


Abstract
Trypanosoma brucei causes human African trypanosomiasis and regularly switches its major surface antigen, VSG, in the bloodstream of its mammalian host to evade the host immune response. VSGs are expressed exclusively from subtelomeric loci, and we have previously shown that telomere proteins TbTIF2 and TbRAP1 play important roles in VSG switching and VSG silencing regulation, respectively. We now discover that the telomere duplex DNA-binding factor, TbTRF, also plays a critical role in VSG switching regulation, as a transient depletion of TbTRF leads to significantly more VSG switching events. We solved the NMR structure of the DNA-binding Myb domain of TbTRF, which folds into a canonical helix-loop-helix structure that is conserved to the Myb domains of mammalian TRF proteins. The TbTRF Myb domain tolerates well the bulky J base in T. brucei telomere DNA, and the DNA-binding affinity of TbTRF is not affected by the presence of J both in vitro and in vivo. In addition, we find that point mutations in TbTRF Myb that significantly reduced its in vivo telomere DNA-binding affinity also led to significantly increased VSG switching frequencies, indicating that the telomere DNA-binding activity is critical for TbTRF's role in VSG switching regulation.
INTRODUCTION
Trypanosoma brucei (T. brucei) is a protozoan parasite that causes human African trypanosomiasis, which can be fatal if left untreated. Current available drugs treating T. brucei infection have severe side effects, and incidents of drug resistance have been increasingly observed (1), making it imperative to develop new generation anti-trypanosome drugs. Bloodstream form (BF) T. brucei regularly switches its major surface antigen, variant surface glycoprotein (VSG), inside its mammalian host to evade the host immune response (2). This antigenic variation is a key pathogenesis mechanism essential for a long-term T. brucei infection.
T. brucei has more than 2000 VSG genes and pseudogenes in its genome, mostly clustered at subtelomeric regions (3,4). However, VSGs are exclusively expressed from VSG expression sites (ESs) in a monoallelic fashion. BF VSG ESs are polycistronic transcription units (PTUs) transcribed by RNA polymerase I (5) and are located immediately upstream of the telomere (6). In the T. brucei Lister 427 strain used in this study, 15 ESs have been identified, all of which have highly conserved DNA sequences (90% identical) and gene organizations (7). VSG is always the last gene in any ES, located within 2 kb from the telomere repeats, while the ES promoter is 40–60 kb upstream (7,8). At any moment, only one ES is fully active, resulting in a single type of VSG being expressed. This monoallelic VSG expression ensures that the previously active VSG is no longer expressed after a VSG switching event, which is essential for the effectiveness of antigenic variation.
VSG switching has several major pathways (9). In in situ switching, the originally active ES is silenced while an originally silent one is expressed without DNA rearrangements. Other major VSG switching pathways are homologous recombination- (HR-) mediated. In crossover (CO) events, part of the active ES, including the VSG gene, changes place with that of a silent ES without losing any genetic information. In gene conversion (GC) events, a silent VSG is copied into the active ES to replace the active VSG gene, resulting in the loss of the originally active VSG and duplication of the newly active VSG. GC is the preferred mechanism of VSG switching in the Lister 427 strain used in this study (9). It can encompass only the VSG gene and its neighboring regions (termed VSG GC) or the entire ES (termed ES GC). In addition, the entire active ES can be lost, and an accompanying in situ switch can give rise to ES loss + in situ switchers (10–12). Several proteins involved in HR have been shown to play important roles in VSG switching: deletion of TbRAD51, TbRAD51-3 or TbBRCA2 leads to decreased VSG switching frequencies (13–15). In contrast, deletion of TOPO3α and RMI1 leads to more frequent VSG switching (11,16).
The T. brucei telomere has been implicated in the regulation of VSG expression and switching (17). We have previously shown that a telomere protein, TbRAP1, is required for silencing subtelomeric VSGs (18,19). We have recently shown that another telomere protein, TbTIF2, is important for maintaining subtelomere/telomere integrity, and depletion of TbTIF2 led to elevated VSG switching frequencies (12). Therefore, telomere proteins are important for regulation of VSG expression and VSG switching. A study by Hovel-Miner et al. showed that T. brucei cells carrying extremely short telomeres (∼1.5 kb) exhibited an ∼10-fold higher VSG switching frequency when compared to cells with longer telomeres of 15 kb on average (20), indicating that the telomere structure indeed plays an important role in VSG switching regulation.
Our previous study identified a duplex telomere DNA-binding factor, TbTRF, which interacts with both TbRAP1 (18) and TbTIF2 (12). While TbTRF is essential for maintaining the terminal telomere structure (21), it does not affect ES-linked VSG silencing in BF T. brucei cells (18). It is intriguing whether TbTRF plays any important role in VSG switching regulation. TbTRF has a similar domain structure as mammalian duplex telomere DNA-binding factors TRF1 and TRF2, with an N-terminal TRF Homology (TRFH) domain and a C-terminal Myb domain based on sequence analysis (21). In mammalian TRF1 and TRF2, the TRFH domain is required for homodimerization of these factors that serve as an interaction platform to bind other telomeric proteins including TIN2 and Apollo (22). The Myb domain specifically binds to telomeric TTAGGG repeats and serves to package both TRF1/2 and their binding partners onto the telomere to form an integrated protective structure (23–25). While our study has shown that TbTRF has a strong duplex TTAGGG repeat binding activity (21), it remains to be confirmed whether its C-terminal Myb domain mediates this direct interaction. Furthermore, whether the Myb-mediated DNA-binding activity of TbTRF exerts any impact on VSG regulation is completely unknown.
Another interesting aspect regarding TbTRF and VSG regulation involves the base J. In kinetoplastids, a heavily modified base β-D-glucosyl(hydroxymethyl)uracil, J, has been identified to replace a small fraction of thymidines in the genome (26,27). J is only present in the BF T. brucei DNA but not in the DNA of procyclic form (PF) T. brucei cells that normally reside in the mid-gut of their insect vector, tsetse (Glossina spp.) (26). A relatively high level of J is found in T. brucei telomeric DNA. Fourteen percent of the T in the (CCCTAA)n strand and 36% of T in the (TTAGGG)n strand is replaced by J (28). Silent VSG ESs contain J, while the active ES does not (29). In addition to its presence in the telomere, J appears to be also enriched at tandem repeat regions of T. brucei genome (30). Furthermore, recent studies also showed that a small amount of J is located at regions flanking RNA polymerase II (RNAP II) PTUs throughout the T. brucei genome (31).
J's function appears to be species-specific in kinetoplastids. In T. brucei, J seems to be involved in regulation of transcription elongation within certain RNAP II PTUs (32). In Trypanosoma cruzi that causes Chagas disease, J is located at the transcription initiation sites of RNAP II PTUs, and loss of J led to elevated RNAP II transcription (33). In Leishmania major that causes leishmaniasis, J is located at the transcription termination sites of RNAP II PTUs, and loss of J led to readthrough of RNAP II-mediated transcription (32,34). With the enrichment of base J in T. brucei telomeric DNA, it is intriguing whether the DNA-binding Myb domain of TbTRF would interact with J-containing TTAGGG repeats any differently from regular sequences. Furthermore, whether such differences would have any implication on VSG regulation is worth investigating.
To address these questions, we conducted a series of structural and functional studies on the Myb domain of TbTRF. The structure of the TbTRF Myb domain was determined by nuclear magnetic resonance (NMR). The DNA-binding activity of TbTRF, particularly the interaction between TbTRF and J-containing TTAGGG sequence, was characterized by in vitro binding assays and in vivo Chromatin Immunoprecipitation (ChIP) experiments. Structure-based mutational analysis identified a few residues critical for Myb domain folding and its binding to telomere. We found that a transient depletion of TbTRF led to a 6-fold increase in VSG switching frequency. More importantly, TbTRF Myb domain mutations that significantly decreased TbTRF's association with the telomeric DNA in vivo also caused significantly more frequent VSG switching events, indicating that the telomere DNA-binding activity of TbTRF plays an important role in VSG switching regulation.
MATERIALS AND METHODS
VSG switching assay
VSG switching frequencies in S/TRFi clones B1, A7 and A8 were first determined by a method without enrichment of switchers through magnetic-activated cell sorting (MACS) column as described in (12).
The improved VSG switching assay enriches switchers by passing cells through MACS column that binds a VSG2 monoclonal antibody (20). Detailed protocol can be found in the supplemental materials.
ChIP
ChIP was performed as described in (35).
Quantitative reverse transcriptase-polymerase chain reaction
Quantitative RT-polymerase chain reaction (qRT-PCR) was performed the same way as described in (12).
Electrophoresis mobility shift assay
Double-stranded oligos were prepared by standard annealing process after the two single-stranded DNA molecules were mixed in equal molar ratio. The annealed double-stranded oligos were then purified by gel electrophoresis before used in electrophoresis mobility shift assay (EMSA) or NMR experiments. The regular or J-containing DNA (25 pmol) was incubated with increasing amount of TbTRF Myb protein (0, 25, 50, 60, 75, 100, 120 or 130 pmol) in buffer containing 20 mM HEPES pH 7.5, 40 mM KCl, 10 mM MgCl2, 10 mM CaCl2, 10 mM β-mercaptoethanol, 0.25 mg/ml bovine serum albumin, 8% (v/v) glycerol at 4°C for 5 h and 37°C for 15 min immediately before subjected to 5% native polyacrylamide gel and run at 150 V in 0.25 × TBE buffer at 4°C for 2 h. Gels were stained by EtBr and scanned by ChemiDoc™ XRS Imager (BioRad).
Protein expression and purification
The Myb domain of TbTRF (residues 281–382) was cloned as a His6-tagged protein in a modified pET32 vector containing the human rhinovirus (HRV) 3C protease cleavage site and without thioredoxin fusion. The mutants (R298K, R298E, Q320S, Q321S, H346R, R348K, R348E and R352K) were generated by site-directed mutagenesis (Finnzyme) and cloned into the same vector as wild-type TbTRF. Wild-type and mutant TbTRF Myb domain constructs were expressed in Escherichia coli BL21 (DE3) cells at 25°C for 8 h after induction by isopropyl-β-D-thiogalactopyranoside at 0.75 mM concentration. Target proteins were purified by affinity chromatography (HisTrap HP, GE Healthcare). The His6 tag was removed by 3C cleavage and the untagged protein was further purified by size-exclusion chromatography (Superdex 75, GE Healthcare).
NMR spectroscopy
The protein samples for NMR studies were concentrated to ∼0.2 mM for titration experiments and ∼1.0 mM for structural determinations in 100 mM potassium phosphate at pH 5.8. NMR spectra were acquired at 10°C on Varian Inova 500 or 750 MHz spectrometers. For titration experiments, DNA oligos at 50 μM were added into the protein sample. Backbone and side chain resonance assignments were achieved by combination of standard heteronuclear correlation experiments including HNCO, HNCACB, CBCA(CO)NH and HCCH-TOCSY using 15N/13C-labeled protein samples, and 1H 2D TOCSY and NOESY experiments (36–38). Approximate interproton distance restraints were derived from 2D1H-NOESY, 3D 15N-seperated NOESY and 3D 13C-seperated NOESY spectra. Structures were calculated using the program CNS (39). Statistics were summarized in Supplementary Table S1. The coordinates of TbTRF Myb domain have been deposited to Protein Data Bank (PDB ID 2M9H). The structure figures were prepared using the CCP4mg package (40) in CCP4.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) was performed using an iTC200 microcalorimeter (MicroCal Inc.) as described previously (41–44). Additional details can be found in Supplemental Materials.
Synthetic J-DNA oligos
The J-containing oligos are purchased from Fidelity Systems Inc. (http://www.fidelitysystems.com/).
Pulsed field gel electrophoresis
Pulsed field gel electrophoresis (PFGE) was performed as described in (12).
RESULTS
Depletion of TbTRF increases VSG switching frequency and changes VSG switching pattern
We previously found that TbTRF plays an essential role in maintaining the telomere terminal structure (21), but does not seem to affect subtelomeric VSG silencing (18). However, whether TbTRF plays any role in VSG switching regulation is unknown. To test this possible function of TbTRF, we introduced the pZJMβ-based (45) TbTRF RNAi construct (21) into the HSTB261 cells developed for VSG switching assays (16) to establish S/TRFi strains (S for switching). As expected, induction of TbTRF RNAi by doxycycline in S/TRFi cells led to a cell growth arrest by 24 h (Figure 1A, Supplementary Figure S1A) and a decrease in TbTRF protein level (Figure 1B, Supplementary Figure S1B).
Figure 1.
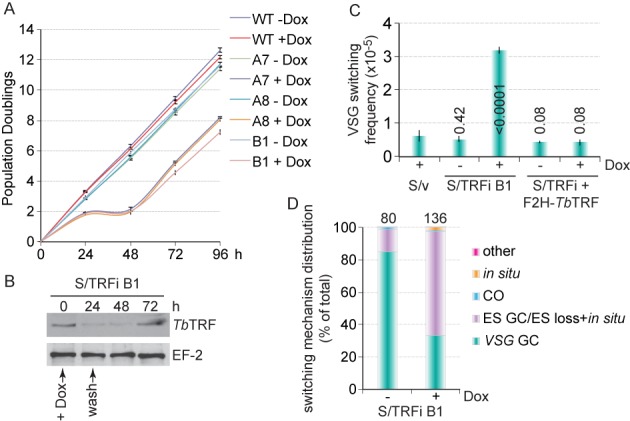
A transient depletion of TbTRF led to an elevated VSG switching frequency. (A) S/TRFi cells experienced a transient growth arrest upon induction of TbTRF RNAi by 100 μg/ml doxycycline for 24 h. Growth curves were calculated from three independent experiments. A7, A8 and B1 are independent S/TRFi clones. In this and other figures, error bars represent standard deviation. (B) TbTRF protein level decreased upon induction of TbTRF RNAi for 24 h. Western blotting was performed using whole cell lysate prepared from S/TRFi clone B1 cells and TbTRF (21) and EF-2 (Santa Cruz Biotechnology, Inc.) antibodies. (C) VSG switching frequencies in S/v, S/TRFi B1 and S/TRFi+F2H-TbTRF cells with or without a transient TbTRF RNAi induction. Average switching frequencies were calculated from at least three independent switching assays. Numbers on columns indicate P values (unpaired t tests) when compared to the switching frequency in S/v cells. A P value less than 0.05 is considered significantly different. (D) VSG switching mechanism distribution in S/TRFi B1 cells under the induced and uninduced conditions. The total number of switchers characterized is indicated above each column. GC, gene conversion; CO, crossover.
In HSTB261, a blasticidin-resistance gene (BSD) was targeted immediately downstream of the active ES promoter, and a puromycin-resistance gene (PUR) fused with a Herpes simplex virus thymidine kinase (TK) gene was inserted between the 70 bp repeats and the VSG2 gene in the same active ES (Supplementary Figure S1C) (16). T. brucei cells expressing TK are sensitive to ganciclovir (GCV), a nucleoside analog (46). Essentially all VSG switchers will lose the PUR-TK expression and become GCV resistant. Therefore, VSG switchers can be easily selected by GCV resistance. Because depletion of TbTRF leads to a cell growth arrest (21), we induced the TbTRF RNAi for a short period of time to enable subsequent recovery of VSG switchers. We found that induction of TbTRF RNAi for 24 h followed by removal of doxycycline led to a depletion of TbTRF protein and a growth arrest for 24 h followed by a recovery of the TbTRF protein level and the growth (Figure 1A and B). The recovered TbTRF RNAi cells exhibited growth arrest again when induced by doxycycline repetitively (Supplementary Figure S1A), indicating that the recovered population retained the ability of RNAi induction.
Before the VSG switching assay, cells were kept under selection with blasticidin and puromycin to ensure uniform expression of VSG2 in the population. At the beginning of the VSG switching assay, blasticidin and puromycin were removed from the medium to allow switchers to arise. Three independent S/TRFi clones were incubated with or without doxycycline for 24 h followed by removal of doxycycline by extensive cell washing and cell recovery. Induced S/TRFi cells typically took ∼48 h to fully recover, while the uninduced cells grew normally. Eventually, all cells were grown for the same number of population doublings to allow a fair comparison of VSG switching frequencies between induced and uninduced cells. A small fraction of cells was plated in the absence of GCV to determine the plating efficiency. Initially, we plated 0.5 million cells after each switching assay and found that a transient depletion of TbTRF led to a significant increase in the VSG switching frequency (Supplementary Figure S1D, S1E), indicating that TbTRF suppresses VSG switching.
We attempted to isolate sufficient switchers from S/TRFi cells to determine the VSG switching mechanisms. However, this original switching method is very inefficient and we could only obtain limited number of switchers in each assay. We therefore adopted the modified switching assay by enriching switchers using MACS beads that bind a monoclonal VSG2 antibody (20). Cells expressing VSG2 were retained by the MACS column, while switchers were enriched in the flow-through fraction and were plated in the presence of GCV for subsequent selection. This modified switching method allowed us to isolate ∼100 switchers under each condition, which greatly increased the assay efficiency. Using the MACS switching assay, we examined the VSG switching frequencies and mechanisms in HSTB261 cells carrying an empty RNAi vector (S/v) (12) and S/TRFi clone B1 cells under induced and uninduced conditions. We grew cells for approximately the same number of population doublings in both the original and the MACS switching assays to ensure that the results were comparable. We confirmed that a transient depletion of TbTRF led to a 6-fold higher VSG switching frequency than that in S/v cells, while uninduced S/TRFi cells exhibited a similar VSG switching frequency as S/v cells (Figure 1C, Supplementary Figure S1F). S/v cells had a similar VSG switching frequency as reported in HSTB261 cells (11,16). Inducing the expression of an ectopic F2H-tagged WT TbTRF allele in S/TRFi cells (Supplementary Figure S1H) also complemented the slow growth (Supplementary Figure S1G) and the elevated VSG switching frequency phenotypes (Figure 1C). Therefore, WT TbTRF suppresses subtelomeric VSG switching. Our previous study showed that TbTRF depletion did not shorten telomere length dramatically (21). We confirmed that telomere length did not change significantly within the short frame of time when TbTRF was depleted (Supplementary Figure S1I), indicating that elevated VSG switching frequency is not due to extremely short telomere lengths. However, extremely short telomere would presumably bind very few TbTRF proteins, which would in turn result in increased VSG switching frequency, as observed in telomerase null cells with short telomeres (20).
Using the MACS switching assay, we obtained ∼100 or more VSG switchers under each assay condition. Analyzing a larger number of VSG switchers allowed us to reveal the typical VSG switching patterns more accurately. By characterizing the genotype and marker expression status, we can determine the VSG switching mechanisms (Supplementary Figure S1C) as described previously (11,12,16). By selecting switchers with various antibiotics and PCR analyses using VSG- or BSD-specific primers, we determined that before TbTRF RNAi induction, most switchers (∼85%) were VSG GC events, while a considerable number of switchers were ES GC or ES loss + in situ events (∼14%) (Figure 1D). In contrast, after a transient depletion of TbTRF, ES GC/ES loss + in situ events became predominant (65%), while only ∼33% of all switchers were VSG GC events (Figure 1D). In addition, in situ switchers were extremely rare in uninduced S/TRFi cells (none in examined VSG switchers), but increased to 1.5% in TbTRF-depleted cells. Therefore, TbTRF influences the VSG switching mechanisms.
The Myb domain of TbTRF adopts the canonical helix-turn-helix structure
TbTRF binds to the duplex TTAGGG telomere repeats directly (21). This affinity has been assigned to its C-terminal region that shares high sequence similarity to the Myb domains in mammalian TRF proteins (47). Myb domains are small structural modules of 50–60 residues that are commonly found in DNA-binding proteins (48). The structures of mammalian TRF Myb domains reveal a conserved fold of three-helix bundle with the classical helix-turn-helix motif as the DNA-binding element (23,49,50).
To further delineate the role of the TbTRF Myb domain in VSG switching regulation, we first solved the NMR structure of this region. While the core region of the Myb domain consists of only 50–60 residues, a longer construct of TbTRF Myb extending about 20 residues more on both the N- and C-termini is necessary to yield soluble protein. The heteronuclear single quantum correlation (HSQC) spectrum of 15N-labeled TbTRF Myb domain showed an excellent dispersion pattern typical of a well-folded protein (Figure 2A). The structure was determined with a conventional multidimensional heteronuclear NMR method with the statistics summarized in Supplementary Table S1. The final 20 structures superimpose well, with root-mean-square deviation (RMSD) of 0.6 Å for the backbone atoms in the core region (Figure 2B). The N- and C-terminal extensions, while critical for solubility of this domain, are highly flexible as they show few medium- and long-range nuclear overhauser effect signals.
Figure 2.
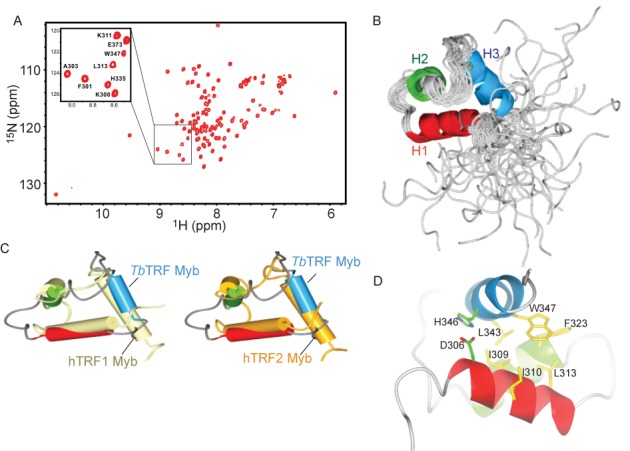
Structure of TbTRF Myb domain. (A) The 1H-15N HSQC spectrum of 15N-labeled TbTRF Myb domain at pH 5.8. A selected region of the spectrum enclosed by a framed rectangle is expanded to show the detail of residue assignments. (B) Ensemble of the final 20 lowest energy structures of TbTRF overlaid on the backbone. The three helices H1–H3 are drawn as ribbons. (C) Structural alignment of TbTRF Myb domain (light blue) with that of human TRF1 (light yellow, PDB ID 1ITY) and TRF2 (gold, PDB ID 1VF9). The TbTRF Myb domain is the energy minimized average structure generated from the ensemble. (D) The hydrophobic core and the D306-H346 salt bridge in the TbTRF Myb structure.
The overall structure of TbTRF Myb domain follows the canonical three-helix bundle architecture, with the second and third helix forming the helix-turn-helix motif for DNA binding. This TbTRF Myb structure is very similar to the human TRF1 and TRF2 Myb structures, especially the three core helices with RMSD of 0.6 and 0.83 Å, respectively (Figure 2C). However, the loops connecting these helices adopt quite different conformations in TbTRF from that of human TRF1 and TRF2, probably due to their intrinsic flexibility (Figure 2C).
TbTRF Myb is stabilized by an extensive hydrophobic core consisting of a few conserved residues including I309, L313, L343 and W347 when compared to mammalian TRFs (Figure 2D). Another interesting observation about TbTRF Myb is the pH sensitivity of its folding. The HSQC spectrum showed significant deterioration when buffer pH was adjusted from 5.8 to 6.5 (data not shown). Close inspection of the structure revealed that H346, the only residue in TbTRF Myb with its imidazole side chain of pKa around 6.5, is likely the major factor for this pH sensitivity. H346, located on the third helix, forms a salt bridge interaction with D306 on the first helix (Figure 2D). This interaction is critical for packing of these two helices to form the three-helix bundle. Deprotonation of H346 when buffer pH arises above 6.5 likely weakens this interaction and may lead to partial unfolding of TbTRF Myb.
TbTRF Myb domain binds to both regular and J-containing telomeric DNA
The direct interaction of the TbTRF Myb domain with telomeric DNA repeats was first characterized by EMSA. Purified TbTRF Myb peptide readily formed a complex with a ‘double-repeat’ DNA oligo that contained two telomeric repeats (5′ GGCGCGCTTAGGGTTAGGGTTACCGCCCG 3′) (Supplementary Figure S2A). We also used ITC to directly measure the binding affinity (Kd) between TbTRF Myb and its DNA substrate. Our data showed that TbTRF Myb bound to single- (5′ GGGGCCTTTAGGGTTATCCGCC 3′) and double-repeat (same sequence as used in EMSA) DNA oligos with comparable affinities at Kd of ∼20 μM (Figure 3A, Supplementary Figure S2B). In summary, these biochemical data confirm that TbTRF Myb can directly bind to telomeric TTAGGG repeats.
Figure 3.
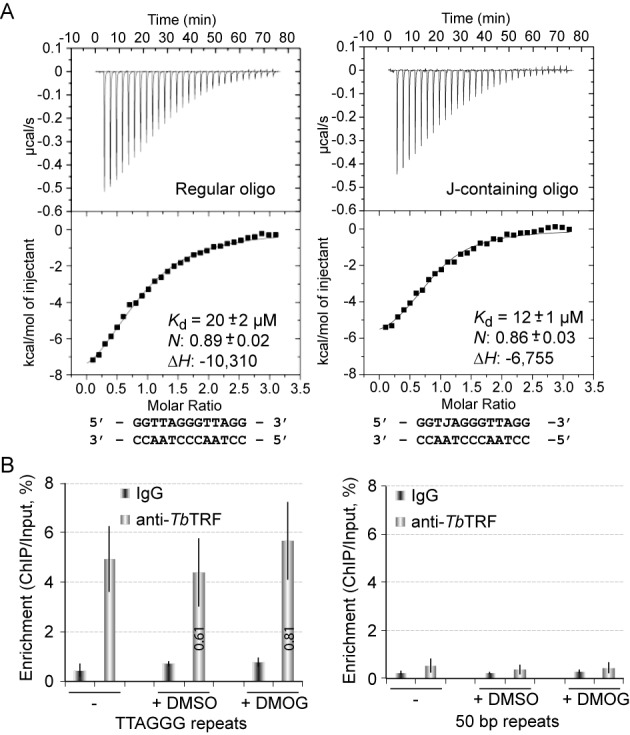
Characterization of TbTRF binding to telomeric DNA. (A) ITC-based measurement of TbTRF Myb–DNA interaction using regular (left) and J-containing oligo (right). Each measurement was repeated three times. Values of binding affinity (Kd), stoichiometry (N) and binding enthalpy (ΔH) with standard deviation are shown. (B) J does not influence the binding of TbTRF on telomeric DNA in vivo. ChIP was performed using TbTRF antibody (21) in cells grown in normal medium, medium with Dimethyl sulfoxide (DMSO) or medium with 0.5 mM Dimethyloxalylglycine (DMOG) dissolved in DMSO. Southern analyses using the TTAGGG repeat (left) or the 50 bp repeat (right) probe were performed to hybridize the ChIP products. Southern blots were exposed to phosphorimager screens and the percentage of enrichment was calculated by dividing the ChIP signal by the input signal. Average values were calculated from at least three independent experiments.
A considerable amount of thymidine in the BF T. brucei telomeric DNA is replaced by the base J (28). J is absent in the PF T. brucei DNA, and TbTRF associates with telomeric DNA in both BF and PF cells (21), indicating that the TbTRF DNA-binding activity is not abolished by J or requires J. However, it is unknown whether the TbTRF Myb domain binds J-containing telomeric DNA the same way as it binds the regular telomeric sequence.
To directly test whether J affects the DNA-binding activity of the TbTRF Myb domain, we first constructed a synthetic J-containing duplex oligo with the sequence of TJAGGG/AATCCC to represent a single-repeat DNA substrate. The presence of J in this oligo was confirmed by mass spectroscopy (Supplementary Figure S3A). ITC analysis revealed that the binding affinity (Kd) of TbTRF Myb to the J-containing oligo is ∼12 μM, comparable to that of the regular TTAGGG oligo (Figure 3A).
Lastly, we performed EMSA analysis using the same J-containing single-repeat DNA as a substrate. The partially purified bacteria-expressed TbTRF myb domain showed a similar DNA-binding activity regardless of whether the telomeric DNA substrate contained J or not (Supplementary Figure S3B). These results confirm that the presence of J in the telomeric DNA does not affect its interaction with the TbTRF Myb domain in vitro.
To better quantify the telomeric DNA-binding activity of TbTRF in vivo, we performed a ChIP analysis in BF cells treated with or without DMOG, a cell-permeable inhibitor of dioxygenase (51). It has been shown that treating BF T. brucei cells with 0.5 mM DMOG in DMSO for 6 days resulted in 80% of decrease in cellular J (52). We performed TbTRF ChIP in the presence and absence of DMOG and found that similar amount of TbTRF was associated with the telomeric DNA under both conditions (Figure 3B). To confirm that treating cells with DMOG removed most telomeric J, we performed a Southern analysis. It has been shown that the active VSG ES does not contain J, while silent ESs do (29), and J modification prevents PstI digestion (53,54). Indeed, we found that in the VSG2-expressor, the active VSG2 gene was fully digested by PstI (Supplementary Figure S3C, S3D). However, in the VSG9-expressor, the same VSG2 gene, now silent, was only partially digested by PstI (Supplementary Figure S3C, S3D). Treating cells with 0.5 mM DMOG for 6 days allowed the silent VSG2 locus to be fully digested with PstI, indicating that DMOG successfully removed the J modification. Treating cells with DMSO (as a control) did not have any effect on J (Supplementary Figure S3C). In addition, as expected, DMOG treatment did not affect cell growth (Supplementary Figure S3E), because J is not essential for T. brucei growth (55). Therefore, J modification does not affect the telomeric DNA-binding activity of TbTRF.
Perturbing the TbTRF Myb domain significantly decreases its DNA-binding activity
NMR titration experiments were performed to identify TbTRF Myb residues critical for DNA binding. A double-repeat DNA oligo with sequence 5′ GGCGCGCTTAGGGTTAGGGTTACCGCCCG 3′ was titrated into 15N-labeled TbTRF Myb and the resulting HSQC spectrum was compared to that of protein alone (Figure 4A). Overall the DNA-titrated HSQC spectrum showed significant difference as compared to the TbTRF Myb-only spectrum, confirming that there was a direct interaction between TbTRF Myb and the double-repeat oligo. The severe broadening of many cross peaks in the titrated spectrum was probably due to the large size of the TbTRF Myb–DNA complex formed (∼80 kDa with two TbTRF Myb domains associated with the double-repeat DNA oligo).
Figure 4.
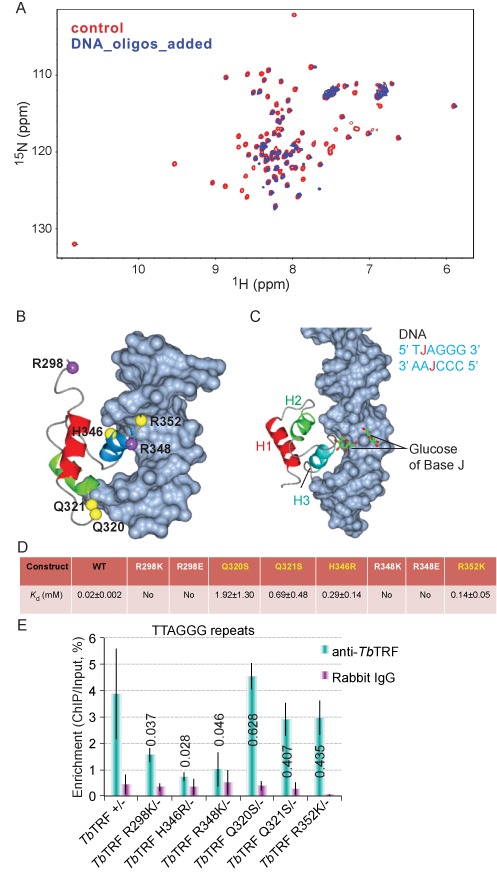
Key DNA-binding residues within TbTRF Myb domain. (A) Superposition plot of the 1H–15N HSQC spectra of TbTRF alone and TbTRF titrated with a double-repeat DNA oligo pair in 1:0.5 molar ratio. (B) Model of TbTRF Myb domain bound to telomeric DNA generated by superimposing TbTRF Myb onto human TRF1 Myb–DNA complex structure (PDB ID 1IV6). (C) Model of TbTRF Myb bound to a J-containing DNA. The J-DNA (PDB ID 308D) is superimposed onto regular DNA in the TbTRF Myb–DNA complex structure of (B). (D) Binding affinity (Kd) of WT and mutant TbTRF Myb domain with a duplex telomeric oligo of sequence 5′ GGCGCGCTTAGGGTTAGGGTTACCGCCCG 3′ as measured by ITC. (E) ChIP analyses of TbTRF myb point mutants. ChIP experiments were performed using TbTRF antibody (21) or normal rabbit immunoglobulin G in cells expressing the TbTRF Myb point mutant as the only TbTRF allele. Southern hybridizations were performed using the TTAGGG repeat probe or the 50 bp repeat probe as a control (Supplementary Figure S5C). Southern blots were exposed to a phosphorimager screen and signal intensities were quantified. The percentage of enrichment was calculated by dividing the ChIP signal by the input signal. The average was calculated from at least three independent experiments. P values (unpaired t tests) of TbTRF ChIP result between each mutant and TbTRF +/- are listed on each column.
While the broadening hindered our effort to pinpoint key DNA-binding residues, we proceeded to generate a model of TbTRF Myb in complex with the telomeric DNA using the human TRF1 Myb-DNA structure as a reference (PDB ID 1W0T) (56). TbTRF Myb was aligned onto human TRF1 Myb with its third helix inserted into the major groove of telomeric DNA, mimicking the canonical Myb–DNA-binding mode (Figure 4B). In this model, the conserved R348 was well positioned to make sequence-specific contact with the G-C triplet in the telomeric repeat. Its correspondence in human TRF1, R425, has been shown to make similar sequence-specific interactions with the telomeric DNA substrate (49). Furthermore, our model showed that Q320 and Q321, two TbTRF Myb-specific residues not highly conserved in the mammalian system, were also in close proximity to make contact with the sugar-phosphate backbone of DNA substrate. Based on our NMR data and modeling observations, we selected these residues (Q320, Q321 and R348) and three additional residues (R298, H346 and R352) for structure-based biochemical and functional studies. H346 is included because the salt bridge interaction it forms with D306 as shown by the NMR structure could play an essential role in stabilizing the folding of TbTRF Myb and facilitating its DNA-binding activity. R298 and R352 were also included because the flexible N- and C-terminal loops have been shown to contact DNA substrates by previous studies (49), although our model cannot precisely confirm this.
Additionally we also constructed a model of the TbTRF Myb domain in complex with the J-containing telomeric DNA (Figure 4C). The J-base was ‘grafted’ onto the TbTRF Myb–DNA model by superimposing a J-containing oligo (PDB ID 308D) onto the existing regular DNA in the model. The base J replacing the T in the TTAGGG strand was located on the surface of the DNA major groove and was in the vicinity of the loop connecting the second and third helix but sufficiently away from the most critical DNA-binding residues on the third helix. The base J replacing the T in the complementary AATCCC strand was positioned completely away from the Myb-binding site and unlikely to exert any influence. Thus, our model supports our binding studies that the presence of J is unlikely to exert any direct influence on the DNA-binding activity of the TbTRF Myb domain.
Focusing on the six residues identified by NMR titration studies and supported by our structural model, we generated a series of mutant TbTRF Myb constructs (Figure 4D). EMSA (Supplementary Figure S4A) and ITC experiments (Supplementary Figure S4B) were performed to characterize the DNA-binding affinities of these mutants in vitro. Indeed, mutations targeting the highly conserved R348 such as R348E and a milder perturbation of R348K completely abolished the DNA binding. Similar effect was also observed for the N-terminal R298 mutations, suggesting that R298 directly participates in DNA binding. However, mutations targeting the TbTRF Myb-specific residues such as Q320, Q321 and H346 did not abolish the DNA binding, although the binding was significantly weakened.
To further characterize the mutant TbTRF DNA-binding activity in vivo, we introduced these point mutations into T. brucei cells. In the TbTRF single knockout (sko) background, we replaced the remaining endogenous WT TbTRF allele with the mutant TbTRF allele carrying an N-terminal Ty1 tag. We could not obtain T. brucei strains that only expressed the TbTRF R298E or TbTRF R348E mutant, suggesting that these strong mutational perturbations completely abolished the in vivo telomeric DNA-binding activity of TbTRF and were not viable. All other TbTRF mutants were expressed equally in western analysis at a level similar to that in TbTRF sko cells (Supplementary Figure S5A), and sequencing analysis confirmed the mutation in each mutant strain. Among TbTRF Myb mutants, H346R exhibited a mild slow growth phenotype, while R298K and R348K had a slow growth phenotype that is even milder (Supplementary Figure S5B).
We performed TbTRF ChIP analysis in the TbTRF mutant strains. H346R had the weakest telomeric DNA-binding activity, while R298K and R348K also exhibited weaker than WT telomeric DNA-binding activities (Figure 4E, Supplementary Figure S5C), and the differences between these three mutants and WT TbTRF are significant. In contrast, Q320S, Q321S and R352K mutants are associated with the telomeric DNA at similar levels to that of the WT (Figure 4E, Supplementary Figure S5C), indicating that the telomeric DNA-binding activities in these TbTRF Myb mutants were not significantly affected in vivo.
To examine whether VSG silencing is affected in TbTRF myb mutants, we performed qRT-PCR to estimate the steady-state mRNA levels of several ES-linked VSGs and several control genes including TbTERT, TbRPS15 and rRNA in TbTRF mutants and sko cells. In most cases, we did not observe higher VSG mRNA levels in TbTRF mutants than in sko cells except that elevated VSG3 and VSG13 mRNA levels were detected in H346R/- and R348K/- cells, respectively (Supplementary Figure S5D), suggesting that decreasing the DNA-binding activity of TbTRF did not affect VSG silencing globally.
TbTRF Myb domain mutants with weakened DNA binding show increased VSG switching frequencies
We found that a transient depletion of TbTRF led to an increased VSG switching frequency. To further test whether the telomeric DNA-binding activity of TbTRF is essential for VSG switching regulation, we introduced TbTRF Myb mutants that significantly reduced the telomere DNA-binding activity (H346R, R298K and R348K) in HSTB261 cells and deleted the remaining WT TbTRF allele. As a control, we also introduced the Q320S mutant that did not affect TbTRF DNA-binding activity into HSTB261. Western analysis confirmed the expression of the TbTRF mutants in these cells (Supplementary Figure S5E). Southern and PCR analyses also confirmed the TbTRF genotype, and sequencing analysis verified each mutant (data not shown).
We performed the VSG switching analysis in TbTRF myb domain mutants using MACS to enrich the VSG switchers. We found that TbTRF sko strain exhibited a similar VSG switching frequency as the S/v cells (Figures 1C and 5A), indicating that a single allele of TbTRF was sufficient for its function in VSG switching regulation. Interestingly, S/H346R had a significantly increased (3.3-fold) VSG switching frequency than S/TRF sko (Figure 5A). In addition, S/R298K and S/R348K cells showed a mild but significant increase in VSG switching frequency (1.6 fold) (Figure 5A). In contrast, S/Q320S behaved similarly as S/TRF sko cells. Therefore, the H346R mutant that significantly reduced the association of TbTRF with the telomeric DNA in vivo led to significantly more VSG switching. R298K and R348K mutants that mildly affected the in vivo DNA-binding activity also mildly increased VSG switching frequency. While the Q320S mutant that did not affect DNA binding in vivo did not affect VSG switching, either, indicating that the telomere DNA-binding activity is critical for the role of TbTRF in VSG switching regulation.
Figure 5.
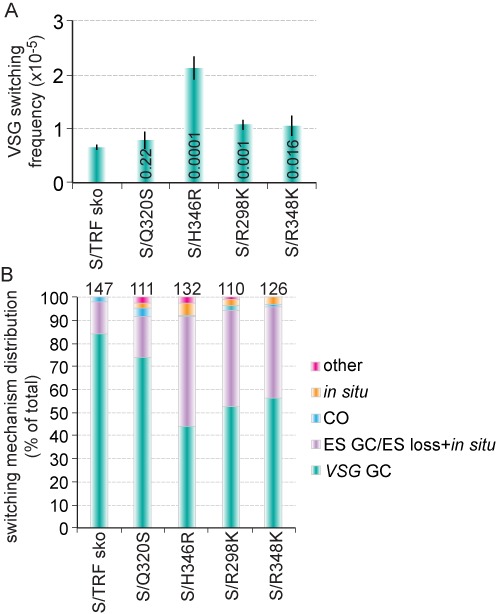
TbTRF myb mutations that decreased the telomere DNA-binding activity also increased the VSG switching frequencies. (A) VSG switching frequencies in S/TRF sko and S/TRF mutants. Average values were calculated from at least three independent experiments. P values (unpaired t tests) comparing S/TRF sko and S/TRF mutants are indicated on each column. (B) VSG switching mechanism distribution in S/TRF sko and S/TRF mutants. The total number of switchers characterized is indicated above each column.
We further characterized the VSG switching mechanisms in S/TRF sko and TbTRF myb mutants. Similar to S/v cells, S/TRF sko cells had predominantly VSG GC events (84%) and a considerable percentage of ES GC/ES loss + in situ events (14%). In TbTRF mutants that exhibited more frequent VSG switching, we observed a decreased percentage of VSG GC events (48–56%) and an increased percentage of ES GC/ES loss + in situ events (40–48%), which was similar to that in TbTRF-depleted cells, although the effects were to a lesser degree (Figures 1D and 5B). In addition, we observed an increase in in situ switchers in TbTRF mutants (from 0% in sko cells to 1.8–5.3% in TbTRF mutants), which is similar to induced S/TRFi cells (Figure 1D). Therefore, the DNA-binding activity of TbTRF is important for its function in VSG switching regulation.
To verify that VSG switchers were correctly assigned with corresponding switching mechanisms, we randomly picked switchers from various TbTRF genetic backgrounds and performed PFGE and Southern analyses using VSG2, BSD and the newly activated VSG probes. Supplementary Figure S6 shows that all tested VSG switchers were correctly classified.
DISCUSSION
TRF is an essential telomeric protein conserved among a number of species from the T. brucei protozoan to human. In mammalian cells, TRF1 and TRF2 are two key components of the Shelterin complex that protects the chromosome end (57). Removal of TRF2 from telomeres led to ATM/p53-dependent ligase IV-mediated NHEJ of telomere ends (58–60). Removal of TRF1 from telomeres led to telomere elongation (61) and fragile telomeres (62). Both TRF1 and TRF2 contain a highly conserved C-terminal Myb domain that directly binds to the telomeric DNA, which enables TRFs to serve as architectural factors and to nucleate the Shelterin complex at the telomere (63). Structural analysis of mammalian TRF myb domains revealed that these structural modules use a conserved helix-turn-helix motif for specific binding to the TTAGGG telomeric sequence (49,56).
T. brucei is a protozoan parasite that undergoes antigenic variation through VSG switching to evade the host immune response (2). Because the repertoire of over 2000 VSG genes in T. brucei are located at subtelomeres (4), the telomere structure and associated proteins have long been speculated to play a role in VSG switching regulation, although limited data have been reported to support this hypothesis (12,20). TbTRF is the first telomere protein identified and found to be essential for maintaining the telomere terminal structure in T. brucei (21). The role of TbTRF in VSG switching has not been investigated, although it is not required for subtelomeric VSG silencing (18).
Here we have conducted structural and functional studies to delineate the role of TbTRF and its Myb domain in VSG switching. First, we found that a transient depletion of TbTRF led to significantly more VSG switchers and most of these arose from ES GC/ES loss + in situ events. This confirms a definitive role of TbTRF in suppression of VSG switching. Although a transient depletion of TbTRF led to a growth arrest, this arrest is short-term and fully reversible, excluding the possibility that the observed more frequent VSG switching is a ‘death phenotype’. RNAi has been used to show that several essential proteins are important in regulation of VSG switching through apparently different mechanisms (12,35,64,65). Furthermore, a simple slow growth is not expected to result in more frequent VSG switching, because deletion of TbMRE11 led to significantly slow growth, but did not affect VSG switching (66).
Second, our structural data show that the TbTRF Myb domain adopts the canonical helix-turn-helix DNA-binding motif shared by all Myb domains. The three core helices are tightly packed together by a cluster of hydrophobic residues. This core region superimposes very well with mammalian TRF1/2 Myb domain structures, suggesting a highly conserved structural module for sequence-specific binding to telomeric DNA repeats. However, the loops connecting the three helices and both the N- and C-termini of Myb region are highly flexible, with little sequence identity between TbTRF and mammalian TRF1/2.
Furthermore, our in vitro data show that the TbTRF Myb domain binds to telomeric sequences containing the bulky J-base with similar affinity as to normal telomeric sequences. Structure-based modeling suggests that although J is a bulky modification, it is not located at the interface of DNA and TbTRF interaction. Thus, J is unlikely to affect TbTRF binding. Our in vivo ChIP analysis further confirmed that TbTRF associated with the telomeric DNA at equal efficiency whether the substrate contained J or not.
There have been no reports on whether mammalian TRF1/2 can bind to J-containing telomeric DNA. A previous study showed that human TRF1 could be expressed in PF T. brucei cells and was located at the telomere that does not have J modification (67). However, human TRF1, when expressed in BF T. brucei cells where the DNA is modified by J, is not located at the telomere (67). Presumably, the presence of J in BF telomeres interfered with efficient binding of human TRF1, although the possibility that other telomere factors are involved in such reduced TRF1 binding cannot be ruled out. Further studies, such as in vitro measurement of the binding affinity of mammalian TRF1/2 Myb to J-DNA and in vivo ChIP assays, are needed to fully address this question.
We generated a series of structure-based mutations within the TbTRF Myb domain to abolish or weaken its DNA-binding affinity. Three assays, including the in vitro EMSA and ITC, together with the in vivo ChIP assay, were used to characterize the effects of individual mutations on the DNA-binding activity of TbTRF. VSG switching assays were also carried out to correlate the DNA-binding affinity of these TbTRF mutations to their VSG regulation function. For all mutations except H346R (discussed below), results from all these assays are consistent or at least show the same trend of effects. Specifically, R298E and R348E abolished in vitro DNA-binding activity, and we could not obtain any cells expressing these mutant alleles, suggesting that they also abolish DNA binding in vivo. R298K and R348K abolished in vitro DNA-binding activity and showed significantly reduced telomere association and increased VSG switching in vivo, while Q320S, Q321S and R348K only weakened in vitro DNA-binding activity and exhibited subtle and insignificant effects on their in vivo telomere association and VSG switching.
Therefore, all tested Myb mutants except H346R showed milder effects in vivo than in vitro. Such mild in vivo effect is likely due to different TbTRF constructs used in in vitro and in vivo methods. TbTRF homodimerizes through its TRFH domain, and this dimerization is critical for its in vivo telomere DNA-binding activity (21), because at least two Myb domains are required for efficient DNA recognition (68). In addition, TbTRF interacts with other telomere proteins in live cells (12,18), which may also influence its in vivo telomere DNA-binding activity. Our in vitro studies were performed with the Myb domain alone, which is not expected to dimerize, while our in vivo assays were performed with full-length TbTRF proteins that are capable of dimerization. Therefore, it is possible that dimerization of TbTRF mutants, as well as its interaction with other telomeric factors, may serve to recover some of the mutants’ DNA-binding activity in vivo.
H346R is unique among the tested mutants, as it showed a stronger phenotype in vivo than in vitro. Its in vitro DNA-binding affinity is stronger than that of R298K and R348K (weakened versus abolished), yet its ChIP enrichment is weaker and its VSG switching frequency higher than the latter two mutants. This is likely because of the unique role of H346 when compared to other residues. As shown by our NMR data, H346 is critical for the proper folding of TbTRF Myb domain, although it does not directly interact with the telomeric DNA. Thus, the stronger in vivo effect of H346R was probably caused by the loss of proper folding of full-length TbTRF when the Myb domain structure was compromised.
In summary, the TbTRF Myb mutations that abolished (R298K and R348K) or weakened (H346R) DNA binding in in vitro analyses led to reduced in vivo DNA binding when analyzed by ChIP, enhanced VSG switching by 1.6–3.3 fold and increased the proportion of ES GC/ES loss + in situ switchers, demonstrating that the function of TbTRF in VSG switching regulation depends on the structural integrity and telomere DNA-binding activity of its Myb domain.
TbTRF plays an essential role in maintaining the telomere terminal structure and is a functional homolog of mammalian TRF2 (21). Similar to loss of TRF2 (58–60), loss of TbTRF could result in telomere fusions and subsequent breakage-fusion-bridge cycle, which often result in large terminal chromosome deletions (69). Since loss of VSG expression is lethal for T. brucei cells (70), it is possible that only switchers that express a different VSG can survive the TbTRF-depletion-induced loss of the active VSG gene. However, chromosome end fusions have not been detected in T. brucei. It is unknown whether NHEJ exists in this protozoan, because no DNA Ligase IV homolog has been identified in T. brucei (71), although Ku homologs have been identified (72,73).
Recent studies showed that increased DNA damages in the active ES lead to greatly increased VSG switching frequencies (10,74), and damages downstream of the active VSG gene often lead to ES loss (10). It is possible that TbTRF has a conserved function in telomere DNA replication as human TRF proteins (62,75). In this case, loss of TbTRF could lead to stalled DNA replication forks in telomere repeats, which would cause increased damages at telomeres that contribute to increased VSG switching. Additional investigations are necessary to examine whether depletion of TbTRF would lead to elevated DNA damages at telomeres and subtelomeres.
TbTRF plays an important role in VSG switching regulation, a critical aspect of antigenic variation in T. brucei. The function of TbTRF is different from that of TbRAP1, which is a TbTRF-interacting factor and is essential for subtelomeric ES-linked and metacyclic VSG silencing in both BF and PF T. brucei cells (18,19). Therefore, although TbTRF interacts with TbRAP1 (18), these two telomere proteins appear to have independent functions in antigenic variation regulation. In contrast, depletion of another telomere protein, TbTIF2, also leads to more frequent VSG switching and more ES GC/ES loss + in situ switching events (12). Because TbTRF is the telomere DNA-binding protein and TbTIF2 is presumably recruited to the telomere by interacting with TbTRF, it is possible that depletion of TbTRF resulted in removal of TbTIF2 from the telomere, which in turn leads to elevated VSG switching frequency (12). Further investigations of how TbTIF2 is recruited to the telomere will help answer this question. TbTRF appears to interact with TbTIF2 more strongly than with TbRAP1 in both yeast 2-hybrid and co-IP experiments (12,18). Hence, it is likely that TbTRF and TbTIF2 function in the same pathway in regulating VSG switching. However, it is necessary to examine VSG switching frequencies and mechanisms in cells depleted of both TbTRF and TbTIF2 to determine definitively whether the two proteins work in the same pathway.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr Hee-Sook Kim and Dr George A. M. Cross for sending us the HSTB261 cells. The Li and Zhao lab members are thanked for their comments on the manuscript. Sandhu Ranjodh and Fei Ye contributed equally.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
These authors contributed equally to the paper as second authors.
FUNDING
National Institute of Health [AI066095, PI to B.L.]; Hong Kong Research Grants Council [PolyU5639/09M, PolyU5644/10M, PolyU5640/11M, HKUST6/CRF/10 and AoE/M-09/12, PI to Y.Z.]; Research Committee of the Hong Kong Polytechnic University; National Natural Science Foundation of China [31200563, PI to X.L.]. Funding for open access charge: National Institute of Health [AI066095, PI to B.L.]; Hong Kong Research Grants Council [PolyU5644/10M, PI to Y.Z.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Babokhov P., Sanyaolu A.O., Oyibo W.A., Fagbenro-Beyioku A.F., Iriemenam N.C. A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis. Pathog. Glob. Health. 2013;107:242–252. doi: 10.1179/2047773213Y.0000000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barry J.D., McCulloch R. Antigenic variation in trypanosomes: enhanced phenotypic variation in a eukaryotic parasite. Adv. Parasitol. 2001;49:1–70. doi: 10.1016/s0065-308x(01)49037-3. [DOI] [PubMed] [Google Scholar]
- 3.Berriman M., Ghedin E., Hertz-Fowler C., Blandin G., Renauld H., Bartholomeu D.C., Lennard N.J., Caler E., Hamlin N.E., Haas B., et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 4.Cross G.A.M., Kim H.S., Wickstead B. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol. Biochem. Parasitol. 2014;195:59–73. doi: 10.1016/j.molbiopara.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Gunzl A., Bruderer T., Laufer G., Schimanski B., Tu L.C., Chung H.M., Lee P.T., Lee M.G. RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei. Eukaryot. Cell. 2003;2:542–551. doi: 10.1128/EC.2.3.542-551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Lange T., Borst P. Genomic environment of the expression-linked extra copies of genes for surface antigens of Trypanosoma brucei resembles the end of a chromosome. Nature. 1982;299:451–453. doi: 10.1038/299451a0. [DOI] [PubMed] [Google Scholar]
- 7.Hertz-Fowler C., Figueiredo L.M., Quail M.A., Becker M., Jackson A., Bason N., Brooks K., Churcher C., Fahkro S., Goodhead I., et al. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One. 2008;3:e3527. doi: 10.1371/journal.pone.0003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crozatier M., van der Ploeg L.H., Johnson P.J., Gommers-Ampt J., Borst P. Structure of a telomeric expression site for variant specific surface antigens in Trypanosoma brucei. Mol. Biochem. Parasitol. 1990;42:1–12. doi: 10.1016/0166-6851(90)90107-w. [DOI] [PubMed] [Google Scholar]
- 9.Morrison L.J., Marcello L., McCulloch R. Antigenic variation in the African trypanosome: molecular mechanisms and phenotypic complexity. Cell. Microbiol. 2009;11:1724–1734. doi: 10.1111/j.1462-5822.2009.01383.x. [DOI] [PubMed] [Google Scholar]
- 10.Glover L., Alsford S., Horn D. DNA break site at fragile subtelomeres determines probability and mechanism of antigenic variation in African trypanosomes. PLoS Pathog. 2013;9:e1003260. doi: 10.1371/journal.ppat.1003260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim H.S., Cross G.A.M. Identification of Trypanosoma brucei RMI1/BLAP75 homologue and its roles in antigenic variation. PLoS One. 2011;6:e25313. doi: 10.1371/journal.pone.0025313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jehi S.E., Wu F., Li B. Trypanosoma brucei TIF2 suppresses VSG switching by maintaining subtelomere integrity. Cell Res. 2014;24:870–885. doi: 10.1038/cr.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Proudfoot C., McCulloch R. Distinct roles for two RAD51-related genes in Trypanosoma brucei antigenic variation. Nucleic Acids Res. 2005;33:6906–6919. doi: 10.1093/nar/gki996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCulloch R., Barry J.D. A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev. 1999;13:2875–2888. doi: 10.1101/gad.13.21.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartley C.L., McCulloch R. Trypanosoma brucei BRCA2 acts in antigenic variation and has undergone a recent expansion in BRC repeat number that is important during homologous recombination. Mol. Microbiol. 2008;68:1237–1251. doi: 10.1111/j.1365-2958.2008.06230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H.S., Cross G.A.M. TOPO3alpha influences antigenic variation by monitoring expression-site-associated VSG switching in Trypanosoma brucei. PLoS Pathog. 2010;6:e1000992. doi: 10.1371/journal.ppat.1000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dreesen O., Li B., Cross G.A.M. Telomere structure and function in trypanosomes: a proposal. Nat. Rev. Microbiol. 2007;5:70–75. doi: 10.1038/nrmicro1577. [DOI] [PubMed] [Google Scholar]
- 18.Yang X., Figueiredo L.M., Espinal A., Okubo E., Li B. RAP1 is essential for silencing telomeric variant surface glycoprotein genes in Trypanosoma brucei. Cell. 2009;137:99–109. doi: 10.1016/j.cell.2009.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pandya U.M., Sandhu R., Li B. Silencing subtelomeric VSGs by Trypanosoma brucei RAP1 at the insect stage involves chromatin structure changes. Nucleic Acids Res. 2013;41:7673–7682. doi: 10.1093/nar/gkt562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hovel-Miner G.A., Boothroyd C.E., Mugnier M., Dreesen O., Cross G.A.M., Papavasiliou F.N. Telomere length affects the frequency and mechanism of antigenic variation in Trypanosoma brucei. PLoS Pathog. 2012;8:e1002900. doi: 10.1371/journal.ppat.1002900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B., Espinal A., Cross G.A.M. Trypanosome telomeres are protected by a homologue of mammalian TRF2. Mol. Cell Biol. 2005;25:5011–5021. doi: 10.1128/MCB.25.12.5011-5021.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y., Yang Y., van Overbeek M., Donigian J.R., Baciu P., de Lange T., Lei M. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science. 2008;319:1092–1096. doi: 10.1126/science.1151804. [DOI] [PubMed] [Google Scholar]
- 23.Bianchi A., Smith S., Chong L., Elias P., de Lange T. TRF1 is a dimer and bends telomeric DNA. EMBO J. 1997;16:1785–1794. doi: 10.1093/emboj/16.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bianchi A., Stansel R.M., Fairall L., Griffith J.D., Rhodes D., de Lange T. TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J. 1999;18:5735–5744. doi: 10.1093/emboj/18.20.5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broccoli D., Smogorzewska A., Chong L., de Lange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 1997;17:231–235. doi: 10.1038/ng1097-231. [DOI] [PubMed] [Google Scholar]
- 26.Gommers-Ampt J., Lutgerink J., Borst P. A novel DNA nucleotide in Trypanosoma brucei only present in the mammalian phase of the life-cycle. Nucleic Acids Res. 1991;19:1745–1751. doi: 10.1093/nar/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gommers-Ampt J.H., Van Leeuwen F., de Beer A.L., Vliegenthart J.F., Dizdaroglu M., Kowalak J.A., Crain P.F., Borst P. beta-D-glucosyl-hydroxymethyluracil: a novel modified base present in the DNA of the parasitic protozoan T. brucei. Cell. 1993;75:1129–1136. doi: 10.1016/0092-8674(93)90322-h. [DOI] [PubMed] [Google Scholar]
- 28.van Leeuwen F., Wijsman E.R., Kuyl-Yeheskiely E., van der Marel G.A., van Boom J.H., Borst P. The telomeric GGGTAA repeats of Trypanosoma brucei contain the hypermodified base J in both strands. Nucleic Acids Res. 1996;24:2476–2482. doi: 10.1093/nar/24.13.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Leeuwen F., Wijsman E.R., Kieft R., van der Marel G.A., van Boom J.H., Borst P. Localization of the modified base J in telomeric VSG gene expression sites of Trypanosoma brucei. Genes Dev. 1997;11:3232–3241. doi: 10.1101/gad.11.23.3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Leeuwen F., Kieft R., Cross M., Borst P. Tandemly repeated DNA is a target for the partial replacement of thymine by beta-D-glucosyl-hydroxymethyluracil in Trypanosoma brucei. Mol. Biochem. Parasitol. 2000;109:133–145. doi: 10.1016/s0166-6851(00)00247-4. [DOI] [PubMed] [Google Scholar]
- 31.Cliffe L.J., Siegel T.N., Marshall M., Cross G.A., Sabatini R. Two thymidine hydroxylases differentially regulate the formation of glucosylated DNA at regions flanking polymerase II polycistronic transcription units throughout the genome of Trypanosoma brucei. Nucleic Acids Res. 2010;38:3923–3935. doi: 10.1093/nar/gkq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reynolds D., Cliffe L., Forstner K.U., Hon C.C., Siegel T.N., Sabatini R. Regulation of transcription termination by glucosylated hydroxymethyluracil, base J, in Leishmania major and Trypanosoma brucei. Nucleic Acids Res. 2014;42:9717–9729. doi: 10.1093/nar/gku714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ekanayake D., Sabatini R. Epigenetic regulation of polymerase II transcription initiation in Trypanosoma cruzi: modulation of nucleosome abundance, histone modification, and polymerase occupancy by O-linked thymine DNA glucosylation. Eukaryot. Cell. 2011;10:1465–1472. doi: 10.1128/EC.05185-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Luenen H.G., Farris C., Jan S., Genest P.A., Tripathi P., Velds A., Kerkhoven R.M., Nieuwland M., Haydock A., Ramasamy G., et al. Glucosylated hydroxymethyluracil, DNA base J, prevents transcriptional readthrough in Leishmania. Cell. 2012;150:909–921. doi: 10.1016/j.cell.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benmerzouga I., Concepcion-Acevedo J., Kim H.S., Vandoros A.V., Cross G.A.M., Klingbeil M.M., Li B. Trypanosoma brucei Orc1 is essential for nuclear DNA replication and affects both VSG silencing and VSG switching. Mol. Microbiol. 2013;87:196–210. doi: 10.1111/mmi.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wuthrich K. NMR—this other method for protein and nucleic acid structure determination. Acta Crystallogr. D Biol. Crystallogr. 1995;51:249–270. doi: 10.1107/S0907444994010188. [DOI] [PubMed] [Google Scholar]
- 37.Wider G. Structure determination of biological macromolecules in solution using nuclear magnetic resonance spectroscopy. Biotechniques. 2000;29:1278–1282. doi: 10.2144/00296ra01. [DOI] [PubMed] [Google Scholar]
- 38.Wishart D. NMR spectroscopy and protein structure determination: applications to drug discovery and development. Curr. Pharm. Biotechnol. 2005;6:105–120. doi: 10.2174/1389201053642367. [DOI] [PubMed] [Google Scholar]
- 39.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 40.Potterton L., McNicholas S., Krissinel E., Gruber J., Cowtan K., Emsley P., Murshudov G.N., Cohen S., Perrakis A., Noble M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 41.Roselin L.S., Lin M.S., Lin P.H., Chang Y., Chen W.Y. Recent trends and some applications of isothermal titration calorimetry in biotechnology. Biotechnol. J. 2010;5:85–98. doi: 10.1002/biot.200900092. [DOI] [PubMed] [Google Scholar]
- 42.Pierce M.M., Raman C.S., Nall B.T. Isothermal titration calorimetry of protein-protein interactions. Methods. 1999;19:213–221. doi: 10.1006/meth.1999.0852. [DOI] [PubMed] [Google Scholar]
- 43.Krell T., Lacal J., Garcia-Fontana C., Silva-Jimenez H., Rico-Jimenez M., Lugo A.C., Darias J.A., Ramos J.L. Characterization of molecular interactions using isothermal titration calorimetry. Methods Mol. Biol. 2014;1149:193–203. doi: 10.1007/978-1-4939-0473-0_16. [DOI] [PubMed] [Google Scholar]
- 44.Ladbury J.E. Application of isothermal titration calorimetry in the biological sciences: things are heating up. Biotechniques. 2004;37:885–887. doi: 10.2144/04376TE01. [DOI] [PubMed] [Google Scholar]
- 45.Wang Z., Morris J.C., Drew M.E., Englund P.T. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 2000;275:40174–40179. doi: 10.1074/jbc.M008405200. [DOI] [PubMed] [Google Scholar]
- 46.Scahill M.D., Pastar I., Cross G.A.M. CRE recombinase-based positive-negative selection systems for genetic manipulation in Trypanosoma brucei. Mol. Biochem. Parasitol. 2008;157:73–82. doi: 10.1016/j.molbiopara.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith S., de Lange T. TRF1, a mammalian telomeric protein. Trends Genet. 1997;13:21–26. doi: 10.1016/s0168-9525(96)10052-4. [DOI] [PubMed] [Google Scholar]
- 48.Oh I.H., Reddy E.P. The myb gene family in cell growth, differentiation and apoptosis. Oncogene. 1999;18:3017–3033. doi: 10.1038/sj.onc.1202839. [DOI] [PubMed] [Google Scholar]
- 49.Nishikawa T., Okamura H., Nagadoi A., Koig P., Rhodes D., Nishimura Y. Solution structure of a telomeric DNA complex of human TRF1. Structure. 2001;9:1237–1251. doi: 10.1016/s0969-2126(01)00688-8. [DOI] [PubMed] [Google Scholar]
- 50.Hanaoka S., Nagadoi A., Nishimura Y. Comparison between TRF2 and TRF1 of their telomeric DNA-bound structures and DNA-binding activities. Protein Sci. 2005;14:119–130. doi: 10.1110/ps.04983705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose N.R., McDonough M.A., King O.N., Kawamura A., Schofield C.J. Inhibition of 2-oxoglutarate dependent oxygenases. Chem. Soc. Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 52.Cliffe L.J., Hirsch G., Wang J., Ekanayake D., Bullard W., Hu M., Wang Y., Sabatini R. JBP1 and JBP2 proteins are Fe2+/2-oxoglutarate-dependent dioxygenases regulating hydroxylation of thymidine residues in trypanosome DNA. J. Biol. Chem. 2012;287:19886–19895. doi: 10.1074/jbc.M112.341974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McClelland M., Nelson M., Raschke E. Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res. 1994;22:3640–3659. doi: 10.1093/nar/22.17.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernards A., van Harten-Loosbroek N., Borst P. Modification of telomeric DNA in Trypanosoma brucei; a role in antigenic variation. Nucleic Acids Res. 1984;12:4153–4170. doi: 10.1093/nar/12.10.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cliffe L.J., Kieft R., Southern T., Birkeland S.R., Marshall M., Sweeney K., Sabatini R. JBP1 and JBP2 are two distinct thymidine hydroxylases involved in J biosynthesis in genomic DNA of African trypanosomes. Nucleic Acids Res. 2009;37:1452–1462. doi: 10.1093/nar/gkn1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Court R., Chapman L., Fairall L., Rhodes D. How the human telomeric proteins TRF1 and TRF2 recognize telomeric DNA: a view from high-resolution crystal structures. EMBO Rep. 2005;6:39–45. doi: 10.1038/sj.embor.7400314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 58.van Steensel B., Smogorzewska A., de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 59.Karlseder J., Broccoli D., Dai Y.M., Hardy S., de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 60.Celli G.B., de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 61.van Steensel B., de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997;385:740–743. doi: 10.1038/385740a0. [DOI] [PubMed] [Google Scholar]
- 62.Sfeir A., Kosiyatrakul S.T., Hockemeyer D., MacRae S.L., Karlseder J., Schildkraut C.L., de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Palm W., de Lange T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 64.Landeira D., Bart J.M., Van Tyne D., Navarro M. Cohesin regulates VSG monoallelic expression in trypanosomes. J. Cell Biol. 2009;186:243–254. doi: 10.1083/jcb.200902119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Povelones M.L., Gluenz E., Dembek M., Gull K., Rudenko G. Histone H1 plays a role in heterochromatin formation and VSG expression site silencing in Trypanosoma brucei. PLoS Pathog. 2012;8:e1003010. doi: 10.1371/journal.ppat.1003010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson N.P., McCulloch R., Conway C., Browitt A., Barry J.D. Inactivation of Mre11 does not affect VSG gene duplication mediated by homologous recombination in Trypanosoma brucei. J. Biol. Chem. 2002;277:26185–26193. doi: 10.1074/jbc.M203205200. [DOI] [PubMed] [Google Scholar]
- 67.Munoz-Jordan J.L., Cross G.A.M. Telomere shortening and cell cycle arrest in Trypanosoma brucei expressing human telomeric repeat factor TRF1. Mol. Biochem. Parasitol. 2001;114:169–181. doi: 10.1016/s0166-6851(01)00259-6. [DOI] [PubMed] [Google Scholar]
- 68.Ogata K., Morikawa S., Nakamura H., Sekikawa A., Inoue T., Kanai H., Sarai A., Ishii S., Nishimura Y. Solution structure of a specific DNA complex of the Myb DNA-binding domain with cooperative recognition helices. Cell. 1994;79:639–648. doi: 10.1016/0092-8674(94)90549-5. [DOI] [PubMed] [Google Scholar]
- 69.Murnane J.P. Telomeres and chromosome instability. DNA Repair (Amst.) 2006;5:1082–1092. doi: 10.1016/j.dnarep.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 70.Smith T.K., Vasileva N., Gluenz E., Terry S., Portman N., Kramer S., Carrington M., Michaeli S., Gull K., Rudenko G. Blocking variant surface glycoprotein synthesis in Trypanosoma brucei triggers a general arrest in translation initiation. PLoS One. 2009;4:e7532. doi: 10.1371/journal.pone.0007532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burton P., McBride D.J., Wilkes J.M., Barry J.D., McCulloch R. Ku heterodimer-independent end joining in Trypanosoma brucei cell extracts relies upon sequence microhomology. Eukaryot. Cell. 2007;6:1773–1781. doi: 10.1128/EC.00212-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Conway C., McCulloch R., Ginger M.L., Robinson N.P., Browitt A., Barry J.D. Ku is important for telomere maintenance, but not for differential expression of telomeric VSG genes, in African trypanosomes. J. Biol. Chem. 2002;277:21269–21277. doi: 10.1074/jbc.M200550200. [DOI] [PubMed] [Google Scholar]
- 73.Janzen C.J., Lander F., Dreesen O., Cross G.A.M. Telomere length regulation and transcriptional silencing in KU80-deficient Trypanosoma brucei. Nucleic Acids Res. 2004;32:6575–6584. doi: 10.1093/nar/gkh991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boothroyd C.E., Dreesen O., Leonova T., Ly K.I., Figueiredo L.M., Cross G.A.M., Papavasiliou F.N. A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature. 2009;459:278–281. doi: 10.1038/nature07982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ye J., Lenain C., Bauwens S., Rizzo A., Saint-Leger A., Poulet A., Benarroch D., Magdinier F., Morere J., Amiard S., et al. TRF2 and apollo cooperate with topoisomerase 2alpha to protect human telomeres from replicative damage. Cell. 2010;142:230–242. doi: 10.1016/j.cell.2010.05.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.