


Abstract
Transcription of hepatitis B virus (HBV) from the covalently closed circular DNA (cccDNA) template is essential for its replication. Suppressing the level and transcriptional activity of cccDNA might have anti-HBV effect. Although cellular transcription factors, such as CREB, which mediate HBV transcription, have been well described, transcriptional coactivators that facilitate this process are incompletely understood. In this study we showed that CREB-regulated transcriptional coactivator 1 (CRTC1) is required for HBV transcription and replication. The steady-state levels of CRTC1 protein were elevated in HBV-positive hepatoma cells and liver tissues. Ectopic expression of CRTC1 or its homolog CRTC2 or CRTC3 in hepatoma cells stimulated the activity of the preS2/S promoter of HBV, whereas overexpression of a dominant inactive form of CRTC1 inhibited HBV transcription. CRTC1 interacts with CREB and they are mutually required for the recruitment to the preS2/S promoter on cccDNA and for the activation of HBV transcription. Accumulation of pregenomic RNA (pgRNA) and cccDNA was observed when CRTC1 or its homologs were overexpressed, whereas the levels of pgRNA, cccDNA and secreted HBsAg were diminished when CRTC1 was compromised. In addition, HBV transactivator protein HBx stabilized CRTC1 and promoted its activity on HBV transcription. Our work reveals an essential role of CRTC1 coactivator in facilitating and supporting HBV transcription and replication.
INTRODUCTION
Hepatitis B virus (HBV), a member in the family of Hepadnaviridae, has a DNA genome 3.2 kb in size which is partially double stranded. Over 350 million people are chronically infected with HBV worldwide, including ∼10% of the population in Hong Kong and China, 15–40% of whom will terminally develop severe liver diseases including hepatocellular carcinoma (HCC) (1). Although multiple factors are involved in the development of HCC in HBV carriers, high level of HBV DNA has been identified as a major risk (2).
HBV infects hepatocytes that express its receptor named sodium taurocholate cotransporting polypeptide (3). Upon viral entry and release of genetic material, covalently closed circular DNA (cccDNA) is generated from relaxed circular DNA (rcDNA) and complexed with different viral proteins, histones, transcription factors and coactivators to form a nuclear minichromosome, which serves as a central template for all HBV transcription (4,5). Transcription of pregenomic RNA (pgRNA) from cccDNA is rate-limiting in genome amplification and replication. cccDNA can be amplified through an unknown mechanism (6). The stability of the cccDNA pool is a major determinant in viral clearance. cccDNA is refractory to antivirals such as nucleoside or nucleotide analogs. cccDNA is also accounted for viral relapse after cessation of anti-HBV therapy (5). cccDNA is therefore an important target for better control and elimination of HBV infection (7,8). In line with this, understanding the mechanism by which HBV transcription from cccDNA is regulated might reveal new strategies for therapeutic intervention.
HBV transcription is mediated by transcription factors recruited to cccDNA including CREB, ATF1, STAT1 and KLF15 (4,9). CREB is essential for HBV replication and it binds to the cAMP response elements (CREs) located at the X and preS2 promoters of the viral genome (10,11). Transcriptional coactivators such as p300, pCAF and CREB binding protein (CBP) are also known to be recruited to cccDNA to support HBV transcription through acetylation of histones and transcription factors (12). In addition, CREB-regulated transcriptional coactivator 2 (CRTC2) is also thought to potentiate HBV transcription through the induction of PGC1α (13). However, the roles of additional CRTC transcriptional coactivators in relation to the function of CREB in HBV transcription have not been characterized.
CRTCs are potent coactivators of CREB-dependent transcription from both canonical cellular CREs and the non-canonical viral CREs found in the long terminal repeats of the oncogenic retrovirus human T-cell leukemia virus type 1 (14,15). There are three isoforms of human CRTCs, namely CRTC1, CRTC2 and CRTC3. They interact with CREB and cooperate with p300/CBP to potentiate CREB-dependent transcription (15). Because various signals converge on CRTCs to exert their effects on the transcription of CREB-regulated genes, they are the targets of pharmaceutical agents such as metformin, a widely prescribed drug for type II diabetes (16). Thus, full characterization of the roles of CRTCs in HBV transcription might reveal new targets for therapeutic intervention.
HBV encodes a multifunctional viral oncoprotein HBx. It acts as a transcriptional activator that modestly activates cellular transcription factors including CREB, NF-κB and AP1 (17). HBx is required for HBV transcription and productive infection in vivo (18,19). HBx does not bind DNA. Its ability to activate HBV promoters is mediated through CREB (10,20). Mechanistically, HBx enhances CREB dimerization and potentiates coactivator function of p300/CBP (20,21). However, the interplay among HBx, CREB and CRTCs in HBV transcription remains to be understood. Particularly, it will be of interest to see whether and how HBx interacts with and modulates CRTC activity.
In this study, we documented the requirement of CRTC1 in HBV transcription. CRTC1 protein was more abundantly detected in liver tissues of HBV-infected individuals. The HBV-induced stabilization of CRTC1 protein was further validated. We provided the first evidence that CRTC1 and its homologs CRTC2 and CRTC3 cooperate with HBx to activate HBV transcription through CREB. Our work suggests a new model in which HBV, through mutual stabilization of HBx and CRTC1, usurps CRTC1 transcriptional coactivator to augment CREB-mediated activation of HBV transcription.
MATERIALS AND METHODS
Human liver tissue samples
Sampling of human liver tissues was performed as previously described (22). Seven Chinese patients who had surgical resection of liver at Queen Mary Hospital of Hong Kong were randomly selected for study. All specimens were obtained immediately after surgical resection, snap-frozen in liquid nitrogen and stored at −70°C. Frozen sections were cut from non-tumorous liver and histological examination was performed to verify homogeneous cell populations of tissues. Hepatitis B surface antigen (HBsAg) was detected in the sera of patients 127, 128, 216 and 229, but not patients 210, 213 and 223. HBV DNA status in liver samples was also verified. The use of human liver tissues was approved by the Joint Institutional Review Board of the University of Hong Kong and Hospital Authority Hong Kong West Cluster.
Cell culture and transfection
HepG2.2.15 cells were cultured in Dulbecco's Modified Eagle Medium supplemented with 10% fetal calf serum, 2 mM L-glutamine and 380 μg/ml G418. Plasmid DNA was transfected into HepG2 cells using GeneJuice transfection reagent (Novagen). siRNA was transfected into HepG2 and HepG2.2.15 cells using Lipofectamine 2000 (Invitrogen).
Plasmids
Reporter plasmids preS2-Luc, X-Luc, preS1-Luc and C-Luc were derived from pGL3-Basic (Promega). preS1 (nucleotides 2710–2834), preS2 (nucleotides 2966–152), X (nucleotides 950–1310) and C (nucleotides 1403–1817) promoter sequences were amplified by polymerase chain reaction (PCR) from the HBV genome of adw2 subtype (Genbank AM282986) in plasmid pHBV1.3D, which has been described elsewhere (9,23). Expression plasmids for CREB, A-CREB, CRTC1, CRTC2, CRTC3, CRTC1-S167A and CRTC1-M1, as well as reporter plasmid pCRE-Luc have also been detailed previously (15,24). The expression plasmid for HBx was based on the pCAGEN vector and the HBx open reading frame (nucleotides 1374–1838) was PCR amplified from pHBV1.3D with the addition of a Flag-HA tag to its 5′ end. The X-deficient pHBV1.3D was made by converting the eighth codon of HBx to a stop codon. The mutagenic primers were 5′-CGCGAAGGAT CCAGTTAGCA GTACAACCTA GCA-3′ and 5′-GCGCTTCCTA GGTCAATCGT CATGTTGGAT CGT-3′. A similar construct has been described elsewhere (25). Expression vector for the lysine-free (K0) mutant of ubiquitin is a kind gift from Dr James Chen (26).
Luciferase reporter assays
Dual luciferase assay was performed as described previously (27,28). Transfection efficiencies were normalized to a control plasmid expressing Renilla luciferase (pSV-RLuc from Promega).
Coimmunoprecipitation and chromatin immunoprecipitation
Coimmunoprecipitation (Co-IP) and chromatin immunoprecipitation (ChIP) assays were performed as previously described (22,29). For Co-IP, HepG2 or HepG2.2.15 cells grown in a 100-mm petri dish were harvested into 1 ml of immunoprecipitation buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.5 mM EDTA, 1% NP-40, 0.1% sodium dodecyl sulfate, 1% sodium deoxycholate, 1 mM dithiothreitol, 20 mM β-glycerophosphate, 1 mM sodium vanadate and 1 mM phenylmethylsulfonyl fluoride). Flag-HBx, CRTC1-V5 and endogenous CREB proteins were immunoprecipitated from the cleared lysate by incubation at 4°C for 2 h. For ChIP, HepG2 cells were cross-linked with 1% formaldehyde for 10 min at room temperature. Cells were washed and then lysed in the presence of protease inhibitor cocktail. The DNA–protein complex was immunoprecipitated. The cross-link was reversed by proteinase K. The DNA was purified by phenol-chloroform extraction. Promoter sequence spanning the two CRE enhancers in the preS2 promoter was analyzed by quantitative PCR using primers 5′-CCCTGCTCCG AATATTGCCT C-3′ and 5′-ACACACGGGT GATCCCCCTA G-3′. For cccDNA ChIP, quantitative PCR was performed with primers 5′-CTCCCCGTCT GTGCCTTCT-3′ and 5′-GCCCCAAAGC CACCCAAG-3′ that selectively amplify cccDNA as previously described (7,12,29).
RNA interference
HepG2 and HepG2.2.15 cells were transfected with 100 nM of siRNA using Lipofectamine 2000 (Invitrogen). RNA interference (RNAi) experiments were performed as described (27,30). siRNAs were listed in Supplementary Table S1.
Real-time RT-PCR
Real-time reverse transcription-PCR (RT-PCR) was performed as described (27,30). Normalization was made to glycerialdehyde-3-phosphate dehydrogenase (GAPDH) mRNA. Quantitation of target mRNA was achieved with the comparative Ct method. Relative expression level of target mRNA was calculated from 2−ΔCt. Primers were listed in Supplementary Table S1.
RESULTS
CRTC1 protein was abundantly detected in HBV-infected cells
To identify proteins that are specifically expressed in HBV-infected liver, we performed a pilot proteomic profiling study using human liver tissues that are infected with HBV. We noted that CRTC1, but not CRTC2 or CRTC3, was more abundantly detected in infected versus uninfected samples (data not shown). We previously showed the requirement of CRTC coactivators in transcriptional activation of human T-cell leukemia virus type 1 (15). We therefore sought to investigate the expression and function of CRTC1 in HBV-infected cells.
To model HBV genome replication, we used HepG2 cells transfected with pHBV1.3D, an HBV molecular clone (9,23). In addition, HepG2.2.15 cells, which constitutively produce HBV (31), were also employed. Expression of endogenous CRTC1 protein was more pronounced in pHBV1.3D-transfected HepG2 and in HepG2.2.15 cells (Figure 1A and B, lanes 2 and 3 or 2–4 compared to 1), whereas the steady-state levels of CREB, CRTC2 and CRTC3 remained largely unchanged. In contrast, the mRNA levels of all three CRTCs were comparable in the presence and absence of HBV (Figure 1C), indicating that the effect on CRTC1 occurs at protein but not mRNA level. To verify CRTC1 expression in natural infection of HBV, we examined a small number of HBV-positive or HBV-negative human liver tissues. CRTC1 protein expression was 6- to 18-fold more abundant in HBV-positive samples than in HBV-negative ones (Figure 1D). One plausible interpretation to these data is that CRTC1 protein is stabilized in HBV-infected cells.
Figure 1.
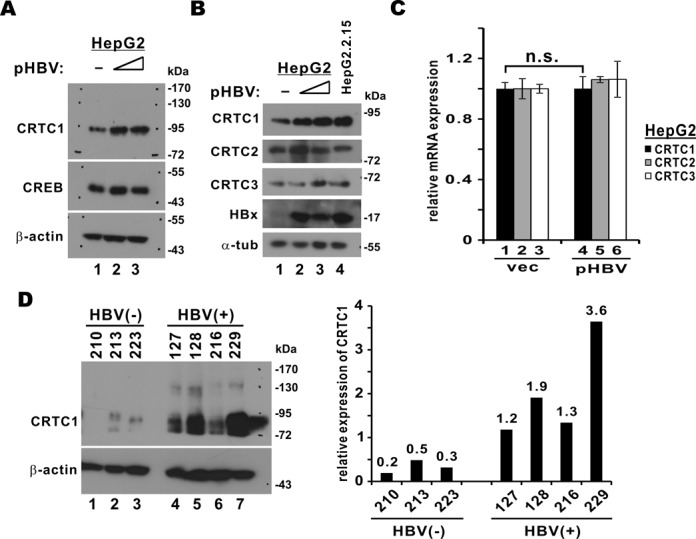
Detection of CRTC1 protein in HBV-positive cells. (A) Detection of CRTC1 protein in pHBV1.3D-transfected HepG2 cells. Cells were seeded into a six-well plate and transfected with escalating amount of pHBV1.3D (1 or 2 μg). Cells were harvested 48 h after transfection. Cell lysates were analyzed by western blotting with rabbit anti-CRTC1 (Cell Signaling), rabbit anti-CREB (Cell Signaling) and mouse anti-β-actin (Sigma) antibodies. (B) Detection of CRTC proteins in HepG2.2.15 cells. Western blotting was performed with rabbit anti-CRTC1/2/3 (Cell Signaling), mouse anti-HBx (Millipore) and anti-α-tubulin (Sigma); α-tub: α-tubulin. (C) Quantitative RT-PCR analysis of CRTC mRNA. HepG2 cells were transfected with empty vector (vec) or pHBV1.3D (2 μg). The difference between bars 1 and 4 is statistically not significant (n.s.) by Student's t test (P = 0.98). (D) Detection of CRTC1 protein in human liver samples. Band intensity was measured by densitometry.
Overexpression of CRTCs promoted HBV transcription and replication
Given that CRTC1 protein is abundant in HBV-infected cells, it will be of interest to assess the influence of CRTC coactivators on HBV transcription. CRTCs potentiate CREB-dependent transcription depending on the promoter context (32,33). Although CRE elements are found in preS2 and X promoters of HBV (10,11), it remains to be determined whether they are activated by CRTCs. With this in mind, we asked how CRTCs might affect transcriptional activity of these two promoters. All three CRTCs were expressed to comparable levels in HepG2 cells (Figure 2A). Whereas all three CRTCs activated preS2 promoter significantly (Figure 2B, groups 2–4 compared to 1), they had minimal or no influence on basal activity of X promoter in the same setting (Figure 2C). Hence, CRTCs are capable of activating HBV transcription from the preS2 promoter.
Figure 2.
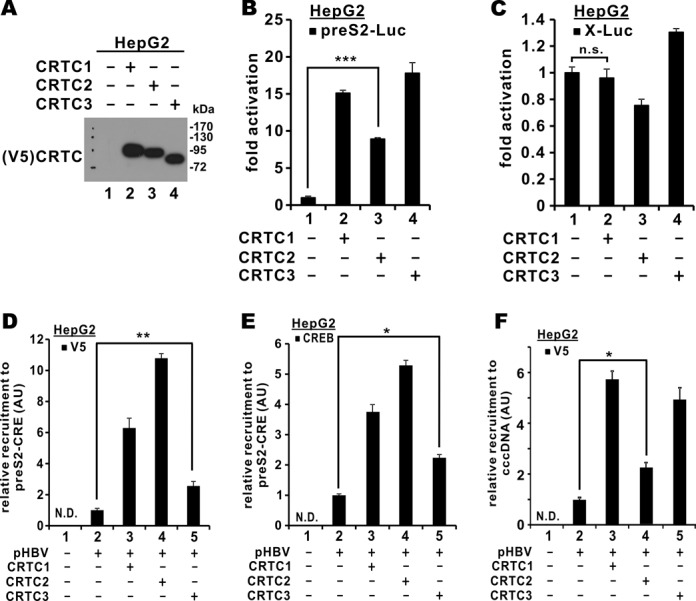
Overexpression of CRTC coactivators augmented HBV transcription. (A) Overexpression of CRTC1/2/3 in HepG2 cells. Western blotting was performed with mouse anti-V5 (Invitrogen). (B) Effect on preS2 promoter activity. HepG2 cells were transfected with preS2-Luc (100 ng) and pcDNA3.1-V5/His-CRTC1/2/3 (100 ng). Dual luciferase assay was performed. Results represent means from three independent experiments and error bars indicate SD. The difference between groups 1 and 3 is statistically significant by Student's t test (P = 0.000024, highlighted with ***). (C) Effect on X promoter activity. The difference between groups 1 and 2 is statistically not significant (n.s.) by Student's t test (P = 0.49). (D) CRTC recruitment to CREs in the preS2 promoter. HepG2 cells were transfected with the indicated plasmids. ChIP was performed to precipitate V5-CRTC1/2/3-preS2 promoter complex using mouse anti-V5 (Invitrogen) and the CRE sequence in the preS2 promoter was analyzed by quantitative PCR. Results represent relative recruitment measured in arbitrary units (AU). The difference between groups 2 and 5 is statistically significant by Student's t test (P = 0.0032, highlighted with **); N.D.: not detected. (E) Impact of CRTC coactivators on CREB recruitment to the preS2 promoter. ChIP was performed to precipitate CREB-preS2 promoter complex using rabbit anti-CREB (Cell Signaling). The difference between groups 2 and 5 is statistically significant by Student's t test (P = 0.029, highlighted with *). (F) CRTC recruitment to cccDNA. ChIP was performed to obtain V5-CRTC1/2/3-DNA complex using mouse anti-V5 and the cccDNA-specific sequence was analyzed by quantitative PCR. The difference between groups 2 and 3 is statistically significant by Student's t test (P = 0.031, highlighted with *).
CRTC2 has recently been shown to promote HBV transcription and replication through the induction of a CREB-regulated gene, PGC1α, which in turn activates the core promoter of HBV modestly (13). Consistent with this, we observed moderate activation of the preS1 and core promoter by all three CRTCs (Supplementary Figure S1A and B, groups 2–4 compared to 1). Because CRTCs activate CRE and preS2 promoter much more potently (Figure 2A and Supplementary Figure S1C) and no CRE was found in preS1 or core promoter, our further analysis will be focused on the preS2 promoter only.
To verify the coactivator function of CRTCs on the preS2 promoter, we performed ChIP assay to assess its recruitment to the CRE elements in this promoter. Indeed, all three CRTCs were recruited to preS2-CRE in pHBV1.3D-transfected HepG2 cells (Figure 2D, groups 3–5 compared to 2). Since CRTCs are thought to work in concert with CREB, we further examined the recruitment of endogenous CREB to preS2-CRE. We found that the HBV-induced recruitment of CREB to preS2-CRE was more pronounced when CRTC1/2/3 was overexpressed (Figure 2E, groups 3–5 compared to 2), indicating that CRTCs and CREB might activate transcription from the preS2 promoter cooperatively. In line with this, all three CRTCs were also recruited to cccDNA (Figure 2F, groups 3–5 compared to 2). Collectively, our results supported the model in which CRTCs facilitate HBV transcription by enhancing the recruitment of CREB to the preS2 promoter.
Activation of HBV transcription might accelerate other steps in viral replication. To verify this, we measured the levels of HBV pgRNA and cccDNA in pHBV1.3D-transfected HepG2 cells. Upon pHBV1.3D transfection, basal expression of pgRNA as well as nuclear and cell-free cccDNA was detected (Figure 3A and B, group 2, and Figure 3C, lane 3). Intriguingly, their expression was boosted when CRTC1/2/3 was overexpressed (Figure 3A and B, groups 3–5 compared to 2 and Figure 3C, lanes 4–6 compared to 3). In other words, overexpression of CRTCs not only stimulates HBV transcription, but also promotes its replication.
Figure 3.
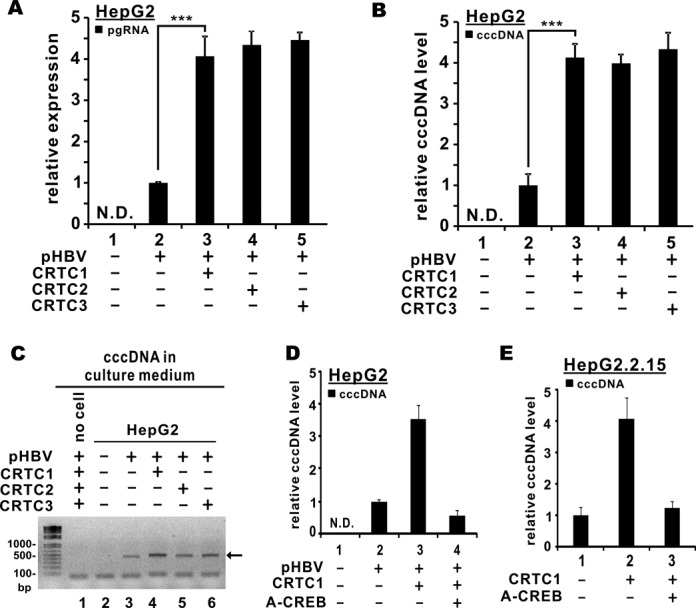
Overexpression of CRTC coactivators promoted HBV replication. (A) CRTC coactivators augmented pgRNA expression. HepG2 cells were co-transfected with pcDNA3.1-V5/His-CRTC1/2/3 and pHBV1.3D. Cells were harvested 48 h post-transfection. Total RNA was extracted and cDNA was synthesized. Quantitative RT-PCR was performed to analyze the relative levels of pgRNA. The difference between bars 2 and 3 is statistically significant by Student's t test (P = 0.0016, highlighted with **); N.D.: not detected. (B) CRTC coactivators promoted cccDNA formation. Nuclear fraction was extracted. DNA was isolated and treated with plasmid-safe DNase I (Epicentre). Treated fraction was analyzed for cccDNA by quantitative PCR. GAPDH transcript was amplified from untreated fraction for normalization. The difference between groups 2 and 3 is statistically significant by Student's t test (P = 0.00029, highlighted with ***). (C) CRTC coactivators increased cccDNA production in cell-free medium. HepG2 cells were co-transfected with pcDNA3.1-V5/His-CRTC1/2/3 and pHBV1.3D for 48 h. Culture medium was harvested and centrifugated to remove debris. Cleared medium was boiled for 15 min, centrifugated at 13 500 rpm for 15 min. Supernatant was collected, treated with plasmid-safe DNase I (Epicentre) and analyzed for cccDNA (highlighted with an arrow) by agarose gel electrophoresis. (D) and (E) CRTC1-induced augmentation of cccDNA formation in HepG2 and HepG2.2.15 cells was ablated by A-CREB. Nuclear cccDNA levels were measured as in (B).
To determine the requirement of CREB in CRTC-induced activation of HBV replication, we employed A-CREB, a previously described dominant inactive form of CREB (34). The expression of A-CREB abrogated CRTC1-induced augmentation of cccDNA formation in pHBV1.3D-transfected HepG2 cells (Figure 3D, group 4 compared to 2 and 3). Consistent with this, the increase in cccDNA level in the presence of CRTC1 in HepG2.2.15 cells was blunted when A-CREB was overexpressed (Figure 3E, group 3 compared to 1 and 2). Thus, CREB is required for CRTC1-induced activation of HBV replication.
Depletion of CRTC1 inhibited HBV replication
Complementary to the gain-of-function analyses described above, loss-of-function experiments were also performed to establish the physiological importance of CRTC1 in HBV transcription and replication. Because only CRTC1 levels were elevated in HBV-infected cells (Figure 1), we chose to focus on CRTC1 in the loss-of-function study. We first utilized M1-CRTC1, a truncated and dominant inactive mutant of CRTC1, which we constructed and successfully used previously (24,27,30). When the dose of M1-CRTC1 was escalated in HepG2 cells, the activity of the preS2 promoter was diminished progressively (Figure 4A, groups 2–4 compared to 1). Moreover, the intracellular cccDNA pool decreased in M1-CRTC1-expressing HepG2 cells (Figure 4B). Accordingly, the pgRNA levels also declined in a dose-dependent manner in these cells (Figure 4C, groups 3 and 4 compared to 2).
Figure 4.
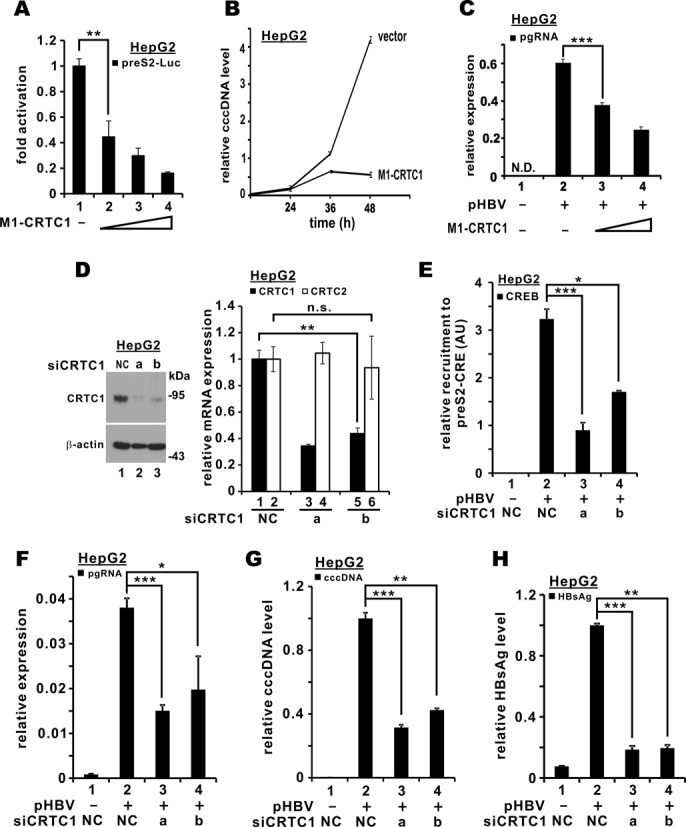
Suppression of CRTC1 inhibited HBV replication. (A) Expression of CRTC1 dominant inactive form (M1-CRTC1) suppressed preS2 promoter activity. HepG2 cells were transfected with preS2-Luc (100 ng) and increasing amount of pGal-M1-CRTC1 (25, 50 and 100 ng). The difference between groups 1 and 2 is statistically significant by Student's t test (P = 0.0077, highlighted with **). (B) Expression of M1-CRTC decreased cccDNA levels. Nuclear DNA was isolated and treated with plasmid-safe DNase I (Epicentre). Treated fraction was analyzed for cccDNA by quantitative PCR at the indicated time points. Relative cccDNA level was derived by normalizing to the amount of GAPDH mRNA amplified from untreated fraction. Data points in the plot are means from three independent experiments and error bars indicate SD. (C) Perturbation of CRTC1 with M1-CRTC1 mitigated pgRNA expression. HepG2 cells were co-transfected with pGal-M1-CRTC1 and pHBV1.3D. Cells were harvested 48 h post-transfection for quantitative RT-PCR analysis of pgRNA. The difference between bars 2 and 3 is statistically significant by Student's t test (P = 0.00045, highlighted with ***); N.D.: not detected. (D) Depletion of endogenous CRTC1 in HepG2 cells by siRNAs. Two independent CRTC1-targeting siRNAs (siCRTC1-a and siCRTC1-b) were used to deplete endogenous CRTC1 at a concentration of 100 nM. siNC was used as a negative control. Endogenous CRTC1 in siRNA-transfected HepG2 cells was analyzed by western blotting at 72 h post-transfection using rabbit anti-CRTC1. Concurrently, total RNA was extracted and cDNA was synthesized. Quantitative RT-PCR was performed to analyze the relative levels of CRTC1 and CRTC2 transcripts. The difference between bars 1 and 5 is statistically significant by Student's t test (P = 0.0018, highlighted with **). The difference between bars 2 and 6 is statistically not significant (n.s.) by Student's t test (P = 0.74). (E) CREB recruitment to pre-S2 promoter was diminished in CRTC1-compromised HepG2 cells. After 24 h of CRTC1 knockdown, pHBV1.3D was transfected into HepG2 cells for another 48 h. ChIP was performed as in Figure 2D. The differences between groups 2 and 3 (P = 0.00023, highlighted with ***) as well as between groups 2 and 4 (P = 0.013, highlighted with *) are statistically significant; AU: arbitrary unit. (F) Knockdown of CRTC1 repressed pgRNA expression. The differences between groups 2 and 3 (P = 0.00031, highlighted with ***) as well as between groups 2 and 4 (P = 0.022, highlighted with *) are statistically significant by Student's t test. (G) Knockdown of CRTC1 dampened cccDNA level. Nuclear cccDNA was analyzed as in Figure 3B. The differences between groups 2 and 3 (P = 0.00012, highlighted with ***) as well as between groups 2 and 4 (P = 0.0089, highlighted with **) are statistically significant by Student's t test. (H) CRTC1 is required for production of cell-free virus. After 24 h of CRTC1 knockdown, pHBV1.3D was transfected into HepG2 cells for another 48 h. Culture medium was centrifugated to remove debris and then detected for HBsAg using MonolisaTM ULTRA kit (BioRad). Relative HBsAg level in siNC-transfected cells was taken as 1. The differences between groups 2 and 3 (P = 0.00071, highlighted with ***) as well as between groups 2 and 4 (P = 0.0047, highlighted with **) are statistically significant by Student's t test.
We next employed RNAi to silence endogenous CRTC1 expression in HepG2 cells. Two independent siRNAs directed against CRTC1 (siCRTC1-a and siCRTC1-b) efficiently depleted its protein expression (Figure 4D, lanes 2 and 3 compared to 1 in the blot). The silencing effect was highly specific since only CRTC1 mRNA was affected, whereas the levels of CRTC2 transcript remained unaltered (Figure 4D, groups 3 and 5 compared to 1, and groups 4 and 6 compared to 2 in the bar chart). In keeping with this, the recruitment of endogenous CREB to preS2-CRE was prevented in CRTC1-depleted cells (Figure 4E, groups 3 and 4 compared to 2), indicating the physiological importance of CRTC1 in mobilizing CREB to the preS2 promoter to activate transcription. Furthermore, the levels of pgRNA, nuclear cccDNA and secreted HBsAg were declined when CRTC1 was compromised (Figure 4F–H, groups 3 and 4 compared to 2). Collectively, our results suggest that CRTC1 is required for HBV transcription and replication.
CREB is required for CRTC activation of HBV transcription
The importance of CREB in HBV transcription prompted us to investigate whether CRTC1 activation of the preS2 promoter requires CREB function. As a first step, we generated additional reporter constructs in which the CRE elements in the preS2 promoter were disrupted. Similar constructs were previously described (10). Indeed, progressive removal of CRE elements effectively blunted CRTC1 activation of the preS2 promoter (Figure 5A, groups 5 and 6 compared to 4). Next, we asked whether functional CREB is critical in supporting CRTC1 activity by overexpressing A-CREB, a dominant inactive form of CREB (34). CRTC1 activation of the preS2 promoter was erased by A-CREB (Figure 5B, groups 3 and 4 compared to 2). Reciprocally, CREB activation of the preS2 promoter was progressively suppressed when we increased M1-CRTC1 expression in HepG2 cells (Figure 5C, groups 3 and 4 compared to 2). In line with this, the recruitment of CRTC1 to preS2-CRE was abrogated when A-CREB was overexpressed in pHBV1.3D-transfected HepG2 cells (Figure 5D, group 4 compared to 3). Reciprocal immunoprecipitation and immunoblotting experiments confirmed the formation of CREB–CRTC1 complex in HepG2 and HepG2.2.15 cells (Supplementary Figure S2A and B). Hence, CRTC1 requires and cooperates with CREB to stimulate HBV transcription.
Figure 5.
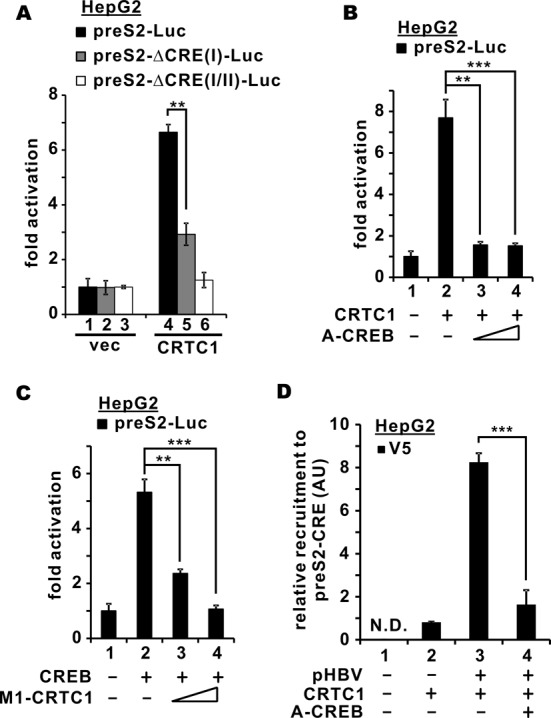
CRTC1 activates preS2 promoter through CREB. (A) Deletion of CRE in the preS2 promoter abrogated CRTC1-mediated activation. HepG2 cells were transfected with preS2-Luc/preS2-ΔCRE(I)-Luc/preS2-ΔCRE(I/II)-Luc (100 ng) and pcDNA3.1-V5/His-CRTC1 (100 ng). The difference between groups 4 and 5 is statistically significant by Student's t test (P = 0.0014, highlighted with **); vec: empty vector. (B) CRTC1 activation of preS2 promoter was blunted by dominant inactive CREB. HepG2 cells were transfected with preS2-Luc (100 ng), a fixed amount of pcDNA3.1-V5/His-CRTC1/2/3 (100 ng) and escalating amounts of A-CREB plasmid (10 and 20 ng). The differences between groups 2 and 3 (P = 0.0010, highlighted with **) as well as between groups 2 and 4 (P = 0.00099, highlighted with ***) are statistically significant by Student's t test. (C) CREB-mediated preS2 promoter activation was ablated in the presence of dominant inactive CRTC1. HepG2 cells were transfected with preS2-Luc (100 ng), a fixed amount of Tag2B-CREB (100 ng) and escalating amounts of M1-CRTC1 plasmids (25 and 50 ng). The differences between groups 2 and 3 (P = 0.0045, highlighted with **) as well as between groups 2 and 4 (P = 0.00040, highlighted with ***) are statistically significant by Student's t test. (D) CRTC1 recruitment to preS2 promoter was prevented when CREB function was compromised. ChIP was performed as in Figure 2D. The difference between groups 3 and 4 is statistically significant by Student's t test (P = 0.00036, highlighted with ***); N.D.: not detected.
HBx promotes CRTC1 activation of HBV transcription
Because HBx also activates CREB and is required for HBV replication (10,19), we investigated the interplay among HBx, CREB and CRTC1 in HBV transcription. We observed that CRTC1 activation of both canonical CREs and the preS2 promoter was more robust when HBx was also expressed (Figure 6A and B, groups 5 and 6 compared to 4). Similar results were also obtained for CRTC2 and CRTC3 (data not shown). In addition, CRTC1 was detected in the HBx immunoprecipitate (Figure 6C, lane 3 compared to 2), indicating their association in cultured cells. Interestingly, the protein bands of both CRTC1 and HBx were more prominent when they were co-expressed (Figure 6C, lane 3 compared to 1 and 2), implicating a mutual stabilization effect. Unexpectedly, an additional slow-migrating HBx species was observed when CRTC1 was co-expressed (Figure 6C, lane 3 compared to 1). Thus, CRTC1 might direct HBx for additional protein modifications.
Figure 6.
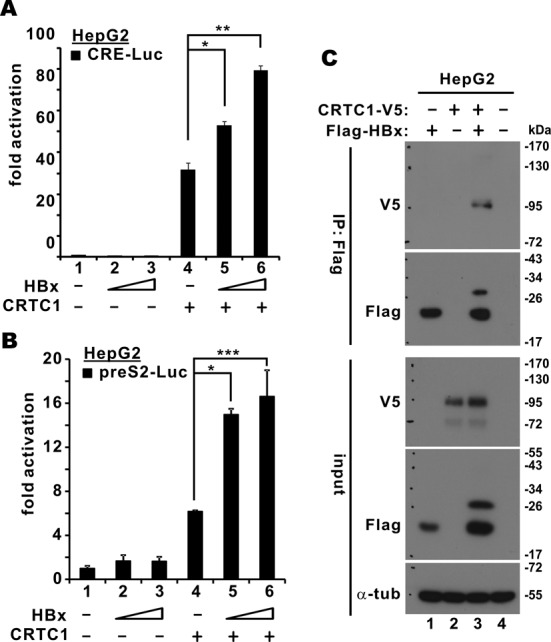
Cooperation between CRTC1 and HBx in the activation of preS2 promoter. (A) and (B) HepG2 cells were transfected with pCRE-Luc or preS2-Luc (100 ng), a fixed amount of pcDNA3.1-V5/His-CRTC1 (100 ng) and escalating amounts (50 and 100 ng) of pCAGEN-Flag-HA-HBx. Fold activation is calculated from preS2-Luc activity normalized to that of pSV-RLuc. The differences between groups 4 and 5 (P = 0.024 for A and P = 0.020 for B, highlighted with *) as well as between groups 4 and 6 (P = 0.0051 for A and P = 0.000051 for B, highlighted with ** and ***, respectively) are statistically significant by Student's t test. (C) Association of CRTC1 with HBx. HepG2 cells were transfected with pcDNA3.1-V5/His-CRTC1 and pCAGEN-Flag-HA-HBx. HBx was precipitated with anti-Flag. The precipitates and the input lysates were probed with mouse anti-Flag, mouse anti-V5 and rabbit anti-CREB; α-tub: α-tubulin.
Phosphorylation of CRTC2 at S171 was thought to trigger polyubiquitination at K628 leading to protein degradation (35). The equivalent site in CRTC1 is S167 (24). To explore whether HBx might affect the stability of CRTC1 through a similar mechanism, we compared the interaction of HBx with CRTC1 and its active non-phosphorylatable form, CRTC1-S167A. When CRTC1 and CRTC1-S167A were co-expressed with HBx in HepG2 cells, similar amounts of CRTC1 protein were found in the HBx immunoprecipitate (Supplementary Figure S3, lane 3 compared to 2). In addition, both forms of CRTC1 enhanced the recruitment of CREB to the HBx-containing complex, but to similar extent (Supplementary Figure S3, lanes 2 and 3 compared to 1). Thus, HBx has no binding preference to CRTC1-S167A. On the other hand, when we overexpressed CREB, the association of HBx and CRTC1 was not affected (Supplementary Figure S4, lane 4 compared to 3). Consistent with results in Supplementary Figure S3, both CRTC1 and CREB were found in the HBx precipitate (Supplementary Figure S4). These data were consistent with the formation of HBx–CRTC1–CREB triple complex.
Mutual stabilization of HBx and CRTC1
To further investigate mutual stabilization of HBx and CRTC1, we expressed in HepG2 cells CRTC1 and its mutant CRTC1-S167A, either individually or together with HBx. Steady-state levels of both CRTC1 and its S167A mutant were increased when HBx was expressed (Figure 7A, lanes 5 and 6 compared to 2 and 3). On the other hand, HBx protein expression was more pronounced and a slow-migrating HBx species appeared when CRTC1 or CRTC1-S167A was expressed (Figure 7A, lanes 5 and 6 compared to 4). Hence, CRTC1 and HBx are mutually required for optimal stability and activity.
Figure 7.
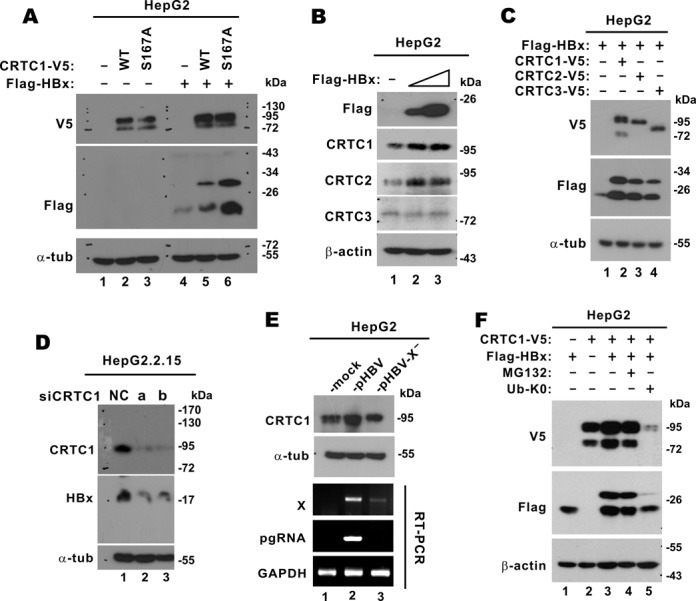
Mutual stabilization of CRTC1 and HBx. (A) HepG2 cells were transfected with pcDNA3.1-V5/His-CRTC1 WT/S167A and pCAGEN-Flag-HA-HBx as indicated. Cell lysates were analyzed by western blotting. (B) Stabilization of endogenous CRTC1/2 by HBx. HepG2 cells were transfected with escalating amounts (0.5 and 1 μg) of pCAGEN-Flag-HA-HBx. Expression of endogenous CRTC1/2/3 was analyzed by western blotting. (C) Stabilization of HBx by CRTC1/2/3. HepG2 cells were transfected with pCAGEN-Flag-HA-HBx and pcDNA3.1-V5/His-CRTC1/2/3. (D) Compromising CRTC1 led to diminution of HBx protein. HepG2.2.15 cells were transfected with siNC or siCRTC1 for 48 h. (E) HBx-dependent stabilization of CRTC1 in HBV-replicating cells. HepG2 cells were transfected with pHBV1.3D or its HBx-deficient mutant pHBV1.3D-X−, in which the expression of HBx protein was abrogated. CRTC1 expression was analyzed by western blotting. RT-PCR was also performed to verify the expression of X transcript and pgRNA. (F) Stabilization of CRTC1 and HBx requires ubiquitination and proteosome. HepG2 cells were transfected with the indicated combinations of pcDNA3.1-V5/His-CRTC1, pCAGEN-Flag-HA-HBx and an expression vector for K0 mutant of ubiquitin. Cells in group 4 were treated with 20 μM of MG132 for 4 h.
We next examined the impact of HBx on the steady-state levels of endogenous CRTC proteins in HepG2 cells. Notably, escalating dose of HBx expression correlated with increased levels of CRTC1 and CRTC2, but not CRTC3 (Figure 7B, lanes 2 and 3 compared to 1). On the other hand, when we overexpressed CRTCs individually, more prominent HBx bands including the slow-migrating species were visible (Figure 7C, lanes 2–4 compared to 1). In other words, CRTC1 and its homologs were capable of stabilizing HBx.
When we depleted endogenous CRTC1 in HepG2.2.15 cells, expression of HBx protein was diminished (Figure 7D, lanes 2 and 3 compared to 1), suggesting a physiological role of CRTC1 in maintaining HBx protein stability. Such destabilization effect on HBx was unlikely due to an effect of CRTC1 on X promoter, which was shown to be unresponsive to CRTC1 (Figure 2B). The stabilization of CRTC1 was also seen in pHBV1.3D-transfected HepG2 cells (Figure 7E, lane 2 compared to 1). In sharp contrast, the steady-state level of CRTC1 remained unchanged (Figure 7E, lane 3 compared to 1 and 2) when HepG2 cells were transfected with pHBV1.3D-X−, an X-deficient molecular clone of HBV (25). Although an X transcript was expressed from this clone at lower level (Figure 7E, lane 3 compared to 2), no functional HBx protein would be produced as shown in the literature (25). Thus, the stabilization of CRTC1 in HBV-replicating cells is HBx dependent.
To shed mechanistic light on the mutual stabilization of HBx and CRTC1, we employed proteosome inhibitor MG132 and a lysine-free (K0) mutant of ubiquitin (26). The K0 mutant of ubiquitin prevents polyubiquitination and subsequent proteasome-mediated degradation, but allows mono- and multi-ubiquitination, which might be linked to lysosome-dependent degradation (36,37). If mutual stabilization of HBx and CRTC1 is mediated solely through polyubiquitination and proteosome, their levels should be unchanged or increased in the presence of MG132 or ubiquitin-K0. Interestingly, the steady-state levels of CRTC1 and HBx decreased only slightly when the cells were treated with MG132 (Figure 7F, lane 4 compared to 3), implicating a role for proteosome in the stabilization effect. However, the expression of ubiquitin-K0 abrogated this stabilization effect almost completely (Figure 7F, lane 5 compared to 1–4). Although there was no simple interpretation to this result, one possibility is that mono- or multi-ubiquitination of HBx and CRTC1 might promote their proteosomal degradation. Plausibly, the stability of HBx and CRTC1 might be governed by multiple mechanisms involving ubiquitination, proteosome and lysosome.
DISCUSSION
In this study we provided the first evidence for the requirement of CRTC1 coactivator for HBV transcription and replication. Our work contributes at least three new messages concerning the role of CRTC1 in HBV infection. First, CRTC1 protein was abundantly expressed in HBV-infected cells (Figure 1). Second, CRTC1 and its homologs (CRTC2 and CRTC3) are recruited to the preS2 promoter and cccDNA to mediate transcriptional activation in cooperation with CREB (Figures 2–5). Finally, HBx functions to stabilize and promote CRTC1 activation of HBV transcription (Figures 6 and 7). Our findings reveal a new cellular accessory factor and a new layer of regulation in HBV gene expression.
CRTC1 is particularly stabilized in HBV-infected cells (Figure 1). This stabilization effect is specific to CRTC1 (Figure 1). Although CRTC2 and CRTC3 expression was not found to be elevated in HBV-infected cells, they were also capable of occupying the preS2 promoter, stabilizing HBx and activating HBV transcription (Figures 2, 3 and 7). In other words, they are fully competent to regulate HBV gene expression. Because they might be induced and activated in response to metabolic stress and hormones (33), further studies are required to elucidate how they modulate HBV transcription under the influence of their metabolic and hormonal activators.
Promoter context is a decisive factor in the requirement of CRTC coactivators for transcriptional activation of cellular CRE-dependent genes (32,33). Our demonstration of CRTC activation of the preS2 but not the X promoter of HBV (Figure 2) supports the notion that the coactivator function of CRTCs in HBV transcription is also promoter context dependent. Although the exact promoter context that governs CRTC requirement is not currently understood, a comparison of the preS2 and X promoters sheds some light on this issue. Noteworthily, there are two consecutive CREs in the preS2 promoter, both of which are required for optimal activation by CRTC1 (Figure 5). In contrast, only one single CRE has been found in the X promoter (11). Further experiments should therefore be performed to determine whether this difference in the number of CREs might be decisive in promoter responsiveness to CRTCs.
Our results supported the model in which CRTCs cooperate with CREB to activate the preS2 promoter (Figure 5). We also showed the recruitment of CRTCs to cccDNA and preS2-CRE in pHBV1.3D-transfected cells (Figure 2). These findings support a more direct involvement of CRTCs in HBV transcription from cccDNA. However, an indirect stimulatory role of CRTC2 on the core promoter mediated through PGC1α has also been suggested (13). We also observed a modest activation of the preS1 and core promoters by all three CRTCs (Supplementary Figure S1). However, since no CRE has been found in these promoters, their general and mild activation by CRTCs is likely mediated indirectly through a CREB-regulated gene such as PGC1α. Thus, the influence of CRTCs on HBV transcription might be exerted through both direct activation as demonstrated in our study and an indirect effect as suggested for CRTC2 (13). Particularly, the stimulatory effect of CRTCs on pgRNA expression (Figure 3) could be mediated through the core promoter.
The activation of the preS2 promoter by CRTC1 was mediated through CREB and CRE (Figures 2 and 5). The structural interface of CRTC2 for recognition and coactivation of CREB has been determined (38). In addition, the regulation of CRTC2 nucleocytoplasmic shuttling by upstream kinases has also been characterized (39). Plausibly, CRTC1 would use a similar interface to recognize CREB and CRTC1 activity would be regulated by the same protein kinases in the context of HBV transcription. It is noteworthy that small molecule activators and inhibitors of LKB1 and its downstream protein kinases, including metformin and salicylate, have been well documented (16,40). We have recently shown that LKB1 and its downstream kinases potently suppress transcriptional activity of CRTC1 and CREB in the context of human T-cell leukemia type 1 virus infection. In addition, the activation of these kinases using small molecule agonists such as metformin exhibits strong antiretroviral effects (27). By the same token, our characterization of the role of CRTCs in HBV life cycle will pave the way for further investigations on pharmacological inhibition of HBV transcription.
HBx activates HBV and cellular gene expression through multiple mechanisms. Particularly, HBx stimulates CREB-dependent transcription by cooperating with p300/CBP coactivators (20), promoting CREB dimerization (21) and inhibiting negative regulators (41). Our documentation of the interaction and mutual stabilization of HBx and CRTC1/2 (Figures 6 and 7) provides a new mechanism by which HBx regulates HBV transcription. Since CRTCs are essential coactivators of CREB-dependent transcription of both HBV and cellular promoters, HBx might also modulate cellular CRE-dependent promoters through its interaction and stabilization of CRTC1/2.
The mutual stabilization of HBX and CRTC1/2 in HBV-infected cells (Figure 7) is attributed at least in part to their interaction (Figure 6). This stabilization unlikely occurs at the mRNA level since CRTC mRNAs remained unchanged in pHBV1.3D-transfected cells (Figure 1C) in which HBx was abundantly expressed (Figure 1B). In addition, the stabilization of HBx was seen not only when it was expressed from a plasmid in transfected HepG2 cells but also when it was expressed in HepG2.2.15 cells from its own promoter in the HBV genome (Figure 7), which is not responsive to CRTCs (Figure 2). The mechanism by which HBx and CRTC1/2 proteins mutually stabilize each other warrants further study. First, the functional domains in HBx and CRTC1/2 that mediate their interaction and stabilization should be determined. CRTC3 is more divergent from CRTC1/2 and smaller in size; comparative analysis might reveal why CRTC3 was not stabilized by HBx (Figure 7). Although CRTC2 level was elevated in HepG2 cells expressing HBx (Figure 7), it was unchanged in HepG2.2.15 or pHBV1.3D-transfected HepG2 cells (Figure 1). Further investigations are required to resolve this discrepancy and to clarify whether and how other viral proteins might affect CRTC2 protein expression. Second, the slow-migrating form of HBx detected in the presence of CRTC1/2/3 (Figure 7) should be further characterized. Particularly, whether it represented mono- or multi-ubiquitinated HBx should be clarified. Finally, exactly how stability of HBx and CRTCs in HBV-infected cells would be regulated through ubiquitination and proteolysis should be clarified. HBx has previously been shown to stabilize AIB1 oncoprotein by preventing its ubiquitination and proteosomal degradation (42). In view of the minimal to mild effect of MG132 on the mutual stabilization of HBx and CRTC1 (Figure 7), a role for polyubiquitination and proteosome was suggested. However, the abrogation of the mutual stabilization effect by ubiquitin-K0 pointed to another direction. This discrepancy should be resolved in future work. The possibility that HBx and CRTC1 might be mono- or multi-ubiquitinated and then degraded by the lysosome should be experimentally validated. Alternatively, K63-linked or other types of polyubiquitination of CRTC1 suppressed by ubiquitin-K0 might be required for its stabilization. Our data cannot exclude that multiple mechanisms might operate in the control of HBx and CRTC stability. Further investigations should be performed to determine the involvement of proteosome, lysosome and different types of ubiquitination in the destruction of HBx and CRTCs in HBV-infected cells.
Results from our gain-of-function and loss-of-function assays consistently supported the requirement of CRTC1 for HBV replication, as indicated in the production of pgRNA, cccDNA and secreted HBsAg (Figures 3 and 4). This is generally consistent with the notion that CRTC1-regulated HBV transcription is influential in viral replication. The decline of cccDNA level upon CRTC1 suppression (Figure 4) is noteworthy. It raises one interesting possibility that compromising CRTC1 might destabilize cccDNA. Histone hypoacetylation is known to regulate cccDNA transcription (43). APOBEC3-mediated cytidine deamination has recently been shown to trigger specific degradation of cccDNA (8). Whether and how CRTC1 recruitment to cccDNA and its post-translational modifications might affect cccDNA stability and function merit further investigations.
CREB and CRTCs have oncogenic potential in various types of cancer (44,45). Our present study reveals a novel function of CRTC1 in HBV transcription and replication. The stabilization and activation of CRTC1 were observed both in HepG2 cells freshly transfected with pHBV1.3D and in HepG2.2.15 cells constitutively harboring HBV genome (Figures 2 and 6). CRTC1 might plausibly play a role in both acute and chronic phase of HBV infection. However, it will still be of importance to characterize in more detail the stabilization of CRTC1 in different phases of HBV infection and HCC development. Particularly, further investigations are required to clarify whether CRTC1 might be critical in HBV reactivation as well as initiation and progression of HCC.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Footnotes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.
FUNDING
Hong Kong Health and Medical Research Fund [12111052, 13121052, 14131162]; Hong Kong Research Grants Council [HKU7/CRF/09, HKU1/CRF/11G, HKU 7869/11]; S.K. Yee Medical Research Fund (2011). Funding for open access charge: Hong Kong Health and Medical Research Fund.
Conflict of interest statement. None declared.
REFERENCES
- 1.Iavarone M., Colombo M. HBV infection and hepatocellular carcinoma. Clin. Liver Dis. 2013;17:375–397. doi: 10.1016/j.cld.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Liaw Y.F. Impact of therapy on the long-term outcome of chronic hepatitis B. Clin. Liver Dis. 2013;17:413–423. doi: 10.1016/j.cld.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H., et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quasdorff M., Protzer U. Control of hepatitis B virus at the level of transcription. J. Viral Hepat. 2010;17:527–536. doi: 10.1111/j.1365-2893.2010.01315.x. [DOI] [PubMed] [Google Scholar]
- 5.Levrero M., Pollicino T., Petersen J., Belloni L., Raimondo G., Dandri M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009;51:581–592. doi: 10.1016/j.jhep.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 6.Coffin C.S., Mulrooney-Cousins P.M., Peters M.G., van Marle G., Roberts J.P., Michalak T.I., Terrault N.A. Molecular characterization of intrahepatic and extrahepatic hepatitis B virus (HBV) reservoirs in patients on suppressive antiviral therapy. J. Viral Hepat. 2011;18:415–423. doi: 10.1111/j.1365-2893.2010.01321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belloni L., Allweiss L., Guerrieri F., Pediconi N., Volz T., Pollicino T., Petersen J., Raimondo G., Dandri M., Levrero M. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Invest. 2012;122:529–537. doi: 10.1172/JCI58847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lucifora J., Xia Y., Reisinger F., Zhang K, Stadler D., Cheng X., Sprinzl M.F., Koppensteiner H., Makowska Z., Volz T., et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou J., Tan T., Tian Y., Zheng B., Ou J.H., Huang E.J., Yen T.S.B. Kruppel-like factor 15 activates hepatitis B virus gene expression and replication. Hepatology. 2011;54:109–121. doi: 10.1002/hep.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tacke F., Liedtke C., Bocklage S., Manns M.P., Trautwein C. CREB/PKA sensitive signalling pathways activate and maintain expression levels of the hepatitis B virus pre-S2/S promoter. Gut. 2005;54:1309–1317. doi: 10.1136/gut.2005.065086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim B.K., Lim S.O., Park Y.G. Requirement of the cyclic adenosine monophosphate response element-binding protein for hepatitis B virus replication. Hepatology. 2008;48:361–373. doi: 10.1002/hep.22359. [DOI] [PubMed] [Google Scholar]
- 12.Belloni L., Pollicino T., De Nicola F., Guerrieri F., Raffa G., Fanciulli M., Raimondo G., Levrero M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. U.S.A. 2009;106:19975–19979. doi: 10.1073/pnas.0908365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian X., Zhao F., Sun W., Zhi X., Cheng Z., Zhou M., Hu K. CRTC2 enhances HBV transcription and replication by inducing PGC1α expression. Virol. J. 2014;11:30. doi: 10.1186/1743-422X-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koga H., Ohshima T., Shimotohno K. Enhanced activation of tax-dependent transcription of human T-cell leukemia virus type I (HTLV-I) long terminal repeat by TORC3. J. Biol. Chem. 2004;279:52978–52983. doi: 10.1074/jbc.M409021200. [DOI] [PubMed] [Google Scholar]
- 15.Siu Y.T., Chin K.T., Siu K.L., Choy E.Y.W., Jeang K.T., Jin D.Y. TORC1 and TORC2 coactivators are required for tax activation of the human T-cell leukemia virus type 1 long terminal repeats. J. Virol. 2006;80:7052–7059. doi: 10.1128/JVI.00103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw R.J., Lamia K.A., Vasquez D., Koo S.H., Bardeesy N., Depinho R.A., Montminy M., Cantley L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neuveut C., Wei Y., Buendia M.A. Mechanisms of HBV-related hepatocarcinogenesis. J. Hepatol. 2010;52:594–604. doi: 10.1016/j.jhep.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 18.Tang H., Delgermaa L., Huang F., Oishi N., Liu L., He F., Zhao L., Murakami S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J. Virol. 2005;79:5548–5556. doi: 10.1128/JVI.79.9.5548-5556.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lucifora J., Arzberger S., Durantel D., Belloni L., Strubin M., Levrero M., Zoulim F., Hantz O., Protzer U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011;55:996–1003. doi: 10.1016/j.jhep.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 20.Cougot D., Wu Y., Cairo S., Caramel J., Renard C.A., Levy L., Buendia M.A., Neuveut C. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J. Biol. Chem. 2007;282:4277–4287. doi: 10.1074/jbc.M606774200. [DOI] [PubMed] [Google Scholar]
- 21.Maguire H.F., Hoeffler J.P., Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science. 1991;252:842–844. doi: 10.1126/science.1827531. [DOI] [PubMed] [Google Scholar]
- 22.Chin K.T., Zhou H.J., Wong C.M., Lee J.M., Chan C.P., Qiang B.Q., Yuan J.G., Ng I.O.L., Jin D.Y. The liver-enriched transcription factor CREB-H is a growth suppressor protein underexpressed in hepatocellular carcinoma. Nucleic Acids Res. 2005;33:1859–1873. doi: 10.1093/nar/gki332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Z., Yen T.S.B., Wu L., Madden C.R., Tan W., Slagle B.L., Ou J.H. Enhancement of hepatitis B virus replication by its X protein in transgenic mice. J. Virol. 2002;76:2579–2584. doi: 10.1128/jvi.76.5.2579-2584.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siu Y.T., Ching Y.P., Jin D.Y. Activation of TORC1 transcriptional coactivator through MEKK1-induced phosphorylation. Mol. Biol. Cell. 2008;19:4750–4761. doi: 10.1091/mbc.E08-04-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z., Protzer U., Hu Z., Jacob J., Liang T.J. Inhibition of cellular proteasome activities enhances hepadnavirus replication in an HBX-dependent manner. J. Virol. 2004;78:4566–4572. doi: 10.1128/JVI.78.9.4566-4572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C., Deng L., Hong M., Akkaraju G.R., Inoue J., Chen Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 27.Tang H.M.V., Gao W.W., Chan C.P., Siu Y.T., Wong C.M., Kok K.H., Ching Y.P., Takemori H., Jin D.Y. LKB1 tumor suppressor and salt-inducible kinases negatively regulate human T-cell leukemia virus type 1 transcription. Retrovirology. 2013;10:40. doi: 10.1186/1742-4690-10-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siu K.L., Yeung M.L., Kok K.H., Yuen K.S., Kew C., Lui P.Y., Chan C.P., Tse H., Woo P.C.Y., Yuen K.Y., et al. Middle East respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in innate antiviral response. J. Virol. 2014;88:4866–4876. doi: 10.1128/JVI.03649-13. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Laras A., Koskinas J., Dimou E., Kostamena A., Hadziyannis S.J. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology. 2006;44:694–702. doi: 10.1002/hep.21299. [DOI] [PubMed] [Google Scholar]
- 30.Chan C.P., Siu Y.T., Kok K.H., Ching Y.P., Tang H.M.V., Jin D.Y. Group I p21-activated kinases facilitate Tax-mediated transcriptional activation of the human T-cell leukemia virus type 1 long terminal repeats. Retrovirology. 2013;10:47. doi: 10.1186/1742-4690-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sells M.A., Chen M.L., Acs G. Production of hepatitis B virus particles in HepG2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. U.S.A. 1987;84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amelio A.L., Caputi M., Conkright M.D. Bipartite functions of the CREB co-activators selectively direct alternative splicing or transcriptional activation. EMBO J. 2009;28:2733–2747. doi: 10.1038/emboj.2009.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altarejos J.Y., Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahn S., Olive M., Aggarwal S., Krylov D., Ginty D.D., Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol. Cell. Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dentin R., Liu Y., Koo S.H., Hedrick S., Vargas T., Heredia J., Yates J., III, Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 36.Piper R.C., Lehner P.J. Endosomal transport via ubiquitination. Trends Cell Biol. 2011;21:647–655. doi: 10.1016/j.tcb.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clague M.J., Urbé S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143:682–685. doi: 10.1016/j.cell.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Luo Q., Viste K., Urday-Zaa J.C., Senthil Kumar G., Tsai W.W., Talai A, Mayo K.E., Montminy M., Radhakrishnan I. Mechanism of CREB recognition and coactivation by the CREB-regulated transcriptional coactivator CRTC2. Proc. Natl. Acad. Sci. U.S.A. 2012;109:20865–20870. doi: 10.1073/pnas.1219028109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J.M., Seo W.Y., Song K.H., Chanda D., Kim Y.D., Kim D.K., Lee M.W., Ryu D., Kim Y.H., Noh J.R., et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J. Biol. Chem. 2010;285:32182–32191. doi: 10.1074/jbc.M110.134890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardie D.G., Ross F.A., Hawley S.A. AMP-activated protein kinase: a target for drugs both ancient and modern. Chem. Biol. 2012;19:1222–1236. doi: 10.1016/j.chembiol.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cougot D., Allemand E., Riviere L., Benhenda S., Duroure K., Levillayer F., Muchardt C., Buendia M.A., Neuveut C. Inhibition of PP1 phosphatase activity by HBx: a mechanism for the activation of hepatitis B virus transcription. Sci. Signal. 2012;5 doi: 10.1126/scisignal.2001906. ra1. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y., Tong Z., Li T., Chen Q., Zhuo L., Li W., Wu R.C., Yu C. Hepatitis B virus X protein stabilizes amplified in breast cancer 1 protein and cooperates with it to promote human hepatocellular carcinoma cell invasiveness. Hepatology. 2012;56:1015–1024. doi: 10.1002/hep.25751. [DOI] [PubMed] [Google Scholar]
- 43.Pollicino T., Belloni L., Raffa G., Pediconi N., Squadrito G., Raimondo G., Levrero M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology. 2006;130:823–837. doi: 10.1053/j.gastro.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Siu Y.T., Jin D.Y. CREB-a real culprit in oncogenesis. FEBS J. 2007;274:3224–3232. doi: 10.1111/j.1742-4658.2007.05884.x. [DOI] [PubMed] [Google Scholar]
- 45.Komiya T., Coxon A., Park Y., Chen W.D., Zajac-Kaye M., Meltzer P., Karpova T., Kaye F.J. Enhanced activity of the CREB co-activator Crtc1 in LKB1 null lung cancer. Oncogene. 2010;29:1672–1680. doi: 10.1038/onc.2009.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.