


Abstract
Multidomain transcription factors, which are especially abundant in eukaryotic genomes, are advantageous to accelerate the search kinetics for target site because they can follow the intersegment transfer via the monkey-bar mechanism in which the protein forms a bridged intermediate between two distant DNA regions. Monkey-bar dynamics highly depends on the properties of the multidomain protein (the affinity of each of the constituent domains to the DNA and the length of the linker) and the DNA molecules (their inter-distance and inter-angle). In this study, we investigate using coarse-grained molecular dynamics simulations how the local conformation of the DNA may affect the DNA search performed by a multidomain protein Pax6 in comparison to that of the isolated domains. Our results suggest that in addition to the common rotation-coupled translation along the DNA major groove, for curved DNA the tethered domains may slide in a rotation-decoupled sliding mode. Furthermore, the multidomain proteins move by longer jumps on curved DNA compared with those performed by the single domain protein. The long jumps originate from the DNA curvature bringing two sequentially distant DNA sites into close proximity with each other and they suggest that multidomain proteins may move on highly curved DNA faster than linear DNA.
INTRODUCTION
Multidomain transcription factors (TFs) are composed of two or more DNA binding protein domains (DBDs) interconnected via linkers of various lengths. Each DBD contains binding motifs, such as short α-helixes, β-sheets or mixed α/β motifs, to recognize their specific DNA binding sites among myriads of nonspecific DNA sequences. Evolutionary, multidomain TFs have advantages over single domain proteins in terms of folding (1), structural stability and functional diversity (2–4), and they are found in abundance in eukaryotic genomes (5,6). The efficiency of TFs in locating their target DNA sites, often known as ‘cognate sites’, is largely dependent on the stability of each domain and the strength of their inter-communications or ‘cross-talks’ (7–13).
In an extensive computational study, we previously investigated the effects of such ‘cross talks’ between the tethered domains of multidomain TFs while they search DNA (14). We found that, even when the domains of a TF are tethered to each other via a flexible linker and there is no direct interface between them, the tethering itself may strongly influence the characteristics of the search mechanism adopted by the TF (15). The tethering may increase the propensity of a domain to interact with DNA and therefore cause it to slide for longer periods. This tether-induced enhancement of sliding is accompanied by a slower linear diffusion compared with that of an isolated domain.
Apart from sliding, hopping and three-dimensional (3D) diffusion, tethering also plays important role in intersegment transfer (16–18), which is an efficient technique for multidomain proteins to scan the nonspecific DNA sequences at a faster rate. It has been identified that during such motion, multidomain proteins can jump between distant DNA regions via a bridged intermediate in which the two tethered domains interact with distant regions of the DNA that might be separated by a long loop. We described this motion as the monkey-bar mechanism, because of the fact that the protein is composed of two domains, which allows it to jump between different DNA regions as a child swings along monkey-bars at a playground. The existence of intersegment transfer has also been confirmed by a number of in vitro experiments (19–23) and found as a likely mode for fast translocation in multidomain proteins on DNA but is only a sparsely populated (<1%) mode of translocation in folded single domain proteins, such as HoxD9 (24).
An essential molecular property for efficient monkey-bar dynamics is that the domains of the multidomain protein possess different DNA binding affinities. Asymmetry in the affinity to nonspecific DNA sequences allows the multidomain protein both to explore different sections of the DNA and to bind DNA with a reasonable affinity while searching for the target site. The linker mediates between domains possessing different properties (e.g. a propensity for jumping versus binding) and thereby allows cross-talk between them. The existence of this asymmetry in binding affinity was illustrated for about 10 DBD proteins (14) and its effect on DNA search was shown in simulations and experiments for Oct-1 (14), Pax6 (14), and zinc-finger proteins (25).
Our previous study (26) showed that, although single domain proteins are intrinsically unable to adopt the monkey-bar mechanism to search DNA, they can approximate it if they possess a tail containing a stretch of disordered positively charged residues. A charged disordered tail acts as a protein subdomain that spontaneously searches DNA and, when it finds the cognate site, adsorbs quickly on the surface of the DNA molecule. Thus, while the tail keeps the protein associated with DNA molecules, the primary domain can quickly dissociate from its site of attachment and re-associate at a distant DNA site, thereby skipping many intervening DNA sites to accelerate the search dynamics. Indeed, the presence of asymmetricity between the bind affinities of a protein and its tail was observed for tailed DNA-binding proteins. The length of the disordered tail, its net charge and the pattern of charge distribution along the tail have enormous impacts on the formation of the bridged complex and on the overall dynamics of the associated protein (15,27).
Despite advances in our understanding of the molecular underpinnings of efficient DNA search by multidomain DNA-binding proteins, the relationship between the molecular properties of DNA and the adoption of the monkey-bar mechanism has been explored very little. Thus, the effect of DNA geometry on the dynamics of a multidomain protein is unknown. We showed that the inter-distance between the two DNA regions affects drastically the probability to jump using the monkey-bar mechanism (26). Similarly, the inter-angle between the two linear DNA molecules affects the jumping location and the overall chance to form bridged-intermediate (28). In addition to the relative orientation between the two DNA molecules (distance and rotation) the local conformation of each DNA molecule may affect the monkey-bar mechanism as well.
In this study, we performed coarse-grained molecular dynamics simulations of a multidomain protein, the paired box protein Pax6, on two different DNA geometries, namely linear and circular DNA. The choice of circular DNA is inspired by the fact that it mimics bent or curved DNA surface and abundant examples of such DNA geometry (e.g. mitochondrial DNA) are found in the genome. Bends and kinks in DNA are often due to an intrinsic property of the DNA sequences but they can also be induced by some proteins (29–31). For example, DNA associated with Hbb protein from Borrelia burgdorferi shows bending of more than 160° (32), whereas, a DNA bound transcription factor A, mitochondrial (TFAM), features a U-turn on the DNA conformation at the point of association (33,34). In this study, we ask whether the DNA-binding multidomain proteins search DNA differently depending on the conformation and curvature of the DNA. Our motivation to understand if the scanning through curved regions is linked to the efficiency of the search will be explored by comparing the dynamics of multidomain proteins on circular and linear DNA. We also identify the role of tethering domains in the translocation of multidomain proteins compared with single domain proteins.
MATERIALS AND METHODS
Simulation protocol
The molecular nature of the protein search dynamics on double-stranded DNA was studied using a coarse-grained model that was thoroughly described in our previous studies (35–40). Here, we briefly describe the salient features of the model and the simulation method. The protein is modeled by a single bead per residue, centered at the Cα position, where Asp and Glu are each assigned a unit negative charge and Arg and Lys are each assigned a unit positive charge. The DNA molecule is represented by three beads per nucleotide (representing the phosphate, sugar and base) that are positioned at the geometric center of the respective groups. All phosphate atoms carry a unit negative charge.
The protein is allowed to interact with the DNA molecule via nonspecific excluded volume and electrostatic interactions. The latter are modeled by the Debye–Hückel potential. This model, which is valid only for dilute solutions, accounts for the ionic strength of a solute immersed in an aqueous solution (26) of dielectric constant 80 at a temperature at which the protein is completely folded, but does not take into account ion condensation on the DNA. Apart from the nonspecific interactions, the conformational energy of the protein molecule is estimated from its native topology, (41,42)which corresponds to a perfectly funneled energy landscape, where all the nonnative interactions are repulsive and native contacts are favored according to the Lennard–Jones potential. The DNA molecule, on the other hand, is kept static during the simulation in order to isolate the effects of its geometry on protein dynamics from the possible contributions originating from fluctuations in its conformation.
The diffusion of a protein on DNA was captured using Langevin dynamics under a wide range of salt conditions (0.02–0.20 M), where the DNA molecule was placed at the center of a simulation box with periodic boundary conditions and the protein was initially placed 50 Å away on the X-axis from the DNA. The dimensions of the box were such that the DNA was 150 Å away from each side of the box. We note that because the DNA is modeled as a rigid molecule in our simulations, the salt is not needed to stabilize the DNA (by screening the charges on the phosphate groups) but to correctly represent the strength of the protein–DNA interactions. Due to the coarse-grained nature of the model, it is difficult to directly compare the modeled salt concentrations with the physiological ones. The distances between the charged beads of the protein and of the DNA are longer compared to the distances in the fully atomistic models. The longer distances lead to typically weaker electrostatic interactions and correspondingly the common physiological salt concentration that allow strong protein–DNA interactions (0.1–0.15 M) is shifted to lower values (∼0.01–0.05 M). Nevertheless, the coarse-grained model has successfully captured salient features of protein search modes on DNA (26,27,35,51) at salt conditions ranging between 0.01 to 0.2 M.
Structural annotation of the studied multidomain transcription factor
Pax6 is a multidomain TF containing a conserved 128 amino acid DNA binding paired domain that plays critical roles in mammalian development and oncogenesis. Overall, Pax6 consists of an amino-terminal domain (the N-domain; residues 1–60), a linker region (residues 61–76) and a carboxy-terminal domain (the C-domain; residues 77–133). As there are no direct protein–protein contacts between the two globular domains, they interact with each other solely through the flexible 15-residue linker (43). Here, we study the dynamics of the isolated N-domain, which we refer to as Pax6N. The crystal structure of a complex containing the Drosophila paired domain with its DNA sequence shows docking of the N-domain (14) with the DNA surface, while the C-domain does not make any DNA contacts. However, other genetic and biochemical studies suggest that the C-domain of Pax6 may also make DNA contacts, but with a slightly lower DNA affinity (14), and that both the domains are required for efficient binding to promoters (10).
Designing DNA conformations with circular and linear geometry
To generate double-stranded DNA structures of different geometries, we used two different tools: the W3DNA (44) web server was used to generate linear ideal B-DNA conformations, whereas circular DNA conformations were designed using Nucleic Acid Builder (NAB) (45) software. Only the sequence of a single DNA strand is required to design a perfect B-DNA geometry in W3DNA, whereas crafting a circular DNA geometry using NAB requires two inputs: (1) the desired number of base pairs; and (2) the number of helical twists. The first parameter needs to be a multiple of 10 so that a uniform rise of 3.38 Å (similar to that found in ideal B-DNA) is maintained. The second parameter, the number of helical twists, is a function of the amount of supercoiling present in a DNA conformation, and can be specified by the change in linking number, Δlk. In the present study, we generated 100, 300 and 500 bp circular DNA molecules with a linking number of zero. This means that destabilizing forces due to supercoiling are absent from all our circular DNA conformations. Throughout this study we use the notations S100, C500, C300 and C100 for 100 bp linear B-DNA, and for 500 bp, 300 bp and 100 bp circular DNA geometries, respectively. The corresponding curvatures, defined as the inverse of the radii of these DNA geometries, are estimated at 0, 0.003, 0.006 and 0.018 Å−1, respectively.
Sliding, hopping, 3D-diffusion
Search modes were classified as sliding, hopping or 3D diffusion on the basis of their structural conformation using a method similar to that described by Givaty and Levy (35). To identify the different modes of interactions of Pax6 and Pax6N, we focused only on the position of the recognition helix located in the N-domain of the respective protein. This enables a direct comparison to be undertaken between the results for Pax6 and Pax6N, and underscores the effects of tethering two protein domains via a flexible linker. A conformation that refers to the 3D diffusion mode is characterized by a distance of at least 30 Å between the center of the recognition helix and the center of the closest DNA base pair. At this distance, average electrostatic energy drops to about 2% of the energy in the sliding conformation at a low salt concentration, signifying an unbound state for the protein–DNA complex. Sliding conformations were identified if at least 70% of the N-domain recognition helix was in contact with the major DNA groove, the distance of the center of mass of the recognition region from the center of the closest DNA base pair was up to 20 Å, and the orientation angle was <90°. Any snapshot that did not meet any of these criteria was defined as a hopping conformation.
RESULTS AND DISCUSSION
Effects of DNA geometry on the search dynamics of isolated and multidomain transcription factors
Our previous study revealed that the search efficiency of a folded single domain protein is largely dependent on the geometry of the nonspecific DNA molecule it searches (40). In particular, the width of the DNA major groove plays a significant role in determining the electrostatic potential of the DNA, and thereby the usage of sliding and hopping during DNA search dynamics. In this study, we go further and ask how the geometry of the DNA molecule affects the search mechanism adopted by multidomain DNA-binding proteins.
To address this question, we simulated Pax6, a two-DBD protein, searching a 100 bp circular DNA molecule at salt concentrations ranging from 0.02–0.20 M. The interaction between the protein and the DNA was modeled by the Debye–Hückel potential. In addition to considering the tethered DBD, we also performed similar coarse-grained simulations for the isolated N-domain of Pax6 (Pax6N). As control simulations, we studied and compared the dynamics of both tethered and isolated Pax6N on 100 bp linear DNA.
In Figure 1A and B, we present the proportions of sliding and hopping, respectively, performed by the N-domain of tethered Pax6 and by its isolated N-domain (Pax6N). The utilization of both search modes gradually decreases with increasing salt concentration for both the tethered and isolated domains because the corresponding electrostatic attraction between the protein and DNA weakens and the protein tends to dissociate more from the DNA surface. Furthermore, Figure 1A suggests that, irrespective of DNA geometry, the variation in sliding proportion with salt concentration for Pax6 and Pax6N is roughly similar, except at very low salt concentrations (0.02–0.06 M), where the proportion of sliding adopted by Pax6N is higher compared with that of Pax6. This is because of the strong electrostatic interaction between the DNA and the tethered N-domain of Pax6 which hinders the free simultaneous diffusion of both its constituent domains along the DNA contour. As a result, Pax6 lacks the ease with which single-domain Pax6N performs a smooth rotation-coupled sliding motion along the DNA major grooves.
Figure 1.
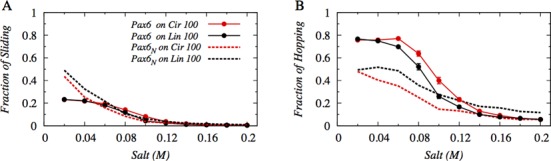
Effect of DNA geometry on the interplay between the DNA search mechanisms adopted by Pax6 (solid lines), which consists of two tethered DNA binding protein domains (N and C), and its isolated N-domain, Pax6N (dotted lines), at different salt concentrations. (A) Sliding and (B) hopping dynamics are shown on a circular 100 bp DNA molecule (red) and on a linear 100 bp B-DNA molecule (black). Sliding and hopping by the tethered Pax6 are analyzed by focusing solely on its N-domain.
A more pronounced effect of tethering can be seen in Figure 1B, where the tethered N-domain of Pax6 shows a significantly higher proportion of hopping events compared with isolated Pax6N over a wide range (0–0.10 M) of salt concentrations. This is related to the increased DNA affinity of Pax6 compared with Pax6N either to linear or circular DNA. It is interesting to note that the proportion of hopping events in the search by tethered N-domain of Pax6 is higher on circular DNA, while Pax6N prefers to hop on linear DNA. This switch in preferences with respect to the hopping search mechanism between different DNA geometries might indicate that proteins may adopt distinctly different search mechanisms on circular and linear DNA when their constituent domains are tethered via a flexible linker.
Circular DNA promotes ‘short sliding’ events in tethered proteins
To probe how the search dynamics on circular and linear DNA differ in the presence or absence of domain tethering via a flexible linker, we investigated the structural details of sliding dynamics performed by Pax6 and Pax6N on circular and linear DNA conformations. In Figure 2, we present the number of sliding events as a function of the number of base pairs traversed along the DNA contour during each event.
Figure 2.
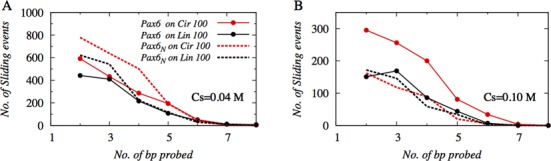
The role of DNA geometry in the distribution of the length of the sliding events at salt concentrations of (A) 0.04 M and (B) 0.10 M on 100 bp linear (black) and circular (red) DNA. The length of each sliding event was measured in terms of the total number of DNA base pairs probed via an uninterrupted sliding motion by the N-domain of Pax6 either when it was tethered to the C-domain (Pax6; solid lines) or as an isolated domain (Pax6N; dotted lines). ‘Uninterrupted’ means sliding that occurred without intervening hopping or 3D diffusion events. Where the protein revisited a previously probed DNA site, it was counted only once.
Our results suggest that at a low salt concentration (0.04 M, Figure 2A), Pax6N performs maximal numbers of short sliding events that typically cover 2–4 DNA base pairs (∼8–16 Å). In comparison, such sliding events are slightly less likely when the protein domains are tethered, as in Pax6, on either linear or circular DNA. This observation is in line with Figure 1A, which shows a lower proportion of sliding performed by Pax6 compared with Pax6N at low salt concentrations. A distinctly different effect, however, emerges at a higher salt concentration (0.10 M, Figure 2B); under such conditions, Pax6 performs almost twice the number of short sliding events on circular DNA compared with linear DNA and almost double the number of sliding events as Pax6N. The enhancement in the numbers of short sliding events for tethered N-domain of Pax6 on circular DNA is linked to the unique geometry of the circular DNA molecule. Our previous study suggested that, due to narrower major grooves inside the circular DNA and the close proximity of negatively charged phosphate atoms, the electrostatic field is stronger inside the DNA circle compared with the outer surface of the molecule (40). Such variation of electrostatic potential in linear DNA is unlikely and, indeed, we found a uniform electrostatic potential (40) along the contour of linear B-DNA. The stronger electrostatic field in the circular DNA aids in promoting more sliding events by Pax6, which has a higher affinity toward DNA than its isolated counterpart, even under high salt conditions. However, the non-uniform major groove width of circular DNA also hinders smooth sliding over many base pairs, which leads to many but short and disrupted sliding events.
To further understand the details of the search mechanism performed by the tethered protein on linear and circular DNA, it is instructive to study its dynamics as a function of DNA curvature, which is 0 Å−1 and 0.018 Å−1 for 100 bp linear and circular B-DBA, respectively. We considered two additional DNA structures; 300 and 500 bp circular DNA that have intermediate curvatures of 0.006 Å−1 and 0.003 Å−1, respectively. To understand the impact of the DNA curvature on the dynamics of tethered N-domain Pax6, we performed coarse-grained molecular dynamics simulations of Pax6 with these four different circular DNA structures and focused only on the conformations where both the N- and C- domains of Pax6 are engaged in sliding dynamics at the DNA major grooves. In Figure 3A–B, we present the profiles of such sliding conformations with respect to the number of DNA base pairs separating the N- and C-domains of Pax6 at 0.04 M and 0.10 M salt concentrations.
Figure 3.
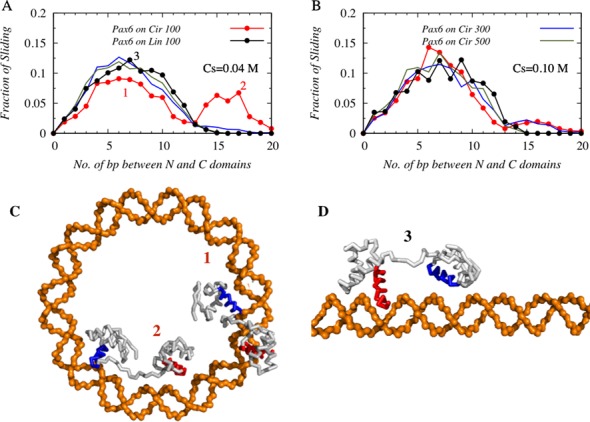
The effects of DNA curvature on the sliding dynamics of Pax6 at a salt concentration of (A) 0.04 M and (B) 0.10 M. Solid red and black lines correspond to the sliding dynamics of the tethered N- and C-domains of Pax6 on 100 bp circular and linear DNA respectively. In addition, the sliding dynamics of Pax6 on 300 bp (blue line) and 500 bp (green line) circular DNA is investigated. The most probable number of DNA base pairs between the N- and C-domains of tethered Pax6 when both domains are engaged in sliding dynamics is estimated from the maxima of the corresponding distributions of the sliding conformations. Snapshots that correspond to the two very different peaks shown for 100 bp circular DNA are presented in (C), while an example of a Pax6 conformation corresponding to the single-peaked distributions on linear DNA geometry is shown in (D).
For DNA structures with the lowest and highest curvatures (i.e. for 100 bp linear and circular DNA), we find single- and double-peaked probability profiles, respectively, at both salt conditions. The first hump of the double-peaked profile and the maximum of the single-peak profile roughly coincide and appear when the distance between the N- and C-domains of Pax6 is ∼7 DNA base pairs (∼24 Å). In contrast, the second peak of the double-peaked probability profile of 100 bp circular DNA appears when the distance between the N- and C-domain is ∼16–17 DNA base pairs (∼54–58 Å). This implies that a common sliding mechanism exists for both 100 bp circular and linear DNA, in which the two recognition helices of the N- and C-domains of Pax6 are typically placed in consecutive major grooves of a DNA structure, as shown in Figure 3C and D (labeled as ‘1’ and ‘3’, respectively). In addition, an alternative, unique mode of sliding dynamics exists for highly curved 100 bp circular DNA, where the two domains of Pax6 simultaneously probe two DNA sites that are far (∼16–17 base pairs) apart from each other and inaccessible otherwise on linear DNA in a single sliding snapshot. An example of such dynamics is shown in the snapshot presented in Figure 3C, labeled ‘2’. These two sliding modes are mechanistically different from each other: the former common sliding technique helps the tethered DBDs to slide along the DNA major grooves and read the DNA base pairs in both linear and circular DNA. Such a sliding motion is strongly coupled with rotation along the DNA major grooves (35,46,47). In the alternative mode of sliding, conformations of tethered DBDs closely resemble the ‘bridged-complex’ formed in the intersegment transfer mechanism, with the difference that, instead of two different DNA segments, the two domains of Pax6 capitalize on the high curvature of 100 bp circular DNA and associate with two sites on the same DNA molecule that are sequentially far apart (separated by ∼16–17 bp). Presumably, the advantage of a curved DNA surface can be attained only when both the tethered domains of Pax6 are inside the circular DNA and consequently this sliding dynamics is decoupled from rotation along the DNA major grooves. With a decrease in DNA curvature from that of the 100 bp circular DNA, the ‘alternative’ sliding mode is only sparsely populated in 300 bp DNA (shown by dotted lines in Figure 3A and B), while on the 500 bp circular DNA, the searching protein behaves in a similar manner as on a 100 bp linear DNA molecule and shows a single-peaked probability distribution. The relative height of the two peaks, however, varies with salt concentration and is lower at the higher salt concentration (0.10 M) because of the weakening of the long range electrostatic attractions.
Circular DNA facilitates ‘jumping’ by tethered proteins
Having shown that the tethered DBDs can adopt two distinct sliding mechanisms on highly curved 100 bp circular DNA, it is interesting to understand how Pax6 switches between these two sliding modes and what other modes of translocation tethered proteins may use while they diffuse along the contour of a highly curved DNA molecule. The intricate details of such complex dynamics of multidomain proteins can only be realized by careful study of the various modes of their nonspecific association with DNA and their relative population depending of the DNA curvature.
Specifically, we examine how the tethered domains move in between two consecutive sliding events on a curved DNA surface? Our analysis suggests that a jumping mechanism operates between two consecutive sliding events, with the result that the N-domain of Pax6 hops many times along the DNA contour, whereas, the C-domain of Pax6 (which has lower DNA affinity than the N-domain), may perform either sliding or hopping dynamics. Any snapshot in which the N-domain of Pax6 performs 3D diffusion and dissociates from the DNA in between two consecutive sliding events is strictly discarded, as DNA geometry will not then have any influence on the positioning of the protein, which is located far from the DNA (see ‘Materials and Methods’ section for the definition of diffusion).
In Figure 4, we estimated the cumulative numbers of jumps performed by Pax6 and Pax6N on 100 bp circular and linear DNA and by Pax6 on 300 bp and 500 bp circular DNA, also. The results on 100 bp linear DNA at both the 0.04 M and 0.10 M salt conditions show fewer jumps by Pax6N (black dotted line in Figure 4.) than by tethered protein Pax6. Also, during a single jump, Pax6N traverses a maximum of ∼3–4 bp (shown by the plateau) on the linear DNA. In comparison, the tethered protein, Pax6, performs longer jumps on linear DNA via its N-domain, which can associate with a new DNA site located even ∼10 DNA base pairs away from the original site of association. The difference in jumping length between isolated and tethered proteins stems from the presence of a tethered C-domain in Pax6, which acts as an additional arm to hook onto the DNA molecule and thereby prevents the N-domain from dissociating, even when it is weakly interacting with the DNA molecule. Unlike the single-domain protein Pax6N, which quickly dissociates once it is slightly off the DNA surface, the N-domain in Pax6 remains close to the DNA because of the interaction between the tethered C-domain and the DNA molecule, and it performs many hopping events that result in jumps that may cover ∼10 DNA base pairs. Another important factor in the ‘jump’ mechanism is the geometry of the DNA structure. Our results suggest that, at a high salt concentration (0.10 M), Pax6 performs almost twice the number of jumps on 100 bp circular DNA (red line with filled circles in Figure 4B) compared with 100 bp linear DNA (black line with filled circles in Figure 4B). The number of jumps on 300 and 500 bp circular DNA descend in decreasing order from the number on 100 bp circular DNA, indicating the role of DNA curvature in facilitating jumping dynamics. The higher curvature, which is coupled with a stronger electrostatic potential, helps the N-domain of Pax6 to jump more frequently between DNA sites that are more distant than it can achieve on linear DNA, so enabling it to traverse 10 or more DNA base pairs in a single jump.
Figure 4.
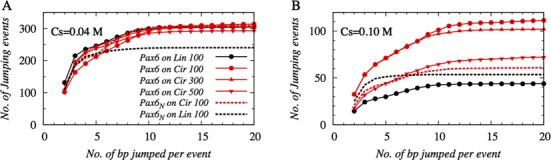
The cumulative distributions of the length of jumping events performed by Pax6 are presented as a function of jumping length, i.e. the number of base pairs jumped per event at salt concentrations of (A) 0.04 M and (B) 0.10 M. The effect of DNA curvature is highlighted by presenting the distributions of jumping lengths on 300 bp (up triangle) and 500 bp (down triangle) circular DNA in addition to the same on 100 bp circular (red circle) and linear (black circle) DNA. The distributions of the length of jumping events performed by isolated Pax6N are also shown in dotted black and dotted red lines, corresponding to 100 bp linear and circular DNA, respectively.
To investigate how the long jumps affect the overall DNA search mechanism of tethered DBDs on circular/curved DNA, we compared the dynamics of Pax6 when interacting with a 100 bp circular or linear DNA. Figure 5A and B show how Pax6 diffuses with time along the DNA contour (blue and red circles corresponds to linear and circular DNA geometries, respectively) and which DNA site are probed using sliding dynamics at salt concentrations of 0.04 and 0.10 M, respectively. When any two sliding events are linked via a jump, this is denoted by a solid black line connecting two circles. The length of the line corresponds to the length of the jump. At a low salt concentration (0.04 M), Pax6 partly scans the 100 bp linear DNA (blue circles are spread over a region that corresponds to DNA base pair indices ∼50 out of 100, Figure 5A). This is also illustrated in Figure 5E, which shows that only about half of the linear DNA is scanned by the rotation-coupled-sliding dynamics (shown as a blue surface on the linear DNA) along the DNA major grooves. The DNA search pattern changes remarkably, however, on 100 bp circular DNA, indicating an efficient translocation of Pax6 that covers roughly the whole contour (Figure 5A and C) of the circular DNA even at a salt concentration of 0.04 M, when 3D diffusion is least probable. This enhanced search ability is achieved by many short sliding events followed by large numbers of long jumps (as discussed in Figures 2 and 4) that allow tethered protein Pax6 to explore a longer DNA contour. At the higher salt concentration of 0.10 M, although the search efficiency of Pax6 on linear DNA increases slightly because of increased proportions of 3D diffusion and hopping, the numbers of sliding and jumping events are still lower than on 100 bp circular DNA.
Figure 5.
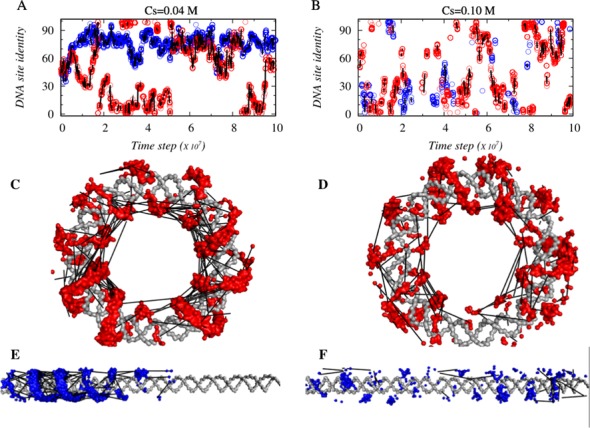
The role of DNA geometry in the sliding and jumping dynamics of Pax6 on 100 bp circular (red circles) and linear (blue circles) DNA at salt concentrations of (A) 0.04 M and (B) 0.10 M. Panels (C–F) demonstrate how the dynamics of Pax6 covers various parts of the circular and linear DNA contours with the aid of connecting jumping events. The adoption of a spiral motion (shown by red and blue surfaces on the gray DNA conformations) along the DNA major groove suggests that Pax6 exhibits helical sliding motions by which it performs one-dimensional diffusion along the DNA contour. In all six panels, every two consecutive sliding events in between which Pax6 jumps are connected via black solid lines, with the length of the line reflecting the length (in terms of the number of DNA base pairs) of the respective jumps.
Correlation between number of long jumps and DNA search efficiency
The ability of the tethered protein to perform more long jumps on 100 bp circular DNA compared with linear DNA is seemingly advantageous to enabling it to probe more DNA base pairs at a faster rate. To verify this, we estimated the number of DNA positions probed, defined as the total number of unique DNA sites visited via a sliding motion, by the N-domain of Pax6 and by Pax6N. The results are presented in Figure 6A as a function of salt concentration for both 100 bp circular and linear DNA.
Figure 6.
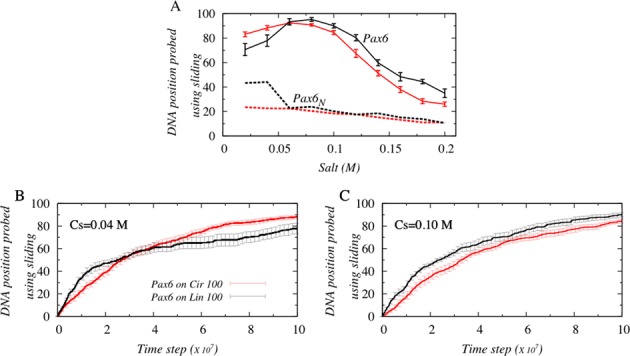
Effect of DNA geometry on the efficiency of DNA search. (A) The number of DNA positions probed using sliding by the DNA binding protein domains Pax6 (solid line) and Pax6N (dotted line) is presented as a function of salt concentration. Red and black colors denote the 100 bp circular and linear B-DNA geometries, respectively. The number of DNA positions probed by Pax6 on 100 bp linear and circular DNA is also studied as a function of time at salt concentrations of (B) at 0.04 M and (C) at 0.10 M.
Figure 6A suggests that DNA scanning efficiency is considerably greater (by up to ∼4–5 fold) when performed by the tethered N-domain Pax6 compared with Pax6N because of the enhanced ability of Pax6 to hop, which allows it to diffuse effectively along the whole contour of circular or linear DNA and the ability to perform long jumps. To investigate whether increased scanning efficiency arising from the more frequent performance of long jumps also leads to faster search kinetics, the number of DNA positions probed was plotted as a function of time (Figure 6B and C). We find that, irrespective of salt conditions, Pax6 reads the first ∼40 DNA sites faster (i.e. fewer molecular dynamics steps are required) on 100 bp linear rather than circular DNA, but thereafter, the efficiency is reversed in the case of low salt concentrations (<0.06 M), and Pax6 probes DNA sites faster on circular DNA. However, the search kinetics of Pax6 remains faster than that of Pax6N on linear DNA under conditions of high salt concentration (>0.06 M). This clearly suggests that long jumps alone do not assure increased DNA scanning efficiency; rather a combination of translocation mechanisms, including sliding, is essential to recognize DNA sites precisely (20). In fact, the performance of many long jumps on circular DNA at high salt concentrations is disadvantageous, partly because Pax6 spends more time in this mode of dynamics and consequently less time utilizing a sliding motion. Also, the protein tends to skip DNA sites lying between the take-off and landing point of a jump (which may include the DNA ‘cognate’ sites as well) in the absence of adequate sliding (i.e. if the proportion of sliding is less than ∼10% of all modes of dynamics, Figure 1A) by the tethered DBDs.
CONCLUSIONS
How TFs rapidly recognize their target sites on DNA is a complex phenomenon to understand. Previous studies have indicated that the monkey-bar mechanism is a key mechanism for boosting the search kinetics of multidomain proteins exposed to high concentrations of DNA molecules. At low DNA concentrations, where only few or no DNA segments are nearby, this mode of search dynamics seems less likely, raising the question of how multidomain TFs perform search dynamics under such conditions. Furthermore, recent studies have established a strong correlation between DNA geometry and the search dynamics adopted by single domain proteins interacting with it (20,26,28,48,49). It is, therefore, interesting to know the nature of the effect of DNA geometry and in particular DNA curvature on the dynamics of multidomain proteins and to understand the possible mechanisms via which a multidomain TF explores the contour of a DNA molecule.
In this study, we investigated these questions by performing coarse-grained molecular dynamics simulations of a multidomain TF Pax6 with DNA geometries that have various curvatures. In our model, the protein is allowed to interact with the DNA molecule via nonspecific electrostatic interactions. We show that, from the mechanistic point of view, the dynamics of Pax6 on 100 bp circular DNA differs from that on linear B-DNA of same length. For example, our results suggest that, in addition to the commonly known rotation-coupled sliding mechanism, another mode of sliding dynamics may exist. This unique mode of sliding originates from the DNA curvature bringing two sequentially distant DNA sites into close proximity with each other, such that the tethered protein Pax6 can associate with two distant DNA sites simultaneously, which is apparently not feasible on linear DNA. The corresponding protein conformations are analogous to the bridged complex formed in the monkey-bar dynamics (15,26). The main difference between the two is that, instead of the multiple DNA segments involved in intersegment transfer, here, multidomain proteins use the DNA curvature to associate with two distant sites on the same DNA molecule. The greater the DNA curvature, the more distant the sites that can be brought spatially close to each other and take part in forming the bridged complex. We also found that such a sliding dynamics is decoupled from the rotation of the protein along the DNA major grooves. While previous nuclear magnetic resonance studies have confirmed the existence of rotation-coupled-sliding dynamics by proteins on DNA (50), we note that using an in silico dynamic model of protein and DNA, Terakawa et. al. has recently reported that the two DBDs of p53 perform rotation-decoupled sliding motion, during which it also causes significant bending in the DNA conformation at the point of p53 association (51,52). This is in line with another experimental study where, using the scanning force microscopy, authors proposed that sharp DNA bending (as much as 60–80°) facilitates the target site location in Cro protein (53). However, what mechanical advantages a curved DNA surface may provide to the interacting multidomain proteins and what is the correlation between rotation-decoupled sliding and the deformation in DNA conformations (bending/curvature) were unknown. Our study based on molecular simulations of a multidomain protein interacting with circular DNA (having curved DNA surface), where the intrinsic flexibility of DNA molecule was ignored to isolate the impacts of DNA curvature on dynamics of tethered proteins is proved to be a potential approach to address these issues. We emphasize that the rotation-decoupled sliding dynamics is feasible only if the protein domains are tethered via a flexible linker and the interacting DNA surface is highly curved, such that the constituent domains of the protein move alternately along the DNA contour through a ‘monkey-bar’-like mechanism (26).
The tethering of protein domains is also advantageous in promoting longer jumps compared with those performed by the single domain protein. When one of two tethered protein domains interacts with DNA, it ensures that the whole protein will remain close to the DNA surface while the second domain may jump to distant DNA sequences. We found that, when Pax6 slides along DNA, the N-domain of the tethered protein performed significantly longer hopping events and jumps to many DNA sites than the isolated N-domain of Pax6N performed. Although, these long jumps boost the speed of the search dynamics and help in global translocation of the multidomain TFs along the DNA contour, they decrease the proportion of the search conducted using the sliding mechanism, which translates into a diminished ability to probe the maximum number of DNA base pairs. This might not be a concern if the cognate sites are not located in highly curved DNA regions. Nevertheless, for a more rugged DNA due to sequence variations, the balance between the search modes of the multidomain TF on a curved DNA can be changed. Our results therefore suggest that, by modulating DNA curvatures, the population of different translocation modes can be altered significantly and only an optimal DNA curvature can provide the necessary fine balance between sliding and jumping dynamics to ensure the most efficient scan of the DNA contour.
Acknowledgments
We are grateful to Richard Lavery for insightful discussions.Y.L. is the Morton and Gladys Pickman professional chair in Structural Biology.
Footnotes
Present address: Arnab Bhattacherjee, Indraprastha Institute of Information Technology (IIIT), New Delhi 110020, India.
FUNDING
Kimmelman Center for Macromolecular Assemblies; United States–Israel Binational Science Foundation [2010424]. Funding for open access charge: Binational Israel–USA [2010424].
Conflict of interest statement. None declared.
REFERENCES
- 1.Han J.-H., Batey S., Nickson A.A., Teichmann S.A., Clarke J. The folding and evolution of multidomain proteins. Nat. Rev. Mol. Cell Bio. 2007;8:319–330. doi: 10.1038/nrm2144. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka T., Kuroda Y., Yokoyama S. Characteristics and prediction of domain linker sequences in multi-domain proteins. J. Struct. Funct. Genomics. 2003;4:79–85. doi: 10.1023/a:1026163008203. [DOI] [PubMed] [Google Scholar]
- 3.Munro A.W., Lindsay J.G., Coggins J.R., Kelly S.M., Price N.C. Analysis of the structural stability of the multidomain enzyme flavocytochrome P-450 BM3. Biochim. Biophys. Acta. 1996;1296:127–137. doi: 10.1016/0167-4838(96)00061-1. [DOI] [PubMed] [Google Scholar]
- 4.Pang A., Arinaminpathy Y., Sansom M.S.P., Biggin P.C. Interdomain dynamics and ligand binding: molecular dynamics simulations of glutamine binding protein. FEBS Lett. 2003;550:168–174. doi: 10.1016/s0014-5793(03)00866-4. [DOI] [PubMed] [Google Scholar]
- 5.Teichmann S.A., Park J., Chothia C. Structural assignments to the Mycoplasma genitalium proteins show extensive gene duplications and domain rearrangements. Proc. Natl. Acad. Sci. U.S.A. 1998;95:14658–14663. doi: 10.1073/pnas.95.25.14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Apic G., Gough J., Teichmann S.A. Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J. Mol. Biol. 2001;310:311–325. doi: 10.1006/jmbi.2001.4776. [DOI] [PubMed] [Google Scholar]
- 7.He H.W., Feng S., Pang M., Zhou H.M., Yan Y.B. Role of the linker between the N- and C-terminal domains in the stability and folding of rabbit muscle creatine kinase. Int. J. Biochem. Cell Biol. 2007;39:1816–1827. doi: 10.1016/j.biocel.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 8.He H.-W., Zhang J., Zhou H.-M., Yan Y.-B. Conformational change in the C-terminal domain is responsible for the initiation of creatine kinase thermal aggregation. Biophys. J. 2005;89:2650–2658. doi: 10.1529/biophysj.105.066142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlicker A., Huthmacher C., Ramírez F., Lengauer T., Albrecht M. Functional evaluation of domain-domain interactions and human protein interaction networks. Bioinformatics. 2007;23:859–865. doi: 10.1093/bioinformatics/btm012. [DOI] [PubMed] [Google Scholar]
- 10.Epstein J.A., Glaser T., Cai J., Jepeal L., Walton D.S., Maas R.L. Two independent and interactive DNA-binding subdomains of the Pax6 paired domain are regulated by alternative splicing. Genes Dev. 1994;8:2022–2034. doi: 10.1101/gad.8.17.2022. [DOI] [PubMed] [Google Scholar]
- 11.Aurora R., Herr W. Segments of the POU domain influence one another's DNA-binding specificity. Mol. Cell. Biol. 1992;12:455–467. doi: 10.1128/mcb.12.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verrijzer C.P., Alkema M.J., van Weperen W.W., Van Leeuwen H.C., Strating M.J., van der Vliet P.C. The DNA binding specificity of the bipartite POU domain and its subdomains. EMBO J. 1992;11:4993–5003. doi: 10.1002/j.1460-2075.1992.tb05606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anand P., Schug A., Wenzel W. Structure based design of protein linkers for zinc finger nuclease. FEBS Lett. 2013;587:3231–3235. doi: 10.1016/j.febslet.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Vuzman D., Polonsky M., Levy Y. Facilitated DNA search by multidomain transcription factors: cross talk via a flexible linker. Biophys. J. 2010;99:1202–1211. doi: 10.1016/j.bpj.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vuzman D., Levy Y. Intrinsically disordered regions as affinity tuners in protein-DNA interactions. Mol. Biosyst. 2012;8:47–57. doi: 10.1039/c1mb05273j. [DOI] [PubMed] [Google Scholar]
- 16.Hu T., Shklovskii B.I. How a protein searches for its specific site on DNA: the role of intersegment transfer. Phy. Rev. E. Stat. Nonlin. Soft Matter Phys. 2007;76:051909. doi: 10.1103/PhysRevE.76.051909. [DOI] [PubMed] [Google Scholar]
- 17.Sheinman M., Kafri Y. The effects of intersegmental transfers on target location by proteins. Phys. Biol. 2009;6:016003. doi: 10.1088/1478-3975/6/1/016003. [DOI] [PubMed] [Google Scholar]
- 18.Loverdo C., Bénichou O., Voituriez R., Biebricher A., Bonnet I., Desbiolles P. Quantifying hopping and jumping in facilitated diffusion of DNA-binding proteins. Phys. Rev. Lett. 2009;102:188101. doi: 10.1103/PhysRevLett.102.188101. [DOI] [PubMed] [Google Scholar]
- 19.Iwahara J., Zweckstetter M., Clore G.M. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc. Natl. Acad. Sci. U.S.A. 2006;103:15062–15067. doi: 10.1073/pnas.0605868103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takayama Y., Clore G.M. Intra- and intermolecular translocation of the bi-domain transcription factor Oct1 characterized by liquid crystal and paramagnetic NMR. Proc. Natl. Acad. Sci. U.S.A. 2011;108:E169–E176. doi: 10.1073/pnas.1100050108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esadze A., Iwahara J. Stopped-flow fluorescence kinetic study of protein sliding and intersegment transfer in the target DNA search process. J. Mol. Biol. 2014;426:230–244. doi: 10.1016/j.jmb.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lieberman B.a., Nordeen S.K. DNA intersegment transfer, how steroid receptors search for a target site. J. Biol. Chem. 1997;272:1061–1068. doi: 10.1074/jbc.272.2.1061. [DOI] [PubMed] [Google Scholar]
- 23.Fried M.G., Crothers D.M. Kinetics and mechanism in the reaction of gene regulatory proteins with DNA. J. Mol. Biol. 1984;172:263–282. doi: 10.1016/s0022-2836(84)80026-1. [DOI] [PubMed] [Google Scholar]
- 24.Iwahara J., Clore G.M. Direct observation of enhanced translocation of a homeodomain between DNA cognate sites by NMR exchange spectroscopy. J. Am. Chem. Soc. 2006;128:404–405. doi: 10.1021/ja056786o. [DOI] [PubMed] [Google Scholar]
- 25.Zandarashvili L., Vuzman D., Esadze A., Takayama Y., Sahu D., Levy Y., Iwahara J. Asymmetrical roles of zinc fingers in dynamic DNA-scanning process by the inducible transcription factor Egr-1. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E1724–E1732. doi: 10.1073/pnas.1121500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vuzman D., Azia A., Levy Y. Searching DNA via a ‘Monkey Bar’ mechanism: the significance of disordered tails. J. Mol. Biol. 2010;396:674–684. doi: 10.1016/j.jmb.2009.11.056. [DOI] [PubMed] [Google Scholar]
- 27.Vuzman D., Levy Y. DNA search efficiency is modulated by charge composition and distribution in the intrinsically disordered tail. Proc. Natl. Acad. Sci. U.S.A. 2010;107:21004–21009. doi: 10.1073/pnas.1011775107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vuzman D., Levy Y. The ‘monkey bar’ mechanism for searching for the DNA target site: the molecular determinants. Isr. J. Chem. 2014;54:1374–1381. [Google Scholar]
- 29.Swinger K.K., Rice P.A. IHF and HU: flexible architects of bent DNA. Curr. Opin. Struct. Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Swinger K.K., Rice P.A. Structure-based analysis of HU-DNA binding. J. Mol. Biol. 2007;365:1005–1016. doi: 10.1016/j.jmb.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sagi D., Friedman N., Vorgias C., Oppenheim A.B., Stavans J. Modulation of DNA conformations through the formation of alternative high-order HU-DNA complexes. J. Mol. Biol. 2004;341:419–428. doi: 10.1016/j.jmb.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 32.Clauvelina N., Olson W.K. The synergy between protein positioning and DNA elasticity: energy minimization of protein-decorated DNA minicircles. 2014 doi: 10.1021/acs.jpcb.0c11612. arXiv: 1405.7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farge G., Laurens N., Broekmans O.D., van den Wildenberg S.M.J.L., Dekker L.C.M., Gaspari M., Gustafsson C.M., Peterman E.J.G., Falkenberg M., Wuite G.J.L. Protein sliding and DNA denaturation are essential for DNA organization by human mitochondrial transcription factor A. Nat. Commun. 2012;3 doi: 10.1038/ncomms2001. doi:10.1038/ncomms2001. [DOI] [PubMed] [Google Scholar]
- 34.Ngo H.B., Lovely G.a., Phillips R., Chan D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014;5 doi: 10.1038/ncomms4077. doi:10.1038/ncomms4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Givaty O., Levy Y. Protein sliding along DNA: dynamics and structural characterization. J. Mol. Biol. 2009;385:1087–1097. doi: 10.1016/j.jmb.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 36.Trizac E., Levy Y., Wolynes P.G. Capillarity theory for the fly-casting mechanism. Proc. Natl. Acad. Sci. U.S.A. 2010;107:2746–2750. doi: 10.1073/pnas.0914727107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khazanov N., Marcovitz A., Levy Y. Asymmetric DNA-Search dynamics by symmetric dimeric proteins. Biochemistry. 2013;52:5335–5344. doi: 10.1021/bi400357m. [DOI] [PubMed] [Google Scholar]
- 38.Marcovitz A., Levy Y. Weak frustration regulates sliding and binding kinetics on rugged protein-DNA landscapes. J. Phys. Chem. B. 2013;117:13005–13014. doi: 10.1021/jp402296d. [DOI] [PubMed] [Google Scholar]
- 39.Marcovitz A., Levy Y. Sliding dynamics along DNA: a molecular perspective. Vol. 10. Cambridge: RSC; 2012. pp. 236–262. [Google Scholar]
- 40.Bhattacherjee A., Yaakov L. Search by proteins for their DNA target site: 1. The effect of DNA conformation on protein sliding. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku932. doi:10.1093/nar/gku932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clementi C., Nymeyer H., Onuchic J.N. Topological and energetic factors: what determines the structural details of the transition state ensemble and ‘en-route’ intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 2000;298:937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 42.Onuchic J.N., Wolynes P.G. Theory of protein folding. Curr. Opin. Struct. Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Xu H.E., Rould M.A., Xu W.Q., Epstein J.A., Maas R.L., Pabo C.O. Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev. 1999;13:1263–1275. doi: 10.1101/gad.13.10.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng G., Lu X.-J., Olson W.K. Web 3DNA–a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res. 2009;37:W240–W246. doi: 10.1093/nar/gkp358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Case T.M.a.D.A. Molecular Modeling of Nucleic Acids. Vol. 24. Washington, DC: American Chemical Society; 1998. Modeling unusual nucleic acid structures; pp. 379–393. [Google Scholar]
- 46.Schurr J.M. One-dimensional diffusion-coefficient of proteins absorbed on DNA - hydrodynamic considerations. Biophys. Chem. 1979;9:413–414. [PubMed] [Google Scholar]
- 47.Marklund E.G., Mahmutovic A., Berg O.G., Hammar P., van der Spoel D., Fange D., Elf J. Transcription-factor binding and sliding on DNA studied using micro- and macroscopic models. Proc. Natl. Acad. Sci. U.S.A. 2013;110:19796–19801. doi: 10.1073/pnas.1307905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010;79:233–269. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rohs R., West S., Sosinnsky A., Liu P., Mann R., Honig B. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–1253. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwahara J., Clore G.M. Detecting transient intermediates in macromolecular binding by paramagnetic NMR. Nature. 2006;440:1227–1230. doi: 10.1038/nature04673. [DOI] [PubMed] [Google Scholar]
- 51.Khazanov N., Levy Y. Sliding of p53 along DNA can be modulated by its oligomeric state and by cross-talks between its constituent domains. J. Mol. Biol. 2011;408:335–355. doi: 10.1016/j.jmb.2011.01.059. [DOI] [PubMed] [Google Scholar]
- 52.Terakawa T., Kenzaki H., Takada S. p53 searches on DNA by rotation-uncoupled sliding at C-terminal tails and restricted hopping of core domains. J. Am. Chem. Soc. 2012;134:14555–14562. doi: 10.1021/ja305369u. [DOI] [PubMed] [Google Scholar]
- 53.Erie D.A., Yang G., Schultz H.C., Bustamante C. DNA bending by Cro protein in specific and nonspecific complexes: implications for protein site recognition and specificity. Science. 1994;266:1562–1566. doi: 10.1126/science.7985026. [DOI] [PubMed] [Google Scholar]