

Abstract
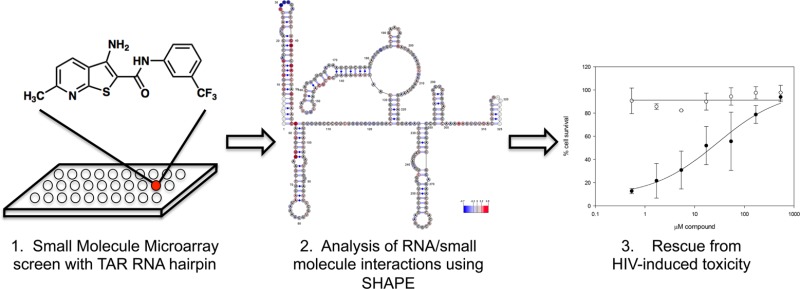
Identifying small molecules that selectively bind to structured RNA motifs remains an important challenge in developing potent and specific therapeutics. Most strategies to find RNA-binding molecules have identified highly charged compounds or aminoglycosides that commonly have modest selectivity. Here we demonstrate a strategy to screen a large unbiased library of druglike small molecules in a microarray format against an RNA target. This approach has enabled the identification of a novel chemotype that selectively targets the HIV transactivation response (TAR) RNA hairpin in a manner not dependent on cationic charge. Thienopyridine 4 binds to and stabilizes the TAR hairpin with a Kd of 2.4 μM. Structure–activity relationships demonstrate that this compound achieves activity through hydrophobic and aromatic substituents on a heterocyclic core, rather than cationic groups typically required. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) analysis was performed on a 365-nucleotide sequence derived from the 5′ untranslated region (UTR) of the HIV-1 genome to determine global structural changes in the presence of the molecule. Importantly, the interaction of compound 4 can be mapped to the TAR hairpin without broadly disrupting any other structured elements of the 5′ UTR. Cell-based anti-HIV assays indicated that 4 inhibits HIV-induced cytopathicity in T lymphocytes with an EC50 of 28 μM, while cytotoxicity was not observed at concentrations approaching 1 mM.
Introduction
The central role of RNA in governing diverse biological processes makes it critical to normal cellular function as well as a variety of disease states.1 For example, the HIV transactivation response (TAR) element RNA has long been considered a target for inhibiting HIV replication due to its complex and pivotal role in facilitating transcription of viral DNA2−4 and critical role in DNA strand transfer.5 The importance of TAR in HIV replication has led to numerous studies of its interactions. Additionally, the challenges associated with identifying TAR-binding small molecules are emblematic of the difficulty in developing RNA-binding molecules in general. This makes TAR an invaluable model system for the discovery and study of RNA-binding molecules as well as novel compounds with anti-HIV activity. As such, a number of small molecules have been reported that bind to TAR, many of which disrupt binding of the HIV-1 transactivating protein Tat to the RNA.6−8 Many of the known TAR-binding small molecules are aminoglycosides (such as neomycin), polymers, or are polycationic and often suffer from poor affinity, physicochemical properties, or poor selectivity. Therefore, the identification of druglike small molecules that bind to TAR remains an important challenge.
Small molecules that interact with RNA are valuable as drugs9−12 and as chemical probes of RNA function and structure.13 However, successful strategies to identify and characterize novel small molecules that bind selectively to structured RNA motifs have substantially lagged behind analogous methods for molecules that interact with proteins. Complicating this area of research is the highly structurally dynamic and chemically unstable nature of RNA, making it challenging to study from both structural and screening standpoints.14,15 Toward this end, a variety of approaches to identify RNA-binding small molecules have been described in several recent reviews.6,7 However, robust technologies capable of screening large, unbiased chemical libraries against specific RNA targets remain relatively rare.
Here we employ a small molecule microarray (SMM) screen of druglike molecules to identify a new chemotype that binds to and stabilizes the TAR hairpin. A combination of biophysical studies and cell-based experiments show that this chemotype binds to TAR selectively, inhibiting HIV-mediated cytopathicity with minimal toxicity. Of three hit structures identified from this screen, one was very similar to a known TAR-binding chemotype. The most active compound, an unprecedented scaffold in this context, was evaluated in detail and found to bind to and stabilize the TAR hairpin. Evaluation of a number of analogues of this scaffold demonstrates that 4 requires hydrophobic and aromatic groups to achieve antiviral activity, as opposed to the cationic substituents typically required to observe affinity to RNA targets. SHAPE analysis of the entire 5′ UTR of the HIV-1 RNA genome was used to map pronounced and remarkably specific effects upon compound binding. Finally, cell models of HIV-1 infectivity indicate that compound 4 rescues T-lymphocytes from HIV-1-mediated toxicity with an EC50 of 28 μM. Remarkably, no cytotoxicity was observed in uninfected cells, even at concentrations up to 1 mM.
Results and Discussion
SMMs are a powerful technology for evaluating large libraries of compounds for their ability to bind to biomolecules.16−18 In this approach, small molecules are printed onto a functionalized glass surface and assessed for their ability to bind to a biomolecule of interest. Large-scale SMM screens have been utilized previously to identify small molecule ligands for a variety of protein targets such as morphogens (sonic hedgehog),19 kinases (aurora kinase),20 transcription factors (Hap3p),21 transcriptional regulators (Ure2p),22 and messenger proteins (calmodulin).23 Similarly, a number of powerful approaches have demonstrated the utility of two-dimensional combinatorial screening and selection- or design-based approaches to identify RNA-binding aminoglycosides, peptoids, or small molecules from focused libraries printed on microarrays.24−31 These approaches have generally identified molecules from focused libraries in which highly charged species and polymeric or Hoechst stain-like molecules32 have been emphasized. However, screening large, unbiased chemical libraries of druglike compounds against specific RNA targets remains a challenge.
As part of an interest in using SMMs to identify ligands for nucleic acids with defined secondary structures, we assembled a library of 20,000 diverse, druglike, commercially available primary alcohols, secondary alcohols, and primary amines (both aromatic and aliphatic). The library was not biased to contain known nucleic acid-binding chemotypes, but rather to resemble “druglike” chemical space33 and to have desirable physical properties that increase the likelihood of cell permeability. The library was arrayed and printed in duplicate onto isocyanate-functionalized glass slides along with appropriate controls to generate the SMM. SMMs were incubated with a Cy5-labeled 29-nt TAR RNA hairpin34 as well as another Cy5-labeled RNA (the miR-21 hairpin35) that was used as a control to rule out promiscuous binders. Array data were analyzed using Axon GenePix software and JMP to generate a composite Z score for each molecule in the library in order to represent the increase in fluorescence of a given array spot upon incubation with the labeled RNA (see Experimental Section). Hit molecules were defined as having a Z score 3 standard deviations from the mean of all compounds in the library as well as the ability to bind to the TAR hairpin and not the control RNA. As a further measure of selectivity, hit compounds were also verified to not interact with three separate DNA sequences that have also been screened against this library, each of which contained well-defined secondary structural elements that do not overlap with the RNA sequences (data not shown).
Using these criteria, three molecules were identified from the screen, corresponding to a hit rate of 0.02% (Figure 1). Each had a composite Z score exceeding 14, representing a robust signal. Molecule 2 exhibited substantial structural similarity to a known class of phenothiazine-derived TAR binders,36 while compound 3 could not be validated as binding to the TAR hairpin. Compound 1 displays the strongest signal and selectivity on the microarray screen, does not bear structural similarity to known TAR-binding small molecules, and does not appear to be chemically reactive or suffer from liabilities commonly associated with RNA-binding small molecules. Thus, scaffold 1 was selected for further study.
Figure 1.

(a) Structure of the HIV TAR hairpin used in this study. U25 is indicated with an asterisk (*). (b) Raw SMM images for each hit structure (compounds are printed in duplicate). The likely site of attachment to the microarray slide is indicated in red for each structure. The behavior of each compound in Negative Control (buffer incubated), Cy5-labeled TAR RNA incubated, and Cy5-labeled miR-21 RNA incubated SMMs are shown for comparison. In each image, brightness and contrast are adjusted by array scanning software, resulting in slight differences in background.
The binding of 1 to the TAR hairpin was next validated and biophysically characterized. Differential scanning calorimetry (DSC) was first used to assess the stability and folding of TAR RNA in the absence of ligand. The TAR hairpin was found to fold reversibly with a Tm of 72 °C (Figure SI-1 in Supporting Information [SI]). Next, effects of compound binding were investigated using differential scanning fluorimetry (DSF). After optimizing concentrations of TAR RNA and the intercalating fluorescent dye SYBR Green II, titration experiments were performed with compound 1, acepromazine, and two other analogues (4 and 5) that had been identified as active in cell-based assays. The effects of compound binding on the thermal stability of TAR RNA were indirectly measured by monitoring the fluorescent signal of the extrinsic dye. In Figure 2B–D, the melting profiles of TAR RNA titrated with compound 1, and two analogues, 4 and 5 (which were not included in the original SMM screen) are shown. Melting temperature (Tm) was measured by recording the maximum of the negative of the first derivative of the fluorescence signal as a function of temperature, −(dF/dT). In all cases, titration produced a decrease in the overall differential fluorescence with a concomitant increase in the melting temperature, indicating an interaction between the small molecule and the hairpin. In the plot of Figure 2E, the change in Tm compared to TAR RNA alone is shown for each compound. Acepromazine, a previously reported TAR RNA binding compound36 similar to one of the hits from the SMM screen (2), is shown for comparison. In this experiment, 1 and acepromazine are nearly identical in their stabilizing effect on TAR RNA. However, evaluation of several closely related analogues of 1 in cell-based assays (see Figure SI-4 in SI) revealed several molecules with improved potency. Compounds 4 and 5, for example, promote a marked and dose-dependent Tm increase compared to both acepromazine and 1.
Figure 2.

(a) Structures of selected TAR-binding compounds investigated in this study. Differential scanning fluorimetry experiments indicating melting temperature changes of the TAR hairpin upon addition of 1 (b), 4 (c), or 5 (d). (e) Graphical representation of the change in melting temperature of the TAR hairpin as a function of the concentration of 1, 4, 5, or acepromazine. (f) 2-Aminopurine fluorescence titration measuring the Kd of 4 with the TAR hairpin.
Direct binding of compounds to the TAR hairpin was evaluated quantitatively using a 2-aminopurine (2-AP) fluorescence titration assay.8,37,38 It is well established that replacing U25 of the TAR hairpin with 2-AP (Figure 1A) leads to fluorescence quenching due to base pair stacking. Upon titration of a small molecule, subtle changes in 2-AP fluorescence intensity can be exploited to derive the Kd of TAR ligands, even in the presence of small quantities of DMSO (<2%). In this assay, a Kd of 2.4 ± 1.1 μM was measured for compound 4 (Figure 2F). In comparison, the Kd of compound 5 was measured to be 0.23 ± 0.02 μM (see Figure SI-5 in SI), while a full binding isotherm could not be measured for 1 due to poor solubility at relevant concentrations (indicative of a substantially weaker Kd). Recent work on ultrafast time-resolved probing of the TAR hairpin has demonstrated that it exists as an ensemble of conformations.39 It is believed that ligands interact with and stabilize one of these pre-existing conformations, thereby influencing the ensemble and modulating other molecular recognition events. In the case of compound 4, the increase in fluorescence intensity indicates that the purine analogue is becoming, on average, less stacked as a result of ligand binding. Thus, it may be concluded that the binding of 4 results in the stabilization of a conformation where U25 is extended and is slightly more solvent exposed. However, the relatively modest 2-fold increase in fluorescence suggests only a subtle conformational shift from the unbound structure. Due to its low molecular weight, superior solubility relative to that of 5 and improved activity relative to that of 1, compound 4 was pursued in further studies.
Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) is a straightforward method of determining RNA secondary structure by examining backbone flexibility, which is directly related to base pairing probability.40 We applied SHAPE to an in vitro-synthesized 365-nt HIV-1 5′ UTR RNA to probe conformational changes that might accompany 4 binding in the context of a larger, native RNA structure. On the basis of the multiple cis-acting regulatory elements located in the 5′ UTR (many of which are structurally distinct hairpins), examining the entire 365-nt sequence also provided a secondary, indirect measure of ligand specificity. Minimal free-energy modeling using SHAPE data as pseudo-free-energy constraints confirmed that the 5′ UTR RNA resembled the previously proposed RNA structure, which includes the trans-activation (TAR) and polyA hairpins, primer binding site (PBS), dimerization initiation site (DIS), and packaging element (Psi).41
Changes in 1-methyl-7-nitroisatoic anhydride (1M7) reactivity values at each nucleotide of the HIV-1 5′ UTR RNA in the presence of 4 are presented in Figure 3 (relative to the analogous experiment in its absence). The increase in nucleotide reactivity upon compound addition is indicated in gradient of red, while the reactivity decrease is represented in gradient of blue, and nucleotide reactivity that was unchanged is in gray. Data shown are an average of three independent experiments. While the overall topology of the 5′ UTR was unchanged upon compound incubation, several substantial changes in chemical reactivity were observed. The most extensive changes were registered within the 5′-TAR structure, including reactivity increase of stem nucleotides U10–G16 and C41–G54, coupled with a strong decrease in reactivity of nucleotides of the apical loop (U31–A35) and the U23–U25 bulge. A small number of alterations in 1M7 reactivity were also observed further downstream, mainly within the very 5′ end of the polyA hairpin (C59–U66) proximal to TAR, and within the U5-IR stem (C144–C147). Although the origin of these effects is unclear, they might reflect the influence of 4 binding on extensive long-range tertiary interactions that have been reported for the HIV-1 5′ UTR region.42,43 A step plot comparing the reactivity profiles obtained for the 5′ UTR region in the absence and presence of 4 is presented in Figure 3B. The homogeneous conformation of HIV-1 RNA 5′ UTR region probed by SHAPE was verified on nondenaturing polyacrylamide gel. This analysis indicated that the RNA migrated as a dimer, and that increasing concentration of 4 did not affect dimer integrity (see Figure SI-3 in SI). Taken together, the shape data indicate a substantial change only in the TAR region of the HIV 5′ UTR, without largely perturbing other features. Thus, effects of compound binding can be mapped directly to the TAR hairpin.
Figure 3.

(a) SHAPE analysis of the HIV-1 5′ UTR in the presence of 4. Nucleotides are color-coded according to normalized SHAPE reactivity values. 1M7 reactivity of the extreme 5′ and 3′ terminal nucleotides (represented in white) are not assigned due to limited resolution in the immediate vicinity of the primer and the final extension product. The TAR hairpin, Poly(A) hairpin, and primer binding site (PBS) are indicated. Nucleotide 1 is vector derived. (b) Step plots for quantitative comparison of 1M7 reactivity values obtained from the HIV-1 RNA 5′ UTR probed in the presence (black) or absence (red) of 4. Boxed region corresponds to nucleotides involved in formation of the 5′ TAR hairpin (nts G2–C58).
To determine if any of the TAR binding compounds showed antiviral activity, we used a whole-cell assay that measures HIV-1-induced cytopathicity in T-lymphoblastic cells (CEM-SS). We evaluated hit molecules 1, 2, and 3, as well as an additional 13 analogues to probe structure–activity relationships on antiviral activity (see SI). Both compound and HIV-induced cytotoxicity were monitored. As shown in Figure 4, 4 displayed good cellular activity (Figure 4C) and 1 also displayed modest protective effects (Figure 4B). A previously reported TAR RNA binding compound, acepromazine maleate,36,44 was not protective in this assay and induced toxicity in control cells (CC50 = 18 μM) (Figure 4A). Analysis of 5 at high concentrations was prevented by modest solubility in the assay medium and DMSO toxicity to CEM-SS cells at a concentration above 1%. Most other analogues assayed were only weakly protective. The most active molecule (4) showed potent activity with a clear dose response and an EC50 of 28 μM. Impressively, no toxicity was observed at concentrations approaching 1 mM (above which the compound was not soluble), resulting in a minimal selectivity index of 36.
Figure 4.

Anti-HIV activity of selected compounds. Uninfected (open circles) and HIV-infected (closed circles) T-lymphoblastic cells were treated with increasing concentrations of acepromazine (a), compound 1 (b), and compound 4 (c). Compounds were assessed for rescue from infection (closed circles) as well as nonspecific toxicity (open circles).
We have identified a new TAR-binding small molecule by screening an unbiased library of 20,000 small molecules in an SMM format. This library was assembled to contain druglike molecules, rather than utilizing preconceived structural elements that favor RNA binding. The relatively low hit rate of 0.02% may reflect that the compound collection was composed of molecules that were selected to generally obey Lipinski’s guidelines,33 and not be biased toward known RNA-binding chemotypes. Furthermore, two out of the three primary hits were validated as binding to the target, resulting in a surprisingly low false positive rate. To the best of our knowledge compound 1 is not structurally similar to any reported TAR-binding molecule, and displays many characteristics of druglike compounds. Of particular interest is the observation that the compound requires hydrophobic and aromatic substituents to achieve antiviral activity (as opposed to requiring cationic groups). This assumes modes of binding not derived from electrostatic interactions, but rather from other forces. We docked compound 4 to six TAR structures from the PDB and identified three plausible binding poses (see Figure SI-6 in SI). These docking simulations indicated that binding in each of these poses was driven primarily by van der Waals forces, followed by hydrogen bonding and hydrophobic interactions, in contrast to the electrostatic interactions that commonly dominate the binding of cationic molecules to RNA. Each of these cell-based assays indicate that analogues lacking the trifluoromethylphenyl group or that lack alkyl groups on the pyridine ring, but retain the aminothienopyridine heterocyclic core, exhibit substantially inferior antiviral activity (see SI). Thus, subtle changes in the structure have dramatic impacts on the antiviral activity of the compound. This is in stark contrast to the broad majority of known TAR-binding compounds that require multiple cationic substituents and bind to RNA through electrostatic interactions. The most active molecule described here (4) has a Kd of 2.4 μM, and a molecular weight of just 351 Da. While other reported ligands to TAR display superior potency, this potency is often associated with a lack of specificity and/or increased toxicity. However, a cell-based model shows that 4 has antiviral activity at a concentration only 10-fold above both the Kd and the concentration at which hairpin stabilization is observed, suggesting that it is selectively engaging its target within the cell.
Importantly, SHAPE analysis shows that 4 binds primarily to TAR even in the context of a larger native RNA sequence. Given that secondary interactions often modulate structure and function in complex RNA sequences, it is particularly useful to have information regarding the effects of ligand binding in a large piece of RNA. However, this valuable information can be difficult to access given the challenges in studying large RNA structures by higher resolution techniques. The data shown here demonstrate minimal global structural perturbations to the 5′ UTR upon incubation with 4. Changes in 1M7 reactivity are noted primarily in the TAR hairpin, suggesting the small molecule binds to this region. Exceptions are a four-nucleotide sequence (C144–C147) of the U5-IR region, which exhibits a moderate change in reactivity, and several residues in the poly-A hairpin (adjacent to TAR in the primary sequence). Although effects outside the TAR hairpin are not large in magnitude, they could represent a long-range destabilizing effect or conformational change on the structure that accompanies 4 binding. The remainder of the 5′ UTR structure was in good agreement with the model of unbound RNA. Thus, the SHAPE analysis shows that the overall structure of the 5′ UTR is not globally perturbed upon ligand binding, and that the ligand exclusively interacts with the TAR hairpin. These results are consistent with the 2-AP titration results, which suggest that only a modest conformational change is induced upon ligand binding. Again, given that there are a number of other hairpins in the 5′ UTR that are not affected, this result also points to a highly specific interaction between 4 and TAR. Perhaps most impressively, nonspecific cytotoxicity was not observed to any degree, even at concentrations nearing 1 mM, indicating that compound 4 is likely not operating through an off-target mechanism.
The nonbiased approach reported here can easily be applied to other biologically relevant RNA structures. The ease with which SMM assays are accomplished enables screening multiple oligonucleotides against large small molecule libraries in relatively short timeframes with minimal assay optimization and small quantities of oligonucleotide. In this work, two RNA hairpins were screened against 20,000 compounds (for a total of 88,000 binding assays) in several days, requiring only ∼12 nmol of fluorescently labeled oligonucleotide. An added but unanticipated benefit of screening in the SMM format is that information is gained about the binding pharmacophore. Because the small molecule is covalently linked to the microarray surface via an amine or alcohol functionality, it is reasonable to hypothesize that tagging identified molecules (e.g., with a fluorescent dye, biotin, NMR spin label, or chemically reactive moiety for footprinting studies) at this functional group will not largely disrupt binding to the target RNA. Thus, this approach may be useful to generate structural probes of more complex RNA sequences in addition to inhibitors of RNA function as described here. Therapeutic applications of such molecules would require optimization of a number of parameters (including potency as well as solubility), and ideally result in submicromolar cellular activity.
Conclusion
In conclusion, our work demonstrates that unbiased small molecule microarrays are a straightforward and viable technology to identify novel, druglike, RNA-binding chemotypes whose biological activity does not depend strictly on a charged, polycationic nature. This work is expected to have broad applications in the identification of small molecules that bind to other RNA targets as well. In this study, SHAPE analysis enabled us to demonstrate the effects of 4 in the context of a large, native RNA structure, as opposed to a small excised hairpin (which was used for the screening assay). Compound 4 binds to TAR and perturbs primarily one structural element of the HIV 5′ UTR, a 365-nucleotide sequence that includes several hairpins and other structural motifs, demonstrating substantial selectivity. Finally, the lack of toxicity and good antiviral activity make 4 a highly attractive candidate for further development.
Experimental Section
Small Molecule Microarray (SMM) Protocol
SMMs were prepared according to Bradner et al.17 Small molecule libraries were acquired from commercial vendors with the requirement that each library member contained at least one primary or secondary amine (aliphatic or aromatic), or one primary or secondary alcohol. Four arrays were printed, each with ∼5000 different molecules printed in duplicate, along with appropriate dyes and control samples for array quality control validation (for a total of 10,800 features per slide). RNA hairpins were purchased from Dharmacon labeled at their 5′ termini with Cy5, and dissolved in DEPC-treated 1 × PBST, pH 7.4 (0.01% Tween-20). RNA was diluted to a concentration of 500 nM, and annealed by heating to 95 °C for 5 min, and cooling to room temperature for 1 h. Microarrays were incubated with the RNA solution at a concentration of 500 nM for 1 h and kept in the dark, after which slides were washed three times with PBST (0.01% Tween-20) and once with water. Slides were dried by centrifugation and immediately imaged.
Analysis of SMM Data
Slides were imaged using an Axon GenePix 4000a array scanner at the appropriate wavelength with a resolution of 5 or 10 μm. The scanned image was then aligned with the GenePix Array List (GAL) file corresponding to the appropriate array, and the resulting GenePix Results (GPR) file was generated. From the GPR file, JMP 9.0 (SAS) was utilized to generate the mean (μ) and standard deviation (σ) for the control (DMSO-printed) spots. For each compound, duplicate spots were averaged, and coefficient of variation (CV) was calculated. A composite Z-score was generated for each compound by the following definition:
Hits were determined using the following criteria: (a) CV for duplicate spots of a compound <100, (b) Average Z score for a compound >3, and (c) [(Z-scoreRNA incubated) – (Z-scoreControl Array)]/Z-scoreControl Array) > 3. Hits were further validated by visual inspection of array images (as shown in Figure 1). In order for a compound to be pursued, it also had to satisfy the hit criteria for the TAR RNA, but not for the control miR-21 RNA. Compounds for further study were purchased from original suppliers.
Differential Scanning Fluorimetry
All differential scanning fluorimetry experiments were conducted on a LightCycler 480 96-well plate Real-Time PCR instrument (Roche Applied Science). A 50 μM solution of TAR RNA in PBS pH 7.4 was heated to 95 °C for 5 min and was allowed to cool to room temperature over 2 h. Annealed RNA was frozen in 1 mL aliquots at −20 °C. A dilution series of TAR RNA with compounds (or with DMSO for negative controls) was incubated for 10 min at room temperature prior to plating 50 μL of the solution into multiwell white plates. Each well was plated with 50 μL of a 0.5 μM solution of TAR RNA (in 1 × PBS buffer, pH 7.4), 0.2 × of SYBR Green II dye, and varying concentrations of compound (1–30 μM). The solution was heated from 30 to 80 °C at a rate of 0.4 °C/s. Fluorescence intensity was measure using excitation and emission wavelengths of 465 and 510 nm, respectively. Melting temperature analysis was performed according to manufacturer’s protocol and corrected for Tm effects caused by DMSO alone.
2-AP Titration
Fluorescence titrations were performed according to literature protocols.8,37,38 TAR RNA where bulge residue U25 was replaced with 2-AP was purchased from Dharmacon (Thermo Fisher). RNA was dissolved in 1 × PBS (pH 7.4) at a concentration of 500 μM, and annealed by heating to 95 °C for 3 min followed by cooling to room temperature over 1 h. RNA was then diluted to a working concentration of 500 nM in PBS. Fluorescence intensity was recorded on a Photon Technology, Inc. Quantamaster fluorimeter at an excitation wavelength of 320 nm and an emission wavelength of 375 nm (the observed emission maximum). Compound was added as a solution in DMSO, and the sample was allowed to equilibrate for 3 min before measuring fluorescence. Fluorescence measurements are reported as the average of 10 measurements at each concentration and were performed in duplicate to ensure reproducibility.
RNA preparation for SHAPE analysis
DNA templates for in vitro transcription were generated by PCR amplification of the HIV-1 molecular clone pNL4-3 using the following primers:
T7L 5′-TAATACGACTCACTATAGGTCTCTCTG-3′ (containing T7 promoter, underlined)
369R 5′-GCTTAATACCGACGCTCTCGC-3′
All PCR experiments were performed using Invitrogen Platinum Taq DNA polymerase High Fidelity. Transcripts were synthesized with the Ambion T7-MEGAscript following the manufacturer’s protocol, and RNAs were fractionated on denaturing 8 M urea/6% polyacrylamide gels, followed by elution and ethanol precipitation. Purified RNAs were dissolved in sterile water and stored at −20 °C.
Renaturation and 1M7 Treatment of RNA
Five picomoles of RNA were heated to 90 °C for 3 min, then immediately placed on ice for 5 min. The volume was adjusted to 10 μL in a final buffer of 50 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5 mM MgCl2. Samples were then divided into 5 μL experimental (+) and control (−) aliquots (2.5 pmol each) and incubated at 37 °C for 15 min. 4 was added to the RNA solution to a final concentration of 5.0 μM and incubated for 10 min at room temperature, while the (−) reaction omitted the compound. Chemical modification was initiated by addition of 1 μL of 1M7 (in anhydrous DMSO) to the (+) RNA solution to a final concentration of 3.5 mM, or DMSO alone to the (−) RNA reaction, followed by incubation at 37 °C for 5 min. Modified RNAs were precipitated at −20 °C with 10 ng/μL glycogen, 0.3 M sodium acetate pH 5.2 and 3 volumes of cold ethanol. RNA was collected by centrifugation, washed once in 70% ethanol and resuspended in 10 μL of water. A parallel experiment lacking 4 was performed as a control.
Primer Extension
For detection of 2′-O-adducts, 2.5 pmol of modified and unmodified RNAs were mixed, respectively, with equimolar amounts of Cy5-labeled (for 1M7-modified samples) or Cy5.5-labeled (for unmodified samples) DNA primer complementary to the 3′ end of the RNA. Samples were incubated at 85 °C for 1 min, 60 °C for 5 min, 35 °C for 5 min, and 50 °C for 2 min to hybridize primers to initiate reverse transcription. cDNA synthesis was performed at 50 °C for 50 min with 100 U RT (Invitrogen Superscript III), 1 × RT buffer (Invitrogen), 5 mM DTT and 500 mM dNTPs (Promega). RNA was hydrolyzed with 200 mM NaOH for 5 min at 95 °C, and samples were neutralized with an equivalent volume of HCl. Sequencing ladders were prepared using the Epicenter cycle sequencing kit according to the manufacturer’s instructions and primers labeled with WellRed D2 and LicorIR-800 dyes. Modified and control samples were mixed with the sequencing ladders, precipitated as above, dried, and resuspended in 40 mL deionized formamide. Primer extension products were analyzed on a Beckman CEQ8000 Genetic Analysis System programmed with the separation method described previously.45
SHAPE Data Analysis
Electropherograms were processed using the SHAPEfinder program, following the software developer’s protocol and included the required precalibration for matrixing and mobility shift for each set of primers as described.39,46 Briefly, the area under each negative peak was subtracted from that of the corresponding positive peak. The resulting peak area difference at each nucleotide position was then divided by the average of the highest 8% of peak area differences, calculated after discounting any results greater than the third quartile plus 1.5× the interquartile range. RNAstructure software version 5.347 was used to predict RNA secondary structure(s) on the basis of pseudo-free energy constraints derived from SHAPE reactivity values. All reactivity data used in 2D structure analysis was averaged from three independent experiments. Varna (ver. 3–7), the visualization applet for RNA secondary structure was used to produce high-quality images.48
Anti-HIV-1 Activity Determination
To determine the antiviral activity of compounds, an XTT-tetrazolium-based assay was used in which HIV-1RF challenged T-lymphoblastic CEM-SS cell viability was measured, as described previously.49 XTT was graciously supplied by the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute. CEM-SS cells were maintained in RPMI 1640 media without phenol red and supplemented with 5% fetal bovine serum (BioWhittaker), 2 mM l-glutamine (BioWhittaker), and 50 μg/mL gentamicin (BioWhittaker) (complete medium). Exponentially growing cells were washed and resuspended in complete medium. Five × 103 cells were added to individual wells of a 96-well microtiter plate containing serial dilutions of test compounds, solubilized in DMSO and brought to a total volume of 100 μL with medium. Stock supernatants of HIV-1RF were diluted in complete medium to yield cytopathicity resulting in 80–90% cell kill after 6 days. A 50 μL aliquot was then added to test wells. A second set of wells with CEM-SS cells only was also treated with serial dilutions of test compounds to determine cellular toxicity. Plates were incubated for 6 days at 37 °C, and then stained for cellular viability using XTT. All experiments were performed in triplicate.
Acknowledgments
This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The work was also partially supported by Yale University. We thank Janie Merkel for helpful discussions regarding the analysis of microarray data. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract HHSN26120080001E. We thank Jennifer Wilson for technical assistance with anti-HIV assays.
Supporting Information Available
DSC studies, minus strand strong stop assay, and complete results for cell-based analysis of analogues are available. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# J.S.-S. and S.R.S. contributed equally to this manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cooper T. A.; Wan L. L.; Dreyfuss G. Cell 2009, 136, 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobovits A.; Smith D. H.; Jakobovits E. B.; Capon D. J. Mol. Cell. Biol. 1988, 8, 2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby M. J.; Bain E. S.; Luciw P. A.; Peterlin B. M. Gene Dev 1989, 3, 547. [DOI] [PubMed] [Google Scholar]
- Richter S.; Cao H.; Rana T. M. Biochemistry 2002, 41, 6391. [DOI] [PubMed] [Google Scholar]
- Berkhout B.; Vastenhouw N. L.; Klasens B. I. F.; Huthoff H. RNA 2001, 7, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J. R.; Hergenrother P. J. Chem. Rev. 2008, 108, 1171. [DOI] [PubMed] [Google Scholar]
- Guan L. R.; Disney M. D. ACS Chem. Biol. 2012, 7, 73. [DOI] [PubMed] [Google Scholar]
- Stelzer A. C.; Frank A. T.; Kratz J. D.; Swanson M. D.; Gonzalez-Hernandez M. J.; Lee J.; Andricioaei I.; Markovitz D. M.; Al-Hashimi H. M. Nat. Chem. Biol. 2011, 7, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann T. Curr. Opin Struct Biol. 2005, 15, 355. [DOI] [PubMed] [Google Scholar]
- Hermann T. Biopolymers 2003, 70, 4. [DOI] [PubMed] [Google Scholar]
- Ogle J. M.; Ramakrishnan V. Annu. Rev. Biochem. 2005, 74, 129. [DOI] [PubMed] [Google Scholar]
- Poehlsgaard J.; Douthwaite S. Nat. Rev. Microbiol 2005, 3, 870. [DOI] [PubMed] [Google Scholar]
- Weeks K. M. Curr. Opin Struct Biol. 2010, 20, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount K. F.; Breaker R. R. Nat. Biotechnol. 2006, 24, 1558. [DOI] [PubMed] [Google Scholar]
- Parsons J.; Castaldi M. P.; Dutta S.; Dibrov S. M.; Wyles D. L.; Hermann T. Nat. Chem. Biol. 2009, 5, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner J. E.; McPherson O. M.; Mazitschek R.; Barnes-Seeman D.; Shen J. P.; Dhaliwal J.; Stevenson K. E.; Duffner J. L.; Park S. B.; Neuberg D. S.; Nghiem P.; Schreiber S. L.; Koehler A. N. Chem. Biol. 2006, 13, 493. [DOI] [PubMed] [Google Scholar]
- Bradner J. E.; McPherson O. M.; Koehler A. N. Nat. Protoc. 2006, 1, 2344. [DOI] [PubMed] [Google Scholar]
- Hergenrother P. J.; Depew K. M.; Schreiber S. L. J. Am. Chem. Soc. 2000, 122, 7849. [Google Scholar]
- Stanton B. Z.; Peng L. F.; Maloof N.; Nakai K.; Wang X.; Duffner J. L.; Taveras K. M.; Hyman J. M.; Lee S. W.; Koehler A. N.; Chen J. K.; Fox J. L.; Mandinova A.; Schreiber S. L. Nat. Chem. Biol. 2009, 5, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H.; Tallarico J. A.; Hayakawa H.; Munger K.; Duffner J. L.; Koehler A. N.; Schreiber S. L.; Lewis T. A. J. Comb Chem. 2007, 9, 245. [DOI] [PubMed] [Google Scholar]
- Koehler A. N.; Shamji A. F.; Schreiber S. L. J. Am. Chem. Soc. 2003, 125, 8420. [DOI] [PubMed] [Google Scholar]
- Kuruvilla F. G.; Shamji A. F.; Sternson S. M.; Hergenrother P. J.; Schreiber S. L. Nature 2002, 416, 653. [DOI] [PubMed] [Google Scholar]
- Barnes-Seeman D.; Park S. B.; Koehler A. N.; Schreiber S. L. Angew. Chem. Int. Ed. 2003, 42, 2376. [DOI] [PubMed] [Google Scholar]
- Tran T.; Disney M. D. Nat. Commun. 2012, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velagapudi S. P.; Pushechnikov A.; Labuda L. P.; French J. M.; Disney M. D. ACS Chem. Biol. 2012, 7, 1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T.; Disney M. D. Biochemistry 2010, 49, 1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney M. D.; Lee M. M.; Pushechnikov A.; Childs-Disney J. L. ChemBioChem 2010, 11, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D. J.; Seedhouse S. J.; Disney M. D. Nucleic Acids Res. 2009, 37, 5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aminova O.; Paul D. J.; Childs-Disney J. L.; Disney M. D. Biochemistry 2008, 47, 12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disney M. D.; Labuda L. P.; Paul D. J.; Poplawski S. G.; Pushechnikov A.; Tran T.; Velagapudi S. P.; Wu M.; Childs-Disney J. L. J. Am. Chem. Soc. 2008, 130, 11185. [DOI] [PubMed] [Google Scholar]
- Childs-Disney J. L.; Wu M. L.; Pushechnikov A.; Aminova O.; Disney M. D. ACS Chem. Biol. 2007, 2, 745. [DOI] [PubMed] [Google Scholar]
- Velagapudi S. P.; Gallo S. M.; Disney M. D. Nat. Chem. Biol. 2014, 10, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Adv. Drug Delivery Rev. 1997, 23, 3. [DOI] [PubMed] [Google Scholar]
- Hwang S.; Tamilarasu N.; Kibler K.; Cao H.; Ali A.; Ping Y. H.; Jeang K. T.; Rana T. M. J. Biol. Chem. 2003, 278, 39092. [DOI] [PubMed] [Google Scholar]
- Chirayil S.; Chirayil R.; Luebke K. J. Nucleic Acids Res. 2009, 37, 5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z. H.; Lind K. E.; James T. L. Chem. Biol. 2002, 9, 707. [DOI] [PubMed] [Google Scholar]
- Bradrick T. D.; Marino J. P. RNA 2004, 10, 1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradrick T. D.; Marino J. P. Biophys. J. 2004, 86, 143A. [Google Scholar]
- Lu J.; Kadakkuzha B. M.; Zhao L.; Fan M.; Qi X.; Xia T. Biochemistry 2011, 50, 5042. [DOI] [PubMed] [Google Scholar]
- Wilkinson K. A.; Gorelick R. J.; Vasa S. M.; Guex N.; Rein A.; Mathews D. H.; Giddings M. C.; Weeks K. M. PLoS Biol. 2008, 6, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deforges J.; Chamond N.; Sargueil B. Biochimie 2012, 94, 1481. [DOI] [PubMed] [Google Scholar]
- Huthoff H.; Berkhout B. RNA 2001, 7, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard C. K.; Andersen E. S.; Knudsen B.; Gorodkin J.; Kjems J. J. Mol. Biol. 2004, 336, 369. [DOI] [PubMed] [Google Scholar]
- Renner S.; Ludwig V.; Boden O.; Scheffer U.; Gobel M.; Schneider G. ChemBioChem 2005, 6, 1119. [DOI] [PubMed] [Google Scholar]
- Mitra S.; Shcherbakova I. V.; Altman R. B.; Brenowitz M.; Laederach A. Nucleic Acids Res. 2008, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badorrek C. S.; Weeks K. M. Biochemistry 2006, 45, 12664. [DOI] [PubMed] [Google Scholar]
- Deigan K. E.; Li T. W.; Mathews D. H.; Weeks K. M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty K.; Denise A.; Ponty Y. Bioinformatics 2009, 25, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulakowski R. J.; Mcmahon J. B.; Staley P. G.; Moran R. A.; Boyd M. R. J. Virol Methods 1991, 33, 87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.