

Abstract

The mechanism for activation of orotidine 5′-monophosphate decarboxylase (OMPDC) by interactions of side chains from Gln215 and Try217 at a gripper loop and R235, adjacent to this loop, with the phosphodianion of OMP was probed by determining the kinetic parameters kcat and Km for all combinations of single, double, and triple Q215A, Y217F, and R235A mutations. The 12 kcal/mol intrinsic binding energy of the phosphodianion is shown to be equal to the sum of the binding energies of the side chains of R235 (6 kcal/mol), Q215 (2 kcal/mol), Y217 (2 kcal/mol), and hydrogen bonds to the G234 and R235 backbone amides (2 kcal/mol). Analysis of a triple mutant cube shows small (ca. 1 kcal/mol) interactions between phosphodianion gripper side chains, which are consistent with steric crowding of the side chains around the phosphodianion at wild-type OMPDC. These mutations result in the same change in the activation barrier to the OMPDC-catalyzed reactions of the whole substrate OMP and the substrate pieces (1-β-d-erythrofuranosyl)orotic acid (EO) and phosphite dianion. This shows that the transition states for these reactions are stabilized by similar interactions with the protein catalyst. The 12 kcal/mol intrinsic phosphodianion binding energy of OMP is divided between the 8 kcal/mol of binding energy, which is utilized to drive a thermodynamically unfavorable conformational change of the free enzyme, resulting in an increase in (kcat)obs for OMPDC-catalyzed decarboxylation of OMP, and the 4 kcal/mol of binding energy, which is utilized to stabilize the Michaelis complex, resulting in a decrease in (Km)obs.
Introduction
The underlying cause for enzymatic catalysis is stabilization of the transition state by interactions with the protein catalyst.1 Interactions between protein catalysts and a nonreacting substrate phosphodianion are utilized to provide ca. 12 kcal/mol stabilization of transition states of a diverse set of enzymatic reactions, including carbon deprotonation (triosephosphate isomerase and orotidine 5′-monophosphate decarboxylase),2−4 decarboxylation (orotidine 5′-monophosphate decarboxylase),5 hydride transfer (l-glycerol phosphate dehydrogenase),6 phosphoryl transfer (phosphoglucomutase),7 and a multistep reaction (1-deoxy-d-xylulose-5-phosphate reductoisomerase).8 This transition state stabilization is due to interactions expressed at the ground-state Michaelis complex, which favor tight substrate binding, and to the utilization of the phosphodianion binding energy to activate the enzyme for catalysis: the latter binding energy is only expressed at the transition state for the catalyzed reaction, and favors a large turnover number kcat.9−12 The use of phosphodianion binding energy for enzyme activation avoids full expression of the large substrate binding energy at the Michaelis complex, and the possibility of effectively irreversible, strongly rate-determining, ligand binding.10,13
Orotidine 5′-monophosphate decarboxylase (OMPDC) employs no metal ions or other cofactors, but yet effects an enormous stabilization of the transition state for the chemically very difficult decarboxylation of orotidine 5′-monophosphate (OMP) to give uridine 5′-monophosphate (UMP),14−18 by a stepwise mechanism through a UMP carbanion reaction intermediate (Scheme 1).4,19−23 OMPDC provides a large 31 kcal/mol stabilization of the transition state for the decarboxylation of OMP,15 and binds this transition state with a much higher affinity than substrate OMP, whose ground-state complex with OMPDC is stabilized by only 8 kcal/mol.24
Scheme 1.

Binding interactions between OMPDC and the phosphodianion of OMP provide 12 of the 31 kcal/mol stabilization of the reaction transition state.5 These interactions do not simply anchor OMP to OMPDC, because the covalent connection between the phosphodianion and the pyrimidine ring is not needed to observe enzyme activation by dianions. This was shown by the estimated 570 000-fold increase in the rate of OMPDC-catalyzed decarboxylation of the truncated substrate 1-(β-d-erythrofuranosyl)orotic acid (EO, Scheme 2A) for a reaction activated by 1.0 M phosphite dianion (HPi).5 This corresponds to an 8 kcal/mol stabilization of the transition state for the decarboxylation reaction by the HPi piece, two-thirds of the 12 kcal/mol intrinsic phosphodianion binding energy.5 The binding of HPi to OMPDC results in a 60 000-fold increase in the second-order rate constant for OMPDC-catalyzed decarboxylation of EO from (kcat/Km)E = 0.026 M–1 s–1 to (kcat/Km)E•HPi = 1600 M–1 s–1.5 This corresponds to a 60 000-fold higher affinity (eq 1 for Scheme 2B) of HPi for binding to the transition state complex [E·EO]⧧ (Kd⧧) as compared to the free enzyme (Kd). Binding interactions between OMPDC and HPi also provide a large 6 kcal/mol stabilization of the transition state for deprotonation of the truncated substrate 1-(β-d-erythrofuranosyl)5-fluorouracil (FEU) in D2O.4,25
| 1 |
Scheme 2.

The strong binding of 6-hydroxyuridine 5′-monophosphate (BMP) to OMPDC induces a protein conformational change (Figure 1).26 This includes closure of the phosphodianion gripper loop (Pro202–Val220) and pyrimidine umbrella (Glu152–Thr165) over the inhibitor, which locks BMP in a protein cage.27 Figure 2 shows interactions of the ligand phosphodianion with the amide side chain of Gln215, the phenol side chain of Tyr217, the guanidine side chain of Arg235, which sits on the protein surface adjacent to the gripper loop (Figure 1) and functions cooperatively with the loop side chains in activating OMPDC for catalysis,28 and with backbone amides from Gly234 and Arg235. We are interested in understanding the role of flexible loops in enzyme catalysis,12 and consider here the mechanism by which ionic and hydrogen-bonding interactions of side chains from the gripper loop, and Arg235, with the phosphodianion of OMP, or with HPi, are utilized in stabilization of the transition state for OMPDC-catalyzed decarboxylation of OMP, deprotonation of UMP, and the corresponding reactions of the phosphodianion truncated substrates EO and FEU,4,23,25,28−34 respectively, at a site 10 Å distant from the gripper loop. The binding of HPi to OMPDC is scarcely detectable, so that the dianion binding energy is not expressed at the OMPDC·HPi complex, but rather provides strong stabilization of the transition state for OMPDC-catalyzed decarboxylation of truncated substrates (eq 1). We have proposed that most or all of the 8 kcal/mol binding energy of HPi is utilized to drive an uphill change in enzyme conformation, which activates OMPDC for catalysis of decarboxylation.10
Figure 1.
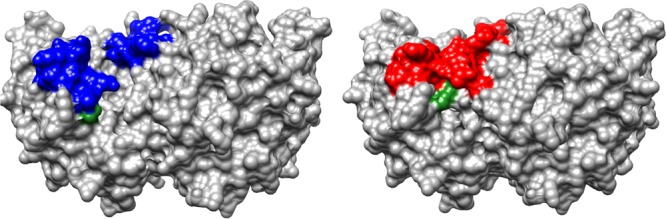
Space-filling models that show the open form of unliganded OMPDC from yeast on the left (PDB entry 1DQW), and the complex to 6-hydroxyuridine 5′-monophosphate on the right (PDB entry 1DQX). Two loops (colored) act to trap the ligand at the enzyme active site. The phosphodianion gripper loop (Pro202–Val220), on the left-hand side, and the pyrimidine umbrella (Glu152–Thr165), on the right-hand side, of each structure close over and trap the ligand in a cage at the enzyme active site. The guanidine side chain of R235, at the base of the phosphodianion gripper loop, is shaded green.
Figure 2.
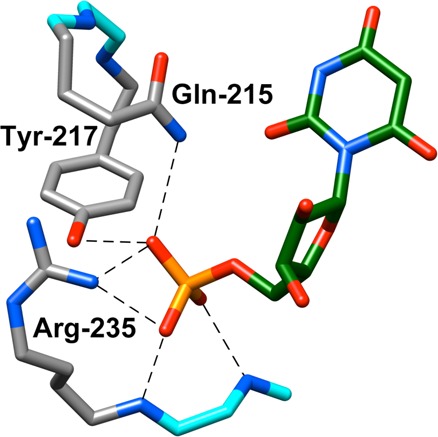
X-ray crystal structure (PDB entry 1DQX) of yeast OMPDC in a complex with 6-hydroxyuridine 5′-monophosphate. This structure shows the important interactions of Gln215, Tyr217, and Arg235 side chains from the phosphodianion gripper loop with the ligand phosphodianion. Hydrogen bonds to the Gly234 and Arg235 backbone amides are also shown. Reprinted with permission from ref (28). Copyright 2012 American Chemical Society.
We previously reported the effects of all single (Q215A, R235A, and Y217F), double (Q215A/Y217F, Q215A/R235A, R235A/Y217F), and triple (Q215A/R235A/Y217F) mutations of amino acid residues that interact with the phosphodianion of OMP (Figure 2), on the kinetic parameters for OMPDC-catalyzed reactions of the substrate pieces EO and HPi.28 Single mutations result in <3-fold decreases in (kcat/Km)E (Scheme 2B) for the catalyzed reaction of the truncated substrate EO, and large decreases in the third-order rate constant for enzyme activation by HPi.28 This shows that interactions between OMPDC and HPi have the sole function of activating OMPDC for catalysis of decarboxylation of EO.
We report here the effect of these same mutations on the kinetic parameters kcat and Km for OMPDC-catalyzed decarboxylation of OMP. The activating nature of enzyme–phosphodianion interactions for catalysis of decarboxylation of the physiological substrate OMP was not detected by standard methods of mechanistic analyses, including X-ray crystallographic analysis, but may be inferred from the results of our experiments using the substrate pieces EO and HPi. We focus on a model that connects the easily quantified activation of OMPDC-catalyzed decarboxylation of a truncated substrate EO by phosphite dianion, with the cryptic activation of OMPDC-catalyzed decarboxylation of OMP by the substrate phosphodianion. We report an unprecedented context dependence of the effect of these single, double, and triple mutations on kcat and Km for OMPDC-catalyzed decarboxylation of OMP, which provides compelling support for a model where the phosphodianion binding interactions are utilized to drive an activating change in the conformation of OMPDC.
Experimental Section
Sodium phosphite (dibasic, pentahydrate), 3-(N-morpholino)propanesulfonic acid (MOPS, ≥99.5%), and ammonium acetate (≥99%) were purchased from Fluka. Water was from a Milli-Q Academic purification system. All other chemicals were reagent grade or better and were used without further purification. The trisodium salt of orotidine 5′-monophosphate (99%) was prepared by adaptation of published enzymatic methods,35 from orotate and phosphoribosylpyrophosphate.25 This synthesis gave OMP that contained small amounts of ammonium bicarbonate, which was removed as described in an earlier publication.25 Solution pH was determined at 25 °C using an Orion model 720A pH meter equipped with a radiometer pHC4006-9 combination electrode that was standardized at pH 7.00 and 10.00 at 25 °C. Stock solutions of OMP were prepared in water, and the concentration of OMP was determined from the absorbance at 267 nm using ε = 9430 M–1 cm–1 of a small aliquot diluted with 0.1 M HCl.36
Wild-Type and Mutant Forms of OMPDC
The plasmid pScODC-15b containing the gene encoding OMPDC from Saccharomyces cerevisiae with a N-terminal His6-tag was available from previous studies.31,33 In all cases, the protein sequence differs from the published sequence for wild-type yeast OMPDC by the following mutations: S2H,37 C155S,38 A160S, and N267D.37 This sequence is the same as that observed in the published crystal structure of wild-type yeast OMPDC,26 except for the C155S mutation, which was introduced to enhance the protein stability.38 The S2H, C155S, A160S, and N267D mutations do not cause a significant change in the kinetic parameters for wild-type OMPDC.
Site-directed mutagenesis on pScODC-15b was carried out using the QuikChange II kit. The procedures for the preparation of the Q215A,33 R235A,34 Y217F,28 Q215A/Y217F,28 Q215A/R235A,28 Y217F/R235A,28 and Q215A/Y217F/R235A28 mutants were described in earlier work. In all cases, the N-terminal-His6 or -His10 tag was removed by the action of thrombin (1 unit/mg mutant OMPDC) at room temperature for ca. 16 h, as described in the Supporting Information to ref (33). Wild-type and mutant forms of OMPDC were stored at −80 °C. The OMPDC was defrosted and dialyzed at 4 °C against 10 mM MOPS (50% free base) at pH 7.1 and 100 mM NaCl. The concentration of stock solutions of wild-type and mutant forms of OMPDC was determined from the absorbance at 280 nm and the extinction coefficient, which was calculated using the ProtParam tool available on the ExPASy server.39,40 A value of ε = 29 900 M–1 cm–1 was determined for wild-type and most mutants of OMPDC, except mutants that contain the Y217F substitution, for which a value of ε = 28 400 M–1 cm–1 was determined.
Enzyme Assays
The decarboxylation of OMP was monitored spectrophotometrically by following the decrease in absorbance at 279, 290, 295, or 300 nm [Δε, λ; −2400 M cm–1, 279 nm; −1620 M cm–1, 290 nm; −842 M s–1, 295 nm; −344 M cm–1, 300 nm]. Care was taken to work at a wavelength where the initial absorbance for OMP is ≤2.0. The following wavelengths were monitored for reactions at different concentrations of OMP: [OMP] ≤ 0.08 mM, 279 nm; [OMP] = 0.1–0.3 mM, 290 nm; [OMP] = 0.3–0.8 mM, 295 nm; [OMP] ≥ 1 mM, 300 nm. Assays for OMPDC-catalyzed decarboxylation of OMP were conducted at 25 °C and pH 7.1 (30 mM MOPS) and at constant ionic strength, which was maintained with NaCl. The initial velocity v (M s–1), for OMPDC-catalyzed decarboxylation of OMP (0.05–2.5 mM), was determined by monitoring the decrease in absorbance at the chosen wavelength for the decarboxylation of 5–10% total OMP. Values of kcat and Km were obtained from the nonlinear least-squares fits of six or more values of v/[E] (s–1) to the Michaelis–Menten equation.
Results
Figure 3 shows plots of v/[E] against [OMP] for decarboxylation catalyzed by the Q215A/Y217F (Figure 3A) and Q215A/R235A (Figure 3B) mutants of OMPDC at 25 °C, pH 7.1 (30 mM MOPS), and different ionic strengths (NaCl). The solid lines in Figure 3 show the nonlinear least-squares fit of these experimental data to the Michaelis–Menten equation. The values of kcat, Km, and kcat/Km for decarboxylation of OMP catalyzed by mutant forms of OMPDC obtained from these fits are reported in Table 1. Table 1 also reports values of kcat, Km, and kcat/Km for decarboxylation of OMP catalyzed by the Y217F mutant of OMPDC [data not shown] and for the R235A and Q215A mutant enzymes reported in earlier work.33,34
Figure 3.

Dependence of v/[E] for decarboxylation of OMP catalyzed by mutant forms of OMPDC on the concentration of OMP for reactions at 25 °C, pH 7.1 (30 mM MOPS) and at different ionic strengths (NaCl). (A) Q215A/Y217F OMPDC: (●) I = 0.105; (▲) I = 0.050. (B) Q215A/R235A: (●) I = 0.105; (B) (▲) I = 0.050.
Table 1. Effect of Mutations of Phosphodianion Gripper Amino Acid Residues on the Kinetic Parameters for OMPDC-Catalyzed Decarboxylation of OMPa.
| OMPDC | I | kcat (s–1)b | Km (M)b | kcat/Km (M–1 s–1) |
|---|---|---|---|---|
| wild type | 0.105 | 15 | 1.4 × 10–6 | 1.1 × 107 |
| Q215Ac | 0.105 | 24 ± 1 | (0.94 ± 0.09) × 10–4 | 2.6 × 105 |
| Y217F | 0.105 | 20 ± 1 | (1.1 ± 0.15) × 10–4 | 1.8 × 105 |
| R235Ad | 0.105 | 1.0 | (1.1 ± 0.15) × 10–3 | 910 |
| Q215A/Y217Fe | 0.105 | 4.8 ± 0.25 | (1.4 ± 0.24) × 10–3 | 3.4 × 103 |
| 0.05 | 5.0 ± 0.32 | (0.29 ± 0.04) × 10–3 | 1.7 × 104 | |
| Q215A/R235Af | 0.105 | 0.020 ± 0.02 | (1.4 ± 0.1) × 10–3 | 14 |
| 0.05 | 0.019 ± 0.05 | (3.5 ± 0.3) × 10–4 | 54 | |
| Y217F/R235Ag | 0.105 | 0.11 ± 0.016 | (27.1 ± 4.4) × 10–3 | 4.1 |
| 0.075 | 0.11 ± 0.016 | (12.9 ± 2.1) × 10–3 | 8.5 | |
| 0.05 | 0.11 ± 0.016 | (6.1 ± 1.1) × 10–3 | 18 | |
| triple mutanth | 0.105 | (4.8 ± 0.8) × 10–4 | (12.9 ± 2.4) × 10–3 | 0.037 |
| 0.05 | (4.8 ± 0.8) × 10–4 | (2.2 ± 0.5) × 10–3 | 0.22 |
For reactions at pH 7.1 (30 mM MOPS), 25 °C, and constant ionic strength (NaCl).
The quoted errors are the standard deviations obtained from the nonlinear least-squares fits of data from Figures 3 and 4 to the Michaelis–Menten equation.
Data from ref (33).
Data from ref (34).
Data from Figure 3A.
Data from Figure 3B.
Data from Figure 4A.
Data from Figure 4B.
Figure 4 shows plots of v/[E] against [OMP] for decarboxylation catalyzed by the Y217F/R235A (Figure 4A) and Q215A/Y217F/R235A (Figure 4B) mutants of OMPDC at 25 °C, pH 7.1 (30 mM MOPS), and different ionic strengths (NaCl). The solid lines in Figure 4 show the nonlinear least-squares fit of the experimental data to the Michaelis–Menten equation. The kinetic parameters obtained from the fits of these plot are listed in Table 1. The deviations from a linear dependence of velocity on [OMP] for Figure 4A and B are consistent with the accumulation of a Michaelis complex, but the curvature is small and the values of Km obtained from the fits of the data to the Michaelis–Menten equation are significantly larger than the highest concentration of OMP used in the determination of Km. Consequently, the uncertainty in the values of the kinetic parameters determined for decarboxylation catalyzed by the Y217F/R235A and Q215A/Y217F/R235A mutants is large.
Figure 4.

Dependence of v/[E] for decarboxylation of OMP catalyzed by mutant forms of OMPDC on the concentration of OMP for reactions at 25 °C, pH 7.1 (30 mM MOPS) and at different ionic strengths (NaCl). (A) Y217F/R235A: (●) I = 0.105; (■) I = 0.075; (▲) I = 0.050. (B) Q215A/Y217F/R235A: (●) I = 0.105; (▲) I = 0.050.
Discussion
The value of kcat/Km = 1.8 × 105 M–1 s–1 for the Y217F mutant is 70-fold larger than kcat/Km = 2.5 × 103 M–1 s–1 for the Y217A mutant determined under similar reaction conditions.41,42 A phenyl ring at position 217 is therefore required for the robust functioning of the dianion gripper loop. We suggest that the aromatic ring serves to optimally position the side chains of Q215 and R235 about the ligand phosphodianion, at a tightly packed enzyme active site, and that shifts in these side chains at the Y217A mutant result in a reduction in their stabilizing interactions with the phosphodianion at the transition state for OMPDC-catalyzed decarboxylation reactions.
By comparison, the Y16F mutation of ketosteroid isomerase (KSI) results in 100 000-fold reduction in kcat/Km for enzyme-catalyzed isomerization of 5(10)-estrene-3,17-dione, while the structurally less more conservative Y16S mutation results in a only a 100-fold reduction in kcat/Km.43 The phenol side chain at position 16 of wild-type KSI is thought to form a stabilizing hydrogen bond to the dienolate intermediate of the KSI-catalyzed reaction. Spectroscopic and structural studies on KSI suggest that the Y16F mutation results in unfavorable interactions beyond the loss of a stabilizing phenol–dienolate hydrogen bond.43
Figures 3 and 4, and Table 1 show that the increase in ionic strength from 0.050 to 0.10 results in similar increases in the value for Km, but has little effect on kcat for the decarboxylation of OMP catalyzed by several mutants of OMPDC. This is in agreement with our earlier report that an increase in ionic strength (I) from 0.020–0.10 results in an increase in Km = 0.16 × 10–3 M to Km = 1.1 × 10–3 M, but little change in kcat for decarboxylation of OMP catalyzed by R235A mutant OMPDC.25 A related increase in Km with increasing I, observed for the triosephosphate isomerase (TIM) catalyzed isomerization of glyceraldehyde 3-phosphate dianion,44 has been attributed to a decreasing activity coefficient for the substrate dianion at increasing ionic strength.45
Phosphodianion Binding Energy for Side Chains of OMPDC
The intrinsic phosphodianion binding energy ((ΔGPi∓)int) is defined as the dianion binding energy utilized in stabilization of the transition state for the wild-type OMPDC-catalyzed reaction. It is calculated from the ratio of second-order rate constants for wild-type OMPDC-catalyzed decarboxylation of OMP and the phosphodianion truncated substrate EO.5,13,46,47 An important goal of this work was to determine the contribution (ΔGPi∓)int of the individual gripper side chains to (ΔGPi∓)int. Scheme 3 compares the kinetic parameters for decarboxylation of the whole substrate OMP (Table 1) and the truncated substrate EO28 catalyzed by wild-type OMPDC and a representative mutant. The intrinsic phosphodianion binding energy for wild-type ((ΔGPi∓)int) and mutant ((ΔGPi∓)int) OMPDC is calculated from the ratio of the values of kcat/Km for the respective enzyme-catalyzed decarboxylation of whole and truncated substrates. The difference between (ΔGPi∓)int and (ΔGPi∓)int is equal to (ΔGPi∓)int, the contribution of the excised side chain to the phosphodianion binding energy for wild-type OMPDC. Table 2 reports values of (ΔGPi∓)int for wild type and different mutants of OMPDC, and(ΔGPi)intX for the phosphodianion gripper side chains, calculated from the effect of the mutation on (ΔGPi)intWT.
| 2 |
Scheme 3.
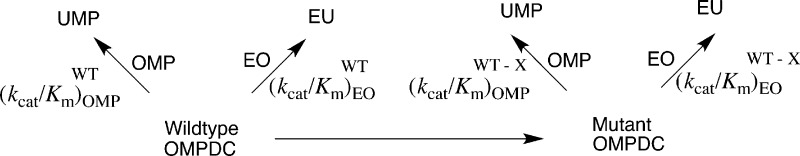
Table 2. Intrinsic Phosphodianion Binding Energies for Wild Type and Mutant Forms of OMPDC and the Effects of Mutations on the Dianion Binding Energya.
| OMPDC | (kcat/Km)OMP/(kcat/Km)EOb | (ΔGPi∓)intc (kcal/mol) | (ΔGPi∓)intd (kcal/mol) |
|---|---|---|---|
| wild type | 4.2 × 108 | 11.7 | |
| Q215A | 2.3 × 107 | 10.0 | 1.7 |
| Y217F | 1.5 × 107 | 9.8 | 1.9 |
| R235A | 3.5 × 104 | 6.2 | 5.5 |
| Q215A/Y217F | 7.4 × 105 | 8.0 | 3.7 |
| Q215A/R235A | 3300 | 4.8 | 6.9 |
| Y217F/R235A | 410 | 3.6 | 8.1 |
| Q215A/Y217F/R235A | 12 | 1.5 | 10.2 |
For OMPDC-catalyzed decarboxylation at 25 °C, pH 7.1 (30 mM MOPS), and ionic strength of 0.105 (NaCl).
Ratio of second-order rate constants for OMPDC-catalyzed decarboxylation of OMP (Table 1) and EO (ref (28)).
RT ln((kcat/Km)OMP/(kcat/Km)EO).
The difference between the 11.7 kcal/mol intrinsic phosphodianion binding energy for wild-type OMPDC and the respective mutant of OMPDC.
The side chains of Q215, Y217, and R235 provide 1.7, 1.9, and 5.5 kcal/mol stabilization, respectively, of the transition state for OMPDC-catalyzed decarboxylation of OMP (Table 2). Each of these apparent interaction energies underestimates the true stabilization of the transition state by ca. 0.3 kcal/mol per side chain. This is because the rate of the wild-type OMPDC-catalyzed reaction is partly limited by substrate binding, so that the side-chain interactions are not fully expressed at the “effective” rate-determining transition state.24 We estimate a total 10 kcal/mol stabilization of the transition state by interactions of the gripper side chains with the substrate phosphodianion, which is ca. 1 kcal/mol larger that the sum of the three interactions energies (9.1 kcal/mol). A similar 10.2 kcal/mol total interaction energy is estimated (Table 2) from the difference in the intrinsic phosphodianion binding energy for wild-type OMPDC (11.7 kcal/mol) and the Q215A/Y217F/R235A mutant (1.5 kcal/mol). The ca. 2 kcal/mol intrinsic dianion binding energy observed for the triple mutant is consistent with a small stabilization of the decarboxylation transition state by the hydrogen bonds from the backbone amides from G234 and R235 to the substrate phosphodianion (Figure 2).
Scheme 4 and Table 2 show that the 12 kcal/mol transition state stabilization by interactions between the phosphodianion and OMPDC represents the sum of a 6 kcal/mol electrostatic interaction from the cationic side chain of R235 and (2 + 2 + 2) = 6 kcal/mol interactions from hydrogen bonds to side chains from Q215 and Y217 and backbone amide −NH– from G234 and R235. Similar analyses of the effect of site-directed mutations on catalysis by 3-ketosteroid isomerase,48 and tryosyl-tRNA synthetase,49,50 have shown that substantial stabilization of the transition states for these enzymatic reactions is obtained from the total, additive, effect of networks of individual stabilizing hydrogen-bonding/electrostatic interactions.
Scheme 4.
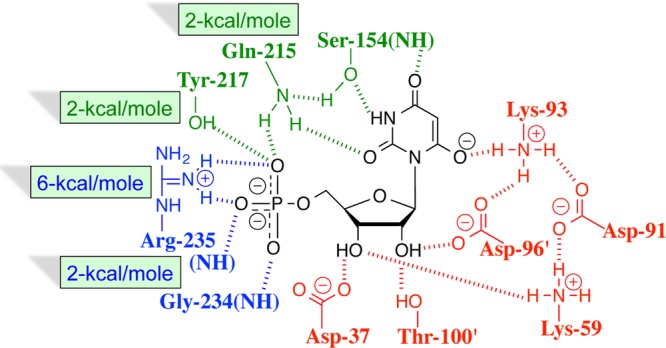
We conclude that the large intrinsic phosphodianion binding energy for OMPDC is obtained from the additive contribution of the network of hydrogen-bonding and electrostatic interactions shown in Scheme 4. We likewise propose that the entire 31 kcal/mol transition state stabilization for OMPDC represents the sum of the binding interactions between the phosphodianion, ribosyl, and pyrimidine fragments of the decarboxylation transition state, which is mimicked by the complex between OMPDC and the transition state analogue BMP (Scheme 4). In other words, the structure of the OMPDC·transition state complex is sufficient to provide a full rationalization of the enzymatic rate acceleration. The conformational change triggered by substrate binding (Figure 1) defines the pathway followed on proceeding from unliganded OMPDC to the active closed enzyme. This is a critical step in the catalytic cycle that deserves close study, to determine the mechanism by which the change in enzyme structure activates OMPDC for catalysis. However, the characterization of the pathway for this conformational change is incidental to the development of an understanding of the magnitude of the stabilization of the decarboxylation transition state, which is determined by the barrier to transition state formation after completion of the enzyme conformational change.
There is strong evidence, for triosephosphate isomerase (TIM)-catalyzed isomerization of d-glyceraldehyde 3-phosphate to dihydroxyacetone phosphate, that enzyme activation results from the utilization of the dianion binding energy to lock TIM into an active conformation, which is present at a low fractional concentration.2,11,51,52 This mechanism is illustrated by Scheme 5 for activation of OMPDC by phosphite dianion, where KC ≪ 1 for the protein conformational change. The open form of OMPDC (EO) is disordered, due to the conformational flexibility of the phosphodianion and pyrimidine gripper loops (Figure 1), and the phosphodianion binding energy is utilized, in part, to organize/position the catalytic side chains at the closed enzyme EC.53,54 The closure of these loops over enzyme-bound OMP requires the sampling of different protein conformations.12,55 This loop closure may partly limit the magnitude of the second-order rate constant kcat/Km for wild-type OMPDC-catalyzed decarboxylation of OMP and 5-fluoro-OMP (Table 1).24,29,31,35,56
Scheme 5.

Reactions of the Whole Substrate and the Substrate in Pieces
A key element of the proposal illustrated by Scheme 5 is that the phosphodianion binding energy is utilized exclusively to drive an unfavorable protein conformational change, but that the dianion does not interact directly with the transition state for the decarboxylation reaction, or affect the transition state structure.57 The barrier for conversion of OMPDC and OMP to the transition state for enzyme-catalyzed decarboxylation [(ΔG⧧)OMP] is defined by the second-order rate constant (kcat/Km)OMP, while the barrier to formation of the transition state for OMPDC-catalyzed decarboxylation of the pieces EO + HPi [(ΔG⧧)EO+HPi] is defined by the third-order rate constant kcat/KHPiKEO (Scheme 6). Figure 5 presents the linear logarithmic free energy relationship between these barriers for wild-type and mutant OMPDC-catalyzed reactions of the whole substrate OMP and the substrate pieces EO + HPi. The unit slope of this correlation (1.05 ± 0.08) requires that these mutations result in the same destabilization of the two transition states for the catalyzed reactions of the whole substrate and substrate pieces, because they exhibit strikingly similar interactions with phosphodianion gripper side chains, and by this criterion are remarkably similar. This is consistent with our proposal that dianion binding energy is utilized for the sole purpose of locking OMPDC into an active conformation.
Scheme 6.
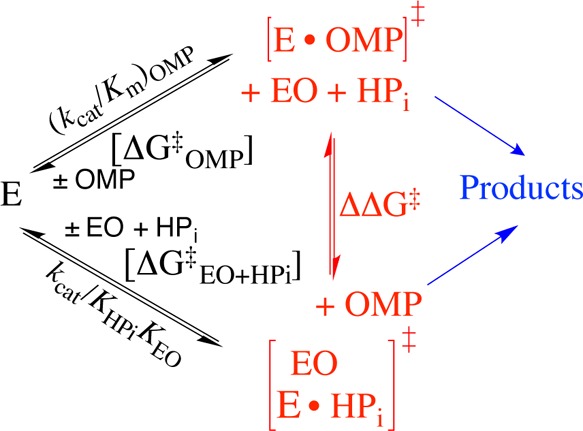
Figure 5.
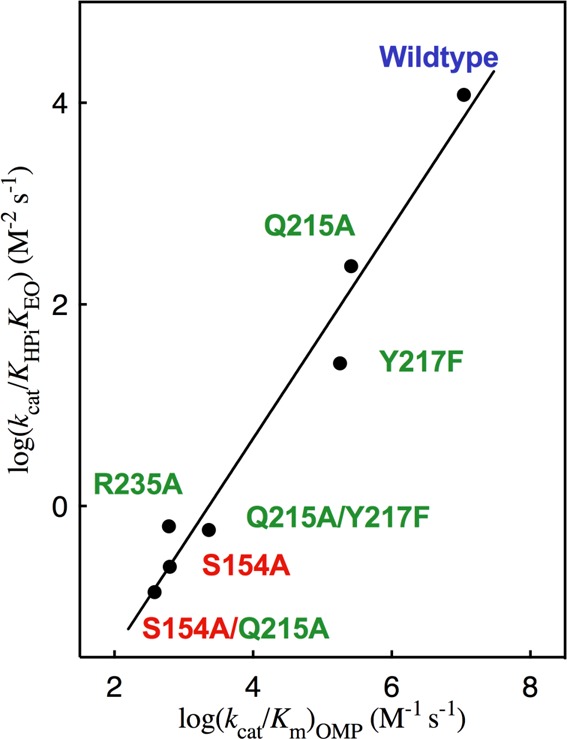
Linear free energy relationship, with slope 1.05 ± 0.08, between the second-order rate constant [log(kcat/Km)OMP] for wild type and mutant OMPDC-catalyzed decarboxylation of OMP and the corresponding third-order rate constant [log(kcat/KHPiKEO)] for the enzyme-catalyzed reactions of the substrate pieces EO and HPi. The residues that interact with the phosphodianion and pyrimidine ring are shown using green and red print, respectively. Literature data are used for the reactions catalyzed by S154A and the S154A/Q215A mutants of OMPDC.33
The unit slope for Figure 5 reflects the constant difference in the activation barriers for the reactions of the whole substrate OMP and the pieces EO + HPi: ΔΔG⧧ = (4.5 ± 0.5) kcal/mol (Scheme 6). This difference is due largely or entirely to the entropic advantage to the binding of the transition state for decarboxylation of OMP compared with the transition state in pieces.58 A larger value of ΔΔG⧧ = 6.6 kcal/mol has been determined in a related study of the effect of site-directed mutations of TIM on the kinetic parameters for enzyme-catalyzed isomerization of the whole substrate d-glyceraldehyde 3-phosphate and the substrate pieces [1-13C]-glycolaldehyde and HPi.57
We conclude that there is a sharp delineation in the function of the catalytic and phosphate dianion binding domains of OMPDC. The catalytic domain carries out decarboxylation of OMP and EO and provides an impressive (31 – 12) = 19 kcal/mol transition state stabilization for the latter substrate. This transition state stabilization is largely independent of any contribution from the dianion binding site, as shown by the similar second-order rate constants for wild type (kcat/Km = 0.026 M–1 s–1) and Q215A/Y217F/R235A triple mutant (kcat/Km = 0.0030 M–1 s–1) OMPDC-catalyzed decarboxylation of EO.28 Dianion binding promotes the chemistry at the distant catalytic domain by providing binding energy, which functions as a “glue” to lock the enzyme into an active conformation. This domain shows a high discrimination between structurally homologous dianions, which has been rigorously characterized for a series of inorganic tetrahedral dianions.59
Interactions Between Protein Side Chains
Point mutations of wild-type OMPDC result in only small changes in kcat/Km for decarboxylation of the phosphodianion truncated substrate EO (ΔΔGX∓)EO,28 which are reflected (eq 3) here as small (≤0.5 kcal/mol) differences between the effect of the mutations on the activation barrier for OMPDC-catalyzed decarboxylation of OMP, (ΔΔGX)OMP (Table 3), and the intrinsic side-chain phosphodianion binding energy, (ΔGPi∓)int (Table 2).
| 3 |
Table 3. Effect of Mutations on the Activation Barrier (ΔG⧧)OMP for Decarboxylation of OMP Catalyzed by Wild-Type OMPDC, and by Previously Mutated OMPDCa.
| (ΔΔGX∓)OMP (kcal/mol)b |
||||
|---|---|---|---|---|
| mutated OMPDC | Q215A | Y217F | R235A | ∑(ΔΔGX∓)OMPc |
| wild type | 2.2 | 2.4 | 5.6 | 10.2 |
| single mutantd | 2.5 (R235A) | 2.5 (Q215A) | 5.8 (Q215A) | 10.8 |
| 2.3 (Y217F) | 3.2 (R235A) | 6.3 (Y217F) | 11.8 | |
| double mutant | 2.8 | 3.5 | 6.8 | 13.4 |
For OMPDC-catalyzed decarboxylation at 25 °C, pH 7.1 (30 mM MOPS), and ionic strength of 0.105 (NaCl).
The effect of the point mutation on (ΔG⧧)OMP for decarboxylation of OMP catalyzed by the precursor enzyme, calculated from the ratio of values of kcat/Km for the precursor and mutant enzymes.
The sum of the effects of the Q215A, Y217F, and R235A mutations on (ΔΔGX∓)OMP.
The enzyme that was mutated is given in parentheses.
A closer examination of the effect of these mutations on (ΔΔGX∓)OMP (Table 3) provides insight into the nature of the interactions between the gripper side chains (Figure 2).60,61 Figure 6 was constructed for mutations of the three amino acid side chains of OMPDC that interact with the phosphodianion, using data from Table 3. Figure 6 and Table 3 show that the effect of a point mutation of residue X on (ΔΔGX)OMP is approximately the same when the mutation is carried out at wild-type OMPDC, singly mutated, or doubly mutated forms of OMPDC. We conclude that the interactions between the phosphodianion gripper side chains are small, but not negligible.
Figure 6.
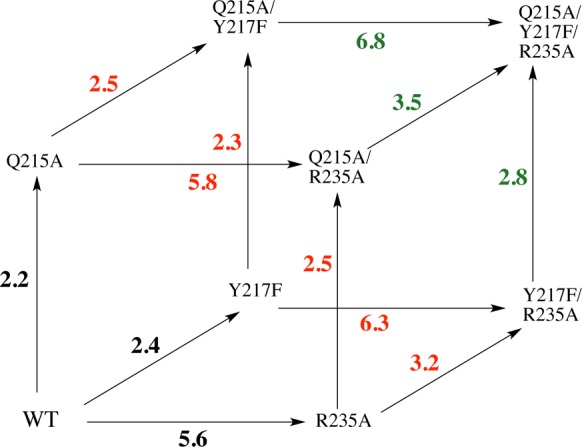
A triple mutant box showing the effects, in kcal/mol, of single amino acid mutations on (ΔG⧧)OMP for reactions catalyzed by wild-type OMPDC (black values), by single mutants of OMPDC (red values), and by double mutants of OMPDC (green values). These effects are reported as (ΔΔGX∓)OMP for the mutations, calculated from the ratio of the kinetic parameters kcat/Km for precursor and mutated OMPDC-catalyzed decarboxylation of OMP, which are reported in Table 3.
Most of the pairwise interaction energies, which can be calculated following procedures described by Horovitz and Fersht, are ≤0.3 kcal/mol.61 However, the effect of the Y217F or R235A mutations on decarboxylation catalyzed by wild-type OMPDC ((ΔΔGX∓)OMP = 2.4 and 5.6 kcal/mol, respectively) is ca. 0.8 kcal/mol smaller than the effect of the corresponding mutations on decarboxylation catalyzed by singly mutated OMPDC ((ΔΔGX)OMP = 3.2 and 6.3 kcal/mol). About 0.3 kcal/mol of this difference results because chemistry is partly rate determining for wild-type OMPDC, but fully rate determining for the relevant mutant enzymes (see above). Even after a 0.3 kcal/mol correction, the effect of the second mutation remains larger than for the initial mutation of wild-type OMPDC. We attribute this difference to a destabilizing interaction between the side chains of Y217 and R235 at wild-type OMPDC, which is relieved at either single mutant.
We note the compact arrangement of gripper side chains around the phosphodianion at wild-type OMPDC (Figure 2) and propose that the loss of the stabilizing interaction between the phosphodianion and the side chains of R235 or Y217 at the single mutants is partly offset by the relief of a small destabilizing steric interaction between the two side chains. By comparison, the effect of any single mutation at wild-type OMPDC is 0.6–1.2 kcal/mol smaller than the effect of the same mutation of the previously doubly mutated enzyme (Table 3). For example, (ΔΔGX∓)OMP = 5.6 and 6.8 kcal/mol, respectively, for an R235A mutation at wild-type OMPDC and at the Q215A/Y217F double mutant. This difference is also consistent with steric crowding of the amino acid side chains around the phosphodianion of wild-type OMPDC (Figure 2), which is relieved by truncation of two amino acid side chains.
Mechanism for Dianion Activation of OMPDC for Decarboxylation of OMP
Interactions between OMPDC and HPi, or the dianion of OMP, are utilized to drive a conformational change that activates OMPDC for catalysis of decarboxylation of EO and OMP, respectively.10 The weak (Kd ≥ 0.1 M)5 binding of HPi to OMPDC (Scheme 5) requires that essentially the entire 8 kcal/mol phosphite dianion binding energy be utilized to drive the activating enzyme conformational change from EO to EC (Scheme 7). Tethering the two pieces in the whole substrate OMP results in an increase in the dianion binding energy to 12 kcal/mol, because of the anchoring effect of the attachment.5,58 This additional ca. 4 kcal/mol of dianion binding energy is available to stabilize the Michaelis complex to OMP, while the same 8 kcal/mol dianion binding energy as for phosphite dianion is needed to drive the activating enzyme conformational change. This predicts that ca. 4 kcal/mol of the dianion binding energy for OMP will be expressed in the value of the observed Michaelis constant and that the 8 kcal/mol of binding energy utilized to drive the enzyme conformational change will be expressed as an increase in (kcat)obs.
Scheme 7.
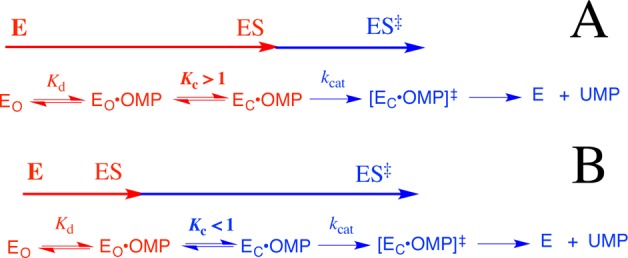
Equations 4–6, derived for Scheme 7, show the relationship between the microscopic rate and equilibrium constants Kd, kcat, and Kc, and the observed kinetic parameters (kcat)obs and (Km)obs for OMPDC-catalyzed decarboxylation of OMP, where Kc is for the enzyme conformational change, Kd is for release of OMP from EO, and kcat is for turnover of EC·OMP.25 If the 12 kcal/mol intrinsic dianion binding energy of OMP is divided between the 8 kcal/mol of binding energy utilized to drive the enzyme conformational change and the 4 kcal/mol expressed as stabilization of the Michaelis complex to OMP, then the closure of OMPDC will be thermodynamically favorable for wild-type OMPDC by 4 kcal/mol (Scheme 7A), so that (Kc ≫ 1), (kcat)obs ≈ kcat, and (Km)obs = Kd/Kc (eqs 4–6). The initial mutation of a gripper residue will cause a decrease in Kc that will be expressed as an increase in (Km)obs but not (kcat)obs, and (Km)obs will increase until the Michaelis complex is destabilized by ca. 4 kcal/mol and Kc = 1. Once Kc < 1, the enzyme conformational change becomes a step on the pathway from the dominant complex EO·OMP to the decarboxylation transition state (Scheme 7B), so that (kcat)obs ≈ kcatKc and (KM)obs≈ Kd (eqs 4–6). Further mutations of gripper residues will now result in a decrease in (kcat)obs but not (Km)obs.
 |
4 |
| 5 |
| 6 |
This analysis predicts that effect of mutations on (Km)obs and (kcat)obs will depend upon the context of the mutation. The mutation of any gripper residue at wild-type OMPDC will be expressed on (Km)obs (Kc ≫ 1), but mutations of the same residue will be expressed on (kcat)obs if previous mutations have resulted in Kc < 1. The predicted context dependence of the effects of these mutations on (kcat)obs and (Km)obs is in good agreement with the observed effects.
(1) The Q215A and Y217F mutations result in (60–70-fold) increases in Km for decarboxylation of OMP, and surprisingly small increases in kcat (Table 1). These changes are consistent with Kc ≫ 1 (eqs 4–6) for wild-type OMPDC, and a small increase in the rate of product release, which limits the value for kcat.24
(2) The overall effect of the R235A mutation is to destabilize the rate-determining transition state for OMPDC-catalyzed decarboxylation by 5.6 kcal/mol (Table 3). About 4 kcal/mol of this effect is expressed as a 700-fold increase in Km to a limiting value of Km ≈ Kd = 1 mM, which corresponds to a 4 kcal/mol destabilization of the Michaelis complex, when Kc ≪ 1. The remaining effect of this mutation (1.6 kcal/mol) is expressed as a 15-fold decrease in kcat (Table 1).
(3) The Q215A mutation of wild-type OMPDC results mainly in an increase in Km. However, the same mutation at the R235A mutant has almost no effect on Km, which is at the limiting value of 1 mM (KC < 1, Scheme 7B). Now, almost the entire effect of the Q215A mutation is expressed as a decrease in kcat (Table 1).
(4) The effects of the Q215A and Y217F single mutations are expressed as changes in Km, while the Q215A/Y217F double mutation results in a limiting Km = 1 mM, and a decrease in the value of kcat to 4.8 s–1 (Table 1).
We note that the values Km for the Y217F/R235A double mutant and the Q215A/Y217F/R235A triple mutants (Table 1) are larger than the value of Kd ≈ 1 mM, which we propose represents the limiting affinity of wild-type EO for OMP. These increases in Km may reflect small second-order effects of extensive mutations on the stability of the unreactive EO·OMP complex (Scheme 7). Finally, the proposed limiting Kd of ∼1 mM for binding of OMP to EO (Scheme 7) is smaller than the Kd ≈ 100 mM estimated for the truncated substrate EO (Scheme 2A).5 This is consistent with stabilization of the EO·OMP complex by nonproductive binding interactions between mutated phosphodianion gripper loops and the phosphodianion of OMP. Such nonproductive binding interactions will result in equal increases in kcat and Km, but will not affect kcat/Km for OMPDC-catalyzed decarboxylation.62
This interpretation of the results from Table 1 unifies the easily quantified utilization of the 8 kcal/mol intrinsic phosphite dianion binding energy in activation of OMPDC for decarboxylation of a truncated substrate EO, and the cryptic utilization of 8 kcal/mol of phosphodianion binding energy in activation of OMPDC for decarboxylation of OMP (Scheme 7). In each case, 8 kcal/mol of dianion binding energy is used to drive an activating change in the conformation of OMPDC from EO to EC. This corresponds to the entire phosphite dianion binding energy for activation of OMPDC-catalyzed decarboxylation of EO. The larger 12 kcal/mol intrinsic dianion binding energy for OMP is divided between the 4 kcal/mol expressed in the observed binding constant (Km)obs, and the 8 kcal/mol that is utilized to drive the unfavorable conversion of inactive EO·OMP to active EC·OMP, and which is expressed as an increase in (kcat)obs.
Summary
We conclude by reviewing what is known about the origin of the extraordinary catalytic clout obtained from OMPDC–dianion interactions.5
(1) There is a fair agreement between the total intrinsic dianion binding energy and the sum of the contributing interactions of the amino acid side chains determined in mutagenesis studies (Scheme 4). This shows that there are only small interactions energies for the dianion gripper side chains (Figure 6).
(2) There is little or no apparent requirement that the binding energy of gripper side chains be “wasted” in immobilizing the rigid loop over the substrate phosphodianion.32 Such an entropic penalty would be a minimum for point mutations at wild-type OMPDC, because the remaining side chains would act to hold the loop in a fixed conformation, and a maximum for point mutations at double mutants to give the Q215A/Y217F/R235A triple mutant. By contrast, point mutations of wild-type OMPDC result in a 0.6–1.2 kcal/mol (Table 3) smaller transition state destabilization than the same point mutation at a double mutant, a difference opposite of that expected if entropic effects were important.
(3) The very small effect of mutations at the dianion gripper loop on kcat/Km for decarboxylation of the truncated substrate EO shows that there is little or no interaction between the amino acid side chains at the dianion binding domain and the pyrimidine ring bound at the catalytic domain.28
(4) The large effect of mutations at the dianion gripper loop on kcat/KHPiKEO for phosphite dianion activation of decarboxylation of EO (Scheme 6) shows that loop dianion interactions act to stabilize the decarboxylation transition state, in the absence of a direct interaction between the loop and the reacting orotate ring.28
(5) Closure of the dianion gripper loop and the smaller movement of the side chain of R235 is just one component of a global conformational change of OMPDC, whose precise role in the catalysis of decarboxylation of OMP remains to be fully determined. A critical element of this enzyme conformational change is formation of a hydrogen bond between the side chains of Ser154 and Gln215 (Scheme 4), which ensures cooperativity in the closure of the phosphodianion gripper loop and the pyrimidine umbrella.33
(6) The enzyme conformational change is expected to result in a tightening of the interactions between OMPDC and the ribosyl and pyrimdine substrate fragments (Scheme 4). It is therefore important to characterize the interactions of these fragments with OMPDC by an experimental protocol similar to that used to examine the role of phosphodianion interactions in the OMPDC-catalyzed decarboxylation reaction.
(7) The separate catalytic and phosphodianion binding domains at OMPDC might be mimicked in the design of an active artificial protein decarboxylase, by first designing a protein that catalyzes decarboxylation at a solvent exposed cleft, and then adding to this protein flexible loop(s), which fold over a nonreacting substrate fragment, such as a phosphodianion, to tightly lock the substrate within a protein cage.12,27 This strategy should result in a catalyst where the tight loop–substrate interactions are expressed as a large turnover number kcat for the decarboxylation reaction.10,13
Acknowledgments
We acknowledge the National Institutes of Health Grant GM39754 (to J.P.R.) and Grant GM65155 (to J.A.G.) for generous support of this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Pauling L. Nature 1948, 161, 707–709. [DOI] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. Biochemistry 2009, 48, 5769–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Biochemistry 2007, 46, 5841–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Amyes T. L.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2011, 133, 6545–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.; Tait J. J. J. Am. Chem. Soc. 2005, 127, 15708–15709. [DOI] [PubMed] [Google Scholar]
- Tsang W.-Y.; Amyes T. L.; Richard J. P. Biochemistry 2008, 47, 4575–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray W. J. Jr.; Long J. W.; Owens J. D. Biochemistry 1976, 15, 4006–4017. [DOI] [PubMed] [Google Scholar]
- Kholodar S. A.; Murkin A. S. Biochemistry 2013, 52, 2302–2308. [DOI] [PubMed] [Google Scholar]
- Zhai X.; Malabanan M. M.; Amyes T. L.; Richard J. P. J. Phys. Org. Chem. 2013, 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Biochemistry 2013, 52, 2021–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. Biochemistry 2012, 51, 2652–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. Curr. Opin. Struct. Biol. 2010, 20, 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 219–410. [DOI] [PubMed] [Google Scholar]
- Miller B. G.; Wolfenden R. Annu. Rev. Biochem. 2002, 71, 847–885. [DOI] [PubMed] [Google Scholar]
- Radzicka A.; Wolfenden R. Science 1995, 267, 90–93. [DOI] [PubMed] [Google Scholar]
- Callahan B. P.; Miller B. G. Bioorg. Chem. 2007, 35, 465–469. [DOI] [PubMed] [Google Scholar]
- Fujihashi M.; Mito K.; Pai E. F.; Miki K. J. Biol. Chem. 2013, 288, 9011–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J.; Cardenas A. M.; Gilbert H. F.; Palzkill T. Protein Sci. 2011, 20, 1891–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2007, 129, 12946–12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2008, 130, 1574–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K.; Gerlt J. A.; Richard J. P. J. Am. Chem. Soc. 2010, 132, 7018–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardi-Kilshtain A.; Doron D.; Major D. T. Biochemistry 2013, 52, 4382–4390. [DOI] [PubMed] [Google Scholar]
- Tsang W.-Y.; Wood B. M.; Wong F. M.; Wu W.; Gerlt J. A.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2012, 134, 14580–14594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D. J. T.; Short S. A. Biochemistry 2000, 39, 11788–11800. [DOI] [PubMed] [Google Scholar]
- Goryanova B.; Goldman L. M.; Amyes T. L.; Gerlt J. A.; Richard J. P. Biochemistry 2013, 52, 7500–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 2011–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. Curr. Opin. Chem. Biol. 2014, 21, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Ming S. A.; Goldman L. M.; Wood B. M.; Desai B. J.; Gerlt J. A.; Richard J. P. Biochemistry 2012, 51, 4630–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Spong K.; Amyes T. L.; Richard J. P. Biochemistry 2013, 52, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood B. M.; Amyes T. L.; Fedorov A. A.; Fedorov E. V.; Shabila A.; Almo S. C.; Richard J. P.; Gerlt J. A. Biochemistry 2010, 49, 3514–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood B. M.; Chan K. K.; Amyes T. L.; Richard J. P.; Gerlt J. A. Biochemistry 2009, 48, 5510–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K.; Amyes T. L.; Wood B. M.; Chan K. K.; Gerlt J. A.; Richard J. P. Biochemistry 2009, 48, 8006–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; Wood B. M.; Gerlt J. A.; Richard J. P. Biochemistry 2008, 47, 7785–7787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; McKay Wood B.; Gerlt J. A.; Richard J. P. Biochemistry 2010, 49, 824–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vleet J. L.; Reinhardt L. A.; Miller B. G.; Sievers A.; Cleland W. W. Biochemistry 2008, 47, 798–803. [DOI] [PubMed] [Google Scholar]
- Moffatt J. G. J. Am. Chem. Soc. 1963, 85, 1118–1123. [Google Scholar]
- Miller B. G.; Smiley J. A.; Short S. A.; Wolfenden R. J. Biol. Chem. 1999, 274, 23841–23843. [DOI] [PubMed] [Google Scholar]
- Sievers A.; Wolfenden R. Bioorg. Chem. 2005, 33, 45–52. [DOI] [PubMed] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. Nucleic Acids Res. 2003, 31, 3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A., Eds. Protein Identification and Analysis Tools on the ExPASy Server; Humana Press Inc.: Totowa, NJ, 2005. [Google Scholar]
- Miller B. G.; Snider M. J.; Short S. A.; Wolfenden R. Biochemistry 2000, 39, 8113–8118. [DOI] [PubMed] [Google Scholar]
- Barnett S. A. Ph.D. Thesis, University at Buffalo, Buffalo, NY, 2009. [Google Scholar]
- Kraut D. A.; Sigala P. A.; Fenn T. D.; Herschlag D. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman F.; Lamuraglia G.; Tomozawa Y.; Wolfenden R. Biochemistry 1975, 14, 5274–5279. [DOI] [PubMed] [Google Scholar]
- Nolte H.-J.; Rosenberry T. L.; Neumann E. Biochemistry 1980, 19, 3705–3711. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; O’Donoghue A. C.; Richard J. P. J. Am. Chem. Soc. 2001, 123, 11325–11326. [DOI] [PubMed] [Google Scholar]
- Morrow J. R.; Amyes T. L.; Richard J. P. Acc. Chem. Res. 2008, 41, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwans J. P.; Sunden F.; Gonzalez A.; Tsai Y.; Herschlag D. J. Am. Chem. Soc. 2011, 133, 20052–20055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fersht A. R. Biochemistry 1987, 26, 8031–8037. [DOI] [PubMed] [Google Scholar]
- Leatherbarrow R. J.; Fersht A. R.; Winter G. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 7840–7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Koudelka A. P.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2012, 134, 10286–10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2011, 133, 16428–16431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Florian J.; Strajbl M.; Villa J. ChemBioChem 2001, 2, 109–111. [DOI] [PubMed] [Google Scholar]
- Warshel A. J. Biol. Chem. 1998, 273, 27035–27038. [DOI] [PubMed] [Google Scholar]
- Schulenburg C.; Hilvert D. Top. Curr. Chem. 2013, 337, 41–67. [DOI] [PubMed] [Google Scholar]
- Rishavy M. A.; Cleland W. W. Biochemistry 2000, 39, 4569–4574. [DOI] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2014, 136, 4145–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 4046–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spong K.; Amyes T. L.; Richard J. P. J. Am. Chem. Soc. 2013, 135, 18343–18346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackers G. K.; Smith F. R. Annu. Rev. Biochem. 1985, 54, 597–629. [DOI] [PubMed] [Google Scholar]
- Horovitz A.; Fersht A. R. J. Mol. Biol. 1990, 214, 613–617. [DOI] [PubMed] [Google Scholar]
- Jencks W. P.Catalysis in Chemistry and Enzymology; McGraw Hill: New York, 1969. [Google Scholar]