

Abstract

Antibiotic-resistant bacteria present an ongoing challenge to both chemists and biologists as they seek novel compounds and modes of action to out-maneuver continually evolving resistance pathways, especially against Gram-negative strains. The dimeric pyrrole–imidazole alkaloids represent a unique marine natural product class with diverse primary biological activity and chemical architecture. This full account traces the strategy used to develop a second-generation route to key spirocycle 9, culminating in a practical synthesis of the axinellamines and enabling their discovery as broad-spectrum antibacterial agents, with promising activity against both Gram-positive and Gram-negative bacteria. While their detailed mode of antibacterial action remains unclear, the axinellamines appear to cause secondary membrane destabilization and impart an aberrant cellular morphology consistent with the inhibition of normal septum formation. This study serves as a rare example of a natural product initially reported to be devoid of biological activity surfacing as an active antibacterial agent with an intriguing mode of action.
Introduction
With an estimated $45 billion in annual treatment costs in the United States, bacterial infections pose a significant burden to the public health system.1 While ongoing efforts are made to combat pathogenic bacteria, the selection pressures that are intrinsic to therapy select for the most fit populations and drive the evolution of resistance to even the most potent antibacterials.2 Rice has described the most dangerous multi-drug-resistant (MDR) bacteria as the “ESKAPE” pathogens.3 While representing a minority of these pathogens, Gram-positive bacteria have been the focus of the majority of development efforts over the past two decades, especially methicillin-resistant Staphylococcus aureus (MRSA).4,5 As a result, treatment options against the majority, and increasingly more dangerous,1,6 MDR Gram-negative pathogens have become limited, in particular due to the diminishing efficacy of last-resort antibiotics: colistin (1, Figure 1A) and polymyxin B (2) (which also have significant liabilities due to nephrotoxicity and neurotoxicity).4,7,8 These MDR pathogens can only be combated by the development of new antibiotics, preferably ones that have novel modes of action to slow the inevitable emergence of resistance. Unfortunately, no new class of antibiotics has been approved for Gram-negative bacteria in over 40 years (Figure 1B).9 Several factors are attributed to this innovation gap: (i) low financial interest by pharmaceutical companies based on returns on investment,10 (ii) lack of novel structures and mechanisms of action, and (11) (iii) antibacterial drug supply problems (since most successful antibiotics come from natural products or natural product-derived scaffolds).9 The combination of natural selection and the above-mentioned factors have left the pursuit of effective antibiotics, especially those with potent activity against MDR Gram-negative pathogens, in a state of emergency.12
Figure 1.

Antibiotic drug discovery. aExamples of drug class not necessarily introduced on the exact date depicted (modified from ref (11)).
With a rich history of primary biological properties ranging from anti-infective to immunosuppressive,13 the bioactive and architecturally complex pyrrole–imidazole alkaloids (PIAs) present a grand opportunity for both chemical synthesis and medicinal chemistry. The challenges toward a scalable production of 4–8, as marine isolates (Figure 1C), are far greater compared to many terrestrially derived natural products, where the producing species can be cultivated. Aside from the traditional challenges such as low isolation yields and non-renewable sources, marine-derived agents suffer from (1) laborious sample isolation (scuba diving/ROVs),14a (2) precarious isolation protocols (desalting water-soluble compounds),14b (3) high-order aquaculture process (non-actinomycetes, symbiosis),14a and (4) marine microbial contamination (community-based microbes).14c Thus, chemical synthesis is ideally suited to the challenge of the material supply15 posed by such natural products.
The axinellamines (4 and 5), isolated by Quinn in 1999, were reported to have virtually no bioactivity (minimum inhibitory concentration (MIC) of 1 mM for axinellamine B and no bioactivity for axinellamine A against Helicobacter pylori).16 A follow-up report showed limited antibiotic activity (MICs of 15–30 μM for axinellamine B against Bacillus subtilis only).17
The first section of this full account reports the biological re-evaluation of synthetic axinellamines (racemic) and the striking finding that this unique structural class in fact has promising activity against both Gram-positive and Gram-negative bacteria via an intriguing mode of action (see Table 1). The second section of this report outlines the synthetic strategy aimed at efficiently solving the stereochemical puzzle embedded in the core of these marine alkaloids. This work ultimately led to an efficient and practical synthesis of these complex marine alkaloids as well as interesting methodological developments of more broad utility.
Table 1. Minimum Inhibitory Concentrations.
| A. MIC (μg/mL) of Axinellamines
for
Gram-Positive and Gram-Negative Bacteria | ||
|---|---|---|
| Strain | Ax A (4) | Ax B (5) |
| E. coli K-12 MG1655 | 4 | 4 |
| Y. pestis KIM6+ | 16 | 32 |
| P. aeruginosa PAO1 | 8 | 8 |
| S. aureus NCTC 8325 | 4 | 4 |
| MRSA N315 | 4 | 4 |
| MRSA USA300 | 2 | 2 |
| S. epidermidis RP62A | 2 | 2 |
| S. pneumoniae D39 | 8 | 8 |
| C. efficiens DSM 44549 | 0.5 | 1 |
| E. faecalis ATCC 33186 | 16 | 8 |
| C. albicans BWP17 | 32 | 32 |
| B. MIC (μg/mL)
of Axinellamines for S. aureus NCTC 8325 as a Function of Added Human Serum | ||||||
|---|---|---|---|---|---|---|
| Serum
(%): |
||||||
| Compd | 0 | 3.1 | 6.3 | 12.5 | 25 | 50 |
| Ax A (4) | 4 | 16 | 16 | 32 | 64 | >64 |
| Ax B (5) | 4 | 16 | 32 | 32 | 64 | >64 |
Results and Discussion
Axinellamines as Broad-Spectrum Antibacterial Agents
Discovery
Despite initial reports of the axinellamines’ limited antibacterial activity,16,17 we found 4 and 5 to be active against the tested bacteria (Table 1A). This activity is conserved across different Gram-positive bacteria, including against both hospital-acquired MRSA (N315) and community-acquired MRSA (USA300), and importantly Gram-negative bacteria. The inhibitory activity is, for the most part, selective for prokaryotic versus eukaryotic cells; the MIC against Candida albicans is at least 2- to 32-fold higher. However, the antibacterial activity of 4 and 5 is progressively decreased by the addition of 3%–50% human serum (Table 1B), suggesting that it is limited by significant protein binding. It should also be noted that 4 and 5 have limited solubility in aqueous media and precipitate significantly at concentrations approaching 60 μg/mL. Because of the identical activities of 4 and 5, we chose to focus further characterization efforts predominantly on variant 4. We first determined the frequency of resistance to 24 μg/mL of 4 (6× MIC) for Escherichia coli K-12 MG1655 and S. aureus NCTC 8325. The frequencies of resistance were 5 × 10–6 and 1 × 10–7, respectively. Unexpectedly, the recovered isolates failed to grow in liquid culture supplemented with the same concentration of 4 and retained parental MICs when recharacterized using the microdilution method. Taken together, these results suggest that the susceptibility to 4 may be highly dependent on the inoculum effect.18 To test this possibility, we repeated the MIC assays for 4 and 5 with varying E. coli and S. aureus inocula, ranging from the CLSI standard of 105 CFU/mL to 108 CFU/mL, and found that the appearance of “resistant” isolates is consistent with the high plating inoculations required for mutant selection on solid media (Table 2). The occurrence of “false mutants” has been observed for daptomycin, for which the occurrence of true resistance is quite low.19,20 These results suggest that the true frequency of resistance for the axinellamines is lower than 5 × 10–6 and 1 × 10–7 for E. coli and S. aureus, respectively.
Table 2. Minimum Inhibitory Concentration (MIC, μg/mL) of Axinellamines as a Function of Bacterial Inoculation.
| Inoculation
(CFU/mL): |
|||||
|---|---|---|---|---|---|
| Compd | Organism | 105 | 106 | 107 | 108 |
| Ax A (4) | E. coli K-12 MG1655 | 4 | 32 | 64 | NTa |
| S. aureus NCTC 8325 | 4 | 4 | 32 | 64 | |
| Ax B (5) | E. coli K-12 MG1655 | 4 | 32 | 64 | NTa |
| S. aureus NCTC 8325 | 4 | 4 | 32 | 64 | |
NT = not tested.
Based on their structure and broad-spectrum activity, we hypothesized that the mechanism of action of the axinellamines may be related to membrane destabilization. In order to test for outer membrane perturbation, we assayed for the cleavage of nitrocefin by periplasmic β-lactamase in E. coli K-12 MG1655 carrying a plasmid-encoded β-lactamase. Cleavage of nitrocefin results in a color change that can be monitored spectrophotometrically. With an intact membrane, nitrocefin is excluded from the periplasm, and thus is not cleaved by the periplasmic enzyme. As a positive control, we examined nitrocefin cleavage after addition of the lytic peptide melittin at a concentration range of 2–128 μg/mL. For compound 4, outer membrane permeabilization was delayed compared to that for melittin (Figure 2), demonstrating that it does not efficiently induce membrane disruption, and that the disruption that does occur may be a secondary effect. Interestingly, the related marine alkaloid sceptrin has been shown to induce peptidoglycan turnover, spheroplast formation, and intracellular potassium ion release in E. coli,21 consistent with membrane damage.
Figure 2.
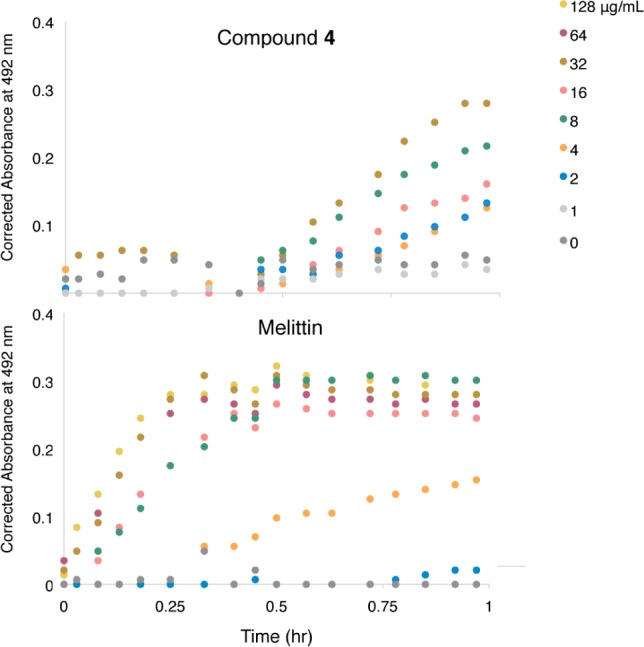
Representative plots showing the outer-membrane permeabilization of E. coli K-12 MG1655 containing pBR322 as a function of [4], μg/mL, using the cleavage of nitrocefin by periplasmic β-lactamase as the reporter. Melittin was used as the positive control. Other runs are shown in Supporting Information, Figure SI-1.
More recently, sceptrin has been shown to bind to MreB,22 the actin homologue in E. coli which plays a key role in elongation and morphology.23,24 Thus, we next sought to further characterize the antibacterial action of 4 by testing its effect on E. coli cellular morphology. After administration of 4 μg/mL of 4 (1× MIC) for 20 min, E. coli takes on an aberrant morphology characterized by mostly elongated and some branching forms (Figure 3). The increase in major axis length is significant compared to the vehicle-only control (Table 3). This phenotype is reminiscent of defects in septation. For example, deletion, mutation, or inhibition of the fts genes, which are essential for cell division, produces cells that grow but do not divide, resulting in a characteristic filamentous phenotype.25,26 Interestingly, after 1 h, the axinellamine-treated cells revert to normal morphology (data not shown). A similar reversion has also been observed for sceptrin under comparable conditions.21 The distinct cellular changes effected by compound 4 suggest that the axinellamines’ antibacterial mode of action may be manifested through the inhibition of normal cellular division. It must be emphasized that the above studies required >100 mg of 4 and 5. In the next section, the chemistry employed to enable these studies will be discussed.
Figure 3.

Cellular morphology of E. coli K-12 MG1655 at 1000× magnification: (A) prior to treatment, (B) treated with DMSO at 20 min, (C,D) treated with Ax A (4) at 20 min. The scale bar (panel A; top right) is equal to 5 μm.
Table 3. Morphological Parametersa (± SEM) Calculated for E. coli Cells Treated with Vehicle vs Axinellamine A, 20 min.
| Treatment group |
||||
|---|---|---|---|---|
| Parameter | DMSO (n = 126) | Ax A (4) (n = 129) | Fold-change | p value |
| perimeter | 135.1 ± 6.7 | 203.2 ± 2.9 | 1.50 | 3.4 × 10–8 |
| major axis | 55.1 ± 2.1 | 86.9 ± 3.0 | 1.58 | 2.5 × 10–12 |
| minor axis | 22.3 ± 1.1 | 26.0 ± 1.8 | 1.16 | 4.7 × 10–2 |
| aspect ratio | 2.6 ± 0.1 | 3.8 ± 0.4 | 1.45 | 3.4 × 10–10 |
The parameters perimeter, major axis, and minor axis are measured in pixels (1 pixel = 0.064 μm).
Retrosynthetic Analysis
Owing to their densely functionalized cores with up to eight contiguous stereocenters, high nitrogen content (ca. 1:2 N:C), and sensitive functional groups, members of the PIA class (4–8) have attained worldwide interest but only very few syntheses have been completed.27 The high degree of molecular complexity of 4–8 can be largely attributed to a single densely functionalized cyclopentyl ring. Guided by the putative biosynthesis, we forged a unified strategy toward the construction of 4–8 from fully functionalized spiroaminoketone 9.28 With this divergent strategy in mind, we accomplished the first successful laboratory syntheses of 4,28b5,28c and 8(28d) on milligram-scale. Although these feasibility-era29 syntheses provided proof of principle for the late-stage strategy, the ultimate challenge of producing these natural products on gram-scale had not been met as the synthetic routes to 4, 5, and 8 suffered from several inefficiencies. The majority of these inefficiencies could be traced back to the route employed to access key spiroaminoketone 9.
The first-generation synthesis of 9 utilized a Diels–Alder/ozonolysis ring contraction strategy to construct the cyclopentyl system and a non-selective aza-Michael addition of the pendant allylic guanidine to set the challenging C7 spirocenter (Scheme 1A). The indirect approach used to access the cyclopentyl ring and the lack of C7 stereoselectivity (1:1) were significant drawbacks, not to mention numerous non-strategic redox manipulations and excessive functional group interchanges. Overall, the synthesis of 9 was accomplished in 20 steps, requiring 12 chromatographic separations to produce 9 in <1% yield from commercially available materials. Considering the poor substrate bias of enone 10 in accessing the correctly configured spirocycle, a new approach that would efficiently set the C7 stereochemistry was deemed essential.
Scheme 1. (A, top) First- and (B, bottom) Second-Generation Retrosynthetic Analysis of Spiroaminoketone (9).
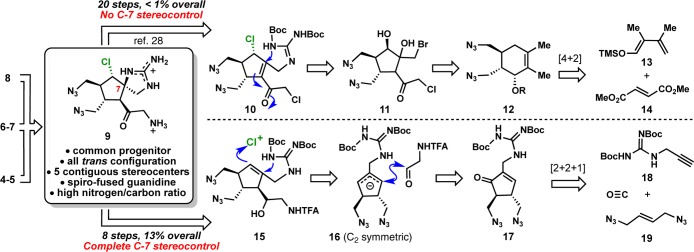
The second-generation route28e envisioned (Scheme 1B) took into account the high stereochemical demand of spiroaminoketone 9 (most importantly the C7 stereochemistry) as well as its all-trans configuration. Thus, a revised retrosynthesis featured a speculative “chloro-guanidylation” of an allylic guanidine 15 by way of stereoselective chloronium formation (presumably due to steric clash between Cl+ and neighboring methylene) followed by anti displacement of the pendant guanidine in a similar fashion to that of iodolactonizations pioneered by Corey.30 Guided by the need for a more efficient synthesis of 9, this key disconnection not only served as an ideal strategy for clearing the two most challenging stereocenters of the fully substituted cyclopentyl framework but also was seen as an opportunity for invention. Building from previous attempts to exploit the hidden symmetry of this class of natural products,27a,31 we proposed allylic guanidine 15 to arise from a stereoselective condensation of a putative C2-symmetric allylic anion (generated from the corresponding allylic chloride) with an appropriate electrophilic side-chain segment. Conversely, one can imagine the same strategic inclusion of a C2-symmetric intermediate using a relevant nucleophile (see Scheme 2C) via an asymmetric allylic functionalization.32 Access to either allylic anion or cation could arise from reduction of cyclopentenone 17, which can be rapidly constructed in a single step via a Pauson–Khand [2+2+1] cycloaddition between propargyl guanidine 18, bis-allylic azide 19, and carbon monoxide.
Scheme 2. Validation and Initial Feasibility Studies.
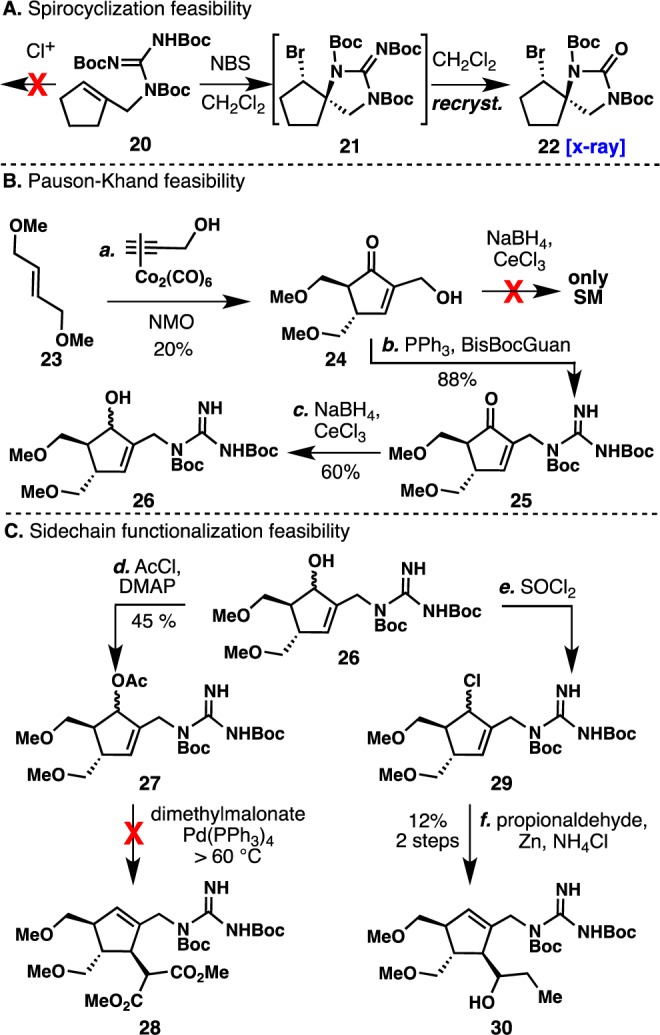
Reagents and conditions: (a) i. propargyl alcohol (1.0 equiv), Co2(CO)8 (1.0 equiv), CH2Cl2, rt, 2 h; ii. (E)-1,4-dimethoxybut-2-ene (3.0 equiv), NMO (6.0 equiv), CH2Cl2, rt, 12 h. (b) PPh3 (1.5 equiv), DIAD (1.5 equiv), bis-Boc-guanidine (1.1 equiv), THF, rt, 2 h. (c) NaBH4 (4.0 equiv), CeCl3·7H2O (1.0 equiv), CH3OH, 0 °C, 12 h. (d) AcCl (1.2 equiv), DMAP (10 mol%), CH2Cl2, rt, 12 h. (e) SOCl2 (1.4 equiv), CH2Cl2, 0 °C, 1 h. (f) Zn (5 equiv), H2O/NH4Cl.
Validation and Initial Forays
Halolactonizations are conducted under mild conditions and have proven to be an effective strategy to build complexity in the synthesis of many natural products.33 Although there has been advances toward the use of unsaturated amides as the nucleophilic partner in iodolactamizations,34 the use of guanidines is still unknown. In addition, current examples involving chlorocyclizations of amides and carbamates are limited to methods involving radical pathways.35 To the best of our knowledge, there is only one example involving an aza-chlorospirocyclization of an unactivated sp2 carbon in high yield,36 and the use of an allylic guanidine as the nucleophilic counterpart is unknown. In order to validate the key transformation to our revised synthetic plan, we synthesized model allylic guanidine 20 and subjected this simple cyclopentene to a variety of chlorinating conditions (Scheme 2A). Not surprisingly, the chlorination of tri-Boc-allylic guanidine 20 was challenging. A range of chlorinating reagents was employed35,37 (CuCl,35a NCS,35b CrCl2,37a SDCS,37btBuOCl,37c Ca(OCl)2,37d TCIA,37e) but all failed to form any chlorospirocyclization product. Alternatively, we were able to achieve halospirocyclization using NBS to furnish bromospirocyclization product 21. Although the crude NMR showed clean conversion, 21 was unstable to silica gel purification. Attempts to directly crystallize intermediate 21 from the crude reaction mixture (CH2Cl2, slow evaporation at room temperature) led to a crystalline urea 22. The success of this unprecedented haloazaspirocyclization was evidence that guanidine may be a viable nucleophilic partner for this type of transformation. Also, it was believed that if a direct chlorination procedure was unsuccessful, the use of NBS could prove useful to access the desired chlorospirocycle via aziridine formation.38
When evaluating strategies toward construction of the cyclopentyl framework, two prerequisites had to be met (preferably in a single step): (i) proper placement of useful functionality for the generation of a C2-symmetric synthon (Scheme 2C) that can be applied toward side-chain installation and (ii) generation of the trans-methylene azide functionality (17) that would serve as the basis for stereochemical control. The venerable Pauson–Khand (P-K) reaction met both of these criteria and pointed to an area in need of improvement since there were no known examples of non-directed intermolecular P-K cycloadditions using unactivated alkenes.39
Due to the intrinsic [3,3]-rearrangement of bis-allylic azide 19(40) and the lack of reactivity displayed by propargyl guanidine 18 in the P-K cycloaddition (a variety of protecting groups under forceful conditions were explored),41 functional group interchanges were deemed unavoidable. Thus, trans-bis-allylic ether (23) and propargyl alcohol were subjected to a variety of P-K reaction conditions, showing cyclopentenone generation only with the use of N-methylmorpholine N-oxide (NMO) (Scheme 2B).42 Although the yield of model cyclopentenone 24 was low, to the best of our knowledge, this NMO-assisted P-K reaction was the first report of a non-directed intermolecular P-K using an unactivated alkene. Allylic alcohol 24 was found to be unreactive toward Luche conditions. Conversely, allylic guanidine 25 (derived from 24), upon treatment with Luche conditions, afforded clean 1,2-reduction and provided alcohol 26 as a viable C2-symmetric precursor.
The strategic inclusion of a C2-symmetric intermediate in the synthetic design not only eliminates the regioselectivity issue during side-chain installation but also renders the stereochemistry of the corresponding allylic alcohol (26) irrelevant. Both electrophilic and nucleophilic side-chain substitution were explored using 27 (Scheme 2C). Although the use of palladium-catalyzed substitution is more attractive since the use of chiral ligands may render the route enantioselective, electrophilic substitution via a Barbier-type reaction was found to be more feasible (use of Pd allylation protocols only gave decomposition upon forceful conditions) and redox economical43 (use of soft nucleophiles such as 1,3-dicarbonyls typical of Tsuji–Trost allylations would require further redox manipulation). With the successful conversion of 29 to 30, we shifted our attention to optimization of the individual steps and finalization of the route to render it both robust and scalable.
Optimization of Key Reactions
Diol N-Oxide-Assisted Pauson–Khand
As discussed above, after many failed attempts, the use of tertiary amine N-oxides in the stoichiometric Co2(CO)8 system originally reported by Schreiber42 was found to be a viable protocol to generate model cyclopentenone 24 from unactivated alkene 23. Discouraged by unsuccessful cycloadditions involving other metal systems in both catalytic and stoichiometric modes and motivated by the vast amount of promoters found in the P-K literature, we redirected our screening efforts toward the optimization of the initial (20% yield; Table 4, entry 4) protocol rather than changing the transition metal.
Table 4. Optimization of N-Oxide-Mediated Pauson–Khand Reaction.


Reproducible yields of 49% were obtained using 4 Å MS or BSA as a dehydrating agent. rsm = recovered starting material.
Optimization of the stoichiometric Co2(CO)8 protocol was carried out in CH2Cl2 with 1.1 equiv of preformed complex (32), to which was directly added an excess of trans-alkene 23 followed by the desired additive and the oxidant (NMO as standard conditions) at room temperature (Table 4). Complex 32 is stable to silica gel chromatography but was used directly from a stock solution (control experiments with silica gel purified 32 compared to in situ generated 32 showed no difference in isolated yields). It was quickly realized that NMO played an enabling role in the reaction, as the use of any other oxidant (I2, Mn(OAc)3, Co(OAc)2, K3Fe(CN)6, Ag2O, FeCl3, and a variety of Cu(II) salts) failed to give any desired product (only two shown in Table 4 for simplicity). Attention then shifted to screening of additives based on extensive literature precedent.44
The majority of known additives are Lewis bases, and it is generally accepted that “hard” Lewis bases, such as amines,44b ethers,44c and sulfides,44d aid in the formation of a more reactive cobalt complex by promoting CO ligand liberation. (Table 4 represents a cross-section of additives screened.) Of these, mercaptans, primary amines, and polyamines (entries 5 and 6) all had detrimental effects on product formation, giving only recovered starting material or decomposition. It was found that the presence of a hydrogen donor (both amines and mercaptans, see entries 5 and 6) is not well tolerated compared to their aprotic counterparts (entries 7 and 8). Interestingly, this is not the case with alcohols (entry 9 compared to entry 10). The presence of a free hydroxyl group does not impede product formation and allows the reaction to proceed with a yield similar to to that obtained with NMO alone (entry 9 compared to entry 4).
The key breakthrough in these studies was the finding that a combination of NMO and unhindered polyols (entry 11) had a dramatic positive effect, providing the desired cyclopentenone 24 in 45–49% yield. Due to reagent practicality, ethylene glycol was selected as the polyol of choice (cost-effective and lower viscosity). Notably, ethylene glycol was evaluated as a promoter on a related catalytic P-K reaction44c and was shown to have no significant effects.
Hypothesizing that both ethylene glycol and NMO are involved in the formation of a more reactive cobalt complex, we designed a hybrid molecule containing components of both reagents. Thus, diol-N-oxide 33 was synthesized from a H2O2 oxidation of 3-diethylaminopropane-1,2-diol in quantitative yield. Although this new reagent proved to be an effective additive for cyclopentenone formation and could be used in reduced quantities compared to the ethylene glycol/NMO mixture, the yield was not substantially better (entry 12). After investigating the effects of oxidant and Lewis base additives on the reaction yield, we determined that an optimal combination of ethylene glycol/NMO (co-solvent and 6 equiv, respectively) or 3 equiv of 33 could be employed in the presence of 4 Å molecular sieves (MS) to reliably produce 24 in excess of 45% isolated yield.
Indium-Mediated Aqueous Barbier Reaction
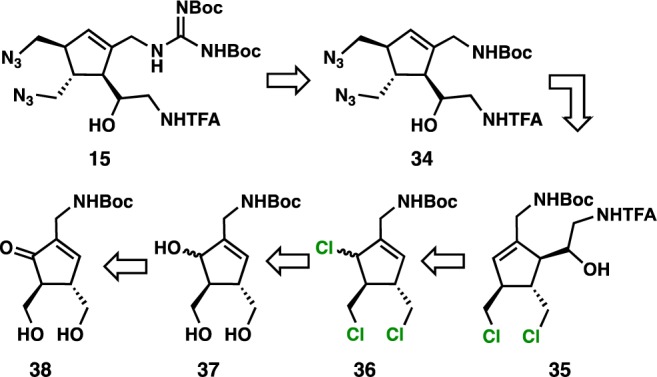
In an effort to offset the synthetic inefficiency caused by the use of unavoidable concession steps (due to P-K incompatibility of 19 and 18), a chemoselective functionalization strategy toward the construction of 15 was envisioned (Scheme 3). In the forward sense, P-K cycloaddition with trans-2-butene-1,4-diol and the cobalt–alkyne complex of N-Boc-propargylamine could give cyclopentenone 38. Selective 1,2-reduction of cyclopentenone 38 to yield triol 37 should be possible using Luche conditions, and conversion of triol 37 to trichloride 36 should be feasible through hydroxyl activation followed by chloride displacement. Chemoselective metal-mediated allylation of an N-protected α-amino aldehyde with trichloride 36 might be possible to furnish homoallylic alcohol 34. Finally, selective N-alkylation of bis-chloride 35 using inorganic azide salts followed by guanidinylation using known reagents45 should provide fully substituted chlorospirocyclization precursor 15. In order to validate the chemoselective allylation, simplified triol 41 was targeted for synthesis and converted to the corresponding trichloride 42.
Scheme 3. Chemoselective Functionalization Strategy to Chlorospirocyclization Precursor 15.
Thus, Co2(CO)8 was used to preform a stable alkyne–cobalt complex with propyne and was subjected to our optimized ethylene glycol/NMO P-K protocol with bis-allylic TMS ether 40 to yield the corresponding bis-TMS-protected cyclopentenone. The protected cyclopentenone was found to have moderate stability to silica gel and could be deprotected in a one-pot operation with CeCl3 in MeOH during the Luche reduction after an extended reaction time. With the successful production of trichloride 42 in good yield (30% overall, three steps from 39), metal-mediated allylation protocols were evaluated using simple propionaldehyde as the model substrate (Table 5).
Table 5. Optimization of Chemoselective Barbier-Type Reactiona.


Reagents and conditions: (a) i. propyne (1.0 equiv), Co(CO)8 (1.0 equiv), CH2Cl2, rt, 2 h; ii. 40 (3.0 equiv), NMO (6.0 equiv), ethylene glycol/CH2Cl2 (1:10, 0.1M) rt, 12 h. (b) CeCl3 (1.0 equiv), NaBH4 (4.0 equiv), MeOH, 0 °C, 12 h. (c) PPh3 (3.4 equiv), NCS (3.4 equiv), THF, rt, 12 h. (d) propionaldehyde (4.0 equiv), Zn (16.0 equiv), In (2 equiv), THF/6% NH4Cl (aq), rt, 3 h.
Initial allylation attempts using Zn in anhydrous THF were discouraging, as they yielded only a trace amount of product (entry 4). After screening a variety of metals in anhydrous THF with no success (entries 1–6), we were prompted to explore the use of aqueous media.46 Although the use of Zn in water alone did not yield any product, the combination of Zn with aqueous NH4Cl47 furnished the desired homoallylic alcohol 43 in 20% yield. The yield of 43 was improved to 50% by varying the solvent choice to a biphasic system (entry 8), which effectively decreased the formation of protodemetalation byproduct 44.
Reviewing the chemical literature involving aqueous organometallic Barbier conditions revealed two important variables: (i) the use of bimetallic systems48 and (ii) the preferential use of indium metal for reactions involving less reactive allyl chlorides (in contrast to allyl bromides) in allylations of ketones and aldehydes.49 Bimetallic conditions are always employed as a redox pair, where one metal serves as an “activator” by reducing the other to a reactive metal species (e.g., Sn–Al,48a InCl3–Zn48b). Although the initial use of indium metal did not show promising results (entry 3), we were delighted to find that its use as a bimetallic mixture with Zn in aqueous NH4Cl provided 43 in 62% yield (entry 13), which could be further optimized to 67% yield by decreasing the concentration of NH4Cl (entry 14). Notably, the previously reported use of InCl3–Zn mixture48b does not mention improved yields relative to In(0) alone. The synergistic effect on yield of a In(0)–Zn(0) combination as a non-redox pair appears to be an unrecognized phenomenon. With all of the key steps toward the construction of 15 validated and partially optimized, we directed our attention to P-K formation of the more complex cyclopentenone 38.
Optimization and Telescoping Process of Key Spirocyclization Precursor
In order to avoid discrete protection/deprotection steps, it was surmised that mild conditions to install and remove TMS ethers could be employed en route to triol 37. Therefore, bis-allylic alcohol 46(50) was protected in situ using 7 equiv of bis(trimethylsilyl)acetamide (BSA) in CH2Cl2. Using our optimized P-K conditions, alkyne–cobalt complex 45 was then added to the same solution with diol-N-oxide 33 as the promoter to produce cyclopentenone 47 in 45% yield (Scheme 4). As in the previous synthesis of 41, 1,2-reduction and TMS deprotection could be carried out in a one-pot operation by extending the reaction time to 12 h. Conversion of the corresponding triol to trichloride 36 using Appel conditions was possible in 95% overall yield (from cyclopentenone 47). Using our optimized bimetallic Zn/In conditions, side-chain installation proceeded smoothly in 64% yield, providing the desired homoallylic alcohol 35 as a single diastereomer. Alkyl chloride displacement of 35 using an excess of sodium azide in DMF (monitored by NMR for full conversion) proceeded cleanly, and after acidic workup, the crude product was of sufficient purity to be taken on directly to the next step. Boc deprotection of 34 and subsequent use of N,N′-di-Boc-N″-triflylguanidine 50 (Goodman’s reagent) in acidic media yielded the corresponding allylic guanidine, which after evaporation of solvent and addition of CH2Cl2 followed by tBuOCl to the crude reaction mixture yielded chlorospirocyclization product 48 in 55% overall yield (three steps from 35). To our surprise, no optimization was needed for the chlorospirocyclization of crude allylic guanidine 48 when using tBuOCl (conditions that failed using model guanidine 20). Interestingly, although clean chlorospirocyclization was observed with crude batches of 48, purified batches or batches using other guanidinylation reagents (49 or 51) varied greatly compared to crude batches (judged by NMR). Assuming from these results that the success of chlorospirocyclization was dependent on TfNH2 generation from the guanidinylation step,45 we pursued an investigation that would help understand the precise role of TfNH2.
Scheme 4. Synthetic Route Finalization and Chlorospirocyclization of Advanced Allylic Guanidine Intermediate 15.
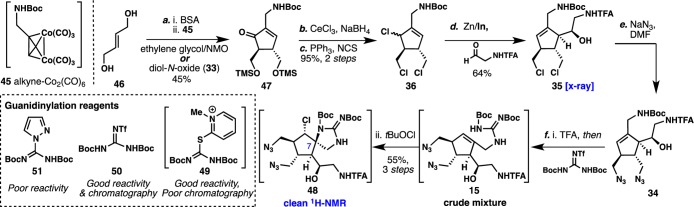
Reagents and conditions: (a) i. BSA (7.0 equiv), CH2Cl2, 40 °C, 3h; ii. 45 (1.0 equiv), diol-N-oxide (3.0 equiv), CH2Cl2, rt, 12 h. (b) CeCl3 (1.0 equiv), NaBH4 (4.0 equiv), MeOH, 0 °C, 12 h. (c) PPh3 (3.4 equiv), NCS (3.4 equiv), THF, rt, 12 h. (d) 2,2,2-Trifluoro-N-(2-oxoethyl)acetamide (10 equiv), zinc (16 equiv), indium (1.9 equiv), THF, NH4Cl (6% aqueous), rt, 3 h. (e) NaN3 (10 equiv), DMF, 85 °C, 16 h. (f) i. TFA (50% in CH2Cl2), rt, 23 h then TEA (5.0 equiv), 50 (1.5 equiv), rt, 12 h; ii. tBuOCl (2.0 equiv), CH2Cl2, 0 °C, 1 h.
Mechanism of TfNH2 in Chlorospirocyclization and Discovery of Stable N-Chloroguanidine 58
Although isolation of desired spirocycle 73 (see Scheme 7, below) was possible using pure batches of 48, the yields were significantly lower compared to yields obtained from crude batches (50% and 98%, respectively). In addition, characterization of undesired products from this batch-dependent variability proved challenging. Therefore, in order to study the effect of TfNH2 on the chlorospirocyclization event, a simplified model 52 displaying the correct protecting group regiochemistry was synthesized (Scheme 5). In this model system, both conditions using tBuOCl (with and without 20 mol% TfNH2) showed chlorospirocyclization product formation; however, multiple intermediate products (53, 54) were detected in batches using TfNH2 as an additive. Interestingly, upon treatment with TFA in CH2Cl2, both of these reaction mixtures led to desired spirocycle 56 in comparable yields. Additionally, the use of 1.0 or 2.0 equiv of tBuOCl did not seem to influence chlorospirocyclization formation but did contribute to variability in intermediate (53, 54) formation.
Scheme 7. Scalability of Final Synthetic Route and Reactivity of 2-Aminoimidazole toward DMDO Oxidation.

Reagents and conditions: (a) i. TFA/CH2Cl2 (1:1, 0.2M), rt, 12 h; ii. CH2Cl2, TEA (5.0 equiv), N,N′-bis-Boc-N″-triflylguanidine (1.5 equiv). (b) i. TfNH2 (0.25 equiv), CH2Cl2, tBuOCl (2.0 equiv), 0 °C, 30 min; ii. DMP (1.2 equiv), rt, 12 h. (c) H2O/TFA (1:1, 0.2 M), 70 °C, 36 h. (d) i. AcOH; ii. NCNH2 (25 equiv), 0.2 M NaOH (pH ∼5.5), 70 °C, 4 h. (e) i. TFA/CH2Cl2 (5:95, 0.1 M), DMDO (1.25 equiv), 0 °C, 1 h; ii. TFA/CH2Cl2 (1:1, 0.1 M), rt, 12 h. (f) H2O/TFA (9:1, 0.07 M), silver(II) picolinate (2.5 equiv), rt, 2 h. (g) i. H2O/TFA (19:1, 0.1 M), PtO2 (0.1 equiv), H2(g); ii. 2,3-dibromo-5-trichloroacetylpyrrole (5.0 equiv), DIPEA (4.5 equiv).
Scheme 5. TfNH2 Effect on Model Chlorospirocyclization.
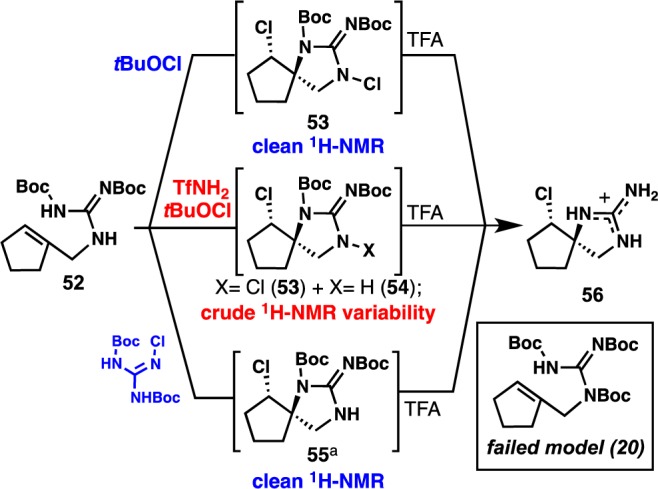
Addition of 1.0 equiv of TfNH2 to 53 produced 55 in quantitative yield.
Realizing that TfNH2 has an effect on intermediate formation but is not essential for attaining high yields of desired spirocycle 56, we directed our attention to a key structural difference between our original model allylic guanidine 20 (triBoc) and revised model 52 (bisBoc). Thus, it was reasoned that N-chloroguanidine formation could be attributed to the success of the chlorospirocyclization event. To test this hypothesis, bis-Boc-guanidine 57 was treated with tBuOCl in CH2Cl2 at room temperature, and after only a few minutes, new product formation was observed by thin-layer chromatography (Scheme 6A). Isolation of this new product led to the characterization of chlorobis(tert-butoxycarbonyl)guanidine (CBBG, 58) as a bench-stable white solid. To our delight, when CBBG (58) was used as the chlorination source in CH2Cl2, clean chlorospirocyclization of 52 was observed (Scheme 5B). Shockingly, when benzenesulfonamide 59 was treated with tBuOCl in CH2Cl2 at room temperature, no reaction took place, but when 59 was mixed with CBBG (58) under identical conditions, N-chlorobenzenesulfonamide 60 was formed in quantitative yield (similar results using N-methylbenzenesulfonamide were observed, and its product was characterized by X-ray analysis) (Scheme 6A).
Scheme 6. Invention of Reactive yet Stable CBMG.
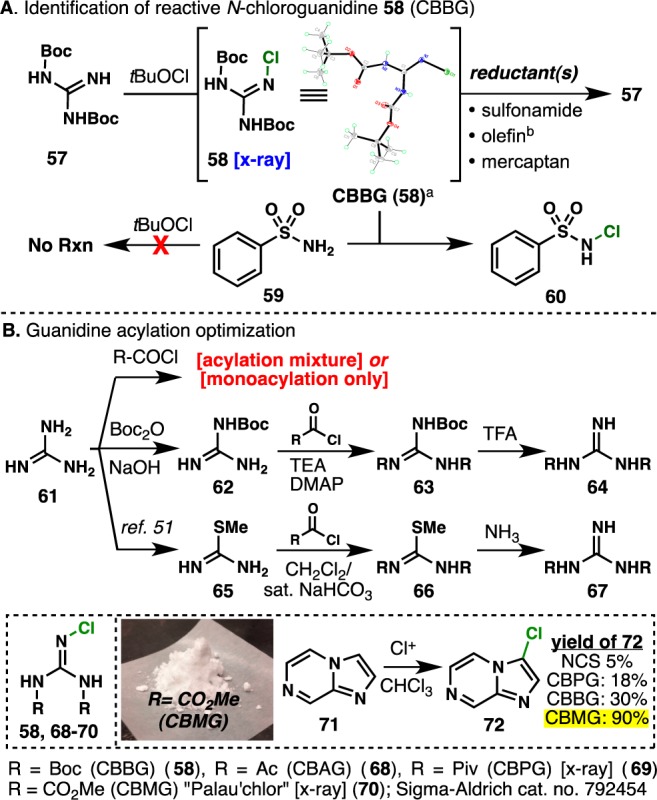
CBBG decomposition was observed when dissolving in acetone.
Chlorinated products were detected from the mixture of CBBG and 2-methylallyl-4-bromobenzonate.
In light of these results, it is proposed that the active chlorinating agent must be an N-chloroguanidine species and the role of TfNH2 is to act as a reductant toward overchlorinated products (such as chlorination of 55). In addition, it is reasoned that conversion of 53 and 54 to 56 is achieved via reduction from isobutene generation from Boc deprotection (2.0 equiv). In the model chlorospirocyclization of 52, TfNH2 proved to be non-essential, as both chlorination conditions (tBuOCl with and without TfNH2) gave the desired spirocycle 56 in high yields. Interestingly, in the case of fully functionalized spirocycle precursor 15, the absence of TfNH2 or the use of an excess of TfNH2 had a detrimental effect on yield.
CBBG (58) was further evaluated as a general chlorinating reagent by comparing its reactivity to existing reagents. With a focus on heteroaromatic chlorination, heterocycle 71 was used as a model substrate for comparing reactivity between NCS and CBBG derivatives. Systematic variation of the pendant acyl groups on the guanidine proved to be a non-trivial process, as direct acylation procedures used on unprotected guanidine 61 only led to acylation mixtures or monoacylation (even under forceful conditions). After extensive experimentation, two viable procedures51 for acyl group variation on guanidine 61 were adopted, and after comparing yields of 72, we identified chloro(bismethoxycarbonyl)guanidine (CBMG or Palau’chlor, 70) as the optimal chlorinating reagent (Scheme 6B). Palau’chlor was found to be an effective chlorinating reagent for a variety of organic substrates and in all cases showed improved yields when compared to other known chlorinating reagents (under identical conditions).52 Palau’chlor proved to be a stable yet reactive chlorinating reagent and has been field-tested at Bristol-Myers Squibb as well as commercialized by Sigma-Aldrich (cat. no. 792454).52
Finalization of Synthetic Route and Scalability
High yields of chlorospirocyclization product 48 were obtained when residual TfNH2 was present in the reaction mixture (Scheme 7). Chlorospirocyclization optimization of 15 led to a process where chromatographically pure 15 is best used in combination with 20 mol% TfNH2 and 2.0 equiv of tBuOCl in CH2Cl2 as the solvent. These conditions provided spirocycle 48 cleanly in near-quantitative yield (judged by NMR) with successful batch reproducibility. Oxidation of homoallylic alcohol 48 to ketone 73 using DMP was found to be compatible under acidic media and was processed in crude form using 10:1 CH2Cl2/TFA as solvent (81% yield from 15). Further treatment of purified 73 with TFA in the presence of H2O furnished spiroaminoketone 9 in a total of eight steps with only four chromatographic purifications and in 13% overall yield from 45. In comparison to our first-generation synthesis of 9, this route reduced the number of steps by more than half and increased the overall yield by >10-fold. Notably, each of the above-mentioned steps was successfully conducted and reproduced on gram-scale, providing 7 g of 9 by one chemist in a single batch from 45 and 46.
Finally, in order to demonstrate the scalability of a final synthetic route to a complex PIA from spiroaminoketone 9 as well as to provide an in-depth follow-up to initial biological activity, the tetracyclic axinellamines were targeted for synthesis. Having to meet a prerequisite of >100 mg of 4 and 5 for biological collaborations, the axinellamines were prepared on gram-scale. Thus, addition of cyanamide to 9 in brine under a controlled pH (pH 5.5–5.7) led to 74 in 75% yield (2.4 g, 2 steps from 9). Oxidation of 2-aminoimidazole 74 with DMDO followed by TFA-mediated dehydration gave tetracycle 75 as the major product in 83% yield from 9 (2 g) and ∼5 mg of tricycle 76, presumably from DMDO oxidation followed by 1,2-shift. Tricycle 76 was characterized by X-ray analysis and was found to share the core carbon skeleton with matching oxidation to that of the donnazoles (6 and 7). In addition, the isolation of tricycle 76 provides evidence for the putative biogenesis of donnazoles from a common pre-axinellamine precursor.53 Notably, the formation of tricycle 76, having the same molecular weight as 75, went undetected by LC-MS in smaller-scale28b oxidation/dehydration batches of 74, and the X-ray analysis of 76 represents the most complex example of a polycyclic PIA intermediate to date. Tetracycle 75 was converted to 77-α and 77-β with silver(II) picolinate in 68% yield (1.4 g, 2.7:1 β:α). Conversion of the azide groups of the acylpyrrole side chains was accomplished using a one-pot operation involving PtO2-mediated reduction followed by acylation with 2,3-dibromo-5-trichloroacetylpyrrole to provide >1 g of 4 and 5 combined in 93% yield over two steps from 77.
Conclusions
Using an N-oxide/ethylene-glycol-assisted intermolecular P-K protocol to enable the use of (E)-alkenes in cyclopentenone generation, a synergistic bimetallic (Zn/In) Barbier-type allylation, and N-chloroguanidine-based chlorospirocyclization to set the two most challenging stereocenters, a robust and stereocontrolled second-generation route to key spiroaminoketone 9 has been developed (20 and 8 steps, 12 and 4 purifications, <1% overall yield and 13% overall yield, respectively). In addition to representing a formal synthesis of palau’amine (8) and massadines,28c the synthesis of spiroaminoketone 9 presented an opportunity for invention. The key chlorospirocyclization not only served as an ideal strategy for clearing the two most challenging stereocenters of the fully substituted cyclopentyl framework but also provided the chemical inspiration for CBBG (58). Optimization of these stable N-chloroguanidines led to the identification of Palau’chlor (70) as a reactive yet practical chlorinating reagent for both C–H and N–H chlorinations, ultimately commercialized by Sigma-Aldrich.52 Finally, scalability of common progenitor 9 to complex members of the PIA family class has been demonstrated on multigram-scale, routinely providing ∼5 g of 73 from a single batch and final elaboration of the axinellamines on gram-scale. This enabled a re-investigation of the biological activity16,17 of 4 and 5, resulting in the surprising finding that they possess promising activity against a broad range of bacterial pathogens, suggesting that their scaffold has the potential for further development.54 This article represents a case study in which the development of a scalable synthesis to a complex natural product enabled discoveries in both chemistry and biology.
Acknowledgments
Financial support for this work was provided by the NIH/NIGMS (GM-073949) and by NSF (predoctoral fellowship to R.A.R.). We are grateful to Dr. A. L. Rheingold and Dr. C. E. Moore for X-ray crystallographic analysis.
Supporting Information Available
Experimental procedures, complete data acquisition, and analytical data for all new compounds, including 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ R.A.R. and D.B.S. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Obrecht D.; Bernardini F.; Dale G.; Dembowsky K. Annu. Rep. Med. Chem. 2011, 46, 245. [Google Scholar]
- Wright G. D. Nat. Rev. Microbiol. 2007, 5, 175. [DOI] [PubMed] [Google Scholar]
- For an editorial commentary describing the ESKAPE pathogens, see:; Rice L. B. J. Infect. Dis. 2008, 197, 1079.and references therein. [DOI] [PubMed] [Google Scholar]
- Vaara M. Curr. Opin. Pharmacol. 2009, 9, 571. [DOI] [PubMed] [Google Scholar]
- Boger D. L. ACS Chem. Biol. 2012, 7, 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander C.; Rietshel E. T. J. Endotoxin Res. 2001, 73167. [PubMed] [Google Scholar]
- Velkov T.; Roberts K. D.; Nation R. L.; Wang J.; Thompson P. E.; Li J. ACS Chem. Biol. 2014, 9, 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Spapen H. D.; Honore P. M.; Gregoire N.; Gobin P.; deRegt J.; Martens G. A.; Pierard D.; Couet W. J. Infect. 2011, 63, 468. [DOI] [PubMed] [Google Scholar]; b Dai C.; Zhang D.; Gao R.; Zhang X.; Li J.; Li J. Environ. Toxicol. Pharmacol. 2013, 36, 659. [DOI] [PubMed] [Google Scholar]; Renal:; c Berlana D.; Llop J. M.; Fort E.; Badia M. B.; Jodar R. Am. J. Health. Syst. Pharm. 2005, 62, 39. [DOI] [PubMed] [Google Scholar]; d Michalopoulos A.; Falagas M. E. Critical Care Clinics 2008, 24, 377. [DOI] [PubMed] [Google Scholar]
- Lewis K.; Ausubel F. M. Nat. Biotechnol. 2006, 24, 1504. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Nat. Rev. Drug Discovery 2007, 6, 29. [DOI] [PubMed] [Google Scholar]
- Walsh C. Nat. Rev. Microbiol. 2003, 1, 65. [DOI] [PubMed] [Google Scholar]
- Becker B.; Cooper M. A. ACS Chem. Biol. 2013, 8, 105. [DOI] [PubMed] [Google Scholar]
- For a review on dimeric, tetrameric, and higher-order PIAs and their corresponding initial biological activity, see:; a Kock M.; Grube A.; Seiple I. B.; Baran P. S. Angew. Chem., Int. Ed. 2007, 46, 6586.and references therein. [DOI] [PubMed] [Google Scholar]; For a more comprehensive review on the PIA family, see:; b Forte B.; Malgesini B.; Piutti C.; Quartieri F.; Scolaro A.; Papeo G. Mar. Drugs 2009, 7, 705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Molinski T. F.; Dalisay D. S.; Lievens S. L.; Saludes P. Nat. Rev. Drug Discovery 2009, 8, 69. [DOI] [PubMed] [Google Scholar]; b Bhakuni D. S.; Raway D. S.. Bioactive Natural Products; Springer: New York, 2005. [Google Scholar]; c Gerwick W. H.; Moore B. S. Chem. Biol. 2012, 19, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen L.; McKerrall S. J.; Kuttruff C. A.; Ungeheuer F.; Felding J.; Baran P. S. Science 2013, 341, 878. [DOI] [PubMed] [Google Scholar]
- Urban S.; de Almeida Leone P.; Carroll A. R.; Fechner G. A.; Smith J.; Hooper J. N. A.; Quinn R. J. J. Org. Chem. 1999, 64, 731. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Khalil Z.; Conte M. M.; Plisson F.; Capon R. J. Tetrahedron Lett. 2012, 53, 3784. [Google Scholar]
- Martinez J. L.; Baquero F. Antimicrob. Agents Chemother. 2000, 44, 1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman J. A.; Oliver N.; Andrew T.; Li T. Antimicrob. Agents Chemother. 2001, 45, 1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn B.; Hussain S.; Malik M.; Drlica K.; Zhao X. J. Antimicrob. Chemother. 2007, 60, 1380. [DOI] [PubMed] [Google Scholar]
- Bernan V. S.; Roll D. M.; Ireland C. M.; Greenstein M.; Maiese W. M.; Steinberg D. A. J. Antimicrob. Chemother. 1993, 32, 539. [DOI] [PubMed] [Google Scholar]
- Rodriguez A. D.; Lear M. J.; La Clair J. J. J. Am. Chem. Soc. 2008, 130, 7255. [DOI] [PubMed] [Google Scholar]
- Kruse T.; Bork-Jensen J.; Gerdes K. Mol. Microbiol. 2005, 55, 78. [DOI] [PubMed] [Google Scholar]
- Varma A.; Young K. D. J. Bacteriol. 2009, 191, 3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker J.; Erickson H. P. J. Bacteriol. 2003, 185, 4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring N. W.; Beckwith J. Curr. Biol. 2005, 15, R514. [DOI] [PubMed] [Google Scholar]
- For a comprehensive list of efforts toward the synthesis of the complex PIAs (axinellamines, massadines, and palau’amine) prior to August 2011, see:; a Seiple I. B.; Su S.; Young I. S.; Nakamura A.; Yamaguchi J.; Jorgensen L.; Rodriguez R. A.; O’Malley D. P.; Gaich T.; Kock M.; Baran P. S. J. Am. Chem. Soc. 2011, 133, 14710.and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; For studies toward the synthesis of axinellamines after August 2011, see:; b Ding H.; Roberts A. G.; Harran P. G. Angew. Chem., Int. Ed. 2012, 51, 4340. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ding H.; Roberts A. G.; Harran P. G. Chem. Sci. 2013, 4, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang X.; Ma Z.; Wang X.; De S.; Ma Y.; Chen C. Chem. Commun. 2014, 50, 8628.and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yamaguchi J.; Seiple I. B.; Young I. S.; O’Malley D. P.; Maue M.; Baran P. S. Angew. Chem., Int. Ed. 2008, 47, 3578. [DOI] [PubMed] [Google Scholar]; b O’Malley D. P.; Yamaguchi J.; Young I. S.; Seiple I. B.; Baran P. S. Angew. Chem., Int. Ed. 2008, 47, 3581. [DOI] [PubMed] [Google Scholar]; c Su S.; Seiple I. B.; Young I. S.; Baran P. S. J. Am. Chem. Soc. 2008, 130, 16490. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Seiple I. B.; Su S.; Young I. S.; Lewis C. A.; Yamaguchi J. Angew. Chem., Int. Ed. 2010, 49, 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Su S.; Rodriguez R. A.; Baran P. S. J. Am. Chem. Soc. 2011, 133, 13922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on scalable synthesis, see:; a Kuttruff C. A.; Eastgate M. D.; Baran P. S. Nat. Prod. Rep. 2014, 31, 419. [DOI] [PubMed] [Google Scholar]; For examples of scalable synthesis of a natural product leading to an antibacterial compound in clinical use, see:; b Brubaker J. D.; Myers A. G. Org. Lett. 2007, 9, 3523. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ronn M.; Zhu Z.; Hogan P. C.; Zhang W.-Y.; Niu J.; Katz C. E.; Dunwoody N.; Gilicky O.; Deng Y.; Hunt D. K.; He M.; Chen C.-L.; Sun C.; Clark R. B.; Xiao X.-Y. Org. Process Res. Dev. 2013, 17, 838. [Google Scholar]
- Corey E. J.; Weinshenker N. M.; Schaaf T. K.; Huber W. J. Am. Chem. Soc. 1969, 20, 5675. [DOI] [PubMed] [Google Scholar]
- For previous bio-inspired, symmetry-based approach toward the PIAs, see ref (27a). For another example of symmetry-based logic toward palau’amine, see:; Li Q.; Hurley P.; Ding H.; Roberts A. G.; Akella R.; Harran P. G. J. Org. Chem. 2009, 74, 5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Vranken D. L. Chem. Rev. 1996, 96, 395. [DOI] [PubMed] [Google Scholar]
- For a review on halonium-induced cyclization reactions, see:Snyder S. A.; Treitler D. S.; Brucks A. P. Aldrichimica Acta 2011, 44, 27.and references therein. [Google Scholar]
- a Knapp S.; Levorse A. T. J. Org. Chem. 1988, 53, 4006. [Google Scholar]; b Yeung Y.-Y.; Hong S.; Corey E. J. J. Am. Chem. Soc. 2006, 128, 6310. [DOI] [PubMed] [Google Scholar]
- For radical chlorocyclizations using CuCl, see:; a Schulte-Wulwer I. A.; Helaja J.; Gottlich R. Synthesis 2003, 12, 1886. [Google Scholar]; For examples of photochemical chlorocyclizations, see:; b Kuehne M. E.; Horne D. A. J. Org. Chem. 1975, 40, 1287. [Google Scholar]
- Wardrop D. J.; Burge M. S. J. Org. Chem. 2005, 70, 10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the use of CrCl2 in the addition of N-haloamides to olefins, see:; a Driguez H.; Paton J. M.; Lessard J. Can. J. Chem. 1977, 55, 700. [Google Scholar]; For an example of direct halonium-induced cyclizations, see:; b Snyder S. A.; Treitler D. S.; Brucks A. P. J. Am. Chem. Soc. 2010, 132, 14303. [DOI] [PubMed] [Google Scholar]; For the use of chlorinating reagents as N-chlorinating agents, see:; c Zimmer H.; Audrieth L. F. J. Am. Chem. Soc. 1954, 76, 3856. [Google Scholar]; d Oleg L. V.; Kozhushkov S. I.; de Meijere A. Synthesis 2003, 12, 1916. [Google Scholar]; e Back T. G.; Chau J. H.-L.; Dyck B. P.; Gladstone P. L. Can. J. Chem. 1991, 1482. [Google Scholar]
- a Grube A.; Immel S.; Baran S. P.; Kock M. Angew. Chem., Int. Ed. 2007, 46, 6721. [DOI] [PubMed] [Google Scholar]; For the first suggestion of such an intermediate in this alkaloid family, see:; b Wang S.; Poullennec K. G.; Dilley A. S.; Romo D. Tetrahedron 2006, 62, 7155. [Google Scholar]
- For an example of a directed P-K reaction using unactivated alkenes, see:; a Krafft M. E.; Juliano C. A.; Scott I. L.; Wright C.; McEachin M. D. J. Am. Chem. Soc. 1991, 113, 1693. [Google Scholar]; For an example of silicon-based tethers for the P-K reaction using an unactivated alkene, see:; b Dobbs A. P.; Miller I. J.; Martinovic S. Beilstein J. Org. Chem. 2007, 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman A. K.; Colasson B.; Sharpless K. B.; Fokin V. V. J. Am. Chem. Soc. 2005, 127, 13444. [DOI] [PubMed] [Google Scholar]
- Under the standard conditions, the corresponding mono-Boc-guanidine produced trace cyclopentenone when using more reactive cis-alkene.
- Shambayati S.; Crowe W. E.; Schreiber S. L. Tetrahedron Lett. 1990, 31, 5289. [Google Scholar]
- For a review on redox economy in organic synthesis, see:; a Burns N. Z.; Baran P. S.; Hoffmann R. W. Angew. Chem., Int. Ed. 2009, 48, 2854. [DOI] [PubMed] [Google Scholar]; For a synthesis example using a redox economic approach, see:; b Richter J. M.; Ishihara Y.; Masuda T.; Whitefield B. W.; Llamas T.; Pohjakallio A.; Baran P. S. J. Am. Chem. Soc. 2008, 17938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a general review on P-K additives, see:; a Sugihara T.; Yamaguchi M.; Nishizawa M. Chem.—Eur. J. 2001, 7, 1589. [DOI] [PubMed] [Google Scholar]; Use of amines, see:; b Dennenberg R. J.; Darensbourg D. J. Inorganic Chem. 1972, 11, 72. [Google Scholar]; Use of ethers, see:; c Sugihara T.; Yamaguchi M. Synlett 1998, 12, 1384. [Google Scholar]; Use of sulfides, see:; d Brown J. A.; Janecki T.; Kerr W. J. Synlett 2005, 13, 2023. [Google Scholar]
- For a comparative study on guanidinylating reagents, see:; a Feichtinger K.; Zapf C.; Sings H. L.; Goodman M. J. Org. Chem. 1998, 63, 38024. [Google Scholar]; For another commercial guanidinylating reagent, see:; b Looper R. E.; Haussener T. J.; Mack B. C. J. Org. Chem. 2011, 76, 6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a general review on organic reactions in aqueous media, see:; a Li C.-J. Chem. Rev. 2005, 105, 3095. [DOI] [PubMed] [Google Scholar]; For an application toward synthesis, see:; b Mendoza A.; Ishihara Y.; Baran P. S. Nat. Chem. 2012, 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mattes H.; Benezra C. Tetrahedron Lett. 1985, 26, 5697. [Google Scholar]; b Zhou J. Y.; Lu G. D.; Wu S. H. Synth. Commun. 1992, 22, 481. [Google Scholar]
- Allylation procedure using SnCl2-Al, see:; a Uneyama K.; Kamaki N.; Moriya A.; Torii S. J. Org. Chem. 1985, 50, 5396. [Google Scholar]; Allylation procedures using InCl3-Zn or InCl3-Al, see:; b Araki S.; Jin S.-J.; Idou Y.; Butsugan Y. Bull. Chem. Soc. Jpn. 1992, 65, 1736. [Google Scholar]; For a recent review on the application of organoindium reagents, see:; c Shen Z.-L.; Wang S.-Y.; Chok Y.-K.; Xu Y.-H.; Loh T.-P. Chem. Rev. 2013, 113, 271. [DOI] [PubMed] [Google Scholar]
- Li C. J.; Chan T. H. Tetrahedron Lett. 1991, 32, 7017. [Google Scholar]
- For the LiAlH4 reduction procedure used to generate 48, see:; Rottger S.; Waldmann H. Eur. J. Org. Chem. 2006, 9, 2093.For a new scalable procedure toward 48, see the Supporting Information.. [Google Scholar]
- For synthesis of N,N′-bismethoxycarbonyl-S-methylisothiourea, see:Skibinski A.; Stec Z.; Januchowski M.; Parys L. Polish J. Appl. Chem. 1993, 37, 291. [Google Scholar]
- Rodriguez R. A.; Pan C–M.; Yabe Y.; Kawamata Y.; Eastgate M. D.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the isolation of donnazoles A and B, see:; Munoz J.; Moriou C.; Gallard J.-F.; Marie P. D.; Al-Mourabit A. Tetrahedron Lett. 2012, 53, 5828. [Google Scholar]
- For a recent insightful review, see:; Wright P. M.; Seiple I. B.; Myers A. G. Angew. Chem., Int. Ed. 2014, 53, 8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.