

Abstract
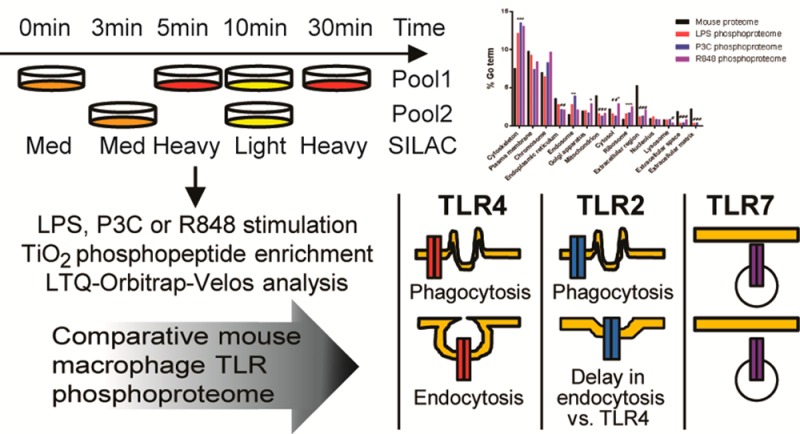
Toll-like receptors (TLRs) are among the first sensors that detect infection and drive immune response. Macrophages encountering a pathogen are usually stimulated not by one TLR, but by a combination of TLRs engaged by distinct microbe ligands. To understand the integrated signaling under complex conditions, we investigated the differences in the phosphoprotein signaling cascades triggered by TLR2, TLR4, and TLR7 ligands using a single responding cell population. We performed a global, quantitative, early poststimulation kinetic analysis of the mouse macrophage phosphoproteome using stable isotope labeling with amino acids coupled to phosphopeptide enrichment and high-resolution mass spectrometry. For each TLR ligand, we found marked elevation of phosphorylation of cytoskeleton components, GTPases of the Rho family, and phospholipase C signaling pathway proteins. Phosphorylation of proteins involved in phagocytosis was only seen in response to TLR2 and TLR4 but not to TLR7 activation. Changes in the phosphorylation of proteins involved in endocytosis were delayed in response to TLR2 as compared to TLR4 ligands. These findings reveal that the phosphoproteomic response to stimulation of distinct TLRs varies both in the major modification targets and the phosphorylation dynamics. These results advance the understanding of how macrophages sense and respond to a diverse set of TLR stimuli.
Keywords: toll-like receptors, phosphoproteomics, macrophage, innate immunity, SILAC, TLR2, TLR4, TLR7
Introduction
Toll-like receptors (TLRs) are part of the innate immune system and play a central and critical role in the elicitation of immune responses to invading pathogens.1 To date, 10 TLRs have been reported in humans and 12 in mice, and each recognizes a specific family of microbial molecules.2,3 Once engaged, TLRs trigger intracellular signaling cascades that orchestrate gene expression programs required for the macrophage to exert its immune function. These gene expression changes of, for example, receptors, signal transducers, and transcription factors, prime the macrophage to mount an immune response.4,5
The cytoplasmic domain of TLRs is highly similar to those of the interleukin-1 (IL-1) receptor family and is therefore referred to as the toll/IL-1 receptor (TIR domain6). In the myeloid differentiation primary response 88 (MyD88) dependent pathway (a component of all TLR pathways except TLR3), the TIR domains of the TLRs interact with the TIR domain of MyD88. MyD88 then recruits IL-1 receptor-associated kinase 4 (IRAK4) through interaction of their respective death domains. IRAK4 is activated by phosphorylation and heterodimerizes with IRAK-1 or -2, which, in turn, allows it to activate the adapter protein TNF-receptor-associated factor 6 (TRAF6). This results in the activation of two distinct signaling pathways, the MAPK and NFκB pathways,5 and, finally, in the activation of JNK and NFκB. In the MyD88 independent pathway, the TIR domain of the TIR-domain-containing adapter-inducing interferon-β (TRIF) interacts with the TIR domain of TLR4 (the only TLR that uses both the MyD88 and TRIF pathways) in response to lipopolysaccharide (LPS), or TLR3, causing the activation of the transcription factor IRF-3 by signaling through IKKε and TBK1. TLR2 signaling has been generally believed to originate from the cell surface (recognizing a variety of bacterial cell wall components),7 but there are reports that it can also signal from the endosome using Tirap (TIR-domain containing adaptor protein)-independent MyD88 signaling pathway distinct from the MyD88 signaling from the plasma membrane, which utilizes Tirap.8,9 TLR7 is not expressed on the cell surface but is located in the late endosomal compartment, from where it exclusively signals in response to its natural ligand, single-stranded DNA.7 TLR4 is the only TLR that has two signaling components: the early MyD88 dependent response signaling component that is initiated from the plasma membrane and the late TRIF dependent response signaling component that is initiated from the early endosomes.7
Protein phosphorylation on serine, threonine, and tyrosine (S/T/Y) is one of the essential regulatory post-translational modifications (PTMs). Because of its highly dynamic nature, S/T/Y phosphorylation serves as a signal transduction mechanism enabling the cells to link extracellular cues to the regulation of many physiological processes, including adaptive and innate immune system activation in response to pathogens, which has been previously studied at the phosphoproteome level.10−13 A comprehensive analysis of the phosphoproteome of cells in which TLR4 was activated by LPS showed that the main phosphorylation events were not restricted to the canonical TLR pathway but that phosphorylation was also present on cytoskeletal proteins and DNA damage-response-associated ATM/ATR kinases.13 One study investigated changes in the kinase profile when macrophages were infected with Staphylococcus aureus,14 while another compared the changes in protein expression with mRNA regulation in response to LPS, with the data indicating crosstalk between multiple pathways.15 Although comparative transcriptome studies of the different TLR pathways have been published,16−18 there are very few phosphoproteomic studies targeted toward any of the other TLRs19 and there have been no studies that compared the global phosphoprotein signatures of different TLRs. As bacterial and other microbial pathogens generally trigger a combination of TLRs,5,20 deciphering the comprehensive signaling pathways of multiple TLRs is a critical step toward characterizing the response of a macrophage to the complex stimuli originating from pathogens during infection.
Progress in mass spectrometry-based proteomics driven by advances in instrument performance21,22 and computational analysis23−25 allows detailed, global, and accurate analysis of proteins and their PTMs. Stable isotope labeling with amino acids in cell culture (SILAC) allows samples to be mixed prior to peptide fractionation and phosphopeptide enrichment and is particularly useful for quantitative comparison of phosphopeptide abundance across a time series or other experimental treatment variation.26−29
In this investigation, SILAC labeling of immortalized macrophages, phosphopeptide enrichment, and high-accuracy mass spectrometry were used to compare phosphoproteome dynamics upon macrophage stimulation with three different TLR ligands (LPS, for TLR4; Pam3C, an analogue of the immunologically active N-terminal portion of bacterial lipoprotein, for TLR2; and an imidazoquinolinamine drug resiquimod, or R848, for TLR7) and to identify differences in the mechanisms of TLR action at five time points. Overall, we identified an average of 500 phosphosites, and about half of these sites showed TLR-responsive changes. Most of the changes were common between the three different TLRs studied, likely reflecting significant overlap in the MyD88 dependent signaling pathways activated. One of the large protein groups responsive to TLR stimuli comprises the proteins involved in cytoskeletal reorganization. Despite many phosphorylation responses being similar between the three TLRs, there were also some key differences that highlight specificity in their mechanisms of action. We detected a significant difference in the dynamics of phosphorylation of the proteins involved in endocytosis by comparing the TLR2 and TLR4 results. We also detected an absence of phosphorylation of proteins involved in phagocytosis in the TLR7 data set. This approach may assist in filling a gap in our understanding of innate immune signaling, and differences in the molecular signatures underlying specific TLR responses to different pathogens might be elucidated.
Experimental Section
Cell Culture
Immortalized macrophages (wild type (WT) and TLR4–/–) derived from C57BL/6 mice (a generous gift from Dr. Eicke Latz)30,31 were grown in Dulbecco’s modified Eagle’s medium (Lonza). For phosphoproteomics, the wild type cells were expanded in SILAC medium for five passages. One day before TLR activation, the nonadherent cells were discarded, and 25 × 106 cells were plated on a 10 cm tissue culture grade plate. Ninety minutes prior to TLR activation, the nonadherent cells were removed and fresh equilibrated SILAC media was added to the cells.
SILAC Medium
Dulbecco’s modified Eagle’s medium with stable glutamine deficient in l-arginine and l-lysine (Cambridge Isotope Laboratories) was supplemented with 10% fetal bovine serum (Atlanta Biologicals) and 20 mM HEPES and then was supplemented with 398 mM l-arginine HCl labeled with 13C6 (Arg6) or 13C615N4 (Arg10) and 798 mM l-lysine 2HCl labeled with 2D4 (Lys4) or 13C615N2 (Lys8) (Cambridge isotope laboratories) or their nonlabeled counterparts (Arg0 and Lys0) (Sigma-Aldrich).
Stimulation and Cell Lysis
The macrophages were allowed to incorporate the labeled amino acid to create the three different labeled media: light (Arg0, Lys0), medium (Arg6, Lys4), and heavy (Arg10, Lys8) media. This was achieved through five passages and confirming label incorporation by analyzing an aliquot of each condition using mass spectrometry (over 95% incorporation was achieved). The cells were stimulated for 3, 5, 10, and 30 min with 1 μg/mL LPS (from Salmonella minnesota R595, Enzo Life Sciences), 1 μM Resquimod (R848, Enzo Life Sciences), 1 μM Pam3Cys (P3C, InvivoGen) or were left untreated according to the following scheme (Figure 1A). For each ligand, macrophages in two 10 cm dishes were stimulated for 10 min in light (unlabeled) media (Arg0, Lys0) to serve as a reference point. Macrophages in medium-labeled media (Arg6, Lys4) in one 10 cm dish were left untreated and in one 10 cm dish stimulated for 3 min. Macrophages in heavy-labeled media (Arg10, Lys8) in one 10 cm dish were stimulated for 5 min and in another 10 cm dish for 30 min. After stimulation, the cells were briefly washed twice with ice cold PBS and lysed with ice-cold 8 M urea containing protease and phosphatase inhibitors (Roche). After 20 min on ice, the lysed cells were centrifuged for 20 min at 4 °C at 14000g, and the supernatants and pellets were frozen at −80 °C. Two biological replicates were prepared and analyzed independently.
Figure 1.
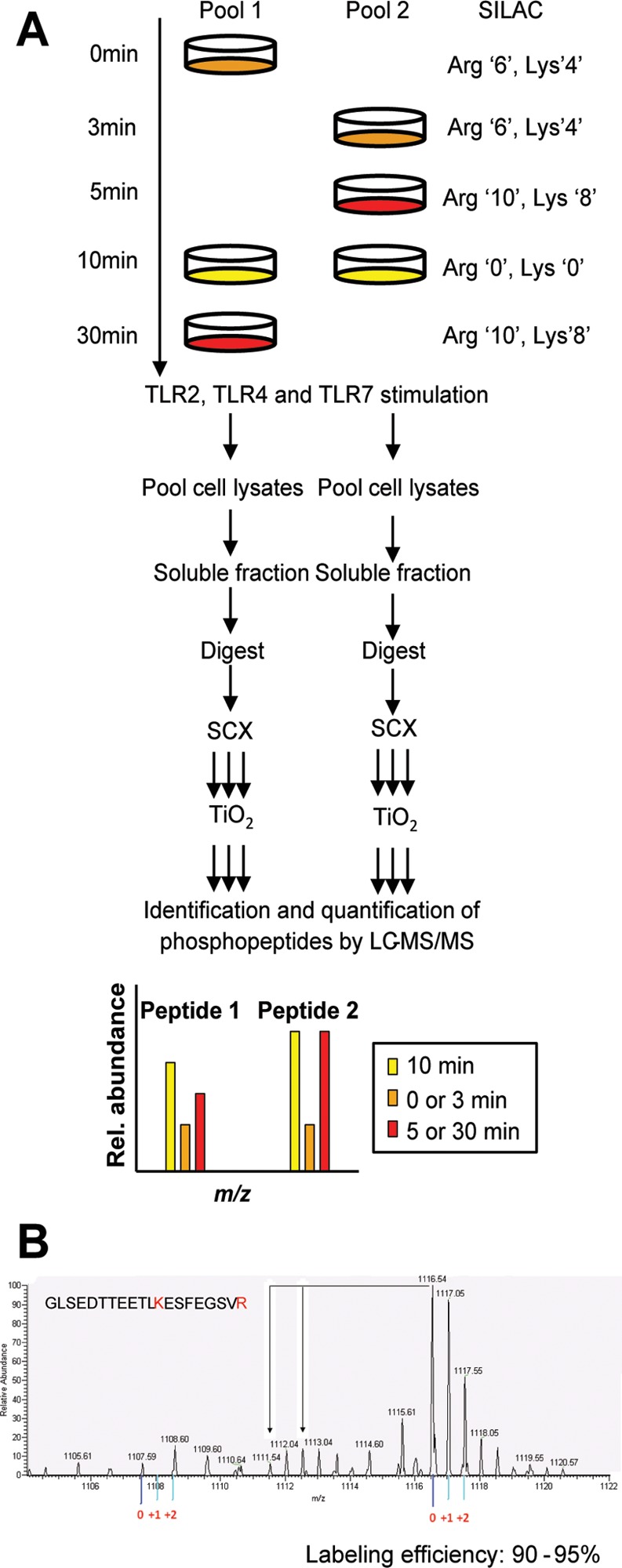
Experimental system and design. (A) Strategy for global and quantitative analyses of LPS-, P3C-, and R848-induced phosphorylation. C57 derived macrophages were SILAC labeled with normal or stable isotope-substituted arginine and lysine amino acids resulting in three states distinguishable by their mass. Each cell population was left untreated or stimulated for 3, 5, 10, or 30 min. The 10 min stimulation time point was included in both pools to serve as a common reference point. Cell lysates to be directly compared were pooled, enzymatically digested, and fractionated by SCX. The phosphopeptides were enriched by TiO2 and analyzed by online LC–MS(/MS). The mass shift introduced by the SILAC amino acids resulted in triplet peaks (i.e., the same peptide from three different time points) with the relative intensities equal to the relative abundance of the peptide. This SILAC approach allows for high-accuracy quantification of phosphopeptides with in most cases localization of the phosphate group with single amino acid accuracy. Two biological replicates were used to perform independent experiments for each ligand stimulation. (B) Labeling efficiency example of a peptide containing a lysine and arginine residue. The arrows indicate the position of partially labeled peptides.
Western Blotting
Activation of the cells was examined using SDS-PAGE and Western blotting. The concentration of the proteins in the supernatant was measured using a BCA assay (Pierce), and 10 μg of total protein for each time point was run on a 10% SDS polyacrylamide gel and then transferred to a nitrocellulose membrane. The membrane was probed for phospho-P38 (Thr180/Tyr182, Cell Signaling), phospho-SAPK/JNK (Thr183/Tyr185, Cell Signaling), phospho-NF-κB P65 (Ser536, Cell Signaling), and phospho-NF-κB P105 (Ser933, Cell Signaling) and RhoGDI (Cell Signaling) as a loading control. These experiments were performed in triplicate.
Strong Cation Exchange (SCX) Chromatography
The supernatant from each time point was aliquoted into samples of 1.3 mg and aliquots from 0, 10, and 30 min were combined and aliquots from 3, 5, and 10 min were combined. The 10 min overlapping time point was used as a reference to enable comparisons across all of the time points between two analyses. The combined samples were reduced with 2 mM DTT at 56 °C for 1 h. The samples were allowed to cool down before being S-carbamidomethylated with 4 mM iodoacetamide for 1 h in the dark. The carbamidomethylated samples were diluted to 2 M urea with 100 mM ammonium acetate pH 8.9, and then trypsin (Promega) was added to a ratio of 1:100 (w/w). The digestion was left to proceed overnight at 37 °C after which it was quenched by adding glacial acetic acid to reduce the pH to below 2. The samples were desalted using an Oasis HLB column (Waters). The column was conditioned with 1 mL of acetonitrile, followed by 0.1% acetic acid. The samples were then loaded on the column and washed with 3 mL of 0.1% acetic acid. The peptides were eluted with 1 mL of 75% acetonitrile in 0.1% acetonitrile and dried in vacuo.
The dried peptides were dissolved in 500 μL of solvent A (5 mM KH2PO4, pH 2.7, 25% acetonitrile) and injected onto a PolySULFOETHYL A SCX column (4.6 mm i.d. × 20 cm length, 5 μm particle size, 200 Å pore size (PolyLC)). SCX chromatography was carried out on an AKTAdesign (GE Healthcare) system at 0.3 mL/min flow rate using the following gradient: 0% B for 2 mL, 0–14% B for 33 mL, 14–100% B for 1 mL, 100% B held for 4 mL (solvent A, 5 mM KH2PO4 pH 2.7, 25% acetonitrile; solvent B, 5 mM KH2PO4 pH 2.7, 500 mM KCl, 25% acetonitrile). UV absorbance was monitored at 214 nm and 2 mL fractions were collected and desalted using Oasis HLB columns prior to phosphopeptide enrichment.
Phosphopeptide Enrichment
Dried SCX fractions were separately resuspended in 100 μL of binding buffer and added to a TiO2 spin tip (Pierce TiO2 phosphopeptide enrichment kit). The spin tip was washed, and then the peptides were eluted first with ammonium hydroxide and then pyrrolidine in a final volume of 200 μL (all according to the manufacturer’s protocol). The eluates were acidified by adding of 100 μL of 2.5% TFA, and then the samples were desalted using the Pierce graphite spin column according to the manufacturer’s protocol. The desalted peptides were finally dried in a SpeedVac.
Mass Spectrometry
Phosphopeptide samples were analyzed by online nanoflow LC–MS/MS. All LC–MS/MS analyses were performed using an Eksigent nano-LC system (ABI Sciex) with a 120 min gradient from 97% A to 60% A (A: 0.1% formic acid, B: 0.1% formic acid in 100% acetonitrile, 200 nL/min flow rate) directly coupled to an Orbitrap Velos mass spectrometer (ThermoFisher Scientific). Reversed phase chromatography was performed using manually prepared packed-tip columns (15 cm column length, 50 μm column inner diameter, Magic C18AQ chromatography media (5 μm diameter, 200 Å pores, Michrom Bioresources)). A top 10 instrument method was used to perform data-dependent acquisition to automatically cycle between Orbitrap full scan MS and LTQ MS/MS. The resulting .RAW files were analyzed using MaxQuant.23 The database searching component of this analysis used the IPI FASTA file of mouse protein sequences (version 3.68, December 18, 2009, 56729 entries, number of MaxQuant contaminants as per Max Planck Institute: 247 entries), and the resulting data were filtered to produce a <1% false discovery rate (FDR). Phosphorylation sites within a peptide sequence were identified using the PTM score algorithm in MaxQuant (FDR < 1%). Phosphopeptide ratios were calculated for each time point in relationship to the signal from unstimulated cells and were normalized so that the median of the all of the ratios of the identified phosphopeptides was equal to 1, to correct for unequal sample mixing. The significance of the quantitation changes was determined in relation to the unstimulated sample: at the basal state, 95% of phosphorylation sites did not change their phosphorylation levels beyond 0.7- and 1.3-fold during the whole time course of measurements (Supplementary Figure 1, Supporting Information). The data were visualized in a heat map using MultiExperiment viewer (MeV: www.tm4.org) with no clustering at this stage. The original mass spectrometry data RAW files have been deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository32 with the data set identifier PXD000761.
Clustering of Phosphorylation Sites
Time series clustering of the different phosphorylation sites for each of the different stimulation conditions was performed using the fuzzy c-means algorithm implemented in the open source R-package GProx.33 Using the average fold change as input, the data were standardized using the standard software function, and a fixed upper threshold of 1.3 and lower threshold of 0.77 were used. A fuzzification parameter of 2 was used with 100 iterations and 5 centers. Changes in phosphorylation were classified as transient if they returned to the baseline within the time series.
GO Analysis
Gene ontology and pathway enrichment analyses were performed using the DAVID Bioinformatics Resources.34,35 The GO terms for cellular components associated with each phosphoprotein identified in our data set were determined using AMIGO (http://amigo.geneontology.org). The GO annotation file for mouse was downloaded from http://www.geneontology.org/GO. Only GO terms with at least three identified phosphoproteins for any treatment were analyzed. GO terms with an odds ratio of ≥1.3 or ≤0.77 and a corrected p-value ≤0.05 were considered significant.
Signaling Pathways
Phosphoproteins were assigned to signaling pathways using Ingenuity Pathway Analysis (IPA, Ingenuity systems; Redwood City, CA) software. The IPA program uses a knowledge database derived from the literature to relate proteins based on function or interaction. Only proteins with phosphosites that were identified in both biological replicates were used for the IPA analysis. The SILAC ratio for each identified phosphosite was converted to fold changes and was uploaded into the IPA software. Ingenuity then created overlapping networks between the candidate proteins for each time point. Associated networks were generated along with a score representing the probability that any such network was generated at random. The top canonical pathways associated with the uploaded phosphoproteins at the different time points were provided by the software along with the p-values (calculated using a right tailed Fisher’s exact test).
Transwell Migration Assay
The cell migration assay was performed in a 24-well plate fitted with a transwell membrane with an 8 μm pore size (Corning, Corning, NY). The bottom chambers were seeded with 105 WT or TLR4–/– immortalized macrophages, and the top chamber was seeded with 104 WT or TLR4–/– immortalized macrophages. The cells were allowed to attach and then treated with 100 ng/mL LPS overnight at 37 °C in media containing 0.01% Hoechst to permit subsequent automated cell counting. The experiments were performed in triplicate. Following incubation, the cells remaining on the top of the membrane were removed, while the cells on the bottom of the membrane were imaged using a Leica DMI6000 SD epifluorescent microscope equipped with an A4 filter cube and a DFC360FX monochrome camera. The membrane in its entirety was captured by taking individual images with a 5× objective. The images were then stitched together using the Leica LAS AF software. The nuclei were counted using the “Spots” feature of the Imaris software (Bitplane). Image collection and data analysis were performed equivalently between all of the samples.
Phagocytosis Assay
Wild type C57/Bl6-derived macrophages were seeded at a density of 50 000 cells/well (100 μL) in 96-well plate and allowed to adhere overnight. Fc-receptor-mediated phagocytosis was measured using the CytoSelect 96-Well Phagocytosis Assay (Cell Biolabs, Inc.). IgG-opsonized erythrocyte suspensions were prepared by mixing and incubating opsonization solution with the sheep erythrocyte suspension at a 1:500 dilution at 37 °C for 30 min. Upon adherence, the cells were either left untreated or were treated with LPS (100 ng/mL), Pam3Cys (1 μM), or R848 (1 μM) for 15 or 30 min, and then the cells were incubated in the presence of 10 μL of opsonized or normal sheep erythrocytes. The culture medium was removed by gentle aspiration, the wells were washed, and the cells were lysed according to the manufacturer’s protocol. The cell lysates were incubated with the substrate solution, and the absorbance was measured at 620 nm in a 96-well plate reader.
Results
Quantitative Phosphoproteome Analysis of TLR Responses in Macrophages
To examine signaling pathway activation by different TLRs, we performed a global and quantitative phosphoproteomic analysis of stimulated C57Bl/6-derived macrophages using established strategies: SILAC quantification, strong cation exchange chromatography (SCX) fractionation, titanium dioxide (TiO2) phosphopeptide enrichment, and high accuracy mass spectrometry.13,29 The macrophage proteins were SILAC labeled with three distinct isotopic forms of both arginine and lysine (Figure 1A). The cells were passaged five times in the SILAC media resulting in a high labeling efficiency (Figure 1B and Supplementary Figure 2, Supporting Information). Pooling the samples from the three different labeling conditions facilitated equal sample treatment and better quantification (Figure 1A). Comparison of more than three conditions was achieved by including a common reference lysate in two pools that allowed for the calculation of phosphopeptide ratios. Here we analyzed the phosphoproteome changes in macrophages in response to LPS (TLR4 ligand), P3C (TLR2/TLR1 ligand), and R848 (TLR7 ligand). Pools of lysates were prepared from unstimulated macrophages and macrophages stimulated for 3, 5, 10, and 30 min (Figure 1A). We chose these time points in order to catch phosphorylation events in the signaling pathway. For example, endocytosis of TLR4 is thought to occur within 15 min of stimulation, and the signaling events leading to it would be lost if we only looked at time points after 15 min.36−38 The concentrations of ligands were chosen so that the response was saturated at 30 min (Supplementary Figure 3, Supporting Information). Effective stimulation of the macrophages was confirmed by measuring the phosphorylation of p38 MAPK, NF-κB p65, NF-APK, NF-κB p105, and JNK for the five time points and the three ligands Supplementary Figures 2 and 3).
Overview of the Phosphoproteome Changes
We investigated the time-dependent changes in phosphorylation state across the phosphoproteome following TLR activation by the three different ligands. All ligands strongly affected protein phosphorylation levels at each investigated time point (Supplementary Figure 4, Supporting Information). For over half of the identified phosphorylated sites, there were dynamic changes in phosphorylation status over the examined time course. The data will be presented separately for each ligand stimulation, and then the similarities and differences between the three ligand stimulations as well as their possible biological significance will be discussed.
Differential Phosphorylation upon Activation of Different TLRs
LPS Stimulation
Following LPS stimulation, we identified 1416 phosphorylation sites on 798 proteins, of which 570 phosphopeptides (398 proteins) were reproducible between the two biological replicates (Figure 2A). Single amino acid phosphosite accuracy was achieved using the PTM score29 and requiring 1% FDR. Most phosphorylation sites were on serine (94%) and threonine residues (4–6%), tyrosine phosphorylation accounting for less than 1% of the sites (Figure 2B). Phosphoproteins from every cellular compartment were detected, but there was an over-representation of phosphoproteins in the cytoskeleton and the endosome compared to the complete mouse proteome and an under-representation of phosphoproteins associated with the mitochondrion, extracellular space, extracellular matrix, and extracellular region (compartments as defined by IPA software) (Figure 2C).
Figure 2.

Global comparison of the phosphorylation events that resulted from different ligand stimulation. (A) Overlap of phosphorylation sites from the two independent experiments for each ligand stimulation. Depicted are the phosphorylation sites that were quantified relative to unstimulated macrophages. The downstream bioinformatics analyses used only the reproducibly identified phosphorylation sites. (B) Distribution of phosphorylated amino acids. The total number of quantified phospho-serine (white), phospho-threonine (gray), and phospho-tyrosine (black) sites for each ligand stimulation is indicated. (C) Distribution of the phosphorylated proteins within cellular compartments. The number of proteins within each cellular compartment (black) was compared to the number of phosphoproteins obtained from each ligand stimulation (red: LPS, blue: P3C, purple: R848). Significantly over-represented GO terms are marked with asterisks, and under-represented GO terms are marked with a hash. (D) The absence of TLR4 expression affects cellular migration. C57 derived macrophages or TLR4 knock out stable transfectants in a C57 derived macrophages background were assayed in the presence or absence of 100 ng/mL LPS. Four different cell combinations were used; C57 cells were seeded in the microchamber in the presence of C57 (C57/C57), or C57 TLR4–/– cells (C57/TLR4–/–) in the well, or C57 TLR4–/– cells were seeded in the microchamber in the presence of C57 (TLR4–/–/C57) or C57 TLR4–/– (TLR4–/–/TLR4–/–) cells in the well. Cell migration was allowed to proceed for 12 h at 37 °C, and cells that transmigrated across the membrane were counted; *p < 0.005 when compared to the wild type C57 derived macrophages. (E) Delay in phosphorylation of phosphoproteins involved in the MyD88 dependent pathway (ERK1, AP-1), which responded earlier to the LPS stimuli compared to the MyD88 independent pathway (TAK1, IRF3), which is activated after TLR4 internalization. NFKBIB is an inhibitor of the MyD88 dependent pathway, and its phosphorylation event coincided with the start of the MyD88 independent pathway. The dotted lines represent significance thresholds (log fold change = ± 0.114). Experiments were performed in duplicate.
To identify pathways and cellular processes that were specifically targeted by the phosphorylation events induced by LPS, we used the Ingenuity software to identify enriched pathways and DAVID (http://david.abcc.ncifcrf.gov) analyses for molecular function and cellular processes analyses. Significant overrepresentation of phosphopeptides with sites that had altered phosphorylation after LPS stimulation was found for ERK/MAPK, Rho family GTPases, and FAK signaling pathway components. These pathways are all known to be activated downstream of the TLRs.39,40 We also found enrichment of modified sites in peptides derived from components of the actin cytoskeleton, integrin-linked kinase (ILK), phospholipase C (PLC), and Fcγ mediated receptor phagocytosis signaling pathways, and of proteins involved in the production of nitric oxide and reactive oxygen species in macrophages and monocytes (Table 1). PLC is associated with the LPS-induced endocytosis of TLR4,41 integrins negatively regulate the TLR response,42,43 and the actin cytoskeleton is involved in the LPS response as the macrophages become motile.44 The latter pathway was also identified as modified in a previously reported phosphoproteomic screen.13 GO analysis revealed enrichment for the terms “cytoskeletal protein binding”, “cytoskeleton organization”, “actin binding”, and “intracellular signaling cascade”, replicating the results obtained by the pathway analysis (Table 1).
Table 1. Signaling Pathways, Biological Processes, and Molecular Functions That Are Targets of LPS-Regulated Phosphorylationa.
| enrichment (p-value) |
|||||
|---|---|---|---|---|---|
| overall | 3 min | 5 min | l0 min | 30 min | |
| (B) Pathway name | |||||
| actin cytoskeleton signaling | **** | * | **** | **** | ** |
| Rho family of GTPases signaling | **** | * | **** | **** | *** |
| FAK signaling | **** | NS | *** | **** | *** |
| Fey receptor mediated phagocytosis | **** | * | **** | **** | *** |
| ERK/MAPK signaling | **** | *** | **** | *** | ** |
| ILK signaling | **** | NS | ** | **** | ** |
| production of NO and ROS | *** | ** | **** | **** | **** |
| phospholipase C signaling | *** | * | *** | *** | ** |
| (B) Gene ontology term | |||||
| cytoskeleton organization | **** | NA | *** | ** | *** |
| cytoskeletal protein binding | **** | ** | *** | **** | **** |
| endocytosis | **** | NA | * | * | **** |
| actin binding | **** | NA | ** | **** | *** |
| intracellular signaling cascade | *** | * | * | ** | * |
****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.1.
Macrophage cell motility enables tissue infiltration from blood vessels and cell migration for antigens. Cytoskeletal rearrangement is necessary for cell motility and requires multiple activation and deactivation steps, which often involve phosphorylation or dephosphorylation.45,46 To confirm that the observed cytoskeletal regulation phosphorylation events directly resulted from TLR signaling, and were not due to secondary cell–cell signaling via a different receptor–ligand interaction, we used the Boyden chamber cell migration assay. Both wild-type and TLR4–/– macrophages were treated with LPS, and cell migration through the membrane was only induced if the inset was seeded with the WT cells and stimulated with LPS irrespective of the type of cells seeded in the well. Stimulating WT cells in the bottom well did not result in the migration of TLR4–/– cells in the inset, indicating that the observed migratory behavior is a direct consequence of the TLR4 stimulation (Figure 2D).
TLR4 can signal through both MyD88-dependent and -independent pathways. Prior studies demonstrated that detectable signaling is seen in the MyD88-independent pathway approximately 15–30 min after TLR4 stimulation and is following CD14 and TLR4 endocytosis.36,37,47 In this survey, phosphoproteins belonging to both the NFκB and IRF3 activation pathways were identified. Their phosphorylation patterns displayed differential dynamics. Within the first 3 min of stimulation, the phosphorylation level of TAK1 at S439 (this site is phosphorylated by PKA, and controls degradation of IκBα and phosphorylation of p38 MAPK) increased and then returned to the basal level at 5 min. Consequently, the phosphorylation levels of Jun (one of the AP-1 transcription factor components) at the S73 activation site and ERK1 at Y205 increased after 5 min of activation and continued increasing for the entire 30 min of the experiment. These results show that the early MyD88 response occurred between 5 and 10 min poststimulation of TLR4 (Figure 2E). This is in agreement with ERK1 and Jun belonging to the MyD88-dependent pathway and thus having an early response to TLR4 stimulation.
P3C Stimulation
As with the LPS stimulation, we examined pathways and GO terms overrepresented with P3C stimulation. We identified a total of 781 phosphosites out of which 543 were reproducible across both biological replicates and corresponded to 404 phosphoproteins (Figure 2A). We discovered overrepresentation of sites on proteins involved in Fcγ receptor mediated phagocytosis and PLC signaling, as for LPS signaling. We also found enrichment of the Rho GTPase signaling pathway (Table 2). The same GO terms as for LPS stimulation were discovered except for the term “intracellular signaling cascade”. The terms “transcription cofactor activity”, “GTPase activator activity”, and “cell activation” were also enriched (Table 2). As with LPS activation, a cytoskeletal regulation response to P3C stimulation was identified (Figure 2C). Some pathways were overrepresented during the entire time course of our experiments (i.e., the phosphorylation level increase or decrease was sustained within the 30 min time window). One of these for P3C was clathrin mediated endocytosis, with p-value (at 3 min) = 0.05 and p-value (at 5 min) = 0.45. Endocytosis was also an enriched GO term at 3 min (p-value 0.01). This is notable because it has been shown that TLR2 might not signal from the membrane, but from the endosome, and that the endocytosis mechanism is mediated by clathrin.8,9,48
Table 2. Signaling Pathways, Biological Processes, and Molecular Functions That Are Targets of P3C-Regulated Phosphorylationa.
| enrichment
(p-value) |
|||||
|---|---|---|---|---|---|
| overall | 3 min | 5 min | 10 min | 30 min | |
| (A) Pathway name | |||||
| Fey receptor mediated phagocytosis | *** | ** | NA | ** | **** |
| phospholipase C signaling | *** | *** | **** | ** | ** |
| Rho family of GTPases signaling | ** | NS | **** | NA | *** |
| (B) Gene ontology term | |||||
| cytoskeletal protein binding | **** | ** | *** | **** | He*** |
| GTPase activator activity | **** | NA | * | * | *** |
| endocytosis | **** | * | ** | ** | *** |
| actin binding | *** | * | *** | **** | **** |
| cytoskeleton organization | *** | * | * | ** | *** |
| transcription cofactor activity | ** | ** | * | NA | **** |
| cell activation | * | * | *** | ** | *** |
| cytoskeletal protein binding | **** | ** | *** | **** | **** |
****p < 0.0001. ***p < 0.001. **p < 0.01. *p < 0.1.
R848 Stimulation
After stimulation of the cells with R848, we identified 1,135 phosphosites, out of which 473 (corresponding to 362 phosphoproteins) were reproducibly identified across the two biological replicates (Figure 2A). Again, as with LPS and P3C stimulation, phospholipase C and the Rho family of GTPases signaling (in particular, Cdc42 signaling) were identified as overrepresented pathways. The FAK (also seen with LPS) and paxillin (associated with FAK signaling) were both overrepresented, as was the actin cytoskeletal regulation signaling. The enriched GO terms for R848 stimulation were “cytoskeletal protein binding”, “cytoskeleton organization”, “actin binding”, and “regulation of Ras protein signal transduction” (Table 3). The first three of these were also observed for LPS and P3C activation.
Table 3. Signaling Pathways, Biological Processes, and Molecular Functions That Are Targets of R848-Regulated Phosphorylationa.
| enrichment
(p-value) |
|||||
|---|---|---|---|---|---|
| overall | 3 min | 5 min | 10 min | 30 min | |
| (A) Pathway name | |||||
| Rho GTPase signaling | **** | * | ** | * | *** |
| actin cytoskeleton signaling | **** | ** | ** | *** | * |
| phospholipase C signaling | *** | NA | ** | ** | * |
| FAK signaling | *** | ** | * | *** | ** |
| paxilin signaling | *** | *** | *** | ** | *** |
| Cdc42 signaling | NS | ** | ** | ** | NS |
| (B) Gene ontology term | |||||
| cytoskeleton organization | **** | ** | *** | **** | **** |
| cytoskeletal protein binding | **** | * | ** | **** | **** |
| actin binding | **** | *** | *** | **** | *** |
| regulation of Ras protein signal transduction | ** | * | **** | ** | **** |
****p < 0.0001. **p < 0.001. **p < 0.01. *p < 0.1.
Ligand-Dependent Dynamics of TLR-Induced Phosphoproteomic Changes Associated with Specific Pathways
Pairwise comparisons of each stimulation time point for the different TLRs resulted in very weak correlations (depicted as a black line on each graph and compared to the gray line showing the correlation of 1) at the early time points (3 and 5 min) and much stronger correlations at the later time points (10 and 30 min) (Supplementary Figure 4, Supporting Information). The correlation increase is very large, for example, R2 (at 3 min) = 0.01 and R2 (at 30 min) = 0.79 for LPS vs P3C. This pattern held true for any given ligand pair. Some of the phosphosites displaying significant differences in the phosphorylation levels (for example, MARCKS S163, NECAP2 S181, Canx S582, Pak2 S141, Vasp S235) are marked with arrows and discussed below.
Phosphosites with similar temporal phosphorylation dynamics profiles were grouped using a fuzzy c-means clustering approach. One of the profiles grouped the sites that did not display any significant changes. The sites that displayed temporal phosphorylation profile changes were grouped into five different profiles independently of the ligand used for stimulation (Figure 3A–E). Figure 3A–J depicts each of the profiles along with an example of a prototypical phosphorylation site. The phosphosite changes identified as resulting from stimulation by more than one ligand were generally assigned to different clusters (example in Figure 3K), but there was a number of common phosphosites assigned to the same cluster (example in Figure 3L) (258 phosphosites, and 167 phosphosites respectively; a complete list of the phosphosites and their resulting cluster assignments is provided in Supplementary Table 1, Supporting Information).
Figure 3.
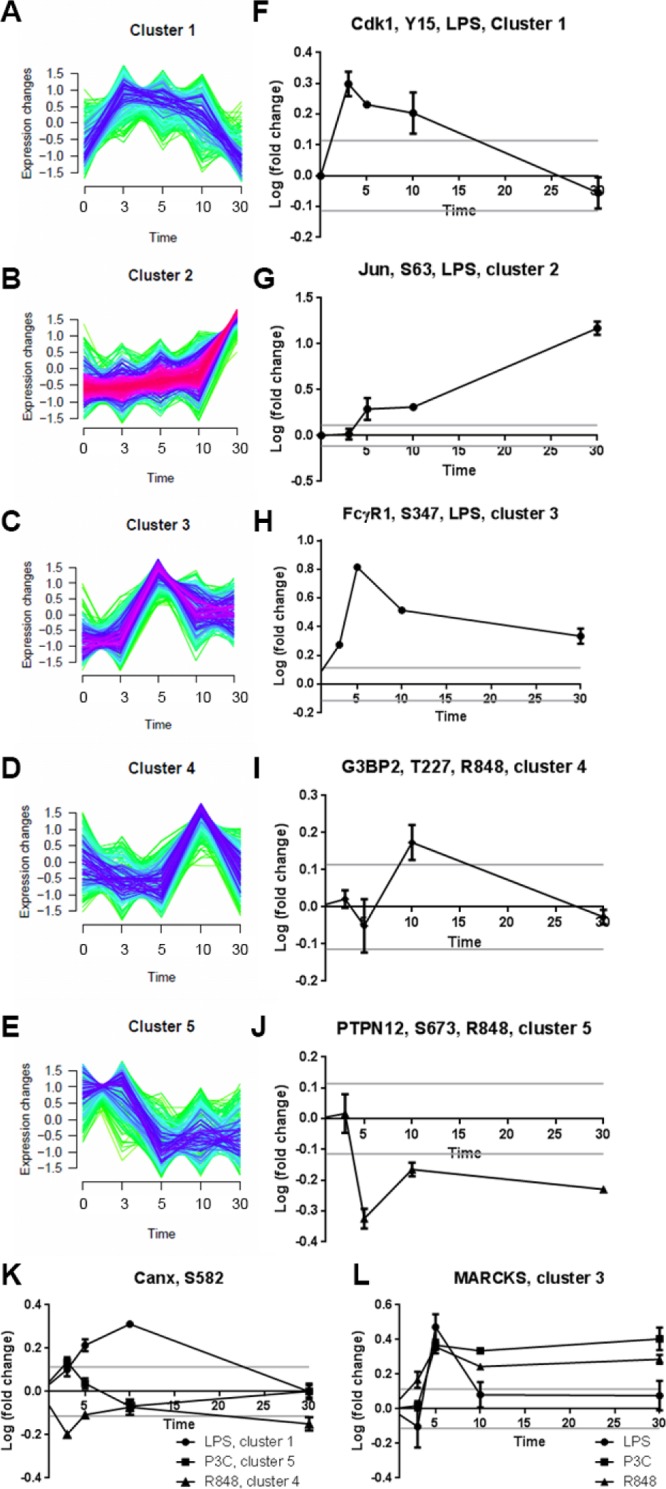
Temporal phosphorylation dynamics profiles. (A–E) Five distinct time-course clusters identified by fuzzy c-means clustering of the time series data for all ligands combined. The five selected phosphopeptides and their respective ligand in F–J are typical of the five clusters found in panels A–E. Some phosphopeptides were found in different clusters after stimulation (an example is depicted in panel K), whereas some phosphopeptides clustered together irrespective of the ligand used for their stimulation (an example is depicted in panel L).
The group of phosphosites that were differentially phosphorylated by the different TLR stimulations is especially valuable as it contains potential leads to the discovery of functional differences in signaling through different TLRs. For example, NECAP2 S181 was regulated by LPS and P3C, but not by R848. PAK2 S141 was regulated more strongly by R848 than by LPS or P3C and so was Vasp S235 (shown in Supplementary Figure 4, Supporting Information). We combined the data for the three receptors, and we identified 145 unique phosphosites corresponding to 123 phosphoproteins. Out of these 145 phosphosites, 42 (on 40 phosphoproteins) were statistically differentially regulated between at least two TLR receptors for one or more time points (log of fold change greater than 0.114 and a p-value < 0.05) (Table 4).
Table 4. Phosphosites Differentially Regulated between the Different TLRs.
| protein | Phosoho site | cellular localization | name |
|---|---|---|---|
| AHNAK2 | S101 | nucleus | AHNAK nucleoprotein 2 |
| Arhgef7 | S228 | focal adhesion | Rho guanine nucleotide exchange factor (GEF) 7 |
| ARRB1 | S412 | Golgi | arrestin, beta 1 |
| ATXN2L | S591 | Penph. mb. protein | ataxin 2-like |
| ATXN2L | S109 | Periph. mb. protein | ataxin 2-like |
| 8CKDK | S31 | mitochondrion | branched chain ketoacld dehydrogenase kinase |
| C130039O16Rik | S456 | nucleus | chromosome 14 open reading frame 43 |
| C77080 | S600 | ER | KIAA1522 |
| Canx | S582 | calnexin | |
| Cdkl8 | S109 | cyclin-dependent kinase 18 | |
| Cdkl8 | S66 | cyclin-dependent kinase 18 | |
| COMT1 | S261 | caffeic acid O-methyltransferase 1 | |
| Corola | T418 | phagocytic cup | coronin, actin binding protein, 1A |
| DIP2B | S99 | nucleus | DIP2 disco-interacting protein 2 homologue B |
| DOCK2 | S1729 | cytoskeleton | dedicator of cytokinesis 2 |
| Histlhlb | S18 | nucleus | histone cluster 1, Hlb |
| IRF2BP2 | S71 | nucleus | interferon regulatory factor 2 binding protein 2 |
| MscU | S15 | mitochondrion | iron–sulfur cluster scaffold homologue |
| KLC4 | S590 | cytoskeleton | kinesin light chain 4 |
| MARCKS | S163 | cytoskeleton | myristoylated alanine-rich protein kinase C substrate |
| Mcm2 | S21 | nucleus | minichromosome maintenance complex component 2 |
| Ncf2 | S324 | cytoplasm | neutrophil cytosolic factor 2 |
| NDRG3 | S344 | cytoplasm | NDRG family member 3 |
| NECAP2 | S181 | clathrin vesicle coat | NECAP endocytosis associated 2 |
| Nop56 | S513 | nucleus | NOP56 ribonucleoprotein homologue |
| TPD52 | S175 | ER | tumor protein D52 |
| NVL | S190 | nucleus | nuclear VCP-like |
| Pak2 | S141 | cytoplasm | p21 protein (Cdc42/Rac)-activated kinase 2 |
| PDPK1 | S244 | cell junction | 3-phosphoinosltide dependent protein kinase-l |
| Pold3 | S306 | nucleus | polymerase (DNA-directed), delta 3, accessory subunit |
| PRKAR1A | S212 | cytosol | protein kinase, cAMP-dependent, regulatory, type 1, alpha |
| Ptprc | S994 | external side of plasma membrane | protein tyrosine phosphatase, receptor type, C |
| PXN | S83 | cytoskeleton | paxillin |
| RAB11FIP5 | S307 | endosome | RAB11 family interacting protein 5 (class 1) |
| SASH3 | S27 | cytoplasm/nucleus | SAM and SH3 domain containing 3 |
| SCAND3 | S45 | nucleus | SCAN domain containing 3 |
| Sdcbp | S6 | cytoplasm | syndecan binding protein (syntenin) |
| Sh3kbpl | S274 | nucleus/cytoplasm | SH3-domaln kinase binding protein 1 |
| SYNRG | S1067 | trans-Golgi | synergin, gamma |
| Vasp | S235 | focal adhesion | vasodilator-stimulated phosphoprotein |
| Zc3hl4 | S515 | nucleus | zinc finger CCCH-type containing 14 |
| Zfml | S606 | nucleus | zinc finder protein 638 |
The same analysis between pairs of TLRs also resulted in few differentially regulated phosphosites. The LPS versus P3C, LPS versus R848, and P3C versus R848 comparisons resulted in 232, 218, and 260 common phosphosites, respectively. Out of these 47, 30, and 58 were statistically differentially regulated at ≥1 time point (Figure 4 A–C, respectively). Among the phosphoproteins that are differentially phosphorylated between the three different ligands were proteins involved in cytoskeletal and focal adhesion regulation (9 out of 40) and in the formation of cytoplasmic vesicles (4 out of 40).
Figure 4.
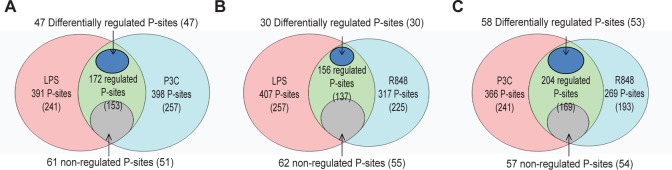
Phosphosites common for LPS and P3C (A), LPS and R848 (B), and P3C and R848 (C). Among the phosphosites shared between pairs of ligand stimulations, the number of phosphosites that are nonregulated, regulated, and differentially regulated is indicated.
As described above, we demonstrated that phosphorylation of proteins in the Fcγ mediated phagocytosis pathway was seen for both TLR4 and TLR2 stimulations but not TLR7 stimulation. FcγR1 T368 phosphorylated peptide was detected for TLR4 and TLR7 stimulation. Phosphorylation of FcγR1 T368 increased 2.5-fold at 5 min after treatment with LPS, whereas the phosphorylation levels on the same site after R848 treatment remained unchanged during the 30 min time course (Figure 5A). Coronin 1A is necessary for an early step of phagosome formation.49 Phosphorylation of coro1A/p57 deregulates its association with F-actin, which in turn allows for the formation of the phagosome.50 We detected a significant increase in the phosphorylation of coro1A T418 during the first 10 min after LPS and P3C stimulation but not after R848 stimulation (Figure 5B).
Figure 5.

Induction of phagocytosis in response to LPS but not to R848 (A, B). Increase in phosphorylation of FcγR1 following the LPS stimulation but not R848 stimulation (A) and of Corola (B) post-LPS and P3C but not post-R848 stimulation (B). Both proteins are involved in phagocytosis. (C, D): Delay in endocytosis after P3C stimulation vs. LPS stimulation. Average of the phosphorylation levels for each time point, for phosphosites in proteins classified as involved in endocytosis by the IPA analysis (C). Representative phosphorylation trend of a phosphosite on NECAP2, a protein involved in endocytosis post LPS, P3C, and R484 stimulation (D). (E) Phagocytosis assays of IgG-coated sheep red blood cells (SRBCs). C57 derived macrophages were cultured in 96-well plates with 2000 IgG treated or nontreated SRBCs for 30 min with either no treatment (black bar) or in the presence of 100 ng/mL LPS (horizontal stripes), 1 μM P3C (vertical stripes) or 1 μM R848 (white bar). Phagocytosis was assessed by measuring the absorbance at 620 nm as described in materials and methods. **p < 0.0001, *p < 0.001 when compared to no treatment.
These receptors have different trafficking patterns and cellular sites of function: TLR4 is initially at the cell surface, and TLR7 signals from the endosome. Therefore, the phagocytosis pathway can be expected to be affected differently by their stimulation, and this was reflected by the differential phosphorylation levels. We performed the Fcγ dependent phagocytosis assays using different TLR ligands, which confirmed that phagocytosis was induced through stimulation of the TLRs present initially at the cell surface (i.e., TLR4 and TLR2), but not by the endosomal TLR7 (Figure 5E).
Endocytosis is another GO term that was enriched for the LPS and P3C stimulation data sets, but not for R848. There were seven phosphosites belonging to seven endocytosis-related phosphoproteins that are common to both the TLR2 and the TLR4 activation data. The average phosphorylation of those seven phosphosites was examined at each time point, and the two different TLR activations had a different endocytosis response (Figure 5C). The TLR4 endocytosis response was relatively quick (present after 5 min stimulation, on average) and returned to near basal level after 30 min. The TLR2 response, in contrast, was less intense, increased relatively slowly, and the maximum observed level was noted at the end of the time course, at 30 min. An example of this difference in phosphorylation kinetics can be seen for NECAP2 at the site S181 (Figure 5D). NECAP2 has been identified in clathrin-coated vesicles,51 though the function of S181 phosphorylation has not yet been elucidated. Maximum phosphorylation for TLR4 stimulation was seen 5 min poststimulation, whereas the maximum phosphorylation post TLR2 stimulation was only reached at 30 min. There was no significant change in the phosphorylation levels of NECAP2 S181 after TLR7 stimulation, reinforcing the result that endocytosis was not a mechanism triggered by R848 stimulation.
Discussion
This study provides the first quantitative and comparative investigation of the macrophage phosphoproteome and its dynamic changes in response to TLR2, TLR4, and TLR7 activation. By monitoring five time points during the first 30 min following TLR activation by three ligands separately, we were able to identify differences between the signaling pathways that might not have come to light otherwise. This constitutes the first phosphoproteomic TLR study of such resolution within the first 30 min following ligand stimulation. The most comprehensive study so far has been the work of Weintz et al.,13 which analyzed the phosphorylation levels at 15 min and 4 h post-LPS stimulation.
As with other studies, activation of the different TLRs resulted in a predominance of serine and threonine phosphorylation.13,29,52,53 The changes in phosphorylation were highly dynamic, which is again in agreement with a previous study,13 even though our study included more and earlier time points. This highlights the importance of phosphorylation within the TLR signaling pathway to transduce the signal from the TLR receptor to transcription factors to stimulate cytokine production.54
The pairwise comparison at each time point and for each ligand revealed a poor correlation at the earlier time points than at the later time points. A possible explanation is that at the earlier time points, many more phosphosites were closer to the basal level of phosphorylation. This is not true: for example, in the LPS vs P3C data, at 3 min poststimulation 68% of the detected phosphosites do not exceed the threshold, whereas 30 min poststimulation the level fell to 61%. This increase in regulated phosphosites is not so dramatic that it can on its own account for the massive increase in correlation. Another explanation is that the early phosphorylation events were at the pathway nodes where the pathways differ the most between the three TLRs. Also, the observed differences were probably partly caused by the location of TLR signaling. Specifically, from the membrane and the endosome (like TLR447) or only from the endosome (like TLR755), and the later time points are when the pathways between the different TLRs likely converge, leading to cytokine production and other effector mechanisms.
As with other TLR phosphoproteomic studies,13,56 this study did not identify many phosphoproteins that belong to the TLR canonical pathway. Most of the TLR pathway phosphoproteins that were identified were transcription factors (e.g., JUN or IRF3) that showed high levels of phosphorylation 30 min poststimulation. We did, however, find many phosphoproteins belonging to pathways linked to cytoskeleton remodeling. In general, the cytoskeleton is not considered part of the TLR signaling pathway,57 although recent studies linked cytoskeleton remodeling to TLR signaling.13,44 Two key features of macrophages are motility and phagocytosis, and both of these depend on cytoskeletal remodeling and are enhanced upon TLR stimulation58,59 through MAPK dependent pathways.58,59 Using Ingenuity pathway enrichment analyses, we discovered two pathways associated with cytoskeletal remodeling for all three ligands tested: the Rho family of GTPases signaling pathway60 and the phospholipase C signaling pathway.61
Phagocytosis and endocytosis were triggered by LPS and P3C but not by R848 stimulation. The most prominent effect was the absence of phosphorylation increase of FcγRI at T368 in the presence of R848. Although the role of T368 phosphorylation is unknown (the phosphorylated peptide has been reported only in large phosphoproteomic screens), further biological investigation of this site may help to elucidate additional mechanisms of phagocytosis control in macrophages stimulated with different pathogen-related ligands. TLR7 responds to single-stranded RNA produced by viruses already internalized by the cell, so, in contrast to the cell-surface TLRs, it does not need to trigger the phagocytic response to an external pathogen.62 Endocytosis was triggered only by LPS and P3C stimulation in our time frame. TLR7 is an endosomal protein and thus does not need the formation of new endosomes to begin signaling. R848 reaches the TLR7 loaded endosome is thought to be transmembrane diffusion, which is a different than virus triggered stimulation, as the virus might have to undergo some form of endocytosis to enter the cell.63 In contrast, both the TLR2 and TLR4 stimulations led to the phosphorylation of proteins with GO terms linked to the endocytic pathway. For example, NECAP2, which is a protein thought to be involved in clathrin mediated endocytosis is phosphorylated at earlier time points after LPS stimulation compared to P3C stimulation. Synergin, a protein that interacts with the gamma subunit of the AP-1 clathrin adaptor complex,64 does not show the same difference in kinetics of phosphorylation between the two TLR stimulations. Whether the difference in kinetics seen with NECAP2 is a true reflection of possible difference in TLR2 endocytosis vs TLR4 endocytosis remains to be determined, especially since the variation between biological replicates of the LPS NECAP2 measurements was quite large.
The prominence of actin binding protein phosphorylation likely indicates that there is a genuine role of the cytoskeleton in TLR signaling. For example, actin-binding proteins may provide a platform for recruitment and spatial targeting of TLR pathway signaling molecules. One of these possible proteins might be MYH9 whose phosphorylation levels on S1943 increased both for LPS and P3C stimulation but not R848 stimulation. Myosins are motor proteins that bind actin and interact with cargo molecules. Phosphorylation of in myosin IIa S1943 was reported to be necessary in NK cells for the linkage of NK-cell lytic granules.65 It is possible that S1943 in myosin IX regulates endosome transport, since phosphorylation of MYH9 S1943 followed that of the endosomal proteins for the LPS and P3C stimulations.
In summary, we performed global analyses of the phosphoproteome changes in response of the three different TLR to their specific ligands. We were not able to identify any specific pathway that was unique to a single receptor and not shared by the others. This is not too surprising, considering that in the canonical TLR pathway, the different receptor responses converge to the same nodes (notably, NF-κB, MAPK, and IRF circuits). We did, however, identify changes to regulators of the cytoskeleton in response to the ligands, and our analysis provided insight into the different responses between the different receptors (e.g., specific phosphoproteins that did or did not respond and phosphoproteins that were delayed in their response.
Collectively, this study provides a new, global perspective on innate immune activation by TLR signaling. We quantitatively detected phosphosites for five different time points and for three different TLRs. The cytoskeleton remodeling emerged as a target of TLR signaling for all three ligands. More interestingly, we found that endocytosis and phagocytosis pathways were up-regulated upon TLR4 and TLR2 stimulations and not upon the TLR7 stimulation, which emphasizes the fact that the pathway signaling by one TLR cannot be generalized to all TLRs.
Acknowledgments
We thank Owen Schwartz at Biological Imaging Section (NIAID/NIH) for help with the imaging. We also thank Iain Fraser and Nathan Manes for advice and discussions, Art Nuccio for technical assistance and Jack Bennink, Marijke Koppenol-Raab, and Jessica Mann for helpful comments on the manuscript. We are also grateful to Ronald N. Germain for his support and advice during the conduct of this study and for help with preparation of the manuscript. This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 2004, 4306996257–63. [DOI] [PubMed] [Google Scholar]
- Kang J. Y.; Lee J. O. Structural biology of the Toll-like receptor family. Annu. Rev. Biochem. 2011, 80, 917–41. [DOI] [PubMed] [Google Scholar]
- Werling D.; Jann O. C.; Offord V.; Glass E. J.; Coffey T. J. Variation matters: TLR structure and species-specific pathogen recognition. Trends Immunol. 2009, 303124–30. [DOI] [PubMed] [Google Scholar]
- Nau G. J.; Richmond J. F.; Schlesinger A.; Jennings E. G.; Lander E. S.; Young R. A. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 2002, 9931503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K.; Akira S. TLR signaling pathways. Semin. Immunol. 2004, 1613–9. [DOI] [PubMed] [Google Scholar]
- Heinig M.; Petretto E.; Wallace C.; Bottolo L.; Rotival M.; Lu H.; Li Y.; Sarwar R.; Langley S. R.; Bauerfeind A.; Hummel O.; Lee Y. A.; Paskas S.; Rintisch C.; Saar K.; Cooper J.; Buchan R.; Gray E. E.; Cyster J. G.; Erdmann J.; Hengstenberg C.; Maouche S.; Ouwehand W. H.; Rice C. M.; Samani N. J.; Schunkert H.; Goodall A. H.; Schulz H.; Roider H. G.; Vingron M.; Blankenberg S.; Munzel T.; Zeller T.; Szymczak S.; Ziegler A.; Tiret L.; Smyth D. J.; Pravenec M.; Aitman T. J.; Cambien F.; Clayton D.; Todd J. A.; Hubner N.; Cook S. A. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature 2010, 4677314460–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan J. C. Signaling organelles of the innate immune system. Cell 2012, 15161168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marre M. L.; Petnicki-Ocwieja T.; DeFrancesco A. S.; Darcy C. T.; Hu L. T. Human integrin alpha(3)beta(1) regulates TLR2 recognition of lipopeptides from endosomal compartments. PLoS One 2010, 59e12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich N.; Lienenklaus S.; Weiss S.; Gekara N. O. Murine toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS One 2010, 54e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V.; Cao L.; Lin J. T.; Hung N.; Ritz A.; Yu K.; Jianu R.; Ulin S. P.; Raphael B. J.; Laidlaw D. H.; Brossay L.; Salomon A. R. A new approach for quantitative phosphoproteomic dissection of signaling pathways applied to T cell receptor activation. Mol. Cell Proteomics 2009, 8112418–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai L. K.; Benoist C.; Mathis D.; White F. M. Quantitative phosphoproteomic analysis of T cell receptor signaling in diabetes prone and resistant mice. J. Proteome Res. 2010, 963135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. E.; White F. M. Quantitative analysis of phosphotyrosine signaling networks triggered by CD3 and CD28 costimulation in Jurkat cells. J. Immunol 2006, 17652833–43. [DOI] [PubMed] [Google Scholar]
- Weintz G.; Olsen J. V.; Fruhauf K.; Niedzielska M.; Amit I.; Jantsch J.; Mages J.; Frech C.; Dolken L.; Mann M.; Lang R. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 2010, 6, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M.; Dreisbach A.; Otto A.; Becher D.; Bernhardt J.; Hecker M.; Peppelenbosch M. P.; van Dijl J. M. Mapping of interactions between human macrophages and Staphylococcus aureus reveals an involvement of MAP kinase signaling in the host defense. J. Proteome Res. 2011, 1094018–32. [DOI] [PubMed] [Google Scholar]
- Du R.; Long J.; Yao J.; Dong Y.; Yang X.; Tang S.; Zuo S.; He Y.; Chen X. Subcellular quantitative proteomics reveals multiple pathway cross-talk that coordinates specific signaling and transcriptional regulation for the early host response to LPS. J. Proteome Res. 2010, 941805–21. [DOI] [PubMed] [Google Scholar]
- Amit I.; Citri A.; Shay T.; Lu Y.; Katz M.; Zhang F.; Tarcic G.; Siwak D.; Lahad J.; Jacob-Hirsch J.; Amariglio N.; Vaisman N.; Segal E.; Rechavi G.; Alon U.; Mills G. B.; Domany E.; Yarden Y. A module of negative feedback regulators defines growth factor signaling. Nat. Genet. 2007, 394503–12. [DOI] [PubMed] [Google Scholar]
- Chevrier N.; Mertins P.; Artyomov M. N.; Shalek A. K.; Iannacone M.; Ciaccio M. F.; Gat-Viks I.; Tonti E.; DeGrace M. M.; Clauser K. R.; Garber M.; Eisenhaure T. M.; Yosef N.; Robinson J.; Sutton A.; Andersen M. S.; Root D. E.; von Andrian U.; Jones R. B.; Park H.; Carr S. A.; Regev A.; Amit I.; Hacohen N. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell 2011, 1474853–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser I. D.; Germain R. N. Navigating the network: signaling cross-talk in hematopoietic cells. Nat. Immunol. 2009, 104327–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y.; Yun D.; Esmon A.; Zou P.; Zuo S.; Yu Y.; He F.; Yang P.; Chen X. Proteomic dissection of agonist-specific TLR-mediated inflammatory responses on macrophages at subcellular resolution. J. Proteome Res. 2008, 783180–93. [DOI] [PubMed] [Google Scholar]
- Kumar H.; Kawai T.; Akira S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 3884621–5. [DOI] [PubMed] [Google Scholar]
- Makarov A.; Denisov E.; Kholomeev A.; Balschun W.; Lange O.; Strupat K.; Horning S. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 2006, 7872113–20. [DOI] [PubMed] [Google Scholar]
- Michalski A.; Damoc E.; Hauschild J. P.; Lange O.; Wieghaus A.; Makarov A.; Nagaraj N.; Cox J.; Mann M.; Horning S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell Proteomics 2011, 109M111 011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26121367–72. [DOI] [PubMed] [Google Scholar]
- Deutsch E. W.; Mendoza L.; Shteynberg D.; Farrah T.; Lam H.; Tasman N.; Sun Z.; Nilsson E.; Pratt B.; Prazen B.; Eng J. K.; Martin D. B.; Nesvizhskii A. I.; Aebersold R. A guided tour of the Trans-Proteomic Pipeline. Proteomics 2010, 1061150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg D.; Deutsch E. W.; Lam H.; Eng J. K.; Sun Z.; Tasman N.; Mendoza L.; Moritz R. L.; Aebersold R.; Nesvizhskii A. I. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell Proteomics 2011, 1012M111 007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen M. V.; Marino G.; Pultz D.; Morselli E.; Faergeman N. J.; Kroemer G.; Andersen J. S. Phosphoproteomic analysis of cells treated with longevity-related autophagy inducers. Cell Cycle 2012, 1191827–40. [DOI] [PubMed] [Google Scholar]
- Gu T. L.; Nardone J.; Wang Y.; Loriaux M.; Villen J.; Beausoleil S.; Tucker M.; Kornhauser J.; Ren J.; MacNeill J.; Gygi S. P.; Druker B. J.; Heinrich M. C.; Rush J.; Polakiewicz R. D. Survey of activated FLT3 signaling in leukemia. PLoS One 2011, 64e19169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger M.; Kratchmarova I.; Blagoev B.; Tseng Y. H.; Kahn C. R.; Mann M. Dissection of the insulin signaling pathway via quantitative phosphoproteomics. Proc. Natl. Acad. Sci. U. S. A. 2008, 10572451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. V.; Blagoev B.; Gnad F.; Macek B.; Kumar C.; Mortensen P.; Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 1273635–48. [DOI] [PubMed] [Google Scholar]
- Bauernfeind F. G.; Horvath G.; Stutz A.; Alnemri E. S.; MacDonald K.; Speert D.; Fernandes-Alnemri T.; Wu J.; Monks B. G.; Fitzgerald K. A.; Hornung V.; Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 1832787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson S. M.; Walker W. S. Immortalization of cloned mouse splenic macrophages with a retrovirus containing the v-raf/mil and v-myc oncogenes. Cell Immunol. 1988, 1162341–51. [DOI] [PubMed] [Google Scholar]
- Vizcaino J. A.; Cote R. G.; Csordas A.; Dianes J. A.; Fabregat A.; Foster J. M.; Griss J.; Alpi E.; Birim M.; Contell J.; O’Kelly G.; Schoenegger A.; Ovelleiro D.; Perez-Riverol Y.; Reisinger F.; Rios D.; Wang R.; Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013, 41Database issueD1063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigbolt K. T.; Vanselow J. T.; Blagoev B. GProX, a user-friendly platform for bioinformatics analysis and visualization of quantitative proteomics data. Mol. Cell Proteomics 2011, 108O110 007450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W.; Sherman B. T.; Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4144–57. [DOI] [PubMed] [Google Scholar]
- Huang da W.; Sherman B. T.; Lempicki R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 3711–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura N.; Saitoh S.; Matsumoto F.; Akashi-Takamura S.; Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 2008, 368194–9. [DOI] [PubMed] [Google Scholar]
- Kagan J. C.; Su T.; Horng T.; Chow A.; Akira S.; Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol 2008, 94361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husebye H.; Halaas O.; Stenmark H.; Tunheim G.; Sandanger O.; Bogen B.; Brech A.; Latz E.; Espevik T. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006, 254683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maa M. C.; Chang M. Y.; Li J.; Li Y. Y.; Hsieh M. Y.; Yang C. J.; Chen Y. J.; Li Y.; Chen H. C.; Cheng W. E.; Hsieh C. Y.; Cheng C. W.; Leu T. H. The iNOS/Src/FAK axis is critical in Toll-like receptor-mediated cell motility in macrophages. Biochim. Biophys. Acta 2011, 18131136–47. [DOI] [PubMed] [Google Scholar]
- Ruse M.; Knaus U. G. New players in TLR-mediated innate immunity: PI3K and small Rho GTPases. Immunol Res. 2006, 34133–48. [DOI] [PubMed] [Google Scholar]
- Zanoni I.; Ostuni R.; Marek L. R.; Barresi S.; Barbalat R.; Barton G. M.; Granucci F.; Kagan J. C. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 2011, 1474868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C.; Jin J.; Xu S.; Liu H.; Li N.; Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat. Immunol. 2010, 118734–42. [DOI] [PubMed] [Google Scholar]
- Wang L.; Gordon R. A.; Huynh L.; Su X.; Park Min K. H.; Han J.; Arthur J. S.; Kalliolias G. D.; Ivashkiv L. B. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity 2010, 324518–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleveta G.; Borzecka K.; Zdioruk M.; Czerkies M.; Kuberczyk H.; Sybirna N.; Sobota A.; Kwiatkowska K. LPS induces phosphorylation of actin-regulatory proteins leading to actin reassembly and macrophage motility. J. Cell Biochem. 2012, 113180–92. [DOI] [PubMed] [Google Scholar]
- Arregui C. O.; Gonzalez A.; Burdisso J. E.; Gonzalez Wusener A. E. Protein tyrosine phosphatase PTP1B in cell adhesion and migration. Cell Adhes. Migr. 2013, 75418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura F. FilGAP and its close relatives: a mediator of Rho-Rac antagonism that regulates cell morphology and migration. Biochem. J. 2013, 453117–25. [DOI] [PubMed] [Google Scholar]
- Ostuni R.; Zanoni I.; Granucci F. Deciphering the complexity of Toll-like receptor signaling. Cell. Mol. Life Sci. 2010, 67244109–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen N. J.; Deininger S.; Nonstad U.; Skjeldal F.; Husebye H.; Rodionov D.; von Aulock S.; Hartung T.; Lien E.; Bakke O.; Espevik T. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J. Leukoc. Biol. 2008, 841280–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M.; Collins R. F.; Grinstein S.; Trimble W. S. Coronin-1 function is required for phagosome formation. Mol. Biol. Cell 2005, 1673077–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku T.; Kaneko Y.; Murofushi K.; Seyama Y.; Toyoshima S.; Tsuji T. Phorbol ester-dependent phosphorylation regulates the association of p57/coronin-1 with the actin cytoskeleton. J. Biol. Chem. 2008, 2834328918–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter B.; Blondeau F.; Denisov A. Y.; Gehring K.; McPherson P. S. Molecular mechanisms in clathrin-mediated membrane budding revealed through subcellular proteomics. Biochem. Soc. Trans. 2004, 32Pt 5769–73. [DOI] [PubMed] [Google Scholar]
- Pan C.; Gnad F.; Olsen J. V.; Mann M. Quantitative phosphoproteome analysis of a mouse liver cell line reveals specificity of phosphatase inhibitors. Proteomics 2008, 8214534–46. [DOI] [PubMed] [Google Scholar]
- Villen J.; Beausoleil S. A.; Gerber S. A.; Gygi S. P. Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U. S. A. 2007, 10451488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T.; Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 1311460–9. [DOI] [PubMed] [Google Scholar]
- Heil F.; Ahmad-Nejad P.; Hemmi H.; Hochrein H.; Ampenberger F.; Gellert T.; Dietrich H.; Lipford G.; Takeda K.; Akira S.; Wagner H.; Bauer S. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur. J. Immunol. 2003, 33112987–97. [DOI] [PubMed] [Google Scholar]
- Sharma K.; Kumar C.; Keri G.; Breitkopf S. B.; Oppermann F. S.; Daub H. Quantitative analysis of kinase-proximal signaling in lipopolysaccharide-induced innate immune response. J. Proteome Res. 2010, 952539–49. [DOI] [PubMed] [Google Scholar]
- Oda K.; Kitano H. A comprehensive map of the toll-like receptor signaling network. Mol. Syst. Biol. 2006, 220060015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander J. M.; Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science 2004, 30456731014–8. [DOI] [PubMed] [Google Scholar]
- West M. A.; Wallin R. P.; Matthews S. P.; Svensson H. G.; Zaru R.; Ljunggren H. G.; Prescott A. R.; Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science 2004, 30556871153–7. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science 1998, 2795350509–14. [DOI] [PubMed] [Google Scholar]
- Saarikangas J.; Zhao H.; Lappalainen P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol. Rev. 2010, 901259–89. [DOI] [PubMed] [Google Scholar]
- De Meyer I.; Martinet W.; Schrijvers D. M.; Timmermans J. P.; Bult H.; De Meyer G. R. Toll-like receptor 7 stimulation by imiquimod induces macrophage autophagy and inflammation in atherosclerotic plaques. Basic Res. Cardiol. 2012, 1073269. [DOI] [PubMed] [Google Scholar]
- Vazquez-Calvo A.; Saiz J. C.; McCullough K. C.; Sobrino F.; Martin-Acebes M. A. Acid-dependent viral entry. Virus Res. 2012, 1672125–37. [DOI] [PubMed] [Google Scholar]
- Page L. J.; Sowerby P. J.; Lui W. W.; Robinson M. S. Gamma-synergin: an EH domain-containing protein that interacts with gamma-adaptin. J. Cell Biol. 1999, 1465993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn K. B.; Mace E. M.; Rak G. D.; Difeo A.; Martignetti J. A.; Pecci A.; Bussel J. B.; Favier R.; Orange J. S. Phosphorylation of the myosin IIA tailpiece regulates single myosin IIA molecule association with lytic granules to promote NK-cell cytotoxicity. Blood 2011, 118225862–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.