

Abstract
The glutamate decarboxylase (GAD) system has been shown to be important for the survival of Listeria monocytogenes in low pH environments. The bacterium can use this faculty to maintain pH homeostasis under acidic conditions. The accepted model for the GAD system proposes that the antiport of glutamate into the bacterial cell in exchange for γ-aminobutyric acid (GABA) is coupled to an intracellular decarboxylation reaction of glutamate into GABA that consumes protons and therefore facilitates pH homeostasis. Most strains of L. monocytogenes possess three decarboxylase genes (gadD1, D2 & D3) and two antiporter genes (gadT1 & gadT2). Here, we confirm that the gadD3 encodes a glutamate decarboxylase dedicated to the intracellular GAD system (GADi), which produces GABA from cytoplasmic glutamate in the absence of antiport activity. We also compare the functionality of the GAD system between two commonly studied reference strains, EGD-e and 10403S with differences in terms of acid resistance. Through functional genomics we show that EGD-e is unable to export GABA and relies exclusively in the GADi system, which is driven primarily by GadD3 in this strain. In contrast 10403S relies upon GadD2 to maintain both an intracellular and extracellular GAD system (GADi/GADe). Through experiments with a murinised variant of EGD-e (EGDm) in mice, we found that the GAD system plays a significant role in the overall virulence of this strain. Double mutants lacking either gadD1D3 or gadD2D3 of the GAD system displayed reduced acid tolerance and were significantly affected in their ability to cause infection following oral inoculation. Since EGDm exploits GADi but not GADe the results indicate that the GADi system makes a contribution to virulence within the mouse. Furthermore, we also provide evidence that there might be a separate line of evolution in the GAD system between two commonly used reference strains.
Introduction
Survival in sometimes harsh environmental conditions is vital for any pathogen en route to infection of the host. The foodborne pathogen Listeria monocytogenes is well noted for an ability to withstand high salt environments [1]–[3], high pressure [4], [5], grow at low temperature [6] and within a broad pH range over which it can survive [3], [7], [8]. This makes it a major concern for the food industry where preservation methods often employ combinations of pH, salinity and temperature controls. In order for L. monocytogenes to survive low pH environments, the bacterium has evolved several mechanisms that allow it to maintain pH homeostasis. These include the arginine deiminase system [9], an F0F1 ATPase [10], the adaptive acid tolerance response (ATR) [11] and the glutamate decarboxylase (GAD) system [12]. The GAD system has been shown in L. monocytogenes to be important for survival in synthetic gastric fluid [12] but not in the presence of organic acids [8] commonly found in foods.
The accepted model for the GAD system (Fig. 1) involves the combined action of a membrane bound antiporter (GadT) and a cytosolic glutamate decarboxylase (GadD). During exposure to low pH, the bacterium can exchange an extracellular molecule of glutamate for an intracellular molecule of γ-aminobutyric acid (GABA) via the GadT antiporter/s. This imported glutamate then undergoes a decarboxylation to form GABA via the GadD enzyme/s. At pH < 4.5, glutamate is imported in a neutral form (Glu0) [13], which allows the removal of intracellular H+ when glutamate is converted to GABA. This consumption of intracellular protons helps to maintain a tolerable intracellular pH. GABA generated via this reaction is expected to exit the cell via the antiporter in exchange for further glutamate, allowing a cycling process to continue (Fig. 1). In previous work we have shown that GAD activity can take place independently of the antiporter, a finding that prompted a revision of the previous model by introducing the concepts of an extracellular GAD system (GADe) i.e. a GAD system relying on the Glu/GABA antiport and an intracellular GAD system (GADi) i.e. a GAD system that relies on intracellular pools of glutamate or glutamate possibly imported into the cell via a glutamate transporter [14].
Figure 1. The standard model for the action of the GAD system.

(a) A membrane bound antiporter carries glutamate into the cell in exchange for GABA. A cytosolic decarboxylase enzyme converts glutamate to GABA, with a consumption of H+. (b) The genomic structure of the genes encoding the GAD system in L. monocytogenes EGD-e.
For L. monocytogenes, the glutamate/GABA antiporter can be encoded by one of two genes (gadT1, T2) while it possesses up to three decarboxylase encoding genes (gadD1, D2 & D3) [15]. Previously, gadD3 has been considered as a putative glutamate decarboxylase encoding gene and some lines of evidence suggest that this glutamate decarboxylase may form an integral part of the GADi [14]. These genes are encoded in three transcriptional units gadD1T1, gadT2D2 and gadD3 [16] (Fig. 1). While all strains possess both gadT2D2 and gadD3, strains from serotype 4 do not possess the gadD1T1 operon [16]. Previously, genetic studies on the GAD system have shown that gadD2 is critical for survival of L. monocytogenes at low pH, while gadD1 is important at a milder pH [12], [16]. This work was carried out in the strain LO28 (serotype 1/2c), however recent work has shown that the GAD system can behave differently depending on both strain and media type. Analysis of strain EGD-e has shown that the antiporters are non functional, which results in a GAD system that produces intracellular GABA from the cytoplasmic pool of glutamate [14]. Furthermore, in a chemically defined media, the strain 10403S also relies solely on the activity of GADi, despite having a functional antiport mechanism [17].
Despite what has been learned so far it is apparent that the accepted model of the GAD system in L. monocytogenes is not complete and certain knowledge gaps remain including elucidating the role of the GadD3 and of the other GAD genes in different environmental conditions as well as the role the GAD system plays in vivo. Therefore, here we compare the GAD system of two important reference strains of L. monocytogenes, EGDm (a murinised form of EGD-e) and 10403S and we describe an extensive genetic and physiological analysis of the GAD system in these strains, including the previously uncharacterised gadD3 gene. Furthermore, while studies have shown that the GAD system is important for survival in porcine gastric juice, the role of this system during pathogenesis in a live animal model remains unknown.
Methods and Materials
Bacterial strains and growth
All strains used in this study are listed in Table 1. Strains were grown on BHI (Lab M) agar plates at 37°C for 24 to 48 h. Cultures grown overnight were set up by inoculating a single colony into 25 ml BHI broth in 250 ml Erlenmeyer flasks and incubating at 37°C with aeration.
Table 1. Bacterial strains and plasmids used in this study.
| Strain or plasmid | Relevant properties | Reference/Source |
| L. monocytogenes strains | ||
| EGDm | Serovar 1/2a, murinised inlA gene | C. Gahan |
| EGDm ΔgadD1 | EGDm with in frame 1326 bp deletion of gadD1 (lmo0047) gene | This study |
| EGDm ΔgadD2 | EGDm with in frame 1365 bp deletion of gadD2 (lmo2363) gene | This study |
| EGDm ΔgadD3 | EGDm with in frame 1275 bp deletion of gadD3 (lmo2434) gene | This study |
| EGDm ΔgadD1D3 | EGDm ΔgadD1 with in frame 1275 bp deletion of gadD3 (lmo2434) gene | This study |
| EGDm ΔgadD2D3 | EGDm ΔgadD3 with in frame 1365 bp deletion of gadD2 (lmo2363) gene | This study |
| 10403S | Serovar 1/2a, Wild-type | K. Boor |
| 10403S ΔgadD1 | 10403S with in frame 1326 bp deletion of gadD1 (lmrg_00139) gene | This study |
| 10403S ΔgadD2 | 10403S with in frame 1365 bp deletion of gadD2 (lmrg_01479) gene | This study |
| 10403S ΔgadD3 | 10403S with in frame 1275 bp deletion of gadD3 (lmrg_01814) gene | This study |
| Plasmid | ||
| pKSV7 | pKSV7 shuttle vector used to carrying the individual deletion cassettes used in mutant generation | [34] |
Generation of knockout mutants
Deletion mutants were generated using the splicing by overlap extension (SOEing) PCR method and allelic replacement, as previously described [18]. All primers used in the process are listed in Table 2. Deletion cassettes were cloned into the shuttle vector pKSV7. Double mutants were generated by introducing the deletion cassette into the confirmed single EGDm ΔgadD3 mutant. PCR amplification of DNA for use in cloning and downstream work was carried out by using the high-fidelity Velocity DNA polymerase (Bioline), while screening was carried out by using Biotaq DNA polymerase (Bioline).
Table 2. Primers used in this study.
| Primer name | Sequence (5' to 3') |
| gadD1 A | CGGAATTCCAAGCAAATTACCAGGTG |
| gadD1 B | GAGTAAAGCCAGAGCCAAACACCGGTACA |
| gadD1 C | CGGTGTTTGGCTCTGGCTTTACTCATTAAG |
| gadD1 D | CGGAATTCATAACGTAAGATAGTGCGCC |
| gadD1 For | ACAAATACGCCACGCATC |
| gadD1 Rev | GGCAAGAACCATAAGAATCCAC |
| gadD2 A | CGGAATTCGTAGTCATTTATTTAGTCGGC |
| gadD2 B | AAGCCGTGACTATATAACATGATTTTTTCCTC |
| gadD2 C | TATATAGTCACGGCTTCACACATTAATAAAAAGGC |
| gadD2 D | CGGAATTCTCGCATATTAATTATTTGACG |
| gadD2 For | TCATTCCTAACTGCCATTTCC |
| gadD2 Rev | TGGAATGAGAATAGTGGACGG |
| gadD3 A | CGGAATTCCTTTATAGTGAAGACGAC |
| gadD3 B | TTGTCATGATACATACAAGCTTCCGAAG |
| gadD3 C | GCTTGTATGTATCATGACAAAGAACGCAAC |
| gadD3 D | TAGAATTCATTTCAGTACGCGAGCCATCAC |
| gadD3 For | GAACCTCCTTATAAGTACCATC |
| gadD3 Rev | GGTGGTTACGGTGCATTC |
Acid survival assays
Cultures of bacteria were grown to mid-exponential (OD600 = 0.35) or stationary phase (16 h) at 37°C in BHI medium. The pH of these cultures was lowered with 3 M HCl to pH 3.5 for mid-exponential or pH 2.5 for stationary phase cultures. Samples were taken every 20 min for 1 h and serially diluted in phosphate-buffered saline (PBS). Dilutions were plated in triplicate onto BHI agar and incubated overnight at 37°C. Colonies were counted to determine the number of surviving cells.
GABase assays
Intracellular GABA (GABAi) and extracellular GABA (GABAe), were measured as previously described [17], [19]. Cultures were grown to mid-exponential phase (OD600 = 0.35) or stationary phase (16 h) at 37°C with aeration in BHI medium. For exponential phase GABA measurements the cultures were reduced to pH 4.0, while for stationary phase GABA measurements, the pH of the cultures was lowered to 4.0 (EGDm) or 3.5 (10403S) with 3 M HCl. Different pH reductions for each strain were necessary to ensure optimal GABA production for each strain. Extractions were made after 1 h of acid treatment. Non HCl-treated cultures were used as negative controls. GABase from Pseudomonas fluorescens (Sigma Aldrich, Steinheim, Germany) was used in the enzymatic assay and increases in OD340 nm were measured using a Tecan Sunrise absorbance plate reader.
Real-time PCR determination of gadD gene transcription
Real-time PCR was used to determine the relative expression of the genes encoding the GAD system, using the 16S rRNA gene as a reference gene. Cells were grown overnight (16 h) in triplicate (biological replicates) in 20 ml BHI at 37°C. Prior to RNA isolation, 1 ml of culture was mixed with 2 ml RNAlater (R) (Sigma) and incubated at room-temperature for 5 min. Subsequently cells were harvested by centrifugation at 8,000×g for 5 min washed once in 1 ml 1 M Tris buffer (pH 7.4) and disrupted by microwaving for 15 s at 700 W. Pellets were resuspended in 350 µl Buffer RLT (Qiagen) and cell debris removed by centrifuging at 12,000×g for 2 min. Total RNA was obtained from each of the biological replicates using the RNeasy kit (Qiagen, Hilden, Germany), quantified using a NanoDrop (Thermo Scientific, Wilmington, DE), treated with DNase (Turbo DNA-free; Ambion, Austin, TX), and then used to synthesize cDNA. To ensure the absence of DNA contamination in the samples of treated RNA, a PCR reaction was carried out on each RNA sample using 16S primers. To obtain cDNA 15 µl of total RNA (2 µg ml−1) was mixed with 1 µl of random primers (3 µg µl−1; Invitrogen, Carlsbad, CA), 1 µl of a 10 mM stock of deoxyribonucleotide (dNTP) mix (Invitrogen, Carlsbad, CA). The mixture was heated to 65°C for 5 min, transferred onto ice, and centrifuged briefly before 4 µl of First Strand buffer and 2 µl or 100 mM dithiothreitol (DTT) was added. Following an incubation of 2 min at 25°C, 1 µl of Superscript II (200 U µl−1) was added. Subsequently, the mixture was incubated at 25°C for 10 min, at 42°C for 50 min, and finally at 70°C for 15 min before the cDNA was stored at −20°C. Relative quantification of the expression of the genes was carried out by fluorometric real-time PCR using the SYBR green master mix and the Quantitect SYBR green PCR kit (Qiagen, Hilden, Germany). Following the instructions of the kit, 2.5 µl of cDNA was mixed with 5 µl QuantiTect SYBR green PCR master mix (containing fast-start Taq DNA polymerase, SYBR green dye, buffer, and MgCl2), and the appropriate set of primers (Table 1) was added to a final concentration of 0.2 µM each. Each set of primers was designed with specificity toward the genome of L. monocytogenes strain 10403S (accession no. AARZ02000000), producing amplicons in the range of 207 to 268 bp (Table 1). Finally, molecular-biology-grade water was added to the mixture, up to a final volume of 10 µl, and placed in a well of the 96-well microtiter plate (LightCycler 480 multiwell plate 384; Roche Diagnostic GmbH, Manheim, Germany). Once the other PCRs were set up in the other wells, the plate was placed in the LightCycler 480 instrument (Roche Diagnostic GmbH, Manheim, Germany), and the PCR was programmed starting with an initial denaturation at 95°C for 10 min, followed by amplification for 40 cycles at 95°C for 1 s, 5 s at various annealing temperatures, depending on the melting temperature of the set of primers (Table 1), and 72°C for 9 to 11 s.
The specificity of amplification for each product was determined by a melting curve analysis at 95°C for 1 s and 65°C for 15 s, followed by a progressive increase of the temperature to 95°C with a ramp rate of 0.11°C s−1, with continued measurement of fluorescence, and finally cooling of the plate at 40°C for 30 s. Alongside each real-time PCR assay, a control reaction without added cDNA was run as a negative control. Relative expression was calculated as a ratio between expression of target genes (gadD1, gadT1, gadD2, gadT2, and gadD3) and the expression of the 16S rRNA gene, which served as reference gene in each cDNA sample. Calculations were carried out following the “advanced relative quantification” settings of the LightCycler 480 software program with PCR efficiency correction.
Animal and infection studies
Eleven week old Balb/c mice were used for infection studies. Animals were housed within the biological services unit, University College Cork and fed 2018S Teklad Global 18% Protein Rodent Diet (Harlan). The EGDm strain used in this study was murinised as previously described [20]. All GAD system mutants were introduced into this strain prior to infection. Overnight cultures of bacteria were grown in BHI medium at 37°C for 16–18 h. Cultures were washed and resuspended in PBS. Mice were inoculated with bacteria to a concentration of 1010 in 200 µl via direct gastric gavage at 5 mice per strain. Three days post infection, mice were sacrificed and the liver, spleen, mesenteric lymph nodes (MLN) and intestinal faeces were harvested. Liver, spleen and MLN were homogenised in PBS. The weight of the intestinal content was determined and then homogenised in PBS. All samples were serially diluted and plated in duplicate onto either BHI agar (Liver & Spleen) or Listeria Selective agar (LSA; Oxoid) (MLN & intestinal content).
Macrophage survival assay
Permanent stocks of a human derived THP-1 macrophage cell line [21] were maintained at a concentration of 1×106 in 1 ml Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma Aldrich) supplemented with 10% (v/v) heat-inactivated foetal bovine serum (FBS; Lonza) and 10% (v/v) DMSO and stored at −80°C. These were recovered by resuspension in 3 ml RPMI supplemented with 10% (v/v) FBS and 100 U ml−1 penicillin/streptomycin (PenStrep; Sigma Aldrich) and incubated at 37°C in 5% CO2 for 2 days. The culture was then centrifuged at 400×g for 5 min and the pellet washed in 5 ml phosphate buffered saline and centrifuged again. The pellet was then resuspended in 5 ml RPMI with 10% FBS and PenStrep and incubated as above for 5–7 days. For survival assays, the macrophages were centrifuged at 400×g for 5 min and washed twice in 5 ml PBS and resuspended in RPMI and 10% FBS supplemented with 0.16 µM phorbol myristate acetate to a concentration of 1×105 cells ml−1. One millilitre was then seeded per well of a 24 well tissue culture plate and the plate was incubated for a further 24 h at 37°C with 5% CO2 to allow the cells to adhere to the surface of the well. After incubation, the media was removed from each well and 1 ml of PBS was added to wash the cells. The plate was shaken gently and the PBS removed. In parallel, L. monocytogenes cultures were grown to stationary phase in BHI where 1 ml of culture was then centrifuged at 10,000×g for 5 min, washed once in 1 ml PBS and resuspended in 1 ml PBS to give an estimated concentration of 1×109 cfu ml−1. Bacterial cells were then diluted to 1×106 in RPMI and 10% FBS. One millilitre of this bacterial suspension was added to each washed well of THP-1 cells to give a multiplicity of infection (MOI) of 10. Plates were incubated at 37°C with 5% CO2 for 1 h to allow for invasion. After the 1 h co-incubation period, the media was removed from the wells and the cells were washed with 1 ml PBS and then 1 ml of RPMI and 10% FBS supplemented with 30 µg ml−1 Gentamicin was added to kill remaining extracellular bacteria. Plates were returned to incubate for 1 h at 37°C with 5% CO2. Bacterial counts were performed by removing the media from each well, washing once in 1 ml PBS and resuspending the cells with 1 ml ice cold sterile dH2O for 1 min. Serial dilutions were performed and the suspension was spread plated in duplicate onto BHI agar plates. All agar plates were incubated for 24 h at 37°C before counting.
Statistical analysis of results
Experiments were carried out with three biological replicates and at least two technical replicates for each sample. Significant differences between samples tested were determined by using either a paired Student t test or one-way ANOVA. Results were considered significant when they possessed a P value of <0.05. Error bars indicating standard deviations from the means are displayed on graphs.
Ethical statement
All animal procedures were approved by the University Animal Experimental Ethics Committee (AEEC) in University College Cork (approval ID 2011/017) and were carried out in a specialized facility. Mice were euthanized by cervical dislocation. Work was carried out under license from the Irish Department of Health.
Results
Deletion of gadD1 or gadD2 increases acid survival in EGDm
To investigate the role of the GAD system in acid tolerance and virulence each of the three decarboxylase genes (gadD1, gadD2, gadD3) were deleted individually from a murinised strain of EGD-e (designated EGDm), generating three deletion mutants. The murinisation process as explained and carried out previously [22], involved the targeted mutagenesis of two key amino acids (Ser192Asn and Tyr369Ser) in the InlA protein of EGD-e. Strains lacking a single gadD gene were grown to stationary phase (at which point the GAD system is known to play a crucial role in the survival of acidic conditions [12], [23]) and tested for survival at low pH. After 20 min the numbers of surviving wild-type EGDm cells began to rapidly reduce as did those of the strain with a deletion in gadD3. Sixty minutes post treatment both the parental EGDm and EGDm ΔgadD3 had reduced by over 4.5 log-cycles (Fig. 2a ). In contrast the numbers of both EGDm ΔgadD1 and EGDm ΔgadD2 strains reduced by only ∼2 log-cycles after 60 min. When the survival of mid-exponential phase cells was tested the gadD1 and gadD2 deletions did not confer any survival advantage compared to the parental control (Fig. S1a ). Measurements of the stationary phase transcript levels of each gadD gene prior to acid exposure showed that gadD3 was expressed by almost 2 orders of magnitude greater than either gadD1 or gadD2 (Fig. 2c ). Deletion of any of the gadD genes did not appear to significantly affect the base levels of any of the remaining two gadD genes although gadD1 in the ΔgadD3 background was reduced but this difference displayed a p-value of 0.05. Thus the increased stationary phase acid resistance observed in the EGDm ΔgadD1 and EGDm ΔgadD2 strains cannot be explained by an increase in the transcription of the remaining gadD genes.
Figure 2. Acid survival of L. monocytogenes gad mutants.

Stationary phase EGDm (a) and 10403S (b) Δgad mutants were challenged at pH 2.5. Cell counts were taken every 20 min. Values are the means of data from three individual cultures, with the cell counts for each culture being the means of counts from three platings. Relative transcript levels of EGDm (c) or 10403S (d) gadD1 (dark grey fill), gadD2 (hatched) and gadD3 (grey) genes to 16S gene prior to acid exposure in each mutant strain. Error bars represent the standard error from the mean value of three individual biological repeats. The numbers over the bar charts (c & d) indicate the p-value for the difference between each gene expression compared to wild-type levels as determined by student’s t-test.
Deletion of gadD2 and gadD3 reduces acid tolerance in 10403S
To determine if the relative contribution of the gadD genes to acid tolerance was conserved in another well-studied reference strain the same gene deletions were introduced into L. monocytogenes 10403S, which belongs to the same lineage (II) as EGD-e, but carries over 30,000 SNPs [24]. Once more the effect of acid treatment was tested on stationary phase cultures for 1 h in BHI. After 60 min wild-type 10403S showed a reduction in cell numbers of over 1.5 log-cycles (Fig. 2b ), substantially less killing than was observed in EGDm over this period (Fig. 2a ). Deletion of gadD1 did not affect survival at this pH compared to the wild-type. For 10403S a steady decrease in cell numbers was observed over 60 min with a final reduction of about 3 log-cycles, 100-fold greater than the parent strain. The increased sensitivity of the ΔgadD2 mutant was also observed in exponentially growing cells (Fig. S1b ). 10403S ΔgadD3 did not show a significant reduction in cell numbers compared to 10403S until after 40 min of acid treatment and reached a final reduction of about 3 log-cycles after 60 min (Fig. 2b ). A profile of the gadD gene transcript levels prior to acid exposure in 10403S showed that gadD2 is the dominant transcript while gadD1 is transcribed at the lowest levels (Fig. 2d ). As in EGDm, deletion of one gadD gene did not significantly affect the expression of the remaining genes apart from gadD2 in the ΔgadD1 background. Together these results suggest that the two strains have evolved different transcriptional controls over the gadD genes and further indicate that gadD gene transcript levels per se don’t determine the intrinsic level of acid tolerance.
EGDm relies on gadD3 for GABA production while 10403S requires gadD2
GABA is the main product of the GAD system and therefore measurement of its production could give insights into how the systems compare in these strains. To this end stationary phase cultures of both strains were challenged to mild acidic pH for one hour and both the extracellular and intracellular GABA (GABAe/GABAi) production was measured. Previous work has indicated that EGD-e and therefore, EGDm does not produce GABAe while 10403S produces both GABAe and GABAi [14]. As expected, GABAe was not produced here by any EGDm strain in response to low pH (Fig. 3a ). After 1 h at pH 4.0 however EGDm produced over 5 mM GABAi. A finding worth documenting was that EGDm produced higher concentrations of GABAi in response to acidification than our previously studied EGD-e strain (data not shown). Since the parent EGD-e strains came from separate laboratories it may be that some mutation had arisen in one of the lines that impacts on the GAD system. Deletion of either gadD1 or gadD2 however did not affect GABAi production compared to the parent strain (Fig. 3b ). GABA production in EGDm ΔgadD3 however was almost abolished, with production just above detectable levels for the assay (0.45 mM; Fig. 3a ). In exponential phase no differences in GABA levels were detected between EGDm and the corresponding gadD mutants; indeed GABA levels were at or below the detection limit of the assay in these cultures (Fig. S2). Unlike EGDm, stationary phase cultures of 10403S produced both GABAi (2.72 mM) and GABAe (3.35 mM) after acid treatment. Deletion of gadD1 or gadD3 did not significantly affect the acid-induced GABA production. 10403S ΔgadD2 did have a significant decrease in both GABAi (0.64 mM; Fig. 3b ) and GABAe (0.75 mM; Fig. 3a ). A similar effect on GABA production was observed for the ΔgadD2 mutant (Fig. S2). Thus the gadD gene with the highest transcript levels in each strain also contributed the most to GABA production in that strain; gadD3 for EGDm and gadD2 for 10403S.
Figure 3. GABA production from L. monocytogenes gad mutants.
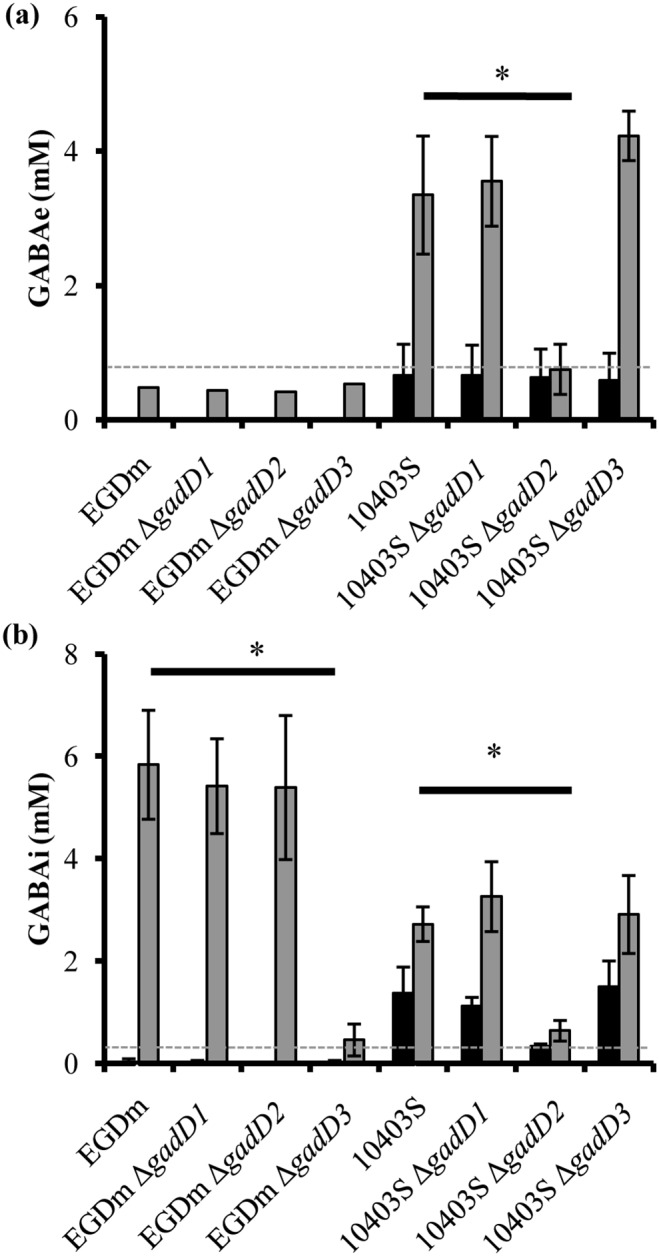
(a) Production of GABAe by EGDm and 10403S gad mutants with (grey) or without (black) 1 h exposure to acid at pH 4.0 (EGDm) or pH 3.5 (10403S). (B) Production of GABAi by EGDm and 10403S gadD mutants with (grey) or without (black) 1 h exposure to acid at pH 4.0 (EGDm) or pH 3.5 (10403S). Dashed horizontal lines indicate the detection limits for GABA in each experiment. Error bars represent the standard deviation from the mean of three individual biological repeats for each sample. An asterix represents signifcant difference of less than 0.05 between a given mutant and respective wild-type as determined by a student’s t-test.
Expression of the GAD system is pH-independent
To determine the transcriptional response of EGDm and 10403S to both low pH exposure and GAD system mutation, the expression levels of all three decarboxylase genes was compared to the expression of 16S rRNA in each strain. Overall expression of the gadD genes did not change in EGDm in response to low pH (Fig. 4a ). The expression of gadD3 remained higher than both gadD1 and gadD2 in the parent EGDm strain and EGDm ΔgadD1 and EGDmΔgadD2 throughout the course of the acid challenge. In 10403S, the relative expression of gadD2 was significantly greater than the other genes at stationary phase (Fig 2. d ). Deletion of any of the gadD genes did not affect the expression of the remaining two in stationary phase. The expression of the gadD genes did not appear to change in response to acid challenge for any of the four 10403S strains (Fig. 4b ). σB activity, which was measured indirectly by recording the levels of the known σB-dependent gene lmo2230 (which encodes a putative arsenate reductase) [2], appeared to remain stable in all strains throughout the acid challenge (Fig. 4a bottom right). Overall it appeared that regardless of pH gadD3 was the dominant GAD transcript in EGDm while for 10403S it was gadD2.
Figure 4. Relative expression of gad genes in response to acid treatment.

Expression of gadD1, gadD2, gadD3 and lmo2230 relative to expression of the 16S rRNA gene prior to, 15 min and 30 min after exposure in BHI broth to pH 4.0 (EGDm (a)) or pH 3.5 (10403S (b)). Error bars represent the standard error in the mean of 3 independent biological repeats. Differences found to be significant between the genes at any time-point for each strain are shown with * Significance was determined where p <0.05 as determined by a Student’s t-test.
Deletion of gadD3 together with either gadD1 or gadD2 reduces acid tolerance seen in single mutants and abolishes GABA production
To account for the increase in acid survival seen in both EGDm ΔgadD1 and EGDm ΔgadD2 (Fig. 2a ) double knockout mutant were constructed and tested for survival to exposure in pH 2.5 in BHI. EGDm ΔgadD1D3 counts reduced at a faster rate than EGDm and was about 1-log cycle lower than the wt after 60 min (Fig. 5a ). The presence of gadD2 on its own did not appear to protect the strain as well as gadD1 alone. Deletion of both gadD2 and gadD3 however followed a pattern of survival similar to the wild-type but reduced compared to the single knockout of gadD1 (Fig. 5a ). Overall it appeared that removing the gadD3 gene from EGDm ΔgadD1 or EGDm ΔgadD2 negatively impacted on the increased acid survival seen with these strains in stationary phase. In exponential phase the ΔgadD1D3 mutant was also found to have increased sensitivity to acid compared to either single deletion alone (Fig. S1a ), further highlighting the importance of gadD3 to this strain.
Figure 5. Acid survival and GABAi production of L. monocytogenes EGDm double GAD system mutants indicates a key role for gadD3.
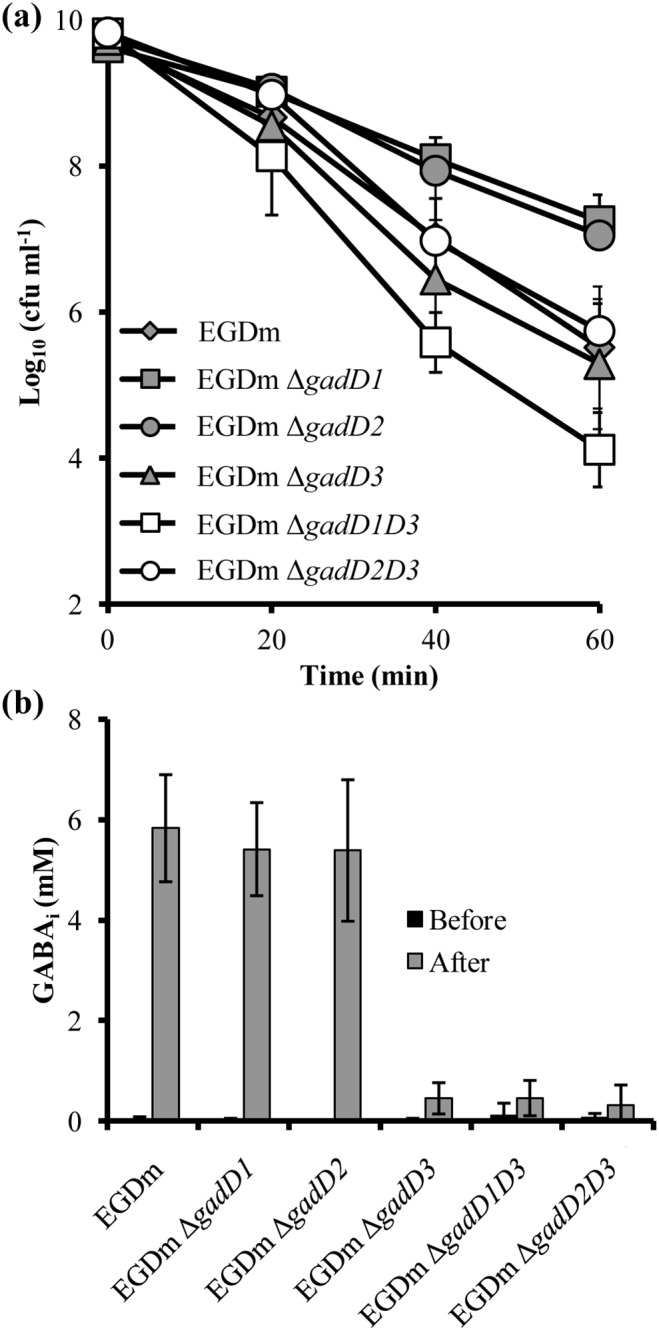
(a) Stationary phase EGDm gadD mutants were acidified to pH 2.5 with 3 M HCl in BHI broth. Cell counts were taken every 20 min. Values are the means of data from three individual cultures, with the cell counts for each culture being the means of counts from three platings. Error bars represent the standard deviation from the mean value for each time-point. (b) Stationary phase EGDm gad mutants were acidified (grey) or not acidified (black) with 3 M HCl to pH 4.0 and GABAi accumulation was quantified. Error bars represent the standard deviation from the mean of three independent biological replicates.
GABAi was measured in stationary phase for each strain after exposure to pH 4.0. The mutants carrying double deletions both failed to produce GABAi in response to exposure to the low pH (Fig. 5b ). Possession of either gadD1 or gadD2 alone did not bestow an ability to produce GABA. As EGDm is unable to utilise the GADe system no GABAe was produced by any of these strains in response to acid treatment (data not shown).
EGDm gadD mutants survive gastric passage in mice
In order to analyse the role that the GADi system plays in vivo, survival through a live animal gastric passage was carried out. Three days post oral inoculation of female Balb/C mice with each of the EGDm gad mutants, the animals were sacrificed and dissemination of the strains was analysed. Counts ranged from 6.50×103 to 3.80×107 cfu ml−1 and were similar in the liver, spleen and intestinal content for all single deletion strains and the respective wild-type. The counts for EGDm ΔgadD1 and EGDm ΔgadD3 however were significantly lower in the mesenteric lymph node (MLN) compared to the wild-type (1-log; Fig. 6). Double deletion of either gadD1 with gadD3 or gadD2 with gadD3 resulted in reduced counts from both the liver and spleens of mice 3 days post infection. These data suggest that the GADi system plays a role within the host during the development of an infection and is essential for full virulence potential following oral infection of mice.
Figure 6. Infection of Balb/C mice with EGDm GAD system mutants.

Plate counts of surviving EGDm GAD system mutants 3 days post infection from female Balb/C mice (n = 5). Isolated from the liver, spleen, mesenteric lymph node (MLN) and faeces. Significant differences (*) between wild-type and mutants were determined using one-way ANOVA.
Growth of L. monocytogenes GAD system mutants in human THP-1 macrophages
In order to cause infection L. monocytogenes must be able to survive inside phagocytic macrophages. The environment that the bacteria encounter inside these cells is reported to be acidic [25] and thus we investigated the role that the GAD system may play in survival. None of the EGDm mutants showed a significant difference in uptake rate by THP-1 cells compared to the wild type after 2 h (Fig. 7a ). Furthermore, there was no significant difference seen in the ability of any strain to grow inside the THP-1 cells over a 7 h time-course. EGDm Δ gadD1 and EGDm ΔgadD2 did however show a significant lag until 5 h but eventually reached similar numbers to both EGDm and the gadD3 mutant (Fig. 7a ). Similarly, 10403S and its isogenic GAD system mutants grew inside THP-1 macrophages with only an apparent lag seen for 10403S ΔgadD2 after 3 h. Otherwise no significant differences were seen between the wild-type and mutants for the duration of the experiment.
Figure 7. Growth of GAD system mutants in THP-1 macrophages.
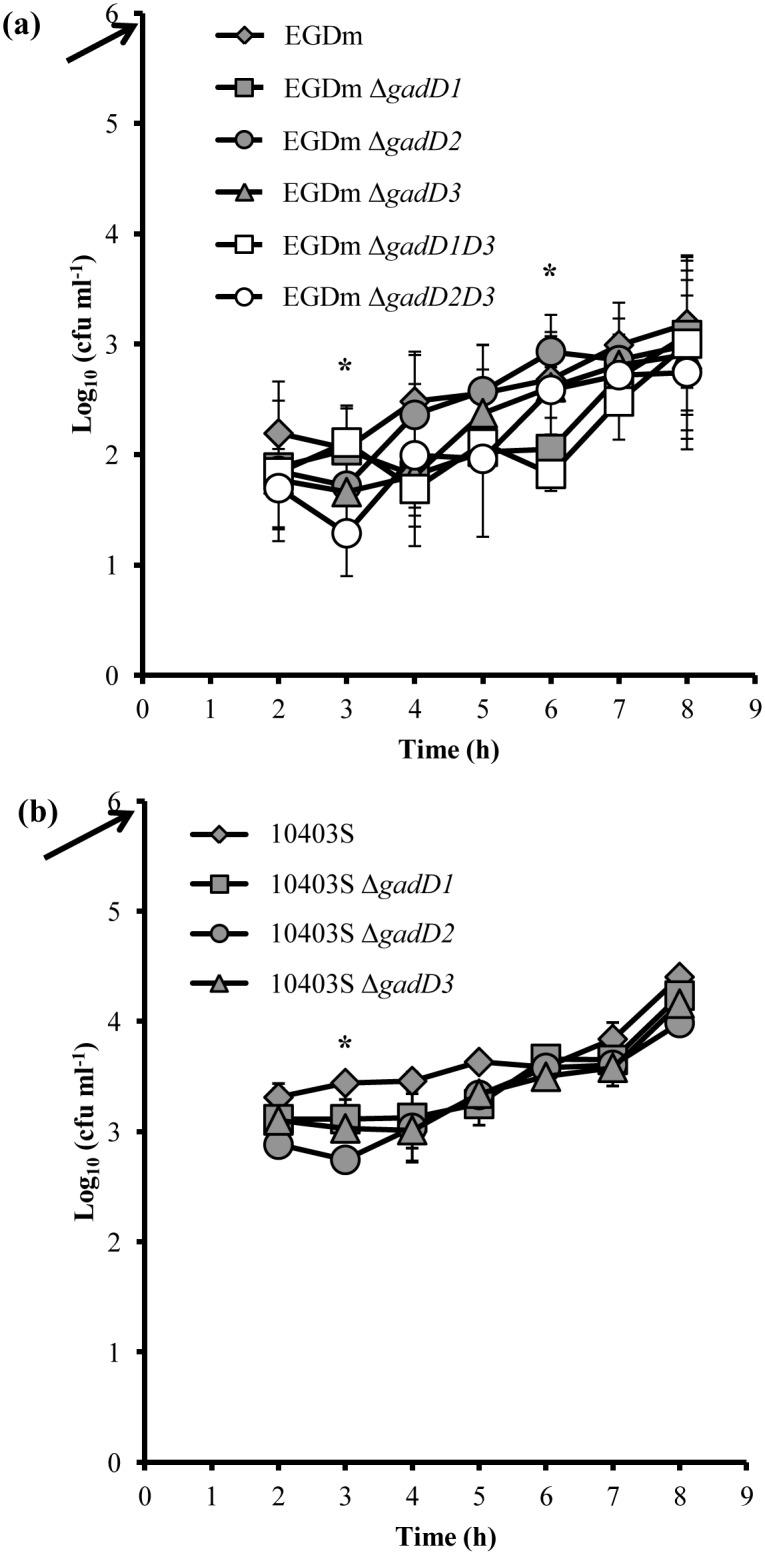
Growth of EGDm (a) and 10403S (b) GAD system mutants inside THP-1 macrophages over 7 h. Counts are recorded 2 h post co-incubation of THP-1 with bacteria at an MOI of 10 (106 bacteria; black arrow). Error bars represent the standard deviation from the mean of at least 4 biological replicates for each strain and time-point. Significant differences (*; p <0.05) were determined using one-way ANOVA.
Discussion
Previous studies with L. monocytogenes have shown that strains can vary greatly in terms of their ability to cope with low pH [12], [14]. Here the GAD system, one of the major mechanisms for acid tolerance was compared between two commonly used reference strains EGDm (murinised EGD-e) and 10403S. The survival of 10403S during exposure to low pH is significantly greater than EGDm (Fig. 2) and this correlates with an ability of 10403S to utilise both GADi and GADe in response to low pH. Despite possessing the genes for a putative glutamate/GABA antiporter, EGDm does not export GABA. This may be due to the fact that the amino acid sequences of GadT2 are different at four positions between 10403S and EGDm (V409I, V419I, M438T, I441M, respectively) unlike GadT1 which varies by only a single amino acid (T196K). However, efforts to clone the gadT2 gene from 10403S into EGDm were unsuccessful in inducing GABAe production in EGDm (data not shown). Further studies will be required to test whether expression of gadT2 together with gadD2 from 10403S (i.e. the entire gadT2D2 operon) can induce GABAe production in an EGD-e background.
As EGDm appears to be solely reliant upon intracellular decarboxylation (GADi), the three genes encoding glutamate decarboxylases (gadD1, gadD2 and gadD3) were deleted. Somewhat surprisingly, deletion of gadD1 or gadD2 improved the ability of the strain to survive low pH during stationary phase (Fig. 2a ), as there was no apparent increase in GABA production, the increased acid resistance may be due to the action of other acid tolerance mechanisms that were not investigated as part of this study. The increase in acid tolerance was comparable to the levels of resistance seen for 10403S, which is one of the most resistant strains of L. monocytogenes [26] (Fig. 2b ). None of the single deletions were seen to reduce the bacterium’s ability to survive at low pH, showing that each is dispensable for wild-type acid tolerance levels in this strain background. However, deletion of both gadD1 and gadD3 together did prevent the increase in survival seen for EGDm ΔgadD1 and this double mutant strain, EGDm ΔgadD1D3, was more acid sensitive than EGDm (Fig. 5a & Fig. S1a ). The deletion of gadD3 along with gadD2 also appeared to prevent the increase in stationary phase acid tolerance seen in the ΔgadD2 mutant however this strain was not as sensitive as EGDm ΔgadD1D3 suggesting that gadD1 has a greater role to play in acid tolerance than gadD2 in this strain. Measurements of GABAi across the strains in response to acid, indicated that only EGDm ΔgadD3 was impaired in an ability to produce GABAi during stationary phase. Taken together with the acid survival data, it would appear that this decarboxylase plays a more important role in the GAD system of this strain compared with the remaining two. Interestingly, a different effect was observed in 104033S after the deletion of the gadD genes. Here deletion of gadD2 negatively impacted on survival during both stationary phase and mid-exponential phase growth (Fig. 2b & Fig. S1b ). In 10403S the gadD2 deletion was also accompanied by a reduction in both GABAi and GABAe (Fig. 3 & Fig. S2). It would appear that failure to produce GABA in this strain impacted negatively on its ability to survive at low pH. As gadD2 gadT2 are part of the same operon it is not surprising that both GABAi and GABAe production was affected. The remaining GadT1D1 system apparently could not compensate for the loss of GadT2D2 activity in this strain.
An examination of the transcriptional response for each of the strains in response to acid confirms the differential importance of either gadD2 or gadD3 for 10403S and EGDm, respectively. The gadD2 transcript was the most abundant of the three in 10403S in stationary phase cultures, whereas gadD3 was dominant in the EGDm background (Fig. 4). Overall, neither strain displayed an alteration in gadD gene expression in response to the pH treatment. This may be due to the fact that the cultures have already reached stationary phase and therefore were expressing each gene to a maximal level. Although little is still known about the transcriptional regulation of the GAD system in L. monocytogenes, it is known that both the gadT2D2 operon and gadD3 are at least partially under the control of σB. Using lmo2230 (encoding a putative arsenate reductase) as a reporter of σB activity [2], [27], there was clearly no change in σB activity over the course of the acid treatment or as a result of the deletions in the gadD genes. As shown previously [2], [27] σB is fully active in stationary phase and additional stress doesn’t enhance its activity beyond that level. This might indicate that the cells already possess functional GAD system proteins prior to acid shock and that regulation of the GAD system occurs post transcriptionally.
While much of the previous work undertaken on the GAD system in L. monocytogenes has focused on in vitro models and synthetic gastric fluid, the role of the system in vivo has not been addressed previously. In fact there is very little evidence in any bacterial pathogen showing that the GAD system is important for survival in the host. Work with Brucella microti has however shown that deletion of the GAD system reduced counts in mice after oral inoculation [28]. In our study we also found a significant role for the GAD system in the virulence of EGDm in a mouse infection model.
Due to the extensive use of the EGD-e strain as a model intracellular pathogen [29]–[31], we focused upon the role of the GADi system in an oral infection model that employs murinised EGDm strain to enhance the progression of invasive disease via the GI tract [20]. A previous study identifying genes expressed by L. monocytogenes in response to Lactobacilli within the mouse gut showed that all three gene systems (gadD1T1, gadD2T2 and gadD3) had significantly increased expression in this environment [32]. This suggests that the GAD system is actively induced within the host GI tract. In our functional study single gadD deletion mutants were not affected in their ability to infect the liver and spleen of mice following oral infection. However the two double mutants displayed significantly reduced levels of infection, indicating that a complete GAD system is required for full infection. This correlates with in vitro survival assays with these mutants, particularly EGDm ΔgadD1D3, which displays a greater sensitivity to low pH than the single mutants and exhibits a significant virulence defect in our model system. It is important to note that the pH in the stomach of Balb/C mice can be as high as pH 4.04 [33] and the gastric acidity of this particular animal host may have implications for the survival of these gad mutant strains. However, the GAD system is likely to play a more important role in hosts that possess stomachs with higher acidity. It will also be important to investigate the role of the GADe system in virulence by using a L. monocytogenes strain capable of producing GABAe.
Subsequently, we investigated another stage in the virulence process of L. monocytogenes; the intracellular cycle. In both EGDm and 10403S, the GAD system does not appear to play a major role in survival within human derived macrophages. This was largely expected since the pH inside bone marrow derived macrophages after compartmentalisation of L. monocytogenes is about pH 5.5 [25] while induction of the GAD system in L. monocytogenes normally occurs below pH 4.5 [17]. Interestingly, although GadD1T1 is known to play a role in growth at mild acidic conditions [16] similar to those occurring within macrophages no major effect of these proteins was documented within macrophages.
Overall, the GAD system in L. monocytogenes appears to show a clear divergence in functionality between these two well-studied strains. The strains appear to have adopted the use of either a GADi system or a combined GADi/GADe system, which likely reflects their unique evolutionary histories. The strain which possessed both GADi and GADe, 10403S, displayed a more acid resistant phenotype suggesting that a functional antiport is highly beneficial. In contrast, the strain which utilised only GADi did not appear to have a sole reliance on any of its three isoforms of decarboxylases for acid tolerance, indicating a more robust system. From comparing these two strains it is clear that functional divergence of physiologically important pathways can readily occur and further highlights the importance of inter-strain comparisons in addressing the biological significance of any pathway.
Supporting Information
Acid survival of L. monocytogenes gad mutants. Mid-log phase EGDm (a) and 10403S (b) Δgad mutants were challenged at pH 3.0. Cell counts were taken every 20 min. Values are the means of data from three individual cultures, with the cell counts for each culture being the means of counts from three platings. Error bars represent the standard error from the mean value of three individual biological repeats.
(TIF)
GABA production from L. monocytogenes gad mutants. (a) Production of GABAe by EGDm and 10403S gad mutants with (grey) or without (black) 1 h exposure to acid at pH 4.0 (EGDm) or pH 3.5 (10403S). (B) Production of GABAi by EGDm and 10403S gadD mutants with (grey) or without (black) 1 h exposure to acid at pH 4.0 (EGDm) or pH 3.5 (10403S). Dashed horizontal lines indicate the detection limits for GABA in each experiment. Error bars represent the standard deviation from the mean of three individual biological repeats for each sample.
(TIF)
Acknowledgments
We are grateful to the Bacterial Stress Response Group and colleagues in Microbiology for helpful discussions. THP-1 macrophages were kindly gifted by, Senthilkumar Alagesan and Serika Naicker from the laboratory of Prof. Matthew Griffin (NUIG).
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
CF, CPOB and KAGK were supported by a Science Foundation Ireland Starting Investigator Research Grant (SIRG; 09/SIRG/B1570), a Marie Curie European Reintegration Grant (ERG 265154) and by the National University of Ireland Galway. PGC, CH and CGMG are supported by Science Foundation Ireland in the form of a centre grant (Alimentary Pharmabiotic Centre; Grant Numbers SFI/12/RC/2273 and 12/RC/2273). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Abram F, Starr E, Karatzas KAG, Matlawska-Wasowska K, Boyd A, et al. (2008) Identification of components of the sigma B regulon in Listeria monocytogenes that contribute to acid and salt tolerance. Appl Environ Microbiol 74: 6848–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Utratna M, Shaw I, Starr E, O’Byrne CP (2011) Rapid, transient, and proportional activation of σB in response to osmotic stress in Listeria monocytogenes. . Appl Environ Microbiol 77: 7841–7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cole MB, Jones M V, Holyoak C (1990) The effect of pH, salt concentration and temperature on the survival and growth of Listeria monocytogenes . J Appl Microbiol 69: 63–72. [DOI] [PubMed] [Google Scholar]
- 4. Karatzas KAG, Wouters JA, Gahan CGM, Hill C, Abee T, et al. (2003) The CtsR regulator of Listeria monocytogenes contains a variant glycine repeat region that affects piezotolerance, stress resistance, motility and virulence. Mol Microbiol 49: 1227–1238. [DOI] [PubMed] [Google Scholar]
- 5. Wemekamp-Kamphuis HH, Wouters JA, Leeuw PPLA De, Hain T, Chakraborty T, et al. (2004) Identification of Sigma Factor B -Controlled Genes and Their Impact on Acid Stress, High Hydrostatic Pressure, and Freeze Survival in Listeria monocytogenes EGD-e. Appl Environ Microbiol 70: 3457–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walker SJ, Archer P, Banks JG (1990) Growth of Listeria monocytogenes at refrigeration temperatures. J Appl Bacteriol 68: 157–162. [DOI] [PubMed] [Google Scholar]
- 7. Young KM FP (1993) Acetic, lactic and citric acids and pH inhibition of Listeria monocytogenes Scott A and the effect on intracellular pH. J Appl Bacteriol 74: 515–520. [PubMed] [Google Scholar]
- 8. Heavin SB, Brennan OM, Morrissey JP, O’Byrne CP (2009) Inhibition of Listeria monocytogenes by acetate, benzoate and sorbate: weak acid tolerance is not influenced by the glutamate decarboxylase system. Lett Appl Microbiol 49: 179–185. [DOI] [PubMed] [Google Scholar]
- 9. Ryan S, Begley M, Gahan CGM, Hill C (2009) Molecular characterization of the arginine deiminase system in Listeria monocytogenes: Regulation and role in acid tolerance. Environ Microbiol 11: 432–445. [DOI] [PubMed] [Google Scholar]
- 10. Cotter PD, Gahan CGM, Hill C (2000) Analysis of the role of the Listeria monocytogenes F0F1 -ATPase operon in the acid tolerance response. Int J Food Microbiol 60: 137–146. [DOI] [PubMed] [Google Scholar]
- 11. Davis MJ, Coote PJ, O’Byrne CP (1996) Acid tolerance in Listeria monocytogenes: the adaptive acid tolerance response (ATR) and growth-phase-dependent acid resistance. Microbiology 142: 2975–2982. [DOI] [PubMed] [Google Scholar]
- 12. Cotter PD, Gahan CGM, Hill C (2001) A glutamate decarboxylase system protects Listeria monocytogenes in gastric fluid. Mol Microbiol 40: 465–475. [DOI] [PubMed] [Google Scholar]
- 13. Tsai M, Mccarthy P, Miller C (2013) Substrate selectivity in glutamate-dependent acid resistance in enteric bacteria. Proc Natl Acad Sci 110: 5898–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karatzas KAG, Suur L, O’Byrne CP (2012) Characterization of the Intracellular Glutamate Decarboxylase System: Analysis of Its Function, Transcription, and Role in the Acid Resistance of Various Strains of Listeria monocytogenes. . Appl Environ Microbiol 78: 3571–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, et al. (2001) Comparative genomics of Listeria species. Science (80- ) 294: 849–852. [DOI] [PubMed] [Google Scholar]
- 16. Cotter PD, Ryan S, Gahan CGM, Hill C (2005) Presence of GadD1 glutamate decarboxylase in selected Listeria monocytogenes strains is associated with an ability to grow at low pH. Appl Environ Microbiol 71: 2832–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karatzas KAG, Brennan O, Heavin S, Morrissey J, O’Byrne CP (2010) Intracellular Accumulation of High Levels of γ-Aminobutyrate by Listeria monocytogenes 10403S in Response to Low pH: Uncoupling of γ-Aminobutyrate Synthesis from Efflux in a Chemically Defined Medium. Appl Environ Microbiol 76: 3529–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, et al. (1993) Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Methods Enzymol 217: 270–279. [DOI] [PubMed] [Google Scholar]
- 19. O’Byrne CP, Feehily C, Ham R, Karatzas KAG (2011) A modified rapid enzymatic microtiter plate assay for the quantification of intracellular γ-aminobutyric acid and succinate semialdehyde in bacterial cells. J Microbiol Methods 84: 137–139. [DOI] [PubMed] [Google Scholar]
- 20. Monk IR, Casey PG, Hill C, Gahan CGM (2010) Directed evolution and targeted mutagenesis to murinize Listeria monocytogenes internalin A for enhanced infectivity in the murine oral infection model. BMC Microbiol 10: 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, et al. (1980) Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer 26: 171–176. [DOI] [PubMed] [Google Scholar]
- 22. Monk IR, Gahan CGM, Hill C (2008) Tools for functional postgenomic analysis of Listeria monocytogenes . Appl Environ Microbiol 74: 3921–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hersh BM, Farooq FT, Barstad DN, Blankenhorn DL, Slonczewski JL (1996) A glutamate-dependent acid resistance gene in Escherichia coli . J Bacteriol 178: 3978–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bécavin C, Bouchier C, Lechat P, Archambaud C, Creno S, et al. (2014) Comparison of Widely Used Listeria monocytogenes Strains EGD, 10403S, and EGD-e Highlights Genomic Differences Underlying Variations in Pathogenicity. MBio 5. [DOI] [PMC free article] [PubMed]
- 25.De Chastellier C, Berche P (1994) Fate of Listeria monocytogenes in murine macrophages: evidence for simultaneous killing and survival of intracellular bacteria. Infect Immun 62. [DOI] [PMC free article] [PubMed]
- 26. Feehily C, O’Byrne CP, Karatzas KAG (2013) Functional γ-Aminobutyrate Shunt in Listeria monocytogenes: role in acid tolerance and succinate biosynthesis. Appl Environ Microbiol 79: 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Utratna M, Cosgrave E, Baustian C, Ceredig R, O’Byrne CP (2014) Effects of Growth Phase and Temperature on σB Activity within a Listeria monocytogenes Population: Evidence for RsbV-Independent Activation of σB at Refrigeration Temperatures. Biomed Res Int. [DOI] [PMC free article] [PubMed]
- 28. Occhialini A, Pilar M, Bagüés J De, Saadeh B, Bastianelli D, et al. (2012) The Glutamic Acid Decarboxylase System of the New Species Brucella microti Contributes to Its Acid Resistance and to Oral Infection of Mice. J Infect Dis 206: 1424–1432. [DOI] [PubMed] [Google Scholar]
- 29. Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, et al. (2009) The Listeria transcriptional landscape from saprophytism to virulence. Nature 459: 950–956. [DOI] [PubMed] [Google Scholar]
- 30. Joseph B, Przybilla K, Stuhler C, Schauer K, Slaghuis J, et al. (2006) Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol 188: 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chatterjee SS, Hossain H, Otten S, Kuenne C, Kuchmina K, et al. (2006) Intracellular Gene Expression Profile of Listeria monocytogenes . Infect Immun 74: 1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Archambaud C, Nahori M, Soubigou G, Bécavin C, Laval L, et al. (2012) Impact of lactobacilli on orally acquired listeriosis. Proc Natl Acad Sci 109: 16684–16689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McConnell EL, Basit AW, Murdan S (2008) Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in vivo experiments. J Pharm Pharmacol 60: 63–70. [DOI] [PubMed] [Google Scholar]
- 34. Smith K, Youngman P (1992) Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spollM gene. Biochimie 74: 705–711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Acid survival of L. monocytogenes gad mutants. Mid-log phase EGDm (a) and 10403S (b) Δgad mutants were challenged at pH 3.0. Cell counts were taken every 20 min. Values are the means of data from three individual cultures, with the cell counts for each culture being the means of counts from three platings. Error bars represent the standard error from the mean value of three individual biological repeats.
(TIF)
GABA production from L. monocytogenes gad mutants. (a) Production of GABAe by EGDm and 10403S gad mutants with (grey) or without (black) 1 h exposure to acid at pH 4.0 (EGDm) or pH 3.5 (10403S). (B) Production of GABAi by EGDm and 10403S gadD mutants with (grey) or without (black) 1 h exposure to acid at pH 4.0 (EGDm) or pH 3.5 (10403S). Dashed horizontal lines indicate the detection limits for GABA in each experiment. Error bars represent the standard deviation from the mean of three individual biological repeats for each sample.
(TIF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.