


Abstract
The Microprocessor plays an essential role in canonical miRNA biogenesis by facilitating cleavage of stem-loop structures in primary transcripts to yield pre-miRNAs. Although miRNA biogenesis has been extensively studied through biochemical and molecular genetic approaches, it has yet to be addressed to what extent the current miRNA biogenesis models hold true in intact cells. To address the issues of in vivo recognition and cleavage by the Microprocessor, we investigate RNAs that are associated with DGCR8 and Drosha by using immunoprecipitation coupled with next-generation sequencing. Here, we present global protein–RNA interactions with unprecedented sensitivity and specificity. Our data indicate that precursors of canonical miRNAs and miRNA-like hairpins are the major substrates of the Microprocessor. As a result of specific enrichment of nascent cleavage products, we are able to pinpoint the Microprocessor-mediated cleavage sites per se at single-nucleotide resolution. Unexpectedly, a 2-nt 3′ overhang invariably exists at the ends of cleaved bases instead of nascent pre-miRNAs. Besides canonical miRNA precursors, we find that two novel miRNA-like structures embedded in mRNAs are cleaved to yield pre-miRNA-like hairpins, uncoupled from miRNA maturation. Our data provide a framework for in vivo Microprocessor-mediated cleavage and a foundation for experimental and computational studies on miRNA biogenesis in living cells.
INTRODUCTION
MicroRNAs (miRNAs) are a large class of ∼22-nt non-coding RNAs. As a component of the RNA-induced silencing complex (RISC), they regulate a large number of genes by translational repression and/or mRNA degradation. The relevance of miRNAs to key biological processes, such as cell-cycle control and differentiation, has been firmly established. In the canonical miRNA biogenesis pathway, imperfect stem-loop structures located in the primary transcripts (pri-miRNA) are sequentially processed by two RNase III family endonucleases, Drosha and Dicer. Genetic ablation of the mouse Drosha and Dicer genes leads to defects in the processing of miRNA precursors, manifesting crucial roles of these two genes in miRNA biogenesis. In the nucleus, Drosha associates with an RNA-binding protein, DGCR8, to form a functional complex, the Microprocessor (1). The Microprocessor cleaves two staggering bonds in the middle of the stem in pri-miRNA to yield a shorter hairpin-structured precursor miRNA (pre-miRNA) with a 2-nt 3′ overhang that is routinely observed in RNase III-mediated cleavage (2). Microprocessor-mediated cleavage is important not only for defining one end of the mature miRNAs, but also for setting the stage for the selection of other cleavage sites by Dicer-mediated cleavage (3,4). Minor miRNAs, or non-canonical miRNAs, bypass the Microprocessor-mediated cleavage stage. In the cytoplasm, Dicer further processes pre-miRNA into double-stranded miRNA. According to the thermodynamic stability model of strand selection, the strand with relative instability at the 5′ end is selected as the mature miRNA to associate with Argonaute (AGO) proteins, and the other strand is degraded (5). Then, the AGO protein-associated miRNA recognizes the target mRNAs by partial base pairing and represses their expression.
The prototypic miRNAs, lin-4 and let-7, were identified in Caenorhabditis elegans by positional cloning of heterochronic genes that control developmental timing. Since then, sequencing of size-fractionated RNA has driven most new discovery of miRNAs. Recently, broad implementation of next-generation sequencing technologies has immensely helped discover miRNAs and other small RNAs expressed at very low levels. Unfortunately, abundant similar-sized non-coding RNAs have hampered applications of next-generation sequencing technologies for profiling miRNA precursors. Consequently, verification of biogenesis and biological functions of individual miRNA still heavily relies on conventional biochemical and molecular genetic approaches that are very low in throughput. To compound the situation, there is no certainty that the information obtained by using current technologies is reflective of miRNA biogenesis in the intact biological setting. Accordingly, distinction of bona fide miRNAs from similar-sized small RNAs has become a significant challenge in the miRNA field.
In contrast to miRNA biogenesis, miRNA-independent functions of the Microprocessor have been controversial. Kim et al. revealed that Microprocessor-mediated cleavage destabilizes the DGCR8 mRNA, and, in turn, DGCR8 stabilizes Drosha by protein–protein interactions to form a regulatory circuit (6). While it was proposed that Microprocessor-mediated cleavage of mRNA is a dedicated regulation mechanism to DGCR8 (7), the Microprocessor was also claimed to directly regulate various mRNAs in a tissue-specific manner (8,9). To our understanding, DGCR8 still remains the only gene supported by multiple lines of solid evidence of mRNA cleavage by the Microprocessor.
In order to clear the ambiguities of recognition and cleavage by the Microprocessor in its biological setting, we focused on the elucidation of Microprocessor-mediated cleavage in human embryonic stem cells (hESC), which have an intact genetic make-up, rather than in cancerous cell lines. Moreover, the relevance of miRNA biogenesis to fundamental biological processes was proved in mammalian ESCs. To obtain comprehensive information on the molecular interactions between the Microprocessor and target RNAs, we unprecedentedly repeated biologically independent experiments of crosslinking and immunoprecipitation (CLIP) for key components of the Microprocessor, DGCR8 and Drosha, followed by next-generation sequencing analysis. In this manuscript, we present a global view of direct protein–RNA interactions with unparalleled sensitivity and specificity. Furthermore, we determine in vivo cleavage sites at single-nucleotide resolution by capturing nascent pre-miRNAs, rather than by drawing inferences from sequences of small RNAs or steady-state pre-miRNAs. The human Microprocessor mainly cleaves canonical pri-miRNAs and, to a lesser degree, miRNA-like stem-loop structures. We find significant discrepancies between authentic Microprocessor-mediated cleavage and common notions, including the characteristic 2-nt-3′-overhang structure of nascent canonical pre-miRNAs and homogeneous cleavage site selection. These findings provide more refined guidelines of bona fide Microprocessor-mediated cleavage.
MATERIALS AND METHODS
Culture and gene targeting of H1 hESC
H1 hESCs were maintained on feeder plate as previously described (10). Gene targeting by BAC-based homologous recombination was described before (11). Briefly, exponentially growing H1 cells pre-treated with 10 μM Y-27632 for 2 h were dissociated to single cells by accutase and washed twice with cold phosphate buffered saline (PBS). Approximately twenty million cells were resuspended in PBS with 50 μg of a linearized targeting construct for Drosha (Supplementary Information and Supplementary Figure S1) and transfected by electroporation with a BioRad Gene Pulser II (320 V, 200 uF). The transfected cells were plated on a feeder layer and selected with 50 μg/ml of G418. After 2 weeks, G418-resistant clones were individually picked and expanded.
As the H1 line is heterozygous at an SNP, rs3805525, which is located just upstream of exon 35, we exploited the loss of heterozygosity caused by gene targeting. A polymerase chain reaction (PCR)-based assay was used to confirm the loss of heterozygosity. PCR primers (i34F/e35R) amplified regions around the single nucleotide polymorphic site only from unmodified loci. Among the two alleles, only the G allele was cut by HpyCH4IV. All works involving hESCs were approved by IRB of Korea University Guro Hospital.
IP of AGO2
Flag-AGO2 H1 cells were lysed in IP buffer containing 1% Empigen BB, 0.1 mM DTT and protease inhibitor cocktail (Roche) in PBS. After centrifugation at 13 000 revolutions per minute (rpm) for 15 min, the supernatant was incubated with an anti-Flag antibody (Wako) or immunoglobulin G (IgG) bound to Protein A/G Dynabeads (Invitrogen). Beads were washed five times with IP buffer. AGO2-bound RNAs were extracted with phenol/chloroform/isoamyl alcohol (Sigma) and precipitated with ethanol.
CLIP experiments
CLIP-seq was based on a previously described protocol with some modifications (12,13). H1 cells were irradiated with 300 mJ/cm2 of UVC light (254 nm) by using the CL-1000 ultraviolet crosslinker (UVP). Two million cells were resuspended in 250 μl lysis buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1 mM MgCl2, 0.1 mM CaCl2, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate (SDS)) containing ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail (Roche) and RNase inhibitor (Ambion) and briefly sonicated by using the Bioruptor. The lysates were treated with 5 μl of Turbo DNase (Ambion) and 10 μl of 10 U/μl or 1 U/μl RNase I (Ambion) at 37°C for 3 min. Cellular debris was removed by centrifugation at 13 000 rpm for 15 min. Protein-RNA complexes were immunopurified with an anti-Flag or anti-DGCR8 antibody (Bethyl laboratories) bound to Protein A/G Dynabeads. Beads were washed twice with high-salt wash buffer (50 mM Tris-HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) followed by two washes with PNK buffer (20 mM Tris-HCl pH 7.4, 10 mM MgCl2 and 0.2% Tween-20). For dephosphorylation of 3′ ends, 1U of FastAP alkaline phosphatase (Fermentas) was added to the beads and incubated at 37°C for 20 min. After washing the beads twice with PNK buffer, protein-bound RNAs were ligated to a pre-adenylylated 3′ adaptor overnight at 16°C. Beads were washed twice with PNK buffer again and labeled with [γ- 32P] adenosine triphosphate for visualization. The labeled RNAs were separated by 4–12% NuPAGE Bis-Tris gel (Invitrogen), transferred to a nitrocellulose membrane and excised from the membrane. The protein was degraded by proteinase K treatment, and RNA was recovered by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation (Supplementary Figure S2).
RNA IP (RIP)
H1 cells were washed twice with PBS and cross-linked with 1% formaldehyde at room temperature for 10 min. After washing twice with PBS, cells were collected and sonicated in 0.2 ml lysis buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS, 0.5% Empigen BB). After centrifugation at 13 000 rpm for 15 min, the supernatant was diluted 2.5-fold in IP buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100) and incubated with an anti-DGCR8 antibody or IgG bound to Protein A/G Dynabeads at 4°C for 2 h. Beads were serially washed with 1 ml washing buffer I (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100), washing buffer II (20 mM Tris-HCl pH 8.0, 2 mM EDTA), washing buffer III (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% NP-40, 1% deoxycholate, 0.25 M LiCl) and TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Beads were then treated with 5 μl of Turbo DNase at 37°C for 3 min and washed twice with PNK buffer. FastAP alkaline phosphatase was added and incubated at 37°C for 20 min. Beads were washed twice with PNK buffer again. DGCR8-bound RNAs were ligated to a pre-adenylylated 3′ adaptor overnight at 16°C and washed twice with PNK buffer. Protein-RNA complexes were removed from the beads through 30 min incubation with 1% SDS. Crosslinked proteins were eliminated by proteinase K treatment at 65°C for 2 h, and RNA was recovered by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation.
Sequencing library construction
Sequencing library preparation was performed according to a previously described protocol (13). Protein-associated RNAs purified by IP, CLIP or RIP were ligated to a pre-adenylylated 3′ adaptor that contains a 2-nt adapter code for de-multiplexing and a 3-nt random sequence to minimize the loss of information after collapsing identical sequences. The RNA-adapter hybrids were converted into cDNAs with reverse transcriptase (RT) primer and SuperScript III RT (Invitrogen). The cDNAs were size-fractionated by electrophoresis on a 12% denaturing polyacrylamide gel and circularized by single-stranded DNA ligase (Epicentre). Circularized cDNAs were amplified with PCR primers, in which one of them contained a 6-nt primer code for de-multiplexing. PCR products were purified by agarose gel elution and subjected to next-generation sequencing. Primer and adaptor sequences are detailed in Supplementary Table S1.
Read processing and mapping
Multiplexed raw sequence reads were de-multiplexed by using adapter and primer codes, and adapter-derived sequences were trimmed before further analysis sequences by using functions in a Bioconductor package, ShortRead (14). CLIP-seq reads were collapsed before mapping to database sequences. The processed reads mapped to abundant non-coding RNA sequences, by using Bowtie short read aligner (version 0.12.7) with parameters -q –m 50 -n 2 -l 44 -5 1 -3 5 -a –best –strata -p 7 –sam. Reads were uniquely mapped to human genome (hg19) and pre-miRNA (miRBase release v20) sequences by changing the -m parameter to 1. Coverage and false discovery rate (FDR) were obtained by using functions in a Bioconductor package, chipseq. Categorization and quantification of reads uniquely mapping to the human genome were conducted by using functions in the GenomicFeatures package. Data visualization was performed by using functions in an R package, ggplot2 and a Bioconductor package, ggbio (15). Prediction of novel miRNAs by the miRDeep2 package was performed according to the manual (16).
RESULTS
Discrepancies between AGO2-associated miRNAs and miRNA annotations
To identify functional miRNAs, we enriched RISC-associated miRNAs in hESCs grown without a feeder layer by IP of flag-AGO2 protein (manuscript submitted). The IP-enriched RNAs were used to construct libraries by using the Single Ligation, Elongation, Circularization method, and the libraries were sequenced by using the Illumina's HiSeq platform (13). Note that 87.5% (3 068 716) of the total reads longer than 18 nt (3 507 476) mapped to pre-miRNAs in the miRBase database (release v20). With two mismatches allowed, 770 228 (25.1%) reads mapping to pre-miRNAs did not map to annotated mature miRNAs. The mapping result suggests that significant proportions of miRNAs in hESCs are substantially heterogeneous, and/or the annotations in miRBase are considerably incomplete or flawed.
Remarkably, we noted that the representative size of miRNAs in hESCs was 23 nt (1 449 405 reads) rather than 22 nt (636 712 reads) (Supplementary Figure S3A). As systematic biases in favor of certain miRNAs have been known as a common feature of different library preparation methods, the anomalistic size distribution could be a consequence of the biases of our library preparation method (17,18). To exclude this possibility, we retrieved publically available sequence reads of a small RNA library from H1 hESCs (http://www.ncbi.nlm.nih.gov/sra, SRA SRX007166), prepared by using the Illumina's small RNA sample preparation kit, to observe similar size distribution of miRNAs (Supplementary Figure S3B). Moreover, the annotated sizes in miRBase were 23 nt for many of the highest expressed miRNAs in hESCs, although 22 nt was the representative size as a whole (Supplementary Figure S3C). Taken together, the observed size distribution of AGO2-associated miRNAs very likely reflects an intrinsic property of miRNAs highly expressed in hESCs.
DGCR8-CLIP-seq from hESCs
Despite the intense investigation of miRNA biogenesis, our understanding has a significant pitfall because biochemical and mutational studies are only based on a handful of model miRNAs. It still remains unanswered whether the suggested biogenesis models are valid in the undisturbed biological context. To address this issue, we captured RNAs directly bound to DGCR8 in hESCs by using a technique, CLIP, in which UV-light irradiation is used to irreversibly crosslink RNAs to proteins (19). Contrary to chemical crosslinking, UV-crosslinking allows for enormous enrichment of transient protein-RNA complexes in the cell, sufficient enough to apply next-generation sequencing analysis (CLIP-seq). Since UV irradiation does not crosslink proteins, the CLIP method utilizes not only stringent IP but also gel electrophoresis to enrich RNAs crosslinked to the protein.
We conducted IP experiments to enrich RNA-DGCR8 complexes from UV-light irradiated H1 hESCs. The IP-enriched RNAs were ligated to a pre-activated DNA linker on protein A/G magnetic beads, followed by gel electrophoresis and membrane transfer. Recovered RNA-linker hybrids from a nitrocellulose membrane were reverse-transcribed to cDNA, circularized, PCR-amplified and sequenced by using the Illumina HiSeq platform to produce 45-nt RNA sequences. Ule et al. have reported a similar procedure, iCLIP, before this manuscript (20). Notably, notwithstanding efficient IP, the radioactivity from enriched RNAs was abnormally but consistently weak (Supplementary Figure S4, lanes 6 and 7), indicating the amount of recovered RNAs were very low. To obtain more comprehensive information of RNAs in direct contact with DGCR8, we conducted three sets of biologically independent CLIP experiments. Each experiment set consisted of at least two biological replicates and an IgG IP negative control. In the two experiment sets, we treated hESC extracts with two different concentrations of RNase I to determine the optimal RNase I-treatment conditions.
We aligned 2 334 407 CLIP-seq reads with non-coding RNAs and human genome sequences (UCSC hg19) after removing identical reads, or collapsing, from individual libraries and combining the collapsed reads. We note that we synthesized pre-activated adapters with randomized 3-nt code to recover independent reads with identical sequences in collapsing and chose minimal PCR amplification cycles to minimize the production of repeated reads from the same amplicons. Because of high read depth of the current deep-sequencing platform, the magnitude of enrichment by the CLIP method, and the limited number of enriched RNA molecules from DGCR8-CLIP, sequencing of independent CLIP-seq libraries was much more informative than deeper sequencing of a single library. In spite of the weak signals after IP, the collapsed reads were uniquely aligned with the human genome to yield highly reproducible stacks with an unprecedented magnitude of coverage.
As expected, the miRNA precursors were the most enriched class of non-coding RNAs by our DGCR8-CLIP experiments. Note that 362 547 (14.4%) reads of the combined collapsed reads (2 515 057) mapped to known pre-miRNA and 60 nt flanking sequences which amounted to ∼0.005% of the human genome. In addition, 440 021 (17.5%) reads uniquely mapped to pre-miRNA and flanking 1 kb segments (Figure 1A). Coverage for certain miRNA precursors was strikingly deep as manifested by the maximum coverage of 41 953 for pre-miR-367 (Figure 1B) and average maximum coverage of 17 929 for the 10 highest pre-miRNAs, which largely correlated with the 10 highest expressed miRNAs in hESCs. In contrast, we barely found any reads identical to the mature miRNA sequences, suggesting an exceptionally high specificity of our CLIP experiments. While stable non-coding RNAs constituted most of the RNAs in the cell, reads mapping to rRNA, tRNA, snRNA and snoRNA accounted for 1.6%, 2.1%, 1.2% and 0.2%, respectively. CLIP-seq reads uniquely mapping to the remaining genomic locations accounted for ∼44.1% (1 107 996) of collapsed reads. Note that 211 546 (19.1%) reads were located in intergenic regions, and 896 447 (80.9%) reads mapped to transcribed regions. Note that 780 147 reads and 166 300 reads were aligned to introns and exons, respectively, which indicate that the primary transcript was highly enriched. Reads uniquely mapping to coding regions, 5′ UTRs, and 3′ UTRs amounted to 65 936, 19 526 and 82 213, respectively. Considering the low maximum coverage, the proportion of reads mapping to transcribed regions is probably overestimated after collapsing (Supplementary Figure S5). Together, our data suggest that miRNA precursors are major binding targets of DGCR8.
Figure 1.
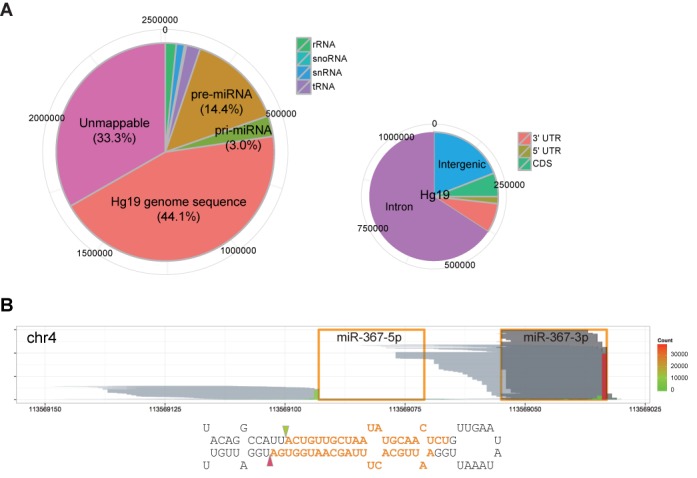
miRNA precursors are a major RNA class interacting with DGCR8. (A) Distribution of DGCR8-CLIP-seq reads on the genome. The larger pie chart shows percentages of collapsed reads mapping to non-coding RNAs and the UCSC human genome (hg19). The smaller pie chart shows distribution of reads uniquely mapping to the hg19 human genome in the intergenic, intronic, coding regions and UTRs. (B) Reads from the DGCR8-CLIP and AGO2-IP libraries aligned to the mir-367 locus. Orange line boxes and orange bold letters denote the annotated mature miR-367–5p and -3p in miRBase. Each pale gray horizontal line represents each collapsed read from the DGCR8-CLIP libraries. Positions of the vertical bars represent the positions of the 3′ terminal nucleotides of DGCR8-CLIP-seq reads, and heights and filled colors of the vertical bars represent counts of the 3′ terminal nucleotides of reads. Note that the pale gray lines map to the miR-367 precursors, but not to the mature miR-367, and the predominant 3′ termini of DGCR8-CLIP-seq reads, denoted by triangles, match the annotated cleavage sites in miRBase. Each dark gray horizontal line represents each read from the AGO2-IP libraries. For better visualization, 6000 reads are sampled from 338 890 AGO2-IP reads after mapping to the mir-367 locus. Note that the uneven 5′ ends of reads from the AGO2-IP libraries are caused by non-templated nucleotide addition to the 3′ end of cDNA.
Nascent canonical pre-miRNAs are specifically enriched by DGCR8-CLIP
Next, we further analyzed CLIP-seq reads mapping to known miRNA loci. From the 1870 annotated human miRNA loci, at least 1 unique read to known 470 pre-miRNAs and maximum coverage was at least 8 for 274 pre-miRNAs (FDR < 0.0001, calculated by using the Poisson-based approach for estimating the noise distribution by using the chipseq package [http://www.bioconductor.org/], Supplementary Table S2). As expected, reads from AGO2-IP-derived libraries were also mapped to 272 miRNA loci that were enriched by DGCR8-CLIP with FDR < 0.0001. Not surprisingly, one of the unmapped mature miRNAs was miR-98, which is post-transcriptionally repressed by LIN28 in hESCs (21). The other unmapped mature miRNA, miR-325, resides in LINE sequences. We ran RepeatMasker (http://www.repeatmasker.org) on 274 loci to reveal that 241 loci reside in non-repeat sequences, and 33 pre-miRNAs contained sequences of genomic repeat elements, such as LINE, SINE, LTR and simple repeats. Interestingly, mature miRNAs derived from repeat elements were expressed at relatively low levels.
Besides canonical miRNAs, DGCR8-independent miRNAs originate from endogenous short hairpin RNAs (shRNAs) and snoRNAs (22,23). In miRBase, 240 mirtrons, which bypass Microprocessor-mediated processing by splicing and debranching from the primary transcripts, are also annotated (24). We compared the read counts from DGCR8-CLIP and AGO2-IP-derived libraries, which we allocated to known or putative DGCR8-independent miRNAs (22). While 4366 and 2727 reads from AGO2-IP-enriched reads mapped to miR-320a and -484, respectively, no DGCR8-CLIP-seq reads mapped to the cognate hairpin structures. The result is consistent with previous reports that mouse miR-320a and -484 derive from endogenous shRNAs in a DGCR8-independent manner. Similarly, while AGO2-IP-derived reads mapped to small RNAs from ACA45 snoRNA, snoRNA36B/miR-664a and mirtrons, no DGCR8-CLIP-seq reads mapped to the cognate hairpin structures. Taken together, these results indicate that DGCR8-CLIP-enriched 274 human miRNAs are processed through the canonical biogenesis pathway.
A peculiarity of our DGCR8-CLIP-seq reads mapping to the miRNA loci was that, in many cases, the positions of the 3′ end exactly matched the annotated Microprocessor-mediated cleavage sites in miRBase. This suggests that CLIP-enriched miRNAs are mostly nascent pre-miRNAs cleaved by the Microprocessor. However, annotations of the Microprocessor-mediated cleavage sites should be taken with caution, since most annotated sites were inferred from mature miRNAs rather than directly determined from nascent pre-miRNAs. Because the guide strand of the miRNA duplex is usually loaded onto the RISC in a highly asymmetric fashion, it is difficult to infer the cleavage sites on the passenger strand. To exacerbate the problem, the 3′ terminal residues of pre-miRNA are frequently trimmed by nucleases and modified by non-templated nucleotide addition. To test whether DGCR8-CLIP-seq reads are reflective of genuine Microprocessor-mediated cleavage sites, we compared the 3′-end positions of CLIP-seq reads with actual in vitro cleavage sites of miR-16–1, miR-30a, let-7a-1 and let-7d, which were directly determined by cloning and sequencing of pre-miRNAs processed in vitro by the purified Microprocessor (25–27). Notably, the reported in vitro cleavage sites of miR-16–1 and let-7d were different from the annotated cleavage sites in miRBase. Mapping to the miRNA loci showed that all the major 3′ ends of CLIP-seq reads were identical to the in vitro Microprocessor-cleavage sites (Figure 2). Based on the cloned pre-miRNA sequences from HeLa cells, Kim et al. reported that the scissile bonds in a subset of let-7 family members are located at an evolutionarily conserved bulged structure to yield pre-miRNA bearing a 1-nt 5′ overhang and a 2-nt 3′ overhang (27). They proposed that the atypical 3′ ends of pre-let-7s are mono-uridylated for efficient Dicer-mediated processing. Consistent with their report, the DGCR8-CLIP-seq reads did not contain an additional uridylate residue at the 3′ end of pre-let-7a, though ∼50% of the reads of AGO2-associated let-7a-3p were mono-uridylated at the 3′ ends (see also Supplementary Figure S6). Taken together, the analyses indicate that we specifically enriched nascent pre-miRNAs, but did not enrich modified pre-miRNAs by non-templated nucleotide addition, which were frequently observed at the 3′ ends of steady-state pre-miRNAs (28). Moreover, we were able to determine the cleavage sites per se by analyzing the 3′-end positions of DGCR8-CLIP reads at single-nucleotide resolution.
Figure 2.
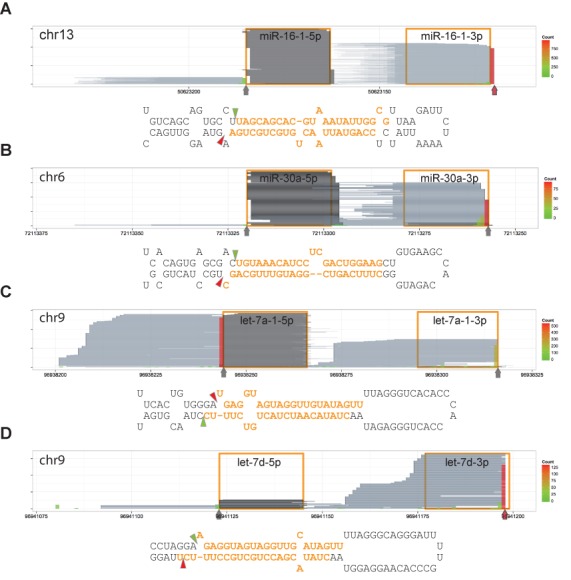
Major 3′ ends of reads from the DGCR8-CLIP libraries are identical to the in vitro cleavage sites. Reads are represented and denoted as the way in Figure 1B. Vertical arrows and triangles represent the predominant 3′ ends of DGCR8-CLIP reads. Predominant 3′ ends of reads that are different from annotated cleavage sites in miRBase are marked by red vertical arrows. (A) mir-16–1 locus. (B) mir-30a locus. (C) let-7a-1 locus. (D) let-7d locus. Note that the predominant 3′ ends of DGCR8-CLIP reads are identical to previously reported in vitro Microprocessor-mediated cleavage sites (25–27) rather than the annotations in miRBase.
Determination of Microprocessor-mediated cleavage sites and end structures of known miRNAs
It was proposed that the molecular determinants for cleavage site selection by the Microprocessor are a ∼11 bp double-stranded RNA stem beyond the cleavage sites and single-stranded flanking segments (29). While it has been presumed that cleavage by the Microprocessor yields homogeneous pre-miRNA bearing a 2-nt 3′ overhang, it is uncertain to what extent the rule holds true in the cell. Intriguingly, we found that ∼30% of selected canonical pre-miRNAs (maximum coverage >200) have other structures than the expected 2-nt 3′ overhang structure on the basis of close inspection of the annotations in miRBase. So, based on our data, we reexamined the Microprocessor-mediated cleavage sites of annotated miRNAs. At the conservative estimate, cleavage sites of 68 miRNAs were incorrectly annotated (Supplementary Table S2). Incorrect annotations were mostly found at the 3′ end of pre-miRNAs, consistent with the observations that the 3′ terminal residues of pre-miRNAs are more susceptible to trimming and modification than the 5′ terminal residues as exemplified by miR-363 and miR-505 (Figure 3A and B). The majority of 3′ terminal uridylate residues were added after the Drosha cleavage stage, epitomizing a pitfall in inferring cleavage sites only from small RNA sequencing data. Consistent with the aforementioned pri-miRNA recognition model, many pri-miRNAs enriched by DGCR8-CLIP experiments were predicted to have a ∼3-turn imperfect stem. While there were annotations of hairpin structures lacking the ∼11-bp helix in miRBase, such as pri-miR-302b and -660, we found alternative hairpin structures bearing longer helixes from sequences encompassing wider regions. However, pri-miR-29a and -92a-1, which apparently lack the ∼11-bp RNA helix, were enriched by DGCR8-CLIP, and cognate mature miRNAs were enriched by AGO2-IP, showing that the ∼3-turn helix is not always predicted from canonical pri-miRNAs.
Figure 3.
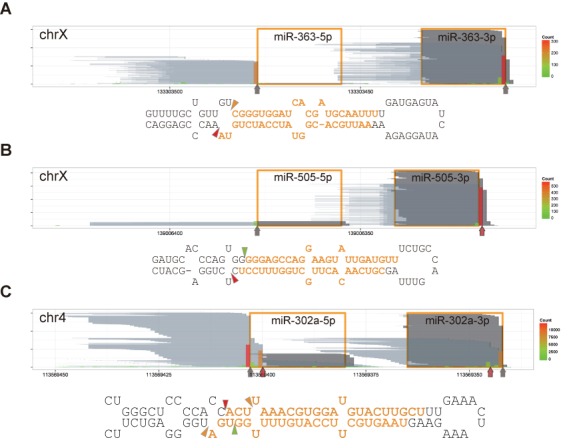
Non-templated nucleotide addition and alternative cleavage. Reads are represented as in Figure 1B. Vertical arrows and triangles indicate the major 3′ ends of DGCR8-CLIP-seq reads. Red vertical arrows mark the major 3′ ends of reads different from the Microprocessor-mediated cleavage site annotations or missing in miRBase. (A) Non-templated nucleotide addition to miR-363. Note that ∼50% of miR-363–3p from the AGO2-IP libraries have additional nucleotide(s) at the 3′ ends. (B) Non-templated nucleotide addition to miR-505. (C) Alternative cleavage sites of miR-302a. Individual short (up to 46 nt) and long (from 51 to 146 nt) reads of miR-302a precursors from the DGCR8-CLIP libraries are represented by pale gray and blue lines in Supplementary Figure S7, respectively. Note that 61- and 55-nt long reads correspond to predominant pre-miRNA isoforms, but miR-302a-5p is derived mainly from the 55-nt isoform, and miR-302a-3p is derived from the 61-nt isoform. Note the discrepancies between annotated miR-302a-5p and AGO2-associated miR-302a-5p.
Although miRNA was initially recognized as a homogeneous population of regulatory small RNAs, deep sequencing data show that mature miRNAs comprise of various isoforms (30). To test whether such heterogeneity originates from Microprocessor-mediated cleavage, we inspected the homogeneity of Microprocessor-mediated cleavage sites. We found that the in vivo cleavage sites of most miRNAs were largely homogeneous, whereas the cleavage sites of certain miRNAs were evidently heterogeneous. Notably, the four major cleavage sites of the second-most miRNA in hESCs, miR-302a, indicated that alternative processing might yield two prominent pre-miRNA isoforms bearing the 2-nt-3′ overhang structure (Figure 3C). We excluded the possibility that the heterogeneity of pre-miR302a was introduced by RNase treatment on the basis that the same heterogeneous 3′ ends were observed with reads from a DGCR8-CLIP library prepared without RNase treatment (Supplementary Figure S7A). We further validated the heterogeneity of pre-miR-302a by cloning and sequencing of the 3′ halves of steady-state pre-miR-302a (Supplementary Figure S8). To verify the presumed pre-miR-302a isoforms from individual entire molecules, we additionally read cDNAs up to 151 bp by using the Illumina's MiSeq platform. We found 1740 collapsed reads mapping to the entire pre-miR-302a isoforms, the majority of which were 61- and 55-nt long (Supplementary Figure S9). Reads from the AGO2-IP-derived libraries showed that both pre-miR-302a isoforms were further processed by Dicer and loaded onto the RISC. While almost all miR-302a-3p originated from the 61-nt pre-miRNA isoform, miR-302a-5p mostly originated from the 55-nt pre-miRNA isoform. The isoform selection was consistent with the thermodynamic stability rule. Similarly, multiple cleavage sites of miR-302c in the same cluster were also detected. Most miR-302c-3p derived from a long pre-miR-302c isoform. Though miR-302c-5p was not as abundant as miR-302a-5p, most of the miR-302c-5p originated from a shorter isoform. The results directly manifest that in vivo Microprocessor-mediated cleavage sites of certain miRNAs are highly heterogeneous, and to some extent heterogeneity of mature miRNAs stems from cleavage by the Microprocessor.
End structures of pre-miRNAs
Although a subset of vertebrate let-7 family members are processed through intermediates having atypical end structures (Supplementary Figure S6), it is uncertain that atypical intermediates are also produced in other miRNA biogenesis. Surprisingly, we found that a significant number of nascent pre-miRNAs in hESCs did not have the 2-nt overhang structure, based on the analysis of DGCR8-CLIP-seq reads.
The major end structure of nascent pre-miR-302d, one of the highest expressed miRNAs, was a 1-nt 3′ overhang (Figure 4A. See also Supplementary Figure S7B), the same end structure that we detected with nascent pre-let-7d, -7g and -7i. Interestingly, most 3′ ends of miR-302d-3p had an additional nucleotide residue, mostly an uridylate. Since the composition of the 3′-end residue of miR-302d-3p was more heterogeneous than the proceeding residues, a substantial portion of the terminal residues might have originated from non-templated nucleotide addition after Drosha cleavage to form the 2-nt overhang structure. Intriguingly, pri-miR-302d had a protruding nucleotide, or a bulge, in the 5′ arm, reminiscent of pri-let-7d and -7i. Similarly, we predicted that pri-miR-138–1 and -138–2 would have a 1-nt bulge in the 5′ arm (Supplementary Figure S10A). Based on the analysis of DGCR8-CLIP-seq reads, nascent pre-miR-138–1 and -138–2 had a 2-nt 5′ overhang and a 3-nt 3′ overhang. The 3′ end of pre-miR-138–1 was likely to be modified after Drosha processing on the basis that the 3′ end residues of miR-138–1–3p had a non-templated uridylate or adenylate residue. Taken together, our data show that, outside the let-7 family members, pre-miRNAs bearing a 1-nt 3′ overhang are also subject to non-templated nucleotide addition. In addition to the miRNAs that have a 1-nt bulge at the 5′ cleavage sites, we found that nascent pre-miR-1915 had a 2-nt 5′ overhang and 3-nt 3′ overhang, which had a 1-nt bulge next to the 5′ cleavage site. Pri-miR-452 hairpin was predicted to have a 2-nt bulge in the 5′ arm. The major end structure of nascent pre-miR-452 was a 3-nt 5′ overhang and a 3-nt 3′ overhang.
Figure 4.
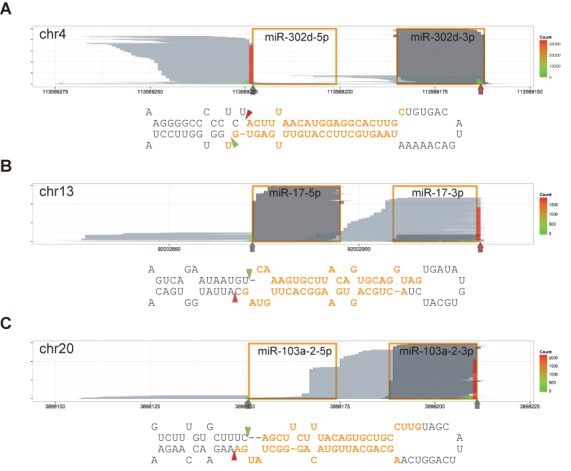
A 2-nt overhang of cleaved base is the hallmark of Microprocessor-mediated cleavage. Reads are represented as in Figure 1B and marked as in Figure 3. Predicted secondary structures are presented as in Figure 2. (A) DGCR8-CLIP-seq reads mapping to the mir-302d locus. Pri-miR-302d bears a 1-nt bulge in the 5′ arm. (B) DGCR8-CLIP-seq reads mapping to the mir-17 locus. Pri-miR-17 bears a 1-nt bulge in the 3′ arm. (C) DGCR8-CLIP-seq reads mapping to the mir-103a-2 locus. Pri-miR-103a-2 bears a 2-nt bulge in the 3′ arm. See also Supplementary Figure S10 for additional information on other bulged miRNAs. Note that the 5′ cleavage sites are located in the bulged structures, and that all the 3′ cleavage sites are in the non-bulged regions to release bases bearing a 2-nt 3′ overhang.
Prototypic oncomiRs, miR-17, -20a, -20b, -93 and -106b, had a 1-nt bulge in the 3′ arm (Figure 4B and Supplementary Figure S10B). We found that the pre-miRNAs had a 2-nt 5′ overhang and a 5-nt 3′ overhang. Cloned sequences of most 3′ ends of pre-miR-17 were consistent with sequences of miR-17 precursors from the DGCR8-CLIP libraries (Supplementary Figure S11). Although miR-106a had a 1-nt bulge beside the cleavage sites, the predicted end structure of pre-miRNA was similar to the bulged oncomiRs. Consistent with the thermodynamic instability rule, miRNAs from the 5′ arm were selected as the guide strand. In contrast, other non-bulged pri-miRNAs from the same oncomiR clusters, such as pri-miR-19a, -19b-1, -19b-2, -25, -92a-1, -92a-2 and -363, were processed to have the typical 2-nt-3′-overhang structure, and miRNAs from the 3′ arms were loaded onto the RISC. The predicted structures of pri-miR-103a-1, -103a-2 and -107 shared the feature of a 2-nt bulge in the 3′ arm. We found that major pre-miRNAs had a 4-nt 3′ overhang (Figure 4C and Supplementary Figure S10C). Contrary to annotations, our prediction indicates that pri-miR-18a and 18b have a 2-nt bulge next to the 3′ cleavage sites to yield pre-miRNAs with a 1-nt 5′ overhang and a 5-nt 3′ overhang (Supplementary Figure S10D). While miR-18a-5p and -18b-5p were predominantly loaded onto the RISC, miRNAs from the 3′ arms were selected from pre-miR-103a-1, -103a-2 and -107. Similarly, pri-miR-29b-1, -29b-2, -190a, -221, -362, -421, -429, -498, -500a and -545 were predicted to have a 1-nt bulge in the 3′ arm. The primary transcripts were cropped into pre-miRNAs bearing a 3-nt 3′ overhang, and miRNAs from the 3′ arms were predominantly selected. Taken together, none of the nascent pre-miRNAs cropped from bulged structures had the 2-nt 3′ overhang. Conversely, the cleaved bases of pri-miRNAs invariably had a 2-nt 3′ overhang. We could not find that the strand selection from atypical pre-miRNAs did not comply with the thermodynamic stability rule. Our data indicate that the end structure of a nascent pre-miRNA molecule can be reliably inferred from a stem-loop structure of pri-miRNA, if the information of either end of a nascent pre-miRNA molecule is available.
Discovery of novel canonical miRNAs
We next analyzed novel miRNAs to verify the biogenesis pathway and cleavage sites. To discover novel miRNAs, we analyzed AGO2-IP-enriched RNAs by using the miRDeep2 software package. The analysis predicted 67 novel miRNA candidates with the estimated signal-to-noise ratio of 4.1 (Supplementary Table S3). In spite of the low coverage of AGO2-IP-derived reads aligned with novel miRNA candidates, DGCR8-CLIP-seq reads mapped to 13 predicted pre-miRNAs. It is highly probable that the 13 miRNA candidates were generated through the canonical biogenesis pathway on the basis that the 3′ end positions of mapped DGCR8-CLIP-seq reads matched the 3′ ends of the predicted pre-miRNAs (Supplementary Figure S12). We note that by analyzing CLIP-seq reads we discovered additional Microprocessor cleavage sites that the miRNA discovery software failed to detect, as exemplified by the hairpin structures in the AURKB and the BRD2 mRNAs.
Microprocessor-mediated mRNA cleavage
It was demonstrated that the Microprocessor cleaves two evolutionarily conserved hairpin structures in the 5′ UTR and the coding region of the DGCR8 mRNA to destabilize the DGCR8 mRNA (6). As expected, 283 and 151 DGCR8-CLIP-seq reads were uniquely mapped to the 5′ UTR and the coding region hairpins, respectively (Figure 5A). Interestingly, the majority of 3′ ends of CLIP reads mapping to the 5′ UTR hairpin exactly matched the staggering sites to form a 2-nt 3′ overhang, the expected end structure yielded by Drosha cleavage, rather than the reported 3-nt overhang (6). To determine the end positions from the individual molecules, we mapped long reads from the DGCR8-CLIP libraries, which were obtained by using the Illumina MiSeq platform, to the DGCR8 mRNA. We detected 39 reads, exactly matching the inferred 58-nt cleavage product from the structure in the 5′ UTR. In addition, we found 24 reads of a 57-nt cleavage product from the coding region. Although these hairpins were not efficiently processed into mature miRNAs, the predicted end structures of the cleaved hairpins had characteristics of canonical pre-miRNA, supporting Microprocessor-mediated cleavage.
Figure 5.
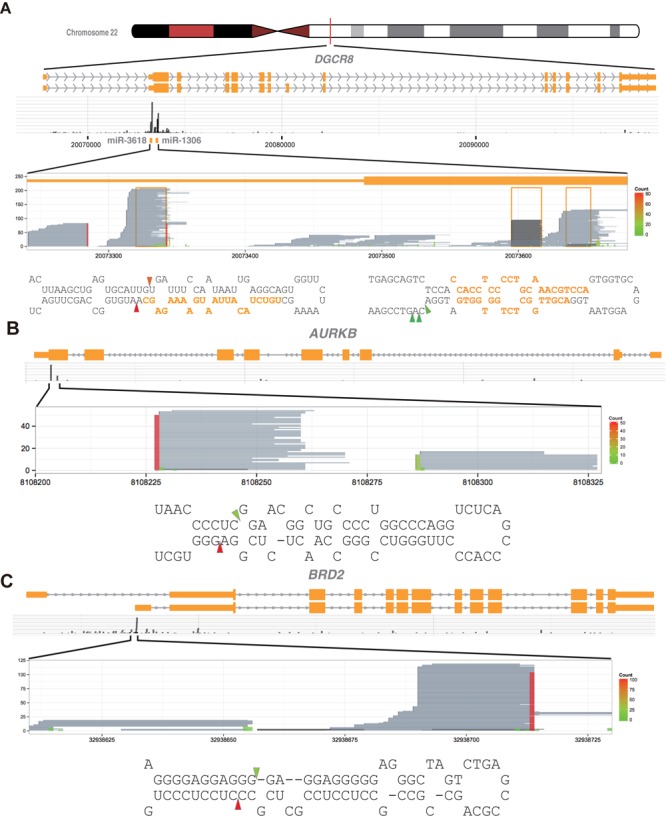
The Microprocessor cleaves miRNA-like structures embedded in mRNAs. (A–C) Shown are DGCR8-CLIP-enriched RNA fragments mapping to (A) the DGCR8, (B) the AURKB and (C) the BRD2 mRNAs. Reads aligned to the entire loci proclaim specificity of the DGCR8-CLIP experiments. Reads from the DGCR8-CLIP libraries and the Microprocessor-mediated cleavage sites in mRNAs are represented as in Figure 1B. Note that the stem-loop structures and cleavage sites have characteristics of typical pri-miRNAs, whereas the pre-miRNA-like hairpins are uncoupled from miRNA maturation.
To discover mRNAs directly cleaved by the Microprocessor, we analyzed DGCR8-CLIP-seq reads, mapping to reference mRNA sequences. We found three additional DGCR8-CLIP-enriched miRNA loci that are embedded in the mRNAs. While 566 DGCR8-CLIP-seq reads were mapped to the miR-671 stem-loop structure in the coding region of CHPF2, 278 AGO2-IP reads were mapped to the mir-671 locus. Similarly, 103 and 68 CLIP-seq reads mapped to the mir-935 and mir-4707 loci, whereas 393 and 26 reads from the AGO2-IP-derived libraries mapped to the mir-935 and mir-4707 loci, located in the coding region of CACNG8 and the 5′ UTR of HAUS4, respectively. Interestingly, we found 73 and 134 DGCR8-CLIP-seq reads uniquely mapped to typical miRNA structures in the coding region of the AURKB mRNA and in the 5′ UTR of the BRD2 mRNA, respectively (Figure 5B and C and Supplementary Figure S13). Moreover, it was predicted that the hairpin structures were processed to yield hairpin-structured RNAs bearing a 2-nt 3′ overhang, the expected end structure of RNase III-mediated cleavage. However, only one and six reads from the AGO2-IP-derived RNA libraries were mapped to the hairpin structures of the AURKB and BRD2 mRNAs, respectively. Together, our data suggest that, besides the DGCR8 mRNA, the Microprocessor cleaves other mRNAs. Our data also suggest that despite a large number of DGCR8-CLIP-seq reads uniquely mapped to mRNAs, on the basis of the fact that maximum coverage for annotated exons was generally low, mRNAs directly cleaved by the Microprocessor are limited to a small set of mRNAs, at least, in the H1 hESC line.
Drosha-CLIP experiments validate DGCR8-CLIP
For validation of the DGCR8-CLIP results, we tried in vain to construct Drosha-CLIP-seq libraries from the cells with the integral genetic make-up. The failure was consistent with previous in vitro experiments that pri-miRNAs were UV-crosslinked only to DGCR8 but not to Drosha (29). We reasoned that low crosslinking efficiency could be overcome by using a high-affinity antibody recognizing the epitope-tagged Drosha. To preserve the regulatory circuit between Drosha and DGCR8, we constructed a knock-in H1 line via homologous recombination that expresses Flag-tagged Drosha under the control of its own endogenous regulatory elements (Supplementary Figure S1). With the Flag-Drosha knock-in cell line, we constructed two independent Drosha-CLIP libraries from two biologically independent cells. Among the 783 476 collapsed reads, 3.6% (28 514 reads) mapped to 113 annotated pre-miRNAs, 110 of which overlapped 274 DGCR8-CLIP enriched miRNAs (Figure 6A and B). Notably, among the 47 canonical pre-miRNAs for which maximum coverage was higher than 7, the 3′ ends of Drosha-CLIP reads mapping to 43 canonical pre-miRNAs were consistent with the cleavage sites determined from DGCR8-CLIP-seq reads. Together, the data from the Drosha-CLIP experiments support that miRNAs enriched by DGCR8-CLIP are canonical miRNAs.
Figure 6.
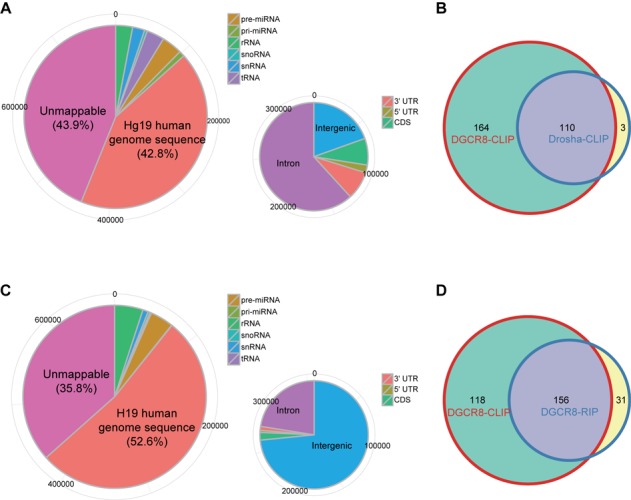
Features of CLIP-seq libraries for Drosha and a RIP-seq library for DGCR8. (A and C) Shown are distributions of locations of (A) Drosha-CLIP-seq and (B) DGCR8-RIP-seq reads on the human genome. Pie charts are presented as in Figure 1A. (B and D) Weighted Venn diagram of annotated miRNA precursors recovered by DGCR8-CLIP, (C) Drosha-CLIP and (D) DGCR8-RIP. Note that although we omit less frequently recovered miRNA precursors from DGCR8-CLIP (maximum coverage <8), miRNA precursors from DGCR8-CLIP overlap 110 (out of 113) and 156 (out of 187) miRNA precursors recovered from Drosha-CLIP and DGCR8-RIP, respectively.
DGCR8-RIP from hESCs
In general, trimming of UV-crosslinked RNAs is a crucial step for the efficient construction of a CLIP-seq library. Since RNase I used for RNA trimming belongs to the endoribonuclease family and cloned sequences of pre-miRNAs were consistent with DGCR8-CLIP-seq reads, it is unlikely that the cleavage sites that we determined by analyzing the DGCR8- and Drosha-CLIP-seq reads were artifacts introduced by RNase I treatments. Moreover, the cleavage sites of 142 miRNAs determined by DGCR8-CLIP without RNase treatment were identical to those determined with RNase I treatment (Supplementary Table S2). Nonetheless, to exclude this possibility further, we enriched DGCR8-associated RNAs by using RIP, a modified chromatin IP, to circumvent the RNase treatment step by using sonication. Because formaldehyde crosslinks proteins in their proximity as well, purification of protein-RNA complexes by gel electrophoresis is not applicable. Transient RNAs enriched by RIP are usually analyzed by RT-PCR for validation of putative interactions rather than by next-generation sequencing for de novo finding due to high vulnerability to contamination by abundant RNAs. As expected, only 4.1% of collapsed reads mapped to 187 miRNA loci (Figure 6C and D), and the reads mapping to 28 miRNA loci reached maximum coverage higher than 7. The major 3′ ends of reads derived from 28 RIP-enriched miRNA precursors were identical to those of DGCR8-enriched RNAs. Moreover, the major 3′ ends of 19 miRNA precursors enriched by DGCR8-RIP experiments were identical to those of Drosha-CLIP-enriched pre-miRNAs, supporting that the 3′ ends of miRNAs determined from DGCR8- and Drosha-CLIP-seq reads were genuine 3′ ends generated by Microprocessor-mediated cleavage (Figure 7A and B and Supplementary Figure S14).
Figure 7.
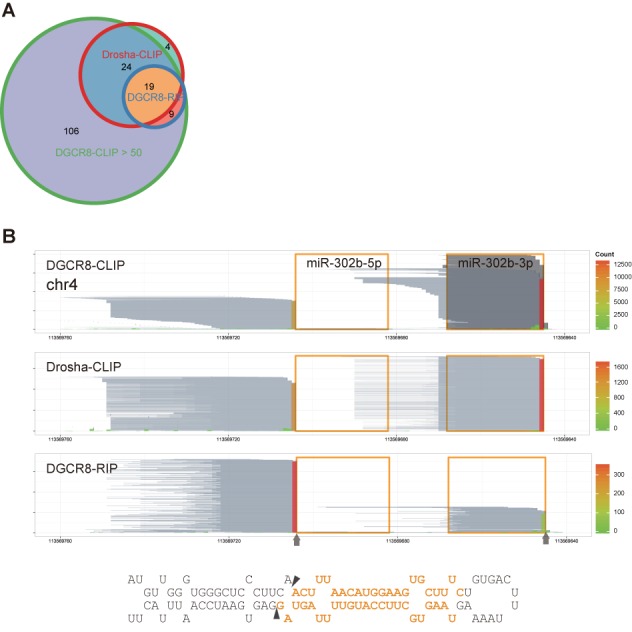
Cleavage sites determined by DGCR8-CLIP-seq reads are supported by Drosha-CLIP-seq and DGCR8-RIP-seq reads. (A) Weighted Venn diagram of cleavage sites on annotated miRNA precursors determined by the reads from DGCR8-CLIP (maximum coverage >50), Drosha-CLIP (>7) and DGCR8-RIP (>7). Cleavage sites determined by DGCR8-CLIP-seq are identical to the cleavages sites on 43 (out of 47) and 28 (out of 28) miRNA precursors determined by Drosha-CLIP-seq and DGCR8-RIP-seq, respectively. The four miRNAs uniquely recovered from Drosha-CLIP have low maximum coverage. (B) Reads from the DGCR8-CLIP, Drosha-CLIP and DGCR8-RIP libraries aligned to the mir-302b locus. Reads are represented as in Figure 1B and marked as in Figure 3. Note that the 5′ and 3′ cleavage sites determined from different experimental approaches are identical.
DISCUSSION
In this study, we present in vivo RNA targets in direct contact with the Microprocessor by using CLIP-seq analyses for DGCR8 and Drosha. Granted the relatively intact genetic make-up of hESCs, the molecular interactions that we provide here may be more pertinent to in vivo functions of the Microprocessor than the information from other cancerous cell lines. We investigated DGCR8-RNA complexes within the context of intact cells, as opposed to ectopically expressed DGCR8 because the forced expression of DGCR8 is likely to perturb the intricate homostatic regulatory circuit for the Microprocessor and aggravate contamination by biologically irrelevant RNAs. By the same token, we constructed a flag-Drosha knock-in cell line for the investigation of Drosha-RNA interaction to minimize disruption of the regulatory circuit. Despite abnormally weak radioactivity from the enriched RNAs, specificity and sensitivity of our data are unprecedentedly high, as proclaimed by the enormous maximum coverage for ESC-specific pre-miRNAs and extremely low coverage for mature miRNAs. Considering the efficient IP of DGCR8 protein, high-affinity DGCR8-binding sites are most likely limited, at least, in H1 hESCs, although we cannot exclude the possibility that certain RNAs are elusive at the crosslinking step.
Binding targets of the Microprocessor
The majority of RNAs enriched by DGCR8-CLIP are miRNA-like RNAs, mostly nascent pre-miRNAs annotated in miRBase. The invariable 2-nt overhang of stem-loop structures indicates RNase III-mediated cleavage, most likely Drosha. It is noteworthy that, given the low maximum coverage for most mapped regions, the count of collapsed reads allocated to transcribed regions may not correlate with the Microprocessor binding to the transcribed regions.
While it is firmly established that the canonical miRNA biogenesis pathway is responsible for most abundant miRNAs, there remain ambiguities as to whether the canonical pathway is used for biogenesis of human miRNAs at modest expression levels. The miRBase database provides the most comprehensive information on miRNAs and is mainly based on information in accepted articles from peer-reviewed journals (31). Because the submitting authors have primary responsibility for the quality of annotations, high-confidence annotations are not guaranteed. Recently added miRNAs discovered by using next-generation sequencing are especially prone to misannotations. Although illegitimate miRNAs have been filtered and removed from miRBase, there remain a significant number of dubious mammalian miRNAs (32,33). The verification of biogenesis pathway for individual miRNA is important to distinguish authentic miRNAs. Our current study attests 274 bona fide canonical miRNAs. Small RNA profiling using DGCR8 and Dicer knockout mESCs and ectopic expression of mouse miRNA hairpins in HEK293T cells have revealed 331 DGCR8-dependent miRNA loci, among which 227 loci are evolutionarily conserved in human (22,32). Since then, no large-scale verification of canonical miRNA biogenesis has been reported. Our list contains 139 human counterparts of reported mouse canonical pre-miRNAs and 24 conserved pre-miRNAs, which were not determined due to technical limitations in previous reports.
In contrast to canonical miRNA biogenesis, there are controversies as to whether Drosha is implicated in the maturation of rRNA, a function of the bacterial RNase III enzymes. Although there are DGCR8- and Drosha-CLIP-seq reads mapping to rRNA, our data support that the Microprocessor is not implicated in the biogenesis of rRNA on the basis of the following observations. First, the read counts mapping to rRNA sequences are very low compared with the counts mapping to miRNAs. Second, the reads are almost exclusively located in the mature rRNAs, and few reads are mapped to external and internal transcribed spacers (Supplementary Figure S15). Considering the abundance of rRNAs in the cells, the reads most likely originated from contaminant rRNAs.
Authentic in vivo cleavage sites of the Microprocessor
One of the distinctions of our data is the information on authentic in vivo cleavage sites and end structures of nascent pre-miRNAs. Inferences of cleavage sites from small RNA sequences are significantly fallible because of extensive modifications after cleavage, as exemplified by miR-302d and -363. Based on our data, we find a significant number of incorrect annotations in miRBase. Moreover, some cleavage sites are too heterogeneous to define major cleavage sites, which are contradictory to the current criterion of miRNA, that is, homogeneous ∼22-nt RNAs.
It is notable that the heterogeneity of miRNAs may play important biological roles. The developmental stage-specific pre-miR-302a and -302c are heterogeneous and extremely abundant in hESCs and mouse epiblast stem cells. Interestingly, alternatively cleaved pre-miRNAs have different strand preferences, and isoform-dependent strand selection seems conserved in the mouse. The strand preferences are consistent with the strand selection rule of thermodynamic stability at the 5′ end of double-stranded miRNA. The isoform-dependent strand selection is reminiscent of a previously proposed model that alternatively processed pre-miRNAs might influence strand preference at the RISC-loading stage, based on the analysis of mouse pre-miR-143 isoforms (34). Change of strand selection was observed in a tissue-dependent manner in mouse ESCs and tissues, conflicting with the thermodynamic stability model (32,35,36). Regulated alternative processing by the Microprocessor may explain part of the puzzling observations. Although strand preference change was not as notable as in miR-302a, it is possible that heterogeneity of the cleavage sites in other miRNAs may influence strand preferences, such as miR-101–1, -101–2, -222, -296, -342, -452, -545, -873, -876 and -1912.
It has been surmised that pre-miRNAs have a 2-nt 3′ overhang on the basis that RNase III cleaves perfect double-stranded RNA into fragments with 2-nt 3′ overhangs. However, a number of pri-miRNAs have a bulge at the Microprocessor-mediated cleavage sites. Since the current miRNA biogenesis models were mainly derived from studies on non-bulged miRNAs, biogenesis from bulged pri-miRNA is poorly understood. Cleavage sites determined in this study indicate that the 2-nt overhang is, although common for pre-miRNAs from non-bulged pri-miRNAs, not a hallmark of all nascent pre-miRNAs. In the present study, we find that bulged pri-miRNAs are processed to yield pre-miRNAs with diverse end structures from no overhang to the 4-nt 3′ overhang.
In spite of this structural diversity, our data reveal common features of bulged miRNAs. First, high expression levels of bulged miRNAs indicate that the bulged pri-miRNAs are adequate substrates for miRNA biogenesis. Unexpectedly, the representative size of miRNAs derived from bulged precursors is 23 nt. Second, the expression of bulged miRNAs is generally tightly controlled in a developmental stage-specific manner. It has been established that the expression of a number of bulged miRNAs are associated with de-differentiation and oncogenesis, as exemplified by oncomiRs. These miRNAs are linked to critical cellular functions, such as cell-cycle control, differentiation and apoptosis. Third, bulged pri-miRNA structures are evolutionarily conserved only in vertebrates, and deep sequencing reads indicate that cleavage site selection by the vertebrate Microprocessor is also conserved, suggesting most miRNAs from bulged structures have evolved after vertebrates split from invertebrates. It explains, at least in part, the observations that the miRNA repertoire of vertebrates is much larger than that of invertebrates. Finally, it is the 5′ cleavage sites that are always found in the distorted structures. In contrast, the 3′ cleavage sites are located in the non-bulged RNA helixes. Interestingly, a 2-nt overhang is always found at the ends of cleaved bases of the stem-loop structures. Biochemical evidence indicates that Drosha has two RNase III domains to form an internal dimer structure (26) that creates a catalytic valley to accommodate double-stranded RNA (37). The N-terminal RNase III domain (RIIIDa) cleaves the 3′ site, and the C-terminal RNase III domain (RIIIDb) cleaves the 5′ site. One possible explanation for the exclusive existence of bulges at the 5′ cleavage sites is that only the catalytic site of RIIIDb in vertebrate Drosha may fit the bulged RNA structures.
mRNA cleavage by the Microprocessor
Since the initial report of cleavage and destabilization of the DGCR8 mRNA by the Microprocessor, there have been suggestions that other mRNAs are regulated by the same mechanism. However, direct evidence of cleavage in the cell is imperative because cleavage by the Microprocessor could be completely uncoupled from later stages for miRNA biogenesis, as exemplified by the hairpin structures in the DGCR8 mRNA. The stem-loop structure in the 5′ UTR of the DGCR8 mRNA is cropped into a pre-miRNA-like structure, that is, an imperfect hairpin with a 2-nt 3′ overhang, but mature miRNA is undetectable from the AGO2-associated small RNAs. The hairpin structure in the coding region is cleaved at multiple sites into multiple hairpin isoforms, and at least an isoform has the 2-nt overhang structure. However, mature miRNAs are mainly derived from a pre-miRNA isoform. In addition to the DGCR8 mRNA, we also show that the Microprocessor cleaves other mRNAs to yield pre-miRNA-like RNAs having a 2-nt overhang. Recently, we reported that cleavage of the hairpin structure in the AURKB mRNA by the Microprocessor is correlated with suppression of Aurora kinase B (AURKB) protein expression in a cell-cycle-dependent manner, and mutations that destabilize the hairpin structure lead to increased AURKB expression levels, which is in line with the notion that the Microprocessor may directly destabilize the mRNAs (38).
Interestingly, although the cleavage products possess structural features of typical pre-miRNAs, the stem-loop structures are not efficiently processed into AGO2-associated miRNAs. One conceivable explanation is that essential elements are missing in the cleaved structures. It is also possible that the negative regulatory elements reside in the pre-miRNA-like structures to hamper further maturation. Granted that we identified the nascent pre-miRNA-like structures, it would be interesting to determine what elements are responsible for the uncoupling and/or efficient coupling of miRNA cropping with further maturation.
Previously, Caceres et al. suggested that several hundred mRNAs are substrates of the Microprocessor on the basis of CLIP-seq for T7 epitope-tagged DGCR8 in HEK293T cells under the control of the cytomegalovirus (CMV) enhancer/promoter (39,40). In contrast, we observed that much smaller mRNAs were cleaved by the endogenous Microprocessor in hESCs. The discrepancies might be reflective of the intrinsic differences between different experimental systems, such as the expression levels of DGCR8. However, it is also possible that their CLIP-seq reads mapping to the mRNAs might originate from non-specific contaminant RNAs on the basis that CLIP experiments are prone to contamination unless conducted properly. Notably, negative controls of immunoprecipitions are missing in their CLIP-seq data sets. To test the latter possibility, we analyzed CLIP-seq reads from epitope-tagged DGCR8 (http://www.ncbi.nlm.nih.gov/sra, SRR518498). Disappointingly, the vast majority of reads mapping to the annotated miRNAs were derived from mature miRNAs and Microprocessor-independent mirtrons rather than from canonical miRNA precursors (Supplementary Figure S16). Moreover, reads mapping to mature rRNAs account for 64.3% of total reads, epitomizing contamination by abundant RNAs (Supplementary Figure S17). Most of all, considering the enormous sequencing depth, maximum coverage of reads mapping to mRNAs was very low except for the overexpressed ectopic RNAs that contained the coding region of epitope-tagged DGCR8 and rabbit β-globin. Together, it is most likely that Caceres et al. significantly overestimated the mRNA targets of the Microprocessor.
Refinement of canonical miRNA annotations and prediction
By definition, miRNAs are ∼22-nt RNA molecules derived from imperfect stem-loop structures and participate in the regulation of target gene expression as a component of the RISC. In practice, miRNAs have been loosely defined as non-coding RNAs that fulfill the following minimal criteria. First, miRNA should be present as a ∼22-nt homogeneous RNA population. Second, miRNA should originate from a characteristic hairpin-structured precursor RNA. Elucidation of the immense biological roles of miRNA and recent advances in next-generation sequencing technologies have led to the rapid expansion of the miRNA list. However, as the miRNA list grows larger, a looser definition, initially intended to distinguish miRNA from siRNA (41), has resulted in blurred boundaries between miRNA and other similar-sized RNAs.
In the present study, we determine the key features of canonical human miRNAs that may help improve annotations and the computational prediction of canonical miRNAs. First, a significant number of miRNAs are longer than 22 nt. Second, the stem-loop structures should not be predicted from presumed pre-miRNAs, but from RNA sequences long enough to encompass the entire ∼3-turn helixes of stem. Our data also suggest that prediction of stem-loop structure may have a pitfall because the most stable structures of pri-miRNA in aqueous solution are not necessarily the most stable structures in the Microprocessor-RNA complex. Third, the cleaved bases rather than the pre-miRNAs have the characteristic 2-nt 3′ overhang structure. Finally, the diversification of mature miRNAs could originate from the heterogeneity of Microprocessor-mediated cleavage sites. The current release of miRBase does not reflect the heterogeneity of the cleavage sites. Microprocessor-mediated cleavage sites may be selected in a tissue-dependent manner, and pre-miRNA isoforms from the same primary transcript may have a different strand preference.
ACCESSION NUMBERS
The raw and processed data are available in GEO (GSE61979).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Matthew Chong, Hee Chul Lee and Soo-Jong Um for their helpful comments on the manuscript.
FUNDING
Ministry of Education, Science and Technology through the National Research Foundation of Korea [NRF-2011–0024962 to Y.-S.K.; NRF-2009–0068994 to Y.-S.K.; 2012R1A1A2003267 to H.S.]. Funding for open access charge: Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology [2012R1A1A2003267].
Conflict of interest statement. None declared.
REFERENCES
- 1.Gregory R.I., Yan K.P., Amuthan G., Chendrimada T., Doratotaj B., Cooch N., Shiekhattar R. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 2.Court D.L., Gan J., Liang Y.H., Shaw G.X., Tropea J.E., Costantino N., Waugh D.S., Ji X. RNase III: genetics and function; structure and mechanism. Ann. Rev. Genet. 2013;47:405–431. doi: 10.1146/annurev-genet-110711-155618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang H., Kolb F.A., Jaskiewicz L., Westhof E., Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 4.Park J.E., Heo I., Tian Y., Simanshu D.K., Chang H., Jee D., Patel D.J., Kim V.N. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature. 2011;475:201–205. doi: 10.1038/nature10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz D.S., Hutvagner G., Du T., Xu Z., Aronin N., Zamore P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 6.Han J., Pedersen J.S., Kwon S.C., Belair C.D., Kim Y.K., Yeom K.H., Yang W.Y., Haussler D., Blelloch R., Kim V.N. Posttranscriptional crossregulation between Drosha and DGCR8. Cell. 2009;136:75–84. doi: 10.1016/j.cell.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shenoy A., Blelloch R. Genomic analysis suggests that mRNA destabilization by the microprocessor is specialized for the auto-regulation of Dgcr8. PLoS ONE. 2009;4:e6971. doi: 10.1371/journal.pone.0006971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chong M.M., Zhang G., Cheloufi S., Neubert T.A., Hannon G.J., Littman D.R. Canonical and alternate functions of the microRNA biogenesis machinery. Genes Dev. 2010;24:1951–1960. doi: 10.1101/gad.1953310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knuckles P., Vogt M.A., Lugert S., Milo M., Chong M.M., Hautbergue G.M., Wilson S.A., Littman D.R., Taylor V. Drosha regulates neurogenesis by controlling neurogenin 2 expression independent of microRNAs. Nat. Neurosci. 2012;15:962–969. doi: 10.1038/nn.3139. [DOI] [PubMed] [Google Scholar]
- 10.Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 11.Song H., Chung S.K., Xu Y. Modeling disease in human ESCs using an efficient BAC-based homologous recombination system. Cell Stem Cell. 2010;6:80–89. doi: 10.1016/j.stem.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z., Tollervey J., Briese M., Turner D., Ule J. CLIP: construction of cDNA libraries for high-throughput sequencing from RNAs cross-linked to proteins in vivo. Methods. 2009;48:287–293. doi: 10.1016/j.ymeth.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 13.Kwon Y.S. Small RNA library preparation for next-generation sequencing by single ligation, extension and circularization technology. Biotechnol. Lett. 2011;33:1633–1641. doi: 10.1007/s10529-011-0611-y. [DOI] [PubMed] [Google Scholar]
- 14.Morgan M., Anders S., Lawrence M., Aboyoun P., Pages H., Gentleman R. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25:2607–2608. doi: 10.1093/bioinformatics/btp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin T., Cook D., Lawrence M. ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol. 2012;13:R77. doi: 10.1186/gb-2012-13-8-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedlander M.R., Mackowiak S.D., Li N., Chen W., Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40:37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linsen S.E., de Wit E., Janssens G., Heater S., Chapman L., Parkin R.K., Fritz B., Wyman S.K., de Bruijn E., Voest E.E., et al. Limitations and possibilities of small RNA digital gene expression profiling. Nat. Methods. 2009;6:474–476. doi: 10.1038/nmeth0709-474. [DOI] [PubMed] [Google Scholar]
- 18.Hafner M., Renwick N., Brown M., Mihailovic A., Holoch D., Lin C., Pena J.T., Nusbaum J.D., Morozov P., Ludwig J., et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 2011;17:1697–1712. doi: 10.1261/rna.2799511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ule J., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 20.Konig J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viswanathan S.R., Daley G.Q., Gregory R.I. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Babiarz J.E., Ruby J.G., Wang Y., Bartel D.P., Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ender C., Krek A., Friedlander M.R., Beitzinger M., Weinmann L., Chen W., Pfeffer S., Rajewsky N., Meister G. A human snoRNA with microRNA-like functions. Mol. Cell. 2008;32:519–528. doi: 10.1016/j.molcel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 24.Ladewig E., Okamura K., Flynt A.S., Westholm J.O., Lai E.C. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res. 2012;22:1634–1645. doi: 10.1101/gr.133553.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Radmark O., Kim S., et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 26.Han J., Lee Y., Yeom K.H., Kim Y.K., Jin H., Kim V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heo I., Ha M., Lim J., Yoon M.J., Park J.E., Kwon S.C., Chang H., Kim V.N. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell. 2012;151:521–532. doi: 10.1016/j.cell.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 28.Newman M.A., Mani V., Hammond S.M. Deep sequencing of microRNA precursors reveals extensive 3′ end modification. RNA. 2011;17:1795–1803. doi: 10.1261/rna.2713611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han J., Lee Y., Yeom K.H., Nam J.W., Heo I., Rhee J.K., Sohn S.Y., Cho Y., Zhang B.T., Kim V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 30.Ameres S.L., Zamore P.D. Diversifying microRNA sequence and function. Nat. Rev. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 31.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiang H.R., Schoenfeld L.W., Ruby J.G., Auyeung V.C., Spies N., Baek D., Johnston W.K., Russ C., Luo S., Babiarz J.E., et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castellano L., Stebbing J. Deep sequencing of small RNAs identifies canonical and non-canonical miRNA and endogenous siRNAs in mammalian somatic tissues. Nucleic Acids Res. 2013;41:3339–3351. doi: 10.1093/nar/gks1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu H., Ye C., Ramirez D., Manjunath N. Alternative processing of primary microRNA transcripts by Drosha generates 5′ end variation of mature microRNA. PLoS ONE. 2009;4:e7566. doi: 10.1371/journal.pone.0007566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ro S., Park C., Young D., Sanders K.M., Yan W. Tissue-dependent paired expression of miRNAs. Nucleic Acids Res. 2007;35:5944–5953. doi: 10.1093/nar/gkm641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jouneau A., Ciaudo C., Sismeiro O., Brochard V., Jouneau L., Vandormael-Pournin S., Coppee J.Y., Zhou Q., Heard E., Antoniewski C., et al. Naive and primed murine pluripotent stem cells have distinct miRNA expression profiles. RNA. 2012;18:253–264. doi: 10.1261/rna.028878.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gan J., Tropea J.E., Austin B.P., Court D.L., Waugh D.S., Ji X. Structural insight into the mechanism of double-stranded RNA processing by ribonuclease III. Cell. 2006;124:355–366. doi: 10.1016/j.cell.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 38.Jung E., Seong Y., Seo J.H., Kwon Y.S., Song H. Cell cycle-dependent regulation of Aurora kinase B mRNA by the Microprocessor complex. Biochem. Biophys. Res. Commun. 2014;446:241–247. doi: 10.1016/j.bbrc.2014.02.104. [DOI] [PubMed] [Google Scholar]
- 39.Macias S., Plass M., Stajuda A., Michlewski G., Eyras E., Caceres J.F. DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat. Struct. Mol. Biol. 2012;19:760–766. doi: 10.1038/nsmb.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cazalla D., Sanford J.R., Caceres J.F. A rapid and efficient protocol to purify biologically active recombinant proteins from mammalian cells. Protein Exp. Purif. 2005;42:54–58. doi: 10.1016/j.pep.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 41.Ambros V., Bartel B., Bartel D.P., Burge C.B., Carrington J.C., Chen X., Dreyfuss G., Eddy S.R., Griffiths-Jones S., Marshall M., et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.