The rest of our findings point to a role of Rad9 not affecting transcription, although coordinated and dependent on the presence of Aft1 protein.
Rad9 localizes to highly active genes and other DNA damage-prone regions of the yeast genome in an Aft1-dependent manner
Our genome-wide analyses showed that under standard growth conditions Rad9 localizes to one-fifth of the yeast genes (t-test P-value = 10−3), predominantly within the protein coding region, displaying a localization bias to the most highly active genes under standard growth conditions. This was further confirmed by genome-wide ChIP analyses of cells grown in galactose instead of glucose or in metal regulon-induced conditions where Rad9 bound to the conditionally induced genes in addition to the highly expressed housekeeping genes. Moreover, overexpressed Rad9 was recruited to a comparable number of genes corroborating selective binding. Although Rad9 seems to bind to ssDNA through its Tudor domain (60), in silico analysis of our data showed no consensus sequence motif that it may recognize (unpublished observations). However, in this work we provide evidence that the detected binding is non-random but follows specific chromatin features in addition or in relation to the highly active genes.
The analysis of Rad9-Aft1 association on specific metal-regulated genes demonstrated that Rad9 was recruited to both their promoter and protein coding regions in an Aft1-dependent quantitative manner. Surprisingly, Aft1 transcriptional regulator was also recruited to the coding region of these genes, but this binding was Rad9 independent. Moreover, aft1Δ mostly and, to a lesser extent, rad9Δ strain displayed a reduced growth phenotype in the 6-AU assay indicating an Aft1 and possibly Rad9 involvement in transcriptional elongation, consistent with their localization to protein coding regions. Genetic studies have already attributed to Aft1 protein functions beyond the transcriptional regulation of iron-responsive genes, such as in chromosome stability, genome integrity, DNA damage, cell wall stability, mitochondrial function and metabolic processes (25–27,47).
Our genome-wide data showed that Rad9 localization bias to the most highly active genes has a strong Aft1 dependency. We found Aft1 also recruited to a significant number of protein coding regions throughout the genome. Moreover, the majority of the coding regions occupied by both Aft1 and Rad9 corresponds to genes that are very highly transcribed. The respective GO enrichment analyses are indicative of a Rad9 functional potential in multiple cellular pathways and clearly reflect the importance of Rad9 dependency on Aft1 since vital gene functional groups are absent or severely underrepresented in aft1Δ cells.
We also demonstrated that, apart from its Aft1-dependent recruitment, Rad9 may also follow specific histone methylation marks for its constitutive recruitment to chromatin. Even though Rad9 can recognize H3K79 methylated histones through its Tudor domain (following a DNA damage event), we found that the relative occupancy pattern of Rad9 on an average gene, in non-DNA damage-inducing conditions, resembled mostly the H3K36me3 and H3K4me but also the H3K79me3 and H3K4me2 patterns as compared to the results of Pokholok et al. (37). This similarity between the distribution patterns of Rad9 and prominent active transcription marks suggest a potential functional link between them in unperturbed conditions.
The strong binding bias of Rad9 to highly active regions under non-DNA damage-induction can be correlated with a strong presence in loci which are prone to DNA damage, even before the actual damage occurs. It is known that the transcriptional activity of a gene is proportionally related to its mutation rate (61) and there is increased accumulation of apurinic and apyrimidinic sites in highly transcribed DNA (62), linking transcription and fidelity of DNA replication. Highly transcribed genes are generally an impediment for replication forks (63), due to often occurring collisions between the DNA replication and transcription machineries (64). This may lead to transcription-associated recombination (dependent on replication) (64–66). In accordance to that, in vegetative cells we found Rad9 localized with increased probability to known meiotic recombination hotspots in an Aft1-dependent manner. These sites are GC rich, likely to comprise regions of open-chromatin (51–53), and thus more prone to DNA damage (50,55–56). Rad9 localization pattern shows similarities to the respective of RNA pol II as compared to other studies (67) (Supplementary Figure S6, for a comparison of Rad9 and RNA polII localization to Chr III), consistent with the strong correlation of Rad9 and high transcription rate. Since most highly active genes can differ depending on the medium, cell phase or strain, we can say that Rad9 exhibits a partly preferential binding which nevertheless predominantly follows the rules of high transcriptional activity, DNA damage sensitivity and high GC content. This means that Rad9 is preferably recruited to the loci where it is most needed at each specific moment.
Consistent with Rad9 performing a constant surveillance of chromatin which is prone to DNA damage, is its recruitment to retrotransposons, centromeres and telomeres. Retrotransposons lie in loci prone to participate in chromosome rearrangements, where replication pauses and chromosomes are more likely to break (68). The transposition event can influence genomic evolution by disrupting coding or transcriptional control elements, or by promoting chromosomal rearrangements via homologous recombination (69). The Aft1-dependence of Rad9 localization to such loci reinforces the proposed role of Aft1 in DNA damage and preservation of genome integrity (26–27,47). We suggest that Rad9 presence on such critical for genomic evolution locations ensures that programmed chromosome rearrangements occur with no alterations by possible random DSB, since Rad9 is already present to rapidly mediate the damage signal.
Rad9 was found in centromeres, also partly dependent on the presence of Aft1 which is known to interact with centromeric proteins (26). It was previously shown that Cbf1 (an Aft1-interacting partner) and Ste12 transcription factors regulate endogenous transcription in centromeres in an RNA pol II-dependent manner and that this may be responsible for proper topology of CEN chromatin (70). In that respect, Rad9 and Aft1 may similarly contribute to local proper chromatin topology or survey the integrity of centromeric DNA which is important for chromosome segregation. The synthetic growth phenotype that we observed in the aft1Δ rad9Δ strain, under physiological unperturbed conditions, is indicative of an important combined effect of the two proteins partially relieved when both proteins are absent. Although explaining synthetic growth phenotypes is not always straightforward, one possible scenario for the aft1Δ rad9Δ strain could be the following. Aft1 has been found to interact with kinetochore proteins and promote (by an undefined yet mechanism) pericentric cohesin association therefore affecting chromosome stability and segregation (26). It is this additional specific role of Aft1 that, together with its role in iron homeostasis regulation, causes the severe aft1Δ slow growth phenotype. It is clear that abnormalities in chromosome segregation/mitosis generate DNA damage [reviewed in (71)]. Therefore, the absence of Aft1 possibly induces a local increase of DNA breaks at specific centromeres which in turn leads cells to enter cell-cycle checkpoint and severe growth retardation. We propose that checkpoint is mediated by Rad9 protein that is recruited on these centromeres at reduced levels (∼50%), due to the absence of Aft1, but sufficient to induce checkpoint activation and growth delay. When additionally to Aft1, Rad9 is also removed, cells fail to enter checkpoint and therefore manage to grow faster, temporarily, disregarding segregation and DNA damage defects. Therefore, in aft1Δ rad9Δ mutants, growth is partially restored. Nonetheless, Rad9 and Aft1 localization to centromeres is in agreement with similar results for DNA damage checkpoint proteins in mammalian systems, including Rad9 partial homologue Brca1 [reviewed in (48)].
Finally, the observed Rad9 localization to telomeres is consistent with several previous studies that include Rad9 in molecular mechanisms which preserve telomere integrity (72–76). According to our data, Rad9 is recruited to telomeres mostly independently of Aft1.
Notably, our analyses of the genome-wide localization maps of Rad9 and Aft1 indicate a subtle interdependency of the two proteins. While Rad9 recruitment to fragile chromatin (coding sequences of very highly expressed genes, GC-rich regions, centromeres, retrotransposons, meiotic hotspots) appears to coincide with Aft1 and depend on its presence, Rad9 overall genomic distribution can occur independently of Aft1. This was indicated by the persistent Rad9 distribution preference for coding regions in the absence of Aft1 (aft1Δ Rad9 in Figures 5A and 7E). On the other hand, Aft1 localization to coding regions seems to be favoured by Rad9, as was indicated by the shift of Aft1 distribution preference to non-coding regions in the absence of Rad9 (rad9Δ Aft1). Nevertheless, the impact of Aft1 on Rad9 localization to fragile sites is more prominent than vice versa.
Surveillance of DNA damage-prone chromatin by Rad9 checkpoint protein and Aft1 transcriptional activator
In conclusion, our analysis shows that Rad9 has a functional potential to recognize chromatin regions prone to DNA damage, before damage occurs. These are often connected with highly transcribed genes related to vital pathways, such as cellular growth, as well as with loci, such as centromeres, meiotic recombination hotspots and retrotransposons. At the most fragile genomic sites, Rad9 attracts Aft1 by favouring interaction between the two proteins. This interaction is critical for Rad9 non-random constitutive dynamic recruitment to chromatin which may facilitate the speed and efficiency of Rad9-dependent response to DNA damage. Aft1 may contribute to an appropriate chromatin state and act with Rad9 in order to stimulate DNA repair possibly by attracting other cofactors which give access to the repair machinery (Figure 8). This is consistent with Aft1 already being connected to DNA damage and genome integrity preservation in yeast, and with mammalian systems where transcriptional activators can stimulate DNA repair in the surroundings of their binding site (77). It seems that cells have developed simple—yet sophisticated—mechanisms to provide their fairly sensitive loci with additional protection. By maintaining DDR proteins in the neighbourhood of fragile sites, possibly recruited by transcriptional activators, they establish a well-orchestrated safeguarding system.
Figure 8.
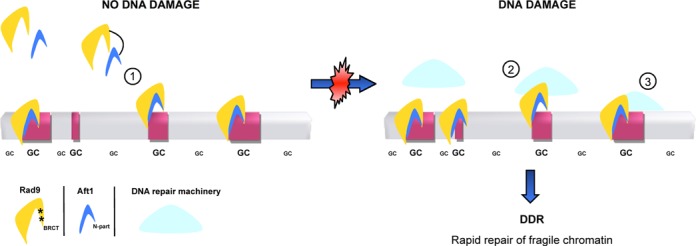
Model for the potential surveillance of DNA damage-prone chromatin by Rad9 and Aft1. Highly active, GC-rich and prone to DNA damage genomic regions are depicted in red rectangles, while non-active correspond to regions in grey. (1) Under non-DNA damage-inducing conditions, Rad9 (in dark yellow) interacts via its BRCT domain (asterisks) with Aft1 (in blue) N-terminal part. Rad9 is recruited to DNA damage-prone chromatin along with Aft1 in an Aft1-dependent manner. It is possible that when a DNA damage event occurs, Aft1 transcription factor could recruit other cofactors. (2) The protein complex at the DNA damage site could facilitate the access of the DNA repair machinery (in light blue), while Rad9 (and possibly more proteins participating in the DDR cascade) are already on the site. (3) The whole DNA repair machinery is recruited to the DNA damage site, ensuring a rapid response to DNA damage leading to an effective repair of the impaired chromatin.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Kalliopi Gkouskou for pDB20-Rad9-flag construct, Rad9–9Myc strain, and for design of CTR1 ChIP primers, Thanasis Margaritis for advice on the microarrays technology, Dimitris Kafetzopoulos for resources, George Garinis for advice and assistance in the analysis of the Affymetrix expression microarray data, George Papagiannakis for technical assistance in the Affymetrix Hybridizations, Nikos Papanikolaou for help with in silico searches, Charalambos Spilianakis, George Garinis, Jürg Bähler and Max Reuter for critical reading and insightful comments on the manuscript, Jürg Bähler for kind permission to use the BioLector technology in his laboratory at UCL and Stephen Kearsey for kind permission to use his laboratory facilities.
Footnotes
Present addresses:
Christos Andreadis, Department of Zoology, University of Oxford, South Parks Road, Oxford, OX1 3PS, United Kingdom.
Georgia Tsiliki, Process Control & Informatics Unit, School of Chemical Engineering, N.T.U.A., 9 Heroon Polytechneiou St., Zografou Campus, Athens 15780, Greece.
FUNDING
Operational Programme «Competitiveness and Entrepreneurship» (OPCE II)-‘Cooperation’ [09SYN-13–901 to D.A.]; National Strategic Reference Framework (NSRF) 2007–2013; EU and the Greek State [THALIS-‘GenAge’-380228 to D.A.]. Institute of Biology and Biotechnology-FORTH and PhD fellowships from Public Welfare Foundation ‘Propondis’ and Alexander S. Onassis Public Benefit Foundation [to C.A.]. Funding for open access charge: EU and the Greek State [THALIS-‘GenAge’-380228 to D.A.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kerzendorfer C., O'Driscoll M. Human DNA damage response and repair deficiency syndromes: linking genomic instability and cell cycle checkpoint proficiency. DNA Repair (Amst) 2009;8:1139–1152. doi: 10.1016/j.dnarep.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 2.Weinert T.A., Hartwell L.H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- 3.Soulier J., Lowndes N.F. The BRCT domain of the S. cerevisiae checkpoint protein Rad9 mediates a Rad9-Rad9 interaction after DNA damage. Curr. Biol. 1999;9:551–554. doi: 10.1016/s0960-9822(99)80242-5. [DOI] [PubMed] [Google Scholar]
- 4.Hammet A., Magill C., Heierhorst J., Jackson S.P. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 2007;8:851–857. doi: 10.1038/sj.embor.7401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nnakwe C.C., Altaf M., Cote J., Kron S.J. Dissection of Rad9 BRCT domain function in the mitotic checkpoint response to telomere uncapping. DNA Repair (Amst) 2009;8:1452–1461. doi: 10.1016/j.dnarep.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Javaheri A., Wysocki R., Jobin-Robitaille O., Altaf M., Cote J., Kron S.J. Yeast G1 DNA damage checkpoint regulation by H2A phosphorylation is independent of chromatin remodeling. Proc. Natl. Acad. Sci. U.S.A. 2006;103:13771–13776. doi: 10.1073/pnas.0511192103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toh G.W., O'Shaughnessy A.M., Jimeno S., Dobbie I.M., Grenon M., Maffini S., O'Rorke A., Lowndes N.F. Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair (Amst) 2006;5:693–703. doi: 10.1016/j.dnarep.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Huyen Y., Zgheib O., Ditullio R.A., Jr, Gorgoulis V.G., Zacharatos P., Petty T.J., Sheston E.A., Mellert H.S., Stavridi E.S., Halazonetis T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 9.Weinert T.A., Hartwell L.H. Characterization of RAD9 of Saccharomyces cerevisiae and evidence that its function acts posttranslationally in cell cycle arrest after DNA damage. Mol. Cell. Biol. 1990;10:6554–6564. doi: 10.1128/mcb.10.12.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paulovich A.G., Margulies R.U., Garvik B.M., Hartwell L.H. RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics. 1997;145:45–62. doi: 10.1093/genetics/145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siede W., Friedberg A.S., Friedberg E.C. RAD9-dependent G1 arrest defines a second checkpoint for damaged DNA in the cell cycle of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7985–7989. doi: 10.1073/pnas.90.17.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emili A. MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol. Cell. 1998;2:183–189. doi: 10.1016/s1097-2765(00)80128-8. [DOI] [PubMed] [Google Scholar]
- 13.Vialard J.E., Gilbert C.S., Green C.M., Lowndes N.F. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 1998;17:5679–5688. doi: 10.1093/emboj/17.19.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toh G.W., Lowndes N.F. Role of the Saccharomyces cerevisiae Rad9 protein in sensing and responding to DNA damage. Biochem. Soc. Trans. 2003;31:242–246. doi: 10.1042/bst0310242. [DOI] [PubMed] [Google Scholar]
- 15.Gilbert C.S., Green C.M., Lowndes N.F. Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol. Cell. 2001;8:129–136. doi: 10.1016/s1097-2765(01)00267-2. [DOI] [PubMed] [Google Scholar]
- 16.Le May N., Egly J.M., Coin F. True lies: the double life of the nucleotide excision repair factors in transcription and DNA repair. J. Nucleic Acids. 2010 doi: 10.4061/2010/616342. doi:10.4061/2010/616342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le May N., Mota-Fernandes D., Velez-Cruz R., Iltis I., Biard D., Egly J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell. 2010;38:54–66. doi: 10.1016/j.molcel.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Mellon I., Spivak G., Hanawalt P.C. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 1987;51:241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 19.Tu Y., Tornaletti S., Pfeifer G.P. DNA repair domains within a human gene: selective repair of sequences near the transcription initiation site. EMBO J. 1996;15:675–683. [PMC free article] [PubMed] [Google Scholar]
- 20.Al-Moghrabi N.M., Al-Sharif I.S., Aboussekhra A. The RAD9-dependent gene trans-activation is required for excision repair of active genes but not for repair of non-transcribed DNA. Mutat. Res. 2009;663:60–68. doi: 10.1016/j.mrfmmm.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Granata M., Lazzaro F., Novarina D., Panigada D., Puddu F., Abreu C.M., Kumar R., Grenon M., Lowndes N.F., Plevani P. Dynamics of Rad9 chromatin binding and checkpoint function are mediated by its dimerization and are cell cycle-regulated by CDK1 activity. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001047. e1001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dion V., Kalck V., Horigome C., Towbin B.D., Gasser S.M. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell. Biol. 2012;14:502–509. doi: 10.1038/ncb2465. [DOI] [PubMed] [Google Scholar]
- 23.Mine-Hattab J., Rothstein R. Increased chromosome mobility facilitates homology search during recombination. Nat. Cell. Biol. 2012;14:510–517. doi: 10.1038/ncb2472. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi-Iwai Y., Ueta R., Fukunaka A., Sasaki R. Subcellular localization of Aft1 transcription factor responds to iron status in Saccharomyces cerevisiae. J. Biol. Chem. 2002;277:18914–18918. doi: 10.1074/jbc.M200949200. [DOI] [PubMed] [Google Scholar]
- 25.Shakoury-Elizeh M., Tiedeman J., Rashford J., Ferea T., Demeter J., Garcia E., Rolfes R., Brown P.O., Botstein D., Philpott C.C. Transcriptional remodeling in response to iron deprivation in Saccharomyces cerevisiae. Mol. Biol. Cell. 2004;15:1233–1243. doi: 10.1091/mbc.E03-09-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamza A., Baetz K. The iron-responsive transcription factor Aft1 interacts with the kinetochore protein Iml3 and promotes pericentromeric cohesin. J. Biol. Chem. 2011;287:4139–4147. doi: 10.1074/jbc.M111.319319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berthelet S., Usher J., Shulist K., Hamza A., Maltez N., Johnston A., Fong Y., Harris L.J., Baetz K. Functional genomics analysis of the Saccharomyces cerevisiae iron responsive transcription factor Aft1 reveals iron-independent functions. Genetics. 2010;185:1111–1128. doi: 10.1534/genetics.110.117531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., Schiebel E. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15:963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 29.Longtine M.S., McKenzie A., 3rd, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 30.Georgatsou E., Mavrogiannis L.A., Fragiadakis G.S., Alexandraki D. The yeast Fre1p/Fre2p cupric reductases facilitate copper uptake and are regulated by the copper-modulated Mac1p activator. J. Biol. Chem. 1997;272:13786–13792. doi: 10.1074/jbc.272.21.13786. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J.F.J., Maniatis T. Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 32.Fragiadakis G.S., Tzamarias D., Alexandraki D. Nhp6 facilitates Aft1 binding and Ssn6 recruitment, both essential for FRE2 transcriptional activation. EMBO J. 2004;23:333–342. doi: 10.1038/sj.emboj.7600043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuo M.H., Allis C.D. In vivo cross-linking and immunoprecipitation for studying dynamic Protein:DNA associations in a chromatin environment. Methods. 1999;19:425–433. doi: 10.1006/meth.1999.0879. [DOI] [PubMed] [Google Scholar]
- 34.Ausubel F.M. Current Protocols in Molecular Biology. New York: Greene Publishing Associates and Wiley-Interscience; pp. 1987–2011. [Google Scholar]
- 35.Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 36.Cline M.S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B., et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pokholok D.K., Harbison C.T., Levine S., Cole M., Hannett N.M., Lee T.I., Bell G.W., Walker K., Rolfe P.A., Herbolsheimer E., et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 38.Li B., Gogol M., Carey M., Pattenden S.G., Seidel C., Workman J.L. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 2007;21:1422–1430. doi: 10.1101/gad.1539307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irizarry R.A., Bolstad B.M., Collin F., Cope L.M., Hobbs B., Speed T.P. Summaries of affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaiseau P.L., Lesuisse E., Camadro J.M. Aft2p, a novel iron-regulated transcription activator that modulates, with Aft1p, intracellular iron use and resistance to oxidative stress in yeast. J. biol. chem. 2001;276:34221–34226. doi: 10.1074/jbc.M104987200. [DOI] [PubMed] [Google Scholar]
- 41.Rutherford J.C., Jaron S., Winge D.R. Aft1p and Aft2p mediate iron-responsive gene expression in yeast through related promoter elements. J. Biol. Chem. 2003;278:27636–27643. doi: 10.1074/jbc.M300076200. [DOI] [PubMed] [Google Scholar]
- 42.Philpott C.C., Protchenko O. Response to iron deprivation in Saccharomyces cerevisiae. Eukaryot. Cell. 2008;7:20–27. doi: 10.1128/EC.00354-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gross C., Kelleher M., Iyer V.R., Brown P.O., Winge D.R. Identification of the copper regulon in Saccharomyces cerevisiae by DNA microarrays. J. Biol. Chem. 2000;275:32310–32316. doi: 10.1074/jbc.M005946200. [DOI] [PubMed] [Google Scholar]
- 44.Mason P.B., Struhl K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol. Cell. 2005;17:831–840. doi: 10.1016/j.molcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 45.Davie J.K., Kane C.M. Genetic interactions between TFIIS and the Swi-Snf chromatin-remodeling complex. Mol. Cell. Biol. 2000;20:5960–5973. doi: 10.1128/mcb.20.16.5960-5973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K., Hannett N.M., Harbison C.T., Thompson C.M., Simon I., et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 47.Measday V., Baetz K., Guzzo J., Yuen K., Kwok T., Sheikh B., Ding H., Ueta R., Hoac T., Cheng B., et al. Systematic yeast synthetic lethal and synthetic dosage lethal screens identify genes required for chromosome segregation. Proc. Natl. Acad. Sci. U.S.A. 2005;102:13956–13961. doi: 10.1073/pnas.0503504102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimada M., Komatsu K. Emerging connection between centrosome and DNA repair machinery. J. Radiat. Res. (Tokyo) 2009;50:295–301. doi: 10.1269/jrr.09039. [DOI] [PubMed] [Google Scholar]
- 49.Lashkari D.A., DeRisi J.L., McCusker J.H., Namath A.F., Gentile C., Hwang S.Y., Brown P.O., Davis R.W. Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13057–13062. doi: 10.1073/pnas.94.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerton J.L., DeRisi J., Shroff R., Lichten M., Brown P.O., Petes T.D. Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2000;97:11383–11390. doi: 10.1073/pnas.97.21.11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dekker J. GC- and AT-rich chromatin domains differ in conformation and histone modification status and are differentially modulated by Rpd3p. Genome Biol. 2007;8:R116. doi: 10.1186/gb-2007-8-6-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yokota H., Singer M.J., van den Engh G.J., Trask B.J. Regional differences in the compaction of chromatin in human G0/G1 interphase nuclei. Chromosome Res. 1997;5:157–166. doi: 10.1023/a:1018438729203. [DOI] [PubMed] [Google Scholar]
- 53.Gilbert N., Boyle S., Fiegler H., Woodfine K., Carter N.P., Bickmore W.A. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell. 2004;118:555–566. doi: 10.1016/j.cell.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 54.Goffeau A., Barrell B.G., Bussey H., Davis R.W., Dujon B., Feldmann H., Galibert F., Hoheisel J.D., Jacq C., Johnston M., et al. Life with 6000 genes. Science. 1996;274:563–547. doi: 10.1126/science.274.5287.546. 546. [DOI] [PubMed] [Google Scholar]
- 55.Robine N., Uematsu N., Amiot F., Gidrol X., Barillot E., Nicolas A., Borde V. Genome-wide redistribution of meiotic double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007;27:1868–1880. doi: 10.1128/MCB.02063-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buhler C., Borde V., Lichten M. Mapping meiotic single-strand DNA reveals a new landscape of DNA double-strand breaks in Saccharomyces cerevisiae. PLoS Biol. 2007;5:e324. doi: 10.1371/journal.pbio.0050324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrison J.C., Haber J.E. Surviving the breakup: the DNA damage checkpoint. Annu. Rev. Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- 58.Longhese M.P., Mantiero D., Clerici M. The cellular response to chromosome breakage. Mol. Microbiol. 2006;60:1099–1108. doi: 10.1111/j.1365-2958.2006.05186.x. [DOI] [PubMed] [Google Scholar]
- 59.Strahl B.D., Grant P.A., Briggs S.D., Sun Z.W., Bone J.R., Caldwell J.A., Mollah S., Cook R.G., Shabanowitz J., Hunt D.F., et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 2002;22:1298–1306. doi: 10.1128/mcb.22.5.1298-1306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lancelot N., Charier G., Couprie J., Duband-Goulet I., Alpha-Bazin B., Quemeneur E., Ma E., Marsolier-Kergoat M.C., Ropars V., Charbonnier J.B., et al. The checkpoint Saccharomyces cerevisiae Rad9 protein contains a tandem tudor domain that recognizes DNA. Nucleic Acids Res. 2007;35:5898–5912. doi: 10.1093/nar/gkm607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim N., Abdulovic A.L., Gealy R., Lippert M.J., Jinks-Robertson S. Transcription-associated mutagenesis in yeast is directly proportional to the level of gene expression and influenced by the direction of DNA replication. DNA Repair (Amst) 2007;6:1285–1296. doi: 10.1016/j.dnarep.2007.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim N., Jinks-Robertson S. dUTP incorporation into genomic DNA is linked to transcription in yeast. Nature. 2009;459:1150–1153. doi: 10.1038/nature08033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Azvolinsky A., Giresi P.G., Lieb J.D., Zakian V.A. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell. 2009;34:722–734. doi: 10.1016/j.molcel.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Prado F., Aguilera A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005;24:1267–1276. doi: 10.1038/sj.emboj.7600602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wellinger R.E., Prado F., Aguilera A. Replication fork progression is impaired by transcription in hyperrecombinant yeast cells lacking a functional THO complex. Mol. Cell. Biol. 2006;26:3327–3334. doi: 10.1128/MCB.26.8.3327-3334.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gottipati P., Cassel T.N., Savolainen L., Helleday T. Transcription-associated recombination is dependent on replication in Mammalian cells. Mol. Cell. Biol. 2008;28:154–164. doi: 10.1128/MCB.00816-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bermejo R., Capra T., Gonzalez-Huici V., Fachinetti D., Cocito A., Natoli G., Katou Y., Mori H., Kurokawa K., Shirahige K., et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell. 2009;138:870–884. doi: 10.1016/j.cell.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 68.Cha R.S., Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297:602–606. doi: 10.1126/science.1071398. [DOI] [PubMed] [Google Scholar]
- 69.Kim J.M., Vanguri S., Boeke J.D., Gabriel A., Voytas D.F. Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res. 1998;8:464–478. doi: 10.1101/gr.8.5.464. [DOI] [PubMed] [Google Scholar]
- 70.Ohkuni K., Kitagawa K. Endogenous transcription at the centromere facilitates centromere activity in budding yeast. Curr. Biol. 2011;21:1695–1703. doi: 10.1016/j.cub.2011.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ganem N.J., Pellman D. Linking abnormal mitosis to the acquisition of DNA damage. J. Cell. Biol. 2012;199:871–881. doi: 10.1083/jcb.201210040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Michelson R.J., Rosenstein S., Weinert T. A telomeric repeat sequence adjacent to a DNA double-stranded break produces an anticheckpoint. Genes Dev. 2005;19:2546–2559. doi: 10.1101/gad.1293805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Enomoto S., Glowczewski L., Lew-Smith J., Berman J.G. Telomere cap components influence the rate of senescence in telomerase-deficient yeast cells. Mol. Cell. Biol. 2004;24:837–845. doi: 10.1128/MCB.24.2.837-845.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.IJpma A.S., Greider C.W. Short telomeres induce a DNA damage response in Saccharomyces cerevisiae. Mol. Biol. Cell. 2003;14:987–1001. doi: 10.1091/mbc.02-04-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lazzaro F., Sapountzi V., Granata M., Pellicioli A., Vaze M., Haber J.E., Plevani P., Lydall D., Muzi-Falconi M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008;27:1502–1512. doi: 10.1038/emboj.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morin I., Ngo H.P., Greenall A., Zubko M.K., Morrice N., Lydall D. Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 2008;27:2400–2410. doi: 10.1038/emboj.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frit P., Kwon K., Coin F., Auriol J., Dubaele S., Salles B., Egly J.M. Transcriptional activators stimulate DNA repair. Mol. Cell. 2002;10:1391–1401. doi: 10.1016/s1097-2765(02)00732-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.