

Abstract
Epitope content plays a critical role in determining T cell and antibody responses to vaccines, biomaterials, and protein therapeutics, but its effects are nonlinear and difficult to isolate. Here, molecular self-assembly was used to build a vaccine with precise control over epitope content, in order to finely tune the magnitude and phenotype of T helper and antibody responses. Self-adjuvanting peptide nanofibers were formed by co-assembling a high-affinity universal CD4+ T cell epitope (PADRE) and a B cell epitope from Staphylococcus aureus at specifiable concentrations. Increasing the PADRE concentration from μM to mM elicited bell-shaped dose-responses that were unique to different T cell populations. Notably, the epitope ratios that maximized T follicular helper and antibody responses differed by an order of magnitude from those that maximized Th1 or Th2 responses. Thus, modular materials assembly provides a means of controlling epitope content and efficiently skewing the adaptive immune response in the absence of exogenous adjuvant; this approach may contribute to the development of improved vaccines and immunotherapies.
Keywords: co-assembly, immunoengineering, MRSA, self-adjuvanting, scaffolds
1. Introduction
Vaccine design is moving away from using whole pathogens in favor of selecting only the most protective antigens for immunization with a suitable adjuvant. The most tailored of these sub-unit vaccines utilize specific B cell or CD8+ T cell epitopes, such as the short peptides and carbohydrates that have been used to elicit antibody or killing responses against malaria pathogens, bacterial infections, or tumors.[1, 2] In addition to the desired target epitope, these vaccines also require one or more CD4+ T cell epitopes to engage CD4+ helper T cells. For example, in the highly successful protein-polysaccharide vaccines, capsular sugars from pneumococcus, Haemophilus influenzae, or Neisseria meningitidis are conjugated to a protein carrier in order to elicit protective T cell-dependent antibody responses.[3] Both professional antigen-presenting cells (APCs), e.g. dendritic cells (DCs), as well as polysaccharide-specific B cells internalize the conjugate and degrade the protein. All of these cells must engage with CD4+ T cells by presenting peptide epitopes from the protein in a class II major histocompatibility complex (MHC II), which binds to the T cell receptor (TCR). Activation signals from DCs and B cells guide T cell differentiation into various effector subsets, while B cells also use TCR-MHC binding to procure essential T cell help. Help from the T follicular helper (Tfh, CXCR5+ PD-1+) subset of CD4+ T cells is particularly important for the induction of class-switched B cells and high-affinity antibodies.[4] Thus, subunit vaccines are effective only if CD4+ T cells differentiate into a suitable effector subset, a process that is mediated in part by the signals they receive through the TCR.
The strength of the TCR signal integrates both the peptide dose and the dwell time of the TCR-peptide-MHC binding event, and it has a nonlinear effect on CD4+ T cell activation and differentiation that is difficult to predict.[5, 6] This uncertain relationship makes it important to develop well-controlled experimental methods to optimize the vaccine composition for the generation of specific CD4+ T cell types. T cells respond to very low doses of epitope with anergy or regulatory T cell induction, but actively suppress regulatory responses at higher doses of antigen.[7] At still higher doses of antigen (e.g., 100 μg protein adjuvanted with alum), CD4+ T cells again become hyporeactive.[8] Recently, it has been shown that intermediate concentrations or dwell times elicit the strongest effector (Th1) responses, while Tfh responses in germinal centers are favored by higher TCR signal strengths,[9, 10] but it can be difficult to predict a priori what epitope concentrations and ratios constitute such concentration regimes.
Despite its importance for T cell activation, precise control over the T cell epitope dose remains challenging to incorporate into vaccine design. Protein carriers, including conjugates and virus-like particles,[3, 11] include many T cell epitopes and raise strong antibody responses, but it is laborious to modify their quantity or composition. A highly precise strategy is to covalently link the target antigen to a synthetic peptide or short protein that comprises only T cell epitopes.[12, 13] Such constructs are usually made at a single molar ratio (e.g. 1:1 or 3:1) between the B cell or CD8+ T cell epitope(s) and the CD4+ T cell epitope, because comparing different ratios requires separate synthesis and purification for each version.[14] Furthermore, for linear constructs or branched multiple-antigen peptides, the maximal ratio of T cell and B cell epitopes is limited by length or branching architecture, respectively.
In this paper, we used non-covalent self-assembly to integrate defined T cell epitopes into a self-adjuvanting vaccine platform. In contrast to covalent conjugation, this approach can incorporate arbitrary concentrations of the desired epitope (here, from the μM to mM range), while keeping the concentrations of other epitopes and the adjuvanting backbone constant. A short assembly domain, called Q11 (acetyl-QQKFQFQFEQQ-amide), was used to form beta-sheet nanofibers in water and physiological buffers and preserve the molar ratio of the precursors in the assembled fibers.[15] We have previously synthesized peptide antigens such as OVA323-339 (pOVA), which contains epitopes for both B cells and CD4+ T cells,[16] in tandem with Q11 (OVAQ11). When assembled into fibers and injected without additional adjuvants into mice, OVAQ11 activated antigen presenting cells in vivo and raised T-cell dependent antibody responses,[17, 18] yet it did not elicit the inflammation typically associated with conventional adjuvants.[19] The absence of inflammation is advantageous in light of evidence that inflammatory adjuvants such as alum and incomplete Freund's adjuvant can diminish the strength of the Tfh response by selecting for T cells with low-affinity receptors.[10, 20] Here we used the Q11 platform to co-assemble separate T cell and B cell epitopes and to isolate the effect of the dose of CD4+ epitope on the T cell and B cell responses.
2. Results
2.1. Nanofibers assembled with a T cell epitope elicit T cell activation
The PADRE peptide (H2N-aKXVAAWTLKAa-amide, where “X” is cyclohexylalanine and “a” is D-alanine) has been shown to bind with high affinity to most common human HLA DR molecules and to the MHC Class II I-Ab molecule expressed by C57Bl/6 mice.[21] It also can provide help for generating antibody and CD8 responses in vivo.[21, 22] We tested whether presentation on beta-sheet nanofibers, without exogenous adjuvants, was sufficient to elicit a functional T cell response to the PADRE epitope. We first synthesized the tandem peptide PADRE-SGSG-Q11, where SGSG serves as a short flexible linker, and then assembled it into Q11 fibers (PADREQ11) (Figure 1a). Subcutaneous immunization of C57Bl/6 mice followed by a boost on day 14 with these fibers induced activated PADRE-specific T cells that secreted IL-4 or IFN-[3], as detected by ELISPOT assays (Figure 1b). We worked with the NIH Tetramer Core facility to develop a PADRE-IAb tetramer that allowed the visualization of the endogenous PADRE-specific CD4+ T cell response in vivo (Figure 1c and Figure S1).
Figure 1. A high-affinity T cell epitope incorporated into Q11 nanofibers induces antigen-specific T cell responses.
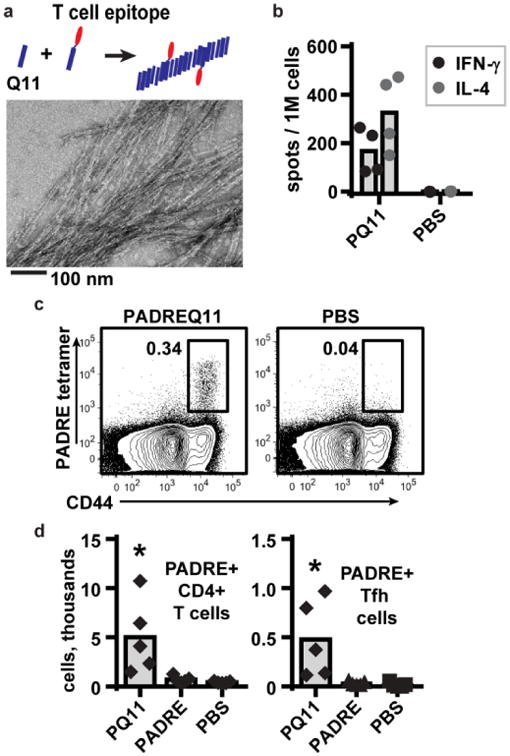
(a) PADRE, a high-affinity CD4+ T cell epitope[21], was synthesized in tandem with the Q11 assembly domain and mixed into unconjugated Q11 to form integrated assemblies observable by TEM (0.05 mM PADRE-Q11, 1.95 mM Q11). (b) Mice were immunized with PADREQ11 (0.05 mM PADREQ11 + 1.95 mM Q11) and boosted on day 14; on day 21, cells from the lymph nodes were stimulated with PADRE peptide, and the IL-4 and IFN-[3] -secreting cells were quantified by ELISPOT. (c) A PADREI-Ab tetramer was validated to detect endogenous PADRE-specific T cells (CD4+ CD44+ PADRE+) by flow cytometry. CD44 is a marker for T cells that have encountered cognate antigen. Data from one PADREQ11-immunized and one PBS-immunized mouse are shown; representative of 3 mice per group, repeated twice. (d) Immunization with PADREQ11 but not free PADRE peptide (0.05 mM PADRE in saline) generated PADRE-specific CD4+ T cells as defined above, as well as PADRE-specific Tfh cells (CD4+ PADRE+ CXCR5+ PD-1+) by 7 days after primary immunization. In b and d, each dot indicates 1 mouse, and cell numbers are quantified from the six draining lymph nodes that were collected from each mouse. Analyzed by 1-way ANOVA. Bars show average values.
A significant increase in the total number of PADRE+ T cells was detected in the lymph nodes at 7 days after primary immunization with PADREQ11 assemblies (Figure 1d). All of these cells expressed CD44, a marker of antigen-experienced cells.[23] Therefore, the CD4+ PADRE+ CD44+ population was used to track the T cells that had responded to PADRE. This population includes all PADRE-specific CD4+ T cells, regardless of their effector polarization (Th1, Tfh, or Th2). We also observed a significant increase in the total number of PADRE-specific Tfh cells (CXCR5+ PD1+) at this time, although they were only d 10 % of all PADRE-specific T cells (Figure 1d). In contrast, no increase in PADRE-specific T cells was observed after immunization with PADRE peptide in buffer, indicating that Q11-dependent fibrillization was required (Figure 1d). These experiments demonstrated that T cells were capable of recognizing and responding to the PADRE epitope presented by I-Ab on APCs.
2.2. Non-covalent assemblies of B cell and T cell epitopes raise modular responses
Next, we tested whether a B cell epitope could be co-assembled with the T-cell epitope to create an integrated material (Figure 2a) capable of raising both T and B cell responses. A model antigen that contained only a B cell epitope was selected based on an initial requirement for conjugation to a carrier protein to raise an antibody response. The E214 peptide (acetyl-KFEGTEDAVETIIQAIEA-amide) from the enolase protein of Staphylococcus aureus, when conjugated to a carrier protein (CRM-mutant diphtheria toxin) and adjuvanted with alum, has been shown to raise antibody responses in mice, rats, and macaques,[24] and it has been investigated as part of a vaccine against methicillin-resistant S. aureus.[24] This E214 peptide was also predicted by the IEDB epitope predictor [25] to contain a B cell epitope but no T cell epitopes for I-Ab (with affinity < 5 mM). We synthesized the tandem peptide E214-SGSG-Q11 (E214Q11), and successfully assembled them into nanofibers. However, immunization of C57Bl/6 mice with E214Q11 alone failed to raise an antibody response, even when adjuvanted with alum (Figure 2b). In contrast, when E214Q11 was co-assembled with PADREQ11, the assemblies were highly immunogenic and raised anti-E214 antibody titers that reached 10[4] after a single boost (Figure 2b).
Figure 2. Co-assembled B and T epitopes raise a high-titer antibodies and a modular T cell response.
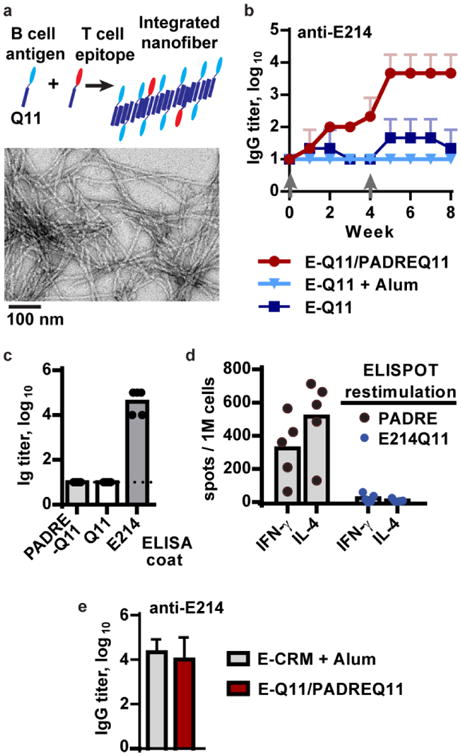
(a) In the co-assembly strategy, B cell and T cell antigens are synthesized separately in tandem with the Q11 assembly domain, then mixed in the specified ratio with unconjugated Q11 to form integrated assemblies that are visible by TEM (shown with 1 mM E214Q11, 0.05 mM PADREQ11, 0.95 mM Q11). (b) Anti-E214 responses to Q11-based vaccines required assembly with a T cell epitope, PADRE. Mice were immunized at week 0 and boosted with a half-dose of peptide at week 4 (grey arrows). E214Q11 (2 mM) failed to raise a response even when adjuvanted with Alum, whereas the co-assembled E214Q11 (1 mM)/PADREQ11 (0.05 mM) vaccine raised a strong response. n = 3 mice per group, representative of at least 2 independent experiments. (c) The antibodies were specific to only the E214 epitope, not PADREQ11 or Q11, in ELISA (serum after two boosts). (d) The T cell IL-4 and IFN-gamma responses were specific to only PADRE, not E214Q11, in ELISPOT assays (lymph nodes collected after two boosts). In (c) and (d), each dot indicates 1 mouse, with 5 mice in each group. (e) Mice immunized with alum-adjuvanted E214-CRM protein or with unadjuvanted E214Q11/PADREQ11 assemblies raised equivalent E214-specific responses. Serum was analyzed at week 9, one week after a second boost. n = 3 mice per group. All error bars show standard deviation.
Consistent with the modular design of this vaccine, serum from immunized mice was reactive in ELISAs only to E214, and not to PADRE-Q11 or SGSG-Q11 alone (Figure 2c). This result indicated that the B cell response was only focused on the E214 epitope. Similarly, cells from the lymph nodes of immunized mice were responsive in IFN-[3] and IL-4 ELISPOT assays only to PADRE, not E214Q11 (Figure 2d), in agreement with the prediction that E214 (and Q11) lacks a T cell epitope. Even though only a single T cell epitope was included, the antibody titers raised by these co-assembled fibers were similar to those raised by our positive control, an alum-adjuvanted E214-CRM protein conjugate (Figure 2e). Thus the Q11 self-assembled vaccine that incorporates PADRE is able to elicit T cell help for antibody responses against antigens that lack an inherent T cell epitope.
2.3. Co-assemblies require T cell - B cell interactions to raise antibody responses
We predicted that the PADRE-Q11 co-assembled vaccine raised antibody responses to the B cell epitope by functioning similarly to a peptide-protein conjugate[3] rather than by acting as an inflammatory adjuvant. In this scenario, B cells would internalize the E214Q11/PADREQ11 nanofiber by engaging E214 through the B-cell receptor. The B cell would then solicit T cell help by presenting the PADRE epitope on I-Ab for recognition by T cell receptors (Figure 3a). Thus, B cells would have to internalize both epitopes together to be able to generate an antibody response. We tested this requirement for cognate T-B interaction by immunizing and boosting mice with either co-assembled E214Q11/PADREQ11 or with separately assembled E214Q11 and PADREQ11. The latter two solutions were delivered in two injections, administered close together to ensure that both peptides drained to the same lymph node. Indeed, only the co-assembled fibers raised an antibody response (Figure 3b), consistent with a requirement for cognate T cell help. The memory B cell response also required T cell help; mice initially immunized with co-assembled E214Q11/PADREQ11 showed an increase in antibody titer only when boosted with co-assembled fibers, not with E214Q11 alone or PBS buffer (Figure S2).
Figure 3. Antibody responses arise from cognate T-B interaction and are not associated with inflammation.
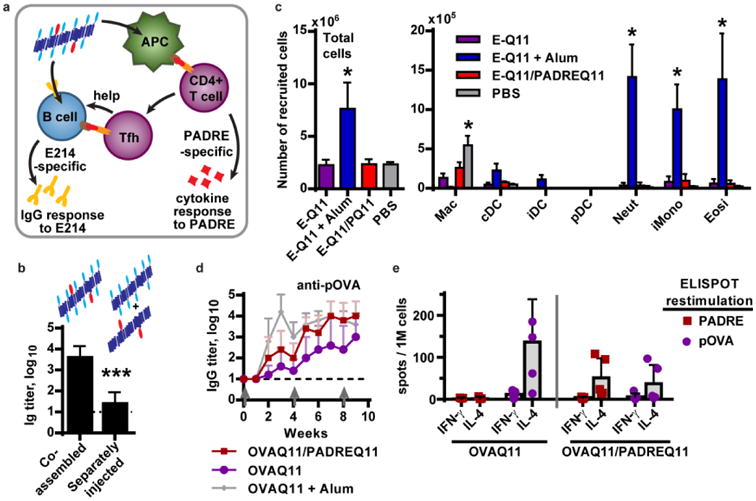
(a) Peptide co-assemblies are proposed to function similar to an adjuvanted peptide-protein conjugate, as described in the text. (b) Antibody responses required co-assembled fibers; mice injected simultaneously with separate solutions of E214Q11 and PADREQ11 fibers failed to raise a response by 7 days after a boost (n = 5 mice per group, two-tailed t-test). (c) Peptide assemblies did not induce recruitment of inflammatory cells after i.p. injection. Cells in the peritoneal lavage fluid were analyzed 20 hr after injection. N = 4 mice per group. *, p <0.05 compared to EQ11 group, by two-way ANOVA with Dunnett correction for multiple comparisons. The immunizations were as follows (100 μL per injection): E-Q11 (1 mM E214-Q11, 1 mM Q11), E-Q11 +Alum (2 mM E214-Q11, 2 mM Q11, mixed 1:1 v/v with Imject Alum), E-Q11/PQ11 (1 mM E214-Q11, 0.95 mM Q11, 0.05 mM PADRE-Q11), PBS (saline). Abbreviations: Macrophages (mac), conventional dendritic cells (cDC), inflammatory DCs (iDC), plasmacytotoid DC (pDC), neutrophils (neut), inflammatory monocytes (iMono), eosinophils (eosi), as defined in ref ([19]). (d,e) C57Bl/6 mice were immunized and boosted with 2 mM OVAQ11 alone, co-assembled with 0.1 mM PADREQ11, or adjuvanted with Imject alum. (e) The pOVA-specific IgG titers were enhanced by co-assembly with PADREQ11 (p = 0.051 by two-way ANOVA over 9 weeks) to levels that equaled the alum-adjuvanted response after one boost. (e) After immunization co-assembled fibers, the T cell response was divided between pOVA and PADRE instead of being focused exclusively on pOVA. Mean + standard deviation is shown; n = 5 mice per group.
We next confirmed that co-assembly with PADRE-Q11 did not alter the non-inflammatory character of the nanofibers (Figure 3c). We previously demonstrated that i.p. immunization of Q11-based vaccines raised comparable antibody responses as s.c. immunization, and that both did so without detectable local inflammation[19]. For ease of analysis and better sensitivity, we chose the i.p. route for these studies. As observed previously for OVAQ11 nanofibers,[19] neither E214Q11 nor co-assembled E214Q11/PADREQ11 fibers elicited recruitment of inflammatory dendritic cells, neutrophils, or eosinophils after i.p. injection. Only an alum-adjuvanted formulation elicited such a response. Immunization with E214Q11 or co-assembled E214Q11/PADREQ11 nanofibers differed from PBS-immunization only in the number of macrophages that remained in the peritoneum, with a 2 – 4-fold reduction in the numbers recovered. The reason for this small reduction in macrophage numbers is not known, but it could reflect a loss of the F4/80 macrophage marker, migration (including adherence to the peritoneal wall), or apoptosis of F4/80+ macrophages following immunization. By comparison, immunization with E214Q11 + alum reduced the numbers of macrophages by greater than 40-fold. Because loss of macrophages from the peritoneal lavage is associated with activation of these cells[26], these observations suggest that the nanofibers may activate macrophages in a nontraditional manner, without recruiting other innate immune cells.
Finally, we tested whether B cells specific for a single B cell epitope could receive help from multiple T cell populations, as predicted by the proposed mechanism of action for these assemblies (Figure 3a). We co-assembled pOVA, which contains both a B cell epitope and a moderate-affinity CD4+ T cell epitope (400 nM IC50), with high-affinity PADRE (94 nM IC50 for IAb).[21] Mice were immunized and boosted with OVAQ11, OVAQ11 adjuvanted with alum, or OVAQ11/PADRE-Q11 co-assemblies, and the antibody responses were measured by ELISA. Co-assembly with PADREQ11 significantly increased the pOVA-specific IgG titers, which were an average of approximately 10-fold higher than with OVAQ11 alone over the entire 9 weeks of the experiment (Figure 3d). Titers were not different between the OVAQ11/PADREQ11 and OVAQ11 + alum groups after one or two boosts, suggesting that inclusion of the extra epitope had increased the anti-pOVA response to its maximum value. When the T cell response was measured by ELISPOT, it was specific for both pOVA and PADRE (Figure 3e). These results suggest that the addition of a second T cell epitope increased the pool of cognate T cells that could interact with OVA-specific B cells, resulting in enhancement of anti-OVA antibody production.
2.4. Different T cell subsets respond differently to the dose of epitope
The dose of T cell epitope can have gross effects on T cell behavior (e.g., high doses inducing anergy) as well as more subtle subset-specific effects. For instance, Tfh differentiation has been recently reported to require higher affinity or longer-lasting interactions between the MHC class II/peptide complex and the T cell receptor than does differentiation into other effector T cell subsets (Th1/Th2).[9, 10] We hypothesized that precise control over the amount of T cell epitope within peptide co-assemblies would enable one to design a vaccine that preferentially favored the Tfh response and therefore maximized the antibody response.
To test this hypothesis, we controlled the amount of PADREQ11 that was mixed with E214Q11 before fiber formation, so that the PADRE concentration ranged from 0.005 to 0.5 mM. The E214 concentration was held constant at 1 mM, and the total peptide concentration (E214Q11 + PADREQ11 + Q11) was maintained at 2 mM by using unconjugated Q11 as filler. Mice were immunized at week 0 and boosted with half-doses at week 4 and week 8. At week 9, the total number of PADRE-specific CD4+ T cells measured by PADRE-tetramer staining (Figure 4a) followed a bell-shaped curve that peaked at the lowest concentration of PADREQ11, 0.005 mM, and dropped steadily at higher concentrations (Figure 4b). This nonlinearity in dose-response, especially hypo-responsiveness at very high doses of antigen, is consistent with previous reports[5, 27] and highlights the importance of selecting a proper dose of the T cell epitope. Bell-shaped profiles were also observed for the PADRE-specific Tfh, Th1, and Th2 responses (Figure 4c), measured in terms of the total numbers of CXCR5+ PD-1+, Tbet+, and Gata3+ cells, respectively, recovered from six draining lymph nodes per mouse. We confirmed the dose response profile for Th1 and Th2 cells by quantifying the frequency of IFN-[3] and IL-4 producing cells in a PADRE-specific ELISPOT assay (Figure 4d).
Figure 4. The activity of Tfh cells follows a different dose-response curve than Th1 or Th2 cells as T cell help is titrated into co-assembled nanofibers.
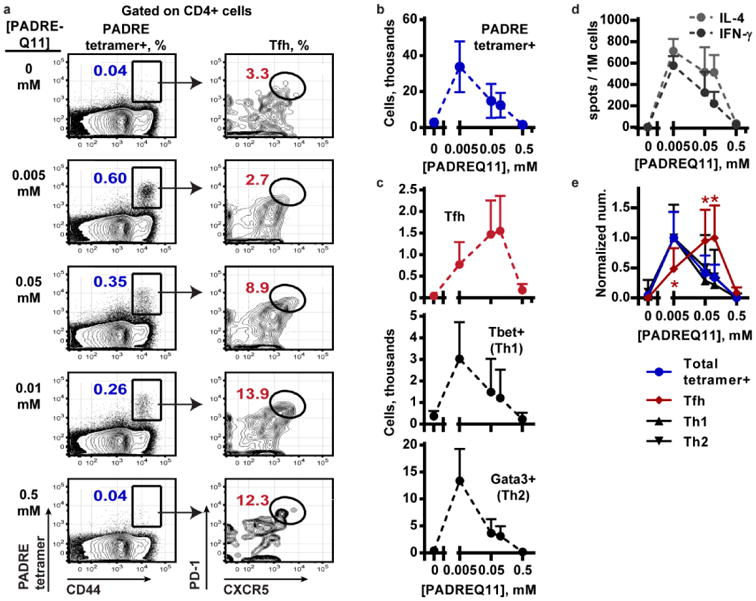
T cell responses were assessed using cells collected from the lymph nodes of mice after immunization and two boosts with E214Q11/PADREQ11. (a) The PADRE-MHC tetramer was used to quantify the PADRE-specific tetramer+ CD4+ T cells (left column) and phenotype them (gating for Tfh shown in right column). A representative result from each dose of PADREQ11 is shown. (b) The numbers of total PADRE tetramer-positive cells and (c) PADRE-specific CXCR5+ PD1+ Tfh cells, Tbet+ Th1, or Gata3+ Th2 cells were quantified. Cell numbers are quantified per mouse, where six draining lymph nodes were collected from each mouse. (d) An ELISPOT quantified the IL-4 and IFN-gamma secreting cells after stimulation with PADRE peptide, which also followed a bell-shaped dose response curve. (e) The response of each cell type was normalized to its maximum, and the responses were compared by 2-way ANOVA. Dunnett's multiple comparisons test was conducted at each dose of PADREQ11, comparing the response of each cell type to the total tetramer+ response. *, p < 0.001. N = 5 mice per group. Mean + standard deviation is shown.
Interestingly, the PADRE-specific Tfh response peaked at a higher dose than the Th1 or Th2 responses did: 0.05 – 0.10 mM PADREQ11 for Tfh, versus 0.005 mM for Th1 and Th2 (Figure 4e). At 0.10 mM PADREQ11, the Tfh cells were 13 ± 5 %, whereas at 0.005 mM, only 2 ± 1 % were Tfh cells (mean and standard deviation, n = 5). No skewing towards Th1 over Th2, or vice versa, was observed over the doses of PADREQ11 examined. Thus, increasing the dose of PADREQ11 in the nanofibers impacted both the magnitude and the quality of the effector CD4 T cells response. Different T cell populations exhibited distinct dose responses when the epitope content was varied, with Tfh development being selected at the higher concentrations of PADREQ11.
2.5. Antibody titers are optimized at the peak of the Tfh response
The magnitude of the antibody response should be controlled by the magnitude of CD4+ T cell response, particularly by the Tfh response.[4] To test whether titration of T cell epitopes could be used to optimize the antibody response to a vaccine, we measured the Ig titers against the E214 peptide using serum collected from the experiment described above (Figure 5a). At each dose of PADREQ11, the titers increased within 1 – 2 weeks after each boost. In the secondary and tertiary responses, the titers followed a bell-shaped dose-response curve that peaked at 0.05 – 0.1 mM PADREQ11 (Figure 5b). This was the same concentration as the maximal Tfh response. At the PADREQ11 dose of 0.5 mM, the anti-E214 titers decreased substantially, paralleling the reduced PADRE-specific Tfh numbers (Figure 4c). We further confirmed this high-dose hypo-responsiveness in a separate experiment using 1 mM PADREQ11 + 1 mM E214Q11, which raised log10 titers of only 2.0 ± 1.0 by week 8 (n = 3 mice). Thus, the development of the antibody titers reflected the development of the Tfh response both in its rise and its fall. Despite this correlation, we cannot exclude the possibility that other non-Tfh subsets also contribute to the antibody response across the entire PADREQ11 dose range.
Figure 5. Optimizing antibody titers to co-assembled fibers using T cell epitope titration.

(a) Fibers were co-assembled with E214Q11 and 5 different concentrations of PADREQ11, and mice were immunized and boosted (half-dose) with these formulations according the schedule shown. (b) The antibody response against E214 was measured over time by ELISA and (d) followed a bell-shaped dose response curve, shown 7 days after the first and second boosts. (c) Antibody isotypes were measured by ELISA and normalized to the total IgG response. Isotypes in responding groups consisted equally of IgG1 and IgG2b at all doses of PADREQ11, with only the lowest dose (0.005 mM) showing a significant polarization to IgG1 (serum from week 9; analyzed for each dose using a t-test; *, p<0.01). Contributions from IgG2c or IgG3 were negligible at all doses. (d) Competitive ELISA was used to compare antibody affinity for E214 in serum samples collected after the second boost. ELISA plates were coated with E214 at high (1 ug/mL) or low (0.1 ug/mL) density to assess lower and higher affinity antibodies, respectively. No significant differences were observed between doses of PADREQ11 or with the CRM+alum group, by 2-way ANOVA. N = 5 mice per group. Mean + standard deviation is shown.
In contrast to the changes in the magnitude of the T cell response and the antibody response relative to the dose of PADREQ11, the IgG isotypes were mostly unaffected. AntiE214 isotypes consisted primarily of a mixture of IgG1 and IgG2b (Figure 5c), except at the lowest dose of PADREQ11, where the IgG1 response was significantly greater than the IgG2b response. The average affinity for E214 was similarly unaffected by dose. Using a competitive ELISA, we observed IC50 values of approximately 0.6 μM and 2 μM IC50 at low and high densities of E214 coated on the ELISA plate, respectively, for all doses of PADREQ11 (Figure 5d). These IC50 values were not significantly different than those of the E214-CRM+alum group (0.2 μM and 0.7 μM, respectively), indicating that the optimized self-adjuvanting vaccine with a single CD4+ T cell epitope raised essentially the same affinity antibodies as the alum-adjuvanted protein conjugate.
3. Discussion
Historically, vaccines have been based on whole pathogens and protein antigens that include their own endogenous CD4+ T cell epitopes. Now, as vaccine design is focusing on ever more highly defined peptide and carbohydrate antigens,[1] CD4+ T cell help may have to be provided to maximize the T and B cell responses, without a priori knowledge of the optimal concentrations of epitopes. Currently antigens that have only B cell epitopes need to be chemically conjugated to protein carriers, usually large inactivated toxins such as tetanus toxoid, diphtheria toxoid, or diphtheria CRM197. While these carriers have been shown to be effective, they are not amenable to the efficient analysis of a wide variation in epitope content. In addition, their production processes can require multiple steps for expression and purification, can employ toxic coupling compounds and/or bacterial expression systems, and are prone to batch-to-batch variability.[28] These processes generate a vaccine product that is often a heterogeneous mix of conjugates, with different numbers of the B cell epitope bound to the protein. In addition, carrier proteins often contain their own B cell epitopes that are the preferential targets of the immune response, thus overriding the response to the antigen (immunodominance).[29] Furthermore, if the same carrier is re-used in a future vaccination, memory responses to the carrier can prevent response to the desired B cell epitope (carrier-specific suppression).[30]
To address some of the technical limitations of peptide/carrier subunit vaccines, here we took advantage of a vaccine system that has modular epitope content and does not require exogenous adjuvants to carefully quantify the effects of T cell antigen dose on the T cell and antibody responses. Our working model is that co-assembled E214Q11/PADREQ11 fibers are taken up by DCs that present PADRE in the context of MHC II molecules to T cells. Additionally, E214-specific B cells take up co-assembled fibers through BCR-mediated endocytosis and then present PADRE in the context of I-Ab to receive co-stimulatory signals from PADRE-specific T cells. The data in Figure 3b, demonstrating that co-assembly of the two epitopes is essential, is consistent with this model.
The ability to integrate epitopes in precise quantities over multiple orders of magnitude into a single construct is a feature that is perhaps unique to self-assembly of small monomers. Covalent conjugation of peptides to intact proteins by chemical linkages would be difficult to regulate over the micromolar to millimolar range. Self-assembly of proteins or polypeptide subunits typically incorporates on the order of tens to hundreds of subunits into spherical or low aspect ratio particles,[31] so if a single epitope is added to each monomer, the available concentration span has at most a hundred-fold range. In contrast, self-assembly of short peptides into long, high aspect ratio, beta-sheet fibers incorporates thousands of monomers per micron length (each monomer is approximately 0.5 nm)[32]. Emulsification of peptides, e.g. in Freund's adjuvant, can achieve this range and has been used to combine B cell or CD8+ T cell with CD4+ T cell epitopes previously,[21, 33] but these adjuvants also induce local inflammation that can alter the helper T cell response. Based on our observations, we anticipate that the construction of other types of modular self-adjuvanting nanoparticles will provide a broad platform for controlled epitope dosage in vaccine design.
As a proof of principle, we focused on using non-covalent self-assembly to specify the dose of CD4+ T cell epitopes, in order to favor the emergence of distinct CD4+ T cell subsets and antibody responses after vaccination. It is generally accepted that the quality of the TCR-peptide-MHCII interactions impact the magnitude of clonal expansion and the acquisition of distinct effector CD4+ T helper cell functions.[20, 34] The quality of interaction is a combination of the TCR affinity for peptide-MHCII,[10] the off rate of TCR binding to peptide-MHCII,[35] abundance of antigen,[36, 37] and the duration of Th-APC contact.[36, 38] Furthermore, in many experiments, the incorporation of adjuvants can obscure the effects of TCR signal strength and alter the clonal composition of the responding T cell population.[10,20] By using non-covalent self-assembly to titrate minimal T cell epitopes across multiple orders of magnitude, we were able to demonstrate that, in the absence of inflammation, the magnitude of the T cell as well as the antibody response also varied by multiple orders of magnitude. Notably, this vaccine system favored the development of Th2 cells, and a ten-fold higher antigen dose was required for optimal Tfh and antibody responses than for optimal Th1 and Th2 responses. These results are consistent with a recent report,[9] where strong TCR signaling inhibited Th1 formation and favored Tfh formation by naïve cells in an infectious model. Finally, the mechanistic basis for a depressed PADRE-specific T cell response at the highest high doses of PADRE is not known, and the possibility of regulatory T cell involvment is under investigation.
The nanofiber co-assembly system based on synthetic peptides has many advantages and also some significant limitations in terms of the range of epitopes can be presented. Whereas CD4+ and CD8+ T cell epitopes are linear, B cell epitopes can be either linear (such as the E214 epitope used here) or conformational (a discontinous sequence requiring proper folding to be recognized by the B cell receptor). Linear peptide epitopes of up to approximately 30 - 40 amino acids in length are ideal candidates for solid-phase synthesis and co-assembly on nanofibers, although it can be challenging to synthesize very hydrophobic sequences in high purity. It may also be possible to synthesize carbohydrate epitopes in tandem with the peptide co-assembly domain. Further, we have recently developed a strategy for incorporating precise combinations of whole proteins into beta-sheet nanofibers by expressing them with a tag that transitions from an alpha-helical to a beta-sheet structure. Antibodies can be raised against such proteins co-assembled into Q11 nanofibers.[39] Alternatively, proteins can also be attached to Q11 nanofibers once the peptide has self-assembled, and these also raise antibody responses.[40] We have not tested whether protein-conjugated nanofibers are processed in the same manner as peptide-displaying fibers, nor whether co-assembly with a precise dose of an epitope such as PADRE would enhance the response to a protein. However, most proteins have abundant CD4+ T cell epitopes, so they may not benefit as much from co-assembly with additional CD4+ T cell epitopes. Thus future experiments will focus on expanding the nanofiber platform to incorporate a wider range of antigen types, and on testing the functionality of the elicited immune response in live infection models in mice.
Precise control over epitope dose to determine the quality and magnitude of the CD4+ T cell and antibody responses contrasts with traditional vaccine design, which relies on the non-specific action of exogenous inflammatory adjuvants to drive the responses to subunit vaccines.[41] Aluminum salts, including aluminum hydroxide, aluminum phosphate, alum or mixed aluminum salts, have been the predominant adjuvant for clinical use. A number of new adjuvants also have been approved, including AS04, which incorporates monophosphoryl lipid A (MPL) in aluminum hydroxide, and AS03 and MF59, which are squalene-based emulsions with or without additional immunomodulators, respectively. Specifically, the incorporation of immune modulatory molecules, such as ligands for Toll-like receptors, has been particularly effective at enhancing the immune response. However, the responses to all epitopes in the vaccine construct are equally enhanced, potentially leading to opportunities for immunodominance and carrier-specific suppression.[29] Furthermore, these adjuvants make the assessment of the safety profile of the vaccine more difficult; for instance, the administration of millions of doses of the AS03-adjuvanted vaccine for the 2009 H1N1 influenza pandemic unexpectedly revealed an increased risk of narcolepsy.[42] The lack of specificity and concerns over unanticipated side effects, in addition to the known effects of adjuvants in inducing inflammation and pain at the site of injection, underscore the need for a new generation of subunit vaccines that work outside of this paradigm. The recent emergence of a new class of chemically defined nanoparticulate vaccines that are effective without exogenous adjuvants or the inclusion of TLR agonists,[11, 13, 18, 19, 43] including the self-assembled peptide nanofibers described here, provides growing evidence that such vaccines may be possible. As such materials and platforms continue to be developed, careful tuning of epitope content may be critical for adjusting the strength and quality of the immune response, as illustrated here. This consideration may be even more important for unadjuvanted particulates than vaccines containing exogenous adjuvants, as the adjuvant may override or mask the effects of epitope content.[10, 20]
4. Conclusion
This study demonstrates that modular assembly can be used to vary the T epitope dose within non-inflammatory materials-based vaccines, enable strong T cell and antibody responses, and specify the resulting CD4 effector types. Maximizing the Tfh response is critical, as the strength of the Tfh response is tightly associated with the magnitude of the antibody responses and protection in human vaccination.[44] This is especially true for vaccines against extracellular bacteria (e.g. S. Pneumoniae and Meningococcus), as well as for influenza, where high titers of neutralizing antibodies and B cell memory have been shown to be correlated with protection.[45] In contrast, the ability to optimize for effector Th1 and Th2 responses would important for the generation of vaccines targeting cancer immunotherapy, viral infections, and parasitic infections. Thus, using modular self-assembly to precisely incorporate B cell and T cell epitopes into vaccine should allow for efficient optimization of vaccines that can elicit the appropriate types of protective immune responses.
5. Experimental Section
Peptide synthesis and characterization
Peptides were synthesized using standard Fmoc solid-phase chemistry, purified by HPLC and MALDI-MS, and lyophilized as previously reported [15]. A list of peptides with sequences and molecular weights is provided in the Supplementary Information (Table S1). C-terminally biotinylated E214 (MW = 2435 g/mol) was prepared by synthesizing E214 on Fmoc-PEG-biotin Novatag resin (Novabiochem #8.55145.8500); preliminary tests showed that N-terminal biotinylation prevented recognition of the E214 epitope in ELISA. For TEM analysis, peptide solutions were prepared as for immunization, diluted to 0.4 mM in 1× PBS immediately before deposition on 400 mesh carbon grids, stained with 1% uranyl acetate, and imaged immediately using a FEI Tecnai F30.
Vaccine preparation
To prepare immunization solutions from PADRE-Q11 co-assembled with Q11 and/or E214-Q11, PADRE-Q11 was first dissolved in sterile water at 0.1 - 0.2 mM and used immediately or frozen at −20 °C until use. The appropriate quantities of lyophilized Q11 and/or E214-Q11 powders were vortexed together for at least 25 min before being dissolved in the PADRE-Q11 solution or in sterile water (4 mM total peptide) and stored overnight at 4 °C. Then, sterile water and 10× PBS (Fisher BP399-500) were added to bring the mixture to the working concentration (2 mM total peptide, 1× PBS), and the solution was incubated for 3- 5 hr at room temperature before immunization. The final pH of the PADREQ11/E214-Q11/Q11 solution was 6.5 at all doses of PADRE-Q11, as measured by pH paper. We verified by TEM imaging that fibers were formed at all doses of PADRE-Q11. For immunizations with Imject Alum, a 4 mM working solution of peptide was mixed 1:1 v/v with Imject Alum (Pierce) and vortexed for 30 – 60 min to allow adsorption. The E214-CRM conjugate was synthesized by Merck by using E214-ahx-Cys to link through the thiol group. Ahx is a linker, 6-aminohexanoic acid.
To prepare OVA-Q11 with or without co-assembled PADRE-Q11, OVA-Q11 powder was dissolved in freshly prepared 0.4 mM PADRE-Q11 or in sterile water to produce 8 mM OVA-Q11. This solution was stored overnight at 4 °C, and then PBS was added to bring the solution to 2 mM OVA-Q11 ± 0.1 mM PADRE-Q11. This solution was incubated for 3- 5 hr at room temperature before immunization.
Endotoxin measurements were conducted on the same peptide solutions that were used for immunization, using the Limulus Amebocyte Lysate chromogenic endpoint assay (Lonza). Endotoxin levels of all peptide solutions used for immunization were < 0.1 EU/mL (< 0.01 EU per 100 μL dose).
Mice and Immunizations
Female C57BL/6 mice were purchased from Harlan laboratory and housed at the animal facility at the University of Chicago. All procedures were approved by the University of Chicago Institutional Animal Care and Use Committee (protocol 71-900). Mice (6 – 12 weeks old; age-matched within each experiment) were randomly assigned in groups of 5 for each condition based on previous findings that this size was sufficient to distinguish responding versus non-responding groups. Anesthetized mice were immunized subcutaneously with the indicated solutions (2 × 50 μL at the shoulders, 200 nmol total peptide) and boosted where indicated with half-doses (2 × 25 μL, 100 nmol total peptide) after 4 weeks and 8 weeks. Serum was collected from the submandibular (cheek) vein for analysis by ELISA. Mice were sacrificed 7 days after the final immunization, and the cells from the draining lymph nodes (axillary, brachial, and inguinal) were collected. Investigator blinding was not used during the experiment.
Testing the requirement for co-assembly
Mice were immunized with co-assembled E214-Q11/ PADRE-Q11/ Q11 (1 mM, 0.05 mM, 0.95 mM, respectively) prepared as described above, or with an injection of E214-Q11 (1 mM) and a separate injection of co-assembled PADRE-Q11/ Q11 (0.05 mM, 0.95 mM, respectively). The separate injections were given at the same time and only approximately 3 mm apart on the shoulder of the animal, to allow drainage to the same lymph nodes. Mice were boosted in the same manner with a half-dose at week 4. Serum was collected at week 5 for analysis by ELISA.
Testing the dose-response to PADRE-Q11
Mice were immunized with co-assembled E214-Q11/ PADRE-Q11/ Q11 (1 mM, 0.005 – 1 mM, 0.995 – 0 mM, respectively) prepared as described above, and boosted with a half-dose at weeks 4 and 8. Serum was collected every 1 – 2 weeks for analysis by ELISA. Antibody affinity and isotypes was measured using serum from week 9. Mice were sacrificed at week 9, lymph nodes were collected, and the T cell response was analyzed by ELISPOT and tetramer-staining.
Antibodies and Flow Cytometry
Antibodies were purchased from eBioscience unless specified. Flow cytometry was performed using LSRII blue (BD). FlowJo (Tree Star Inc.) was used in the analysis of flow data. For analysis of cell recruitment to the peritoneal cavity, cells isolated from i.p. lavage fluid were stained and analyzed as described [19].
PADRE-specific T cell staining
PADRE tetramers were obtained as a custom order from the NIH Tetramer Core Facility (I-Ab MHC class II tetramer against sequence AKFVAAWTLKAA). Draining lymph nodes were collected and processed into a single cell suspension. Then, 2 to 5 million cells were resuspended in 50 μL of flow staining buffer (PBS containing 2 % FCS and 0.01% sodium azide) and pre-blocked with 2.4G2 antibody for blocking non-specific FCR binding site for 5 min at 4 °C. PE-conjugated PADRE tetramer was added at a final concentration of 2.5 μg/mL, and the suspension was incubated at 37 °C in a cell culture incubator for 1 hour. Cells were washed with 2 mL of flow cytometry buffer and then stained with other antibodies as needed for each cell type. For Tfh staining, cells were stained with CD44-APC-Cy7 (IM7, BD Biosciences), CXCR5-APC (2G8, BD Biosciences), PD-1-PE-CY7 (J43), CD4-FITC (GK1.5, Biolegend), and lineage antibodies (i.e. a dump channel) including CD8, B220, Ter119, DX5 and F4/80. For T-bet and GATA-2 intracellular staining, cells were stained with LIVE/DEAD Fixable Violet Dead Cell Staining regent (L34955, Molecular probes) to discriminate live and dead cells according to the manufacturer instructions, then stained with PE-conjugated PADRE tetramer, then with CD4-FITC and CD44-APC-CY7. Foxp3 / Transcription Factor Staining Buffer Set (00-5523-00, eBioscience) was then used for T-bet-PerCP-Cy5.5 (eBio4B10) and GATA3-PE-CY7 (L50-823, BD Biosciences) staining according to its product information.
T cell ELISPOT
T cell ELISPOTs were performed as described [19]. Briefly, 0.5 million cells from the draining lymph nodes were plated in each well of a 96-well ELISPOT plate (Millipore, MSIPS4510), in 200 μL per well. The cells were then stimulated with peptide or left untreated as negative controls. Preliminary experiments showed that the response to PADRE saturated at concentrations above 0.5 μM. Therefore, stimulation was performed with 1 μM PADRE; when E214-Q11 or pOVA were used, they were included at 5 μM. IL-4 (551818) or INF-[3] (551881) ELISPOT Pairs were from BD. Streptavidin-alkaline phosphatase (3310-10) was purchased from Mabtech. Spots were developed using substrate Sigmafast BCIP/NBT (Sigma, B5655). Plates were imaged and enumerated using an ELISPOT reader (Cellular Technology, Ltd).
ELISA for serum antibodies
Serum was analyzed for antigen-specific Ig (Anti-IgG (H+L), Jackson Immuno Research, Cat #115-035-003) or IgG (gamma-specific, #115-035-071) by ELISA, as previously described (Rudra, Tian et al. 2010). To detect E214-specific antibodies, the plate was coated with 5 μg/mL E214-ahx-C (provided by Merck) in PBS or with 5 μg/mL streptavidin (Sigma #85878) followed by 10 μg/mL E214-PEG-biotin in PBS. To detect antibodies specific for Q11 or PADRE-Q11, the plate was coated with 20 μg/mL SGSG-Q11 or PADRE-Q11 in PBS. Isotyping was conducted similarly except that alkaline-phosphataseconjugated antibodies for total IgG (#155-055-071) or IgG1, IgG2b, IgG2c, or IgG3 (#155-055-205, 155-055-207, 155-055-208, 155-055-209, respectively) were used (diluted 1:5000) along with SigmaFast pNPP substrate.
Competitive ELISA
To assess antibody affinity, high-binding ELISA plates were coated overnight with streptavidin (5 μg/mL) and then with E214-PEG-biotin. Preliminary experiments showed that the signal saturated at E214-PEG-biotin concentrations above 0.2 μg/mL. Thus, 0.1 or 1 μg/mL E214-PEG-biotin was used as a low-density and high-density coating, respectively. Lower-density coatings select for higher-affinity antibodies in this assay. Plates were blocked for 1 hr with 1% BSA. Equal volumes of sera from the 5 mice in each group were pooled to create a single sample per group. Each pooled serum sample was used at a single dilution that gave approximately half the maximal possible ELISA signal (e.g., approximately 1:104 for a serum with a titer of 104). After blocking, E214 peptide solution was added to the wells in a dilution series starting at 10-4 M, with seven 10-fold or 5-fold steps plus a buffer control. The diluted serum sample was added immediately, mixed, and the plate was incubated for 2 hr. Plate-bound IgG (gamma-specific) was detected as described above. To calculate the IC50, the ELISA absorbance was plotted against the log of the E214 concentration (in Molar) and fitted with a sigmoidal curve in Prism 6 (log(inhibitor) vs. response (three parameters)) to determine the concentration that gave half-maximal inhibition. The assay was performed twice, and the average and standard deviation reported for each serum sample.
Supplementary Material
Figure S1. Validation of the specificity of PADRE-tetramer staining. Mice were immunized with PADREQ11 (0.05 mM co-assembled with 1.95 mM Q11), and the draining lymph nodes were collected after 7 days. Cells were stained for CD4, lineage markers (i.e. a “dump” channel for CD8 T cells, B cells, dendritic cells, and macrophages) and either the PADRE-MHC tetramer (a) or the control CLIP tetramer (b). Only the PADRE-MHC tetramer stained the cells.
Figure S2. A T cell epitope is required for the boost. Mice were initially immunized with co-assembled E214-Q11 (E-Q11)/PADRE-Q11 (1 mM E214-Q11 + 0.05 mM PADRE-Q11 + 0.95 mM Q11), and then boosted with either E-Q11/PADRE-Q11 co-assemblies (same concentrations as in primary immunization), EQ11 assembled alone (1 mM E214-Q11 + 1 mM Q11), or phosphate-buffered saline. Only the group that received PADRE-Q11 in the booster solution showed a booster response. N = 5 mice per group, analyzed by 2-way ANOVA. Error bars show standard deviation.
Table S1. Peptides used in this paper. All peptides were purified by HPLC, and their masses were confirmed by MALDI (within 2 m/z of the predicted molecular weight + 1 H+).
Acknowledgments
We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for provision of the MHC class II tetramer for PADRE. This publication was made possible by grant numbers AI094444 (NIH/NIAID), EB009701 (NIH/NIBIB), and OPP1061315 (Bill & Melinda Gates Foundation), and by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of these agencies.
Footnotes
Supporting Information: Supporting Information is available from the Wiley Online Library or from the author.
The authors have no conflicting financial interests.
Contributor Information
Rebecca R. Pompano, Email: rpompano@uchicago.edu, Department of Surgery, Committee of Immunology, University of Chicago, 5841 S. Maryland Avenue, MC5032, Chicago, IL 60637 USA.
Jianjun Chen, Department of Surgery, Committee of Immunology, University of Chicago, 5841 S. Maryland Avenue, MC5032, Chicago, IL 60637 USA.
Emily A. Verbus, Department of Surgery, Committee of Immunology, University of Chicago, 5841 S. Maryland Avenue, MC5032, Chicago, IL 60637 USA
Huifang Han, Department of Surgery, Committee of Immunology, University of Chicago, 5841 S. Maryland Avenue, MC5032, Chicago, IL 60637 USA.
Arthur Fridman, Merck Research Labs, West Point, PA 19486 USA.
Tessie McNeely, Merck Research Labs, West Point, PA 19486 USA.
Joel H. Collier, Email: jcollier@surgery.bsd.uchicago.edu, Department of Surgery, Committee of Immunology, University of Chicago, 5841 S. Maryland Avenue, MC5032, Chicago, IL 60637 USA.
Anita S. Chong, Email: achong@surgery.bsd.uchicago.edu, Department of Surgery, Committee of Immunology, University of Chicago, 5841 S. Maryland Avenue, MC5032, Chicago, IL 60637 USA.
References
- 1.Purcell AW, McCluskey J, Rossjohn J. Nat Rev Drug Discov. 2007;6:404. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 2.Chentoufi A, Nesburn A, BenMohamed L. Arch Immunol Ther Exp. 2009;57:409. doi: 10.1007/s00005-009-0049-2. [DOI] [PubMed] [Google Scholar]
- 3.Rappuoli R, De Gregorio E. Nat Med. 2011;17:1551. doi: 10.1038/nm.2587. [DOI] [PubMed] [Google Scholar]
- 4.Crotty S. Annu Rev Immunol. 2011;29:621. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 5.Stone JD, Cochran JR, Stern LJ. Biophys J. 2001;81:2547. doi: 10.1016/S0006-3495(01)75899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogers PR, Croft M. J Immunol. 1999;163:1205. [PubMed] [Google Scholar]
- 7.Mirshahidi S, Huang CT, Sadegh-Nasseri S. J Exp Med. 2001;194:719. doi: 10.1084/jem.194.6.719. [DOI] [PMC free article] [PubMed] [Google Scholar]; Turner MS, Kane LP, Morel PA. J Immunol. 2009;183:4895. doi: 10.4049/jimmunol.0901459. [DOI] [PMC free article] [PubMed] [Google Scholar]; Molinero LL, Miller ML, Evaristo C, Alegre ML. J Immunol. 2011;186:4609. doi: 10.4049/jimmunol.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barwig C, Raker V, Montermann E, Grabbe S, Reske-Kunz AB, Sudowe S. Clin Exp Allergy. 2010;40:891. doi: 10.1111/j.1365-2222.2010.03476.x. [DOI] [PubMed] [Google Scholar]; Sakai, Yokoyama, Kohno, Hiwada Clin Exp Immunol. 1999;118:9. doi: 10.1046/j.1365-2249.1999.01036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tubo Noah J, Pagán Antonio J, Taylor Justin J, Nelson Ryan W, Linehan Jonathan L, Ertelt James M, Huseby Eric S, Way Sing S, Jenkins Marc K. Cell. 2013;153:785. doi: 10.1016/j.cell.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. Nat Immunol. 2009;10:375. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jennings GT, Bachmann MF. Biol Chem. 2008;389:521. doi: 10.1515/bc.2008.064. [DOI] [PubMed] [Google Scholar]
- 12.Mahajan B, Berzofsky JA, Boykins RA, Majam V, Zheng H, Chattopadhyay R, de la Vega P, Moch JK, Haynes JD, Belyakov IM, Nakhasi HL, Kumar S. Infect Immun. 2010;78:4613. doi: 10.1128/IAI.00533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; Calvo-Calle JM, Oliveira GA, Watta CO, Soverow J, Parra-Lopez C, Nardin EH. Infect Immun. 2006;74:6929. doi: 10.1128/IAI.01151-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bettahi I, Dasgupta G, Renaudet O, Chentoufi A, Zhang X, Carpenter D, Yoon S, Dumy P, BenMohamed L. Cancer Immunol Immunother. 2009;58:187. doi: 10.1007/s00262-008-0537-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kaba SA, McCoy ME, Doll T, Brando C, Guo Q, Dasgupta D, Yang YK, Mittelholzer C, Spaccapelo R, Crisanti A, Burkhard P, Lanar DE. PLoS One. 2012;7:e48304. doi: 10.1371/journal.pone.0048304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaba SA, Brando C, Guo Q, Mittelholzer C, Raman S, Tropel D, Aebi U, Burkhard P, Lanar DE. J Immunol. 2009;183:7268. doi: 10.4049/jimmunol.0901957. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fifis T, Mottram P, Bogdanoska V, Hanley J, Plebanski M. Vaccine. 2004;23:258. doi: 10.1016/j.vaccine.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Tam JP, Clavijo P, Lu YA, Nussenzweig V, Nussenzweig R, Zavala F. J Exp Med. 1990;171:299. doi: 10.1084/jem.171.1.299. [DOI] [PMC free article] [PubMed] [Google Scholar]; Falugi F, Petracca R, Mariani M, Luzzi E, Mancianti S, Carinci V, Melli ML, Finco O, Wack A, Tommaso AD, Magistris MTD, Costantino P, Giudice GD, Abrignani S, Rappuoli R, Grandi G. Eur J Immunol. 2001;31:3816. doi: 10.1002/1521-4141(200112)31:12<3816::AID-IMMU3816>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 15.Jung JP, Nagaraj AK, Fox EK, Rudra JS, Devgun JM, Collier JH. Biomaterials. 2009;30:2400. doi: 10.1016/j.biomaterials.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang M, Mine Y. Biochem Biophys Res Comm. 2009;378:203. doi: 10.1016/j.bbrc.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 17.Rudra JS, Mishra S, Chong AS, Mitchell RA, Nardin EH, Nussenzweig V, Collier JH. Biomaterials. 2012;33:6476. doi: 10.1016/j.biomaterials.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; Rudra JS, Sun T, Bird KC, Daniels MD, Gasiorowski JZ, Chong AS, Collier JH. ACS Nano. 2012;6:1557. doi: 10.1021/nn204530r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudra JS, Tian YF, Jung JP, Collier JH. Proc Natl Acad Sci U S A. 2010;107:622. doi: 10.1073/pnas.0912124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, Pompano RR, Santiago FW, Maillat L, Sciammas R, Sun T, Han H, Topham DJ, Chong AS, Collier JH. Biomat. 2013;34:8776. doi: 10.1016/j.biomaterials.2013.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malherbe L, Mark L, Fazilleau N, McHeyzer-Williams LJ, McHeyzer-Williams MG. Immunity. 2008;28:698. doi: 10.1016/j.immuni.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, Snoke K, Serra HM, Kubo RT, Sette A, Grey HM. Immunity. 1994;1:751. doi: 10.1016/s1074-7613(94)80017-0. [DOI] [PubMed] [Google Scholar]
- 22.delGuercio MF, Alexander J, Kubo RT, Arrhenius T, Maewal A, Appella E, Hoffman SL, Jones T, Valmori D, Sakaguchi K, Grey HM, Sette A. Vaccine. 1997;15:441. doi: 10.1016/s0264-410x(97)00186-2. [DOI] [PubMed] [Google Scholar]
- 23.Dutton RW, Bradley LM, Swain SL. Ann Rev Immunol. 1998;16:201. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- 24.Cope L, Fridman A, Joshi A, Mcneely TB, Pak I, Smith S. WO2012065034 A1. 2012
- 25.Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B. Nucleic Acids Res. 2010;38:D854. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kool M, Soullie T, van Nimwegen M, Willart MAM, Muskens F, Jung S, Hoogsteden HC, Hammad H, Lambrecht BN. J Exp Med. 2008;205:869. doi: 10.1084/jem.20071087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aichele P, Brduscha-Riem K, Zinkernagel RM, Hengartner H, Pircher H. J Exp Med. 1995;182:261. doi: 10.1084/jem.182.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dintzis HM, Dintzis RZ, Vogelstein B. Proc Natl Acad Sci U S A. 1976;73:3671. doi: 10.1073/pnas.73.10.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.C. C. Peeters, P. R. Lagerman, O. Weers, L. A. Oomen, P. Hoogerhout, M. Beurret, J.T. Poolman, K. M. Reddin, Vol. 87, 2003, 153.
- 29.Sette A, Fikes J. Curr Opin Immunol. 2003;15:461. doi: 10.1016/s0952-7915(03)00083-9. [DOI] [PubMed] [Google Scholar]
- 30.Dagan R, Poolman J, Siegrist CA. Vaccine. 2010;28:5513. doi: 10.1016/j.vaccine.2010.06.026. [DOI] [PubMed] [Google Scholar]; Herzenberg LA, Tokuhisa T, Hayakawa K. Annu Rev Immunol. 1983;1:609. doi: 10.1146/annurev.iy.01.040183.003141. [DOI] [PubMed] [Google Scholar]; Jegerlehner A, Wiesel M, Dietmeier K, Zabel F, Gatto D, Saudan P, Bachmann MF. Vaccine. 2010;28:5503. doi: 10.1016/j.vaccine.2010.02.103. [DOI] [PubMed] [Google Scholar]
- 31.Kratz PA, Böttcher B, Nassal M. Proc Natl Acad Sci. 1999;96:1915. doi: 10.1073/pnas.96.5.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]; Raman S, Machaidze G, Lustig A, Aebi U, Burkhard P. Nanomedicine: NBM. 2006;2:95. doi: 10.1016/j.nano.2006.04.007. [DOI] [PubMed] [Google Scholar]; Tissot AC, Renhofa R, Schmitz N, Cielens I, Meijerink E, Ose V, Jennings GT, Saudan P, Pumpens P, Bachmann MF. PLoS One. 2010;5:e9809. doi: 10.1371/journal.pone.0009809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aggeli A, Bell M, Boden N, Keen JN, McLeish TCB, Nyrkova I, Radford SE, Semenov A. J Mater Chem. 1997;7:1135. [Google Scholar]
- 33.Sarobe P, Lasarte JJ, Golvano J, Gullón A, Civeira MP, Prieto J, Borrás-Cuesta F. Eur J Immunol. 1991;21:1555. doi: 10.1002/eji.1830210633. [DOI] [PubMed] [Google Scholar]
- 34.Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, Killeen N, Orange JS, Russell SM, Weninger W, Reiner SL. Science. 2007;315:1687. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]; Lanzavecchia A, Sallusto F. Nat Rev Immunol. 2002;2:982. doi: 10.1038/nri959. [DOI] [PubMed] [Google Scholar]; Reiner SL, Sallusto F, Lanzavecchia A. Science. 2007;317:622. doi: 10.1126/science.1143775. [DOI] [PubMed] [Google Scholar]
- 35.Savage PA, Boniface JJ, Davis MM. Immunity. 1999;10:485. doi: 10.1016/s1074-7613(00)80048-5. [DOI] [PubMed] [Google Scholar]
- 36.Vanguri V, Govern CC, Smith R, Huseby ES. Proc Nat Acad Sci. 2013;110:288. doi: 10.1073/pnas.1208328110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rees W, Bender J, Teague TK, Kedl RM, Crawford F, Marrack P, Kappler J. Proc Natl Acad Sci U S A. 1999;96:9781. doi: 10.1073/pnas.96.17.9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iezzi G, Karjalainen K, Lanzavecchia A. Immunity. 1998;8:89. doi: 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]; Miller MJ, Safrina O, Parker I, Cahalan MD. J Exp Med. 2004;200:847. doi: 10.1084/jem.20041236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hudalla GA, Sun T, Gasiorowski JZ, Han H, Tian YF, Chong AS, Collier JH. Nat Mat. 2014 doi: 10.1038/nmat3998. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudalla GA, Modica JA, Tian YF, Rudra JS, Chong AS, Sun T, Mrksich M, Collier JH. Adv Healthc Mater. 2013;2:1114. doi: 10.1002/adhm.201200435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reed SG, Orr MT, Fox CB. Nat Med. 2013;19:1597. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 42.Nohynek H, Jokinen J, Partinen M, Vaarala O, Kirjavainen T, Sundman J, Himanen SL, Hublin C, Julkunen I, Olsen P, Saarenpaa-Heikkila O, Kilpi T. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bershteyn A, Hanson MC, Crespo MP, Moon JJ, Li AV, Suh H, Irvine DJ. J Control Release. 2012;157:354. doi: 10.1016/j.jconrel.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ulery BD, Kumar D, Ramer-Tait AE, Metzger DW, Wannemuehler MJ, Narasimhan B. PLoS One. 2011;6:e17642. doi: 10.1371/journal.pone.0017642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, Flano E, Mejias A, Albrecht RA, Blankenship D, Xu H, Pascual V, Banchereau J, Garcia-Sastre A, Palucka AK, Ramilo O, Ueno H. Sci Trans Med. 2013;5:176ra32. doi: 10.1126/scitranslmed.3005191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plotkin SA. Pediatr Infect Dis J. 2001;20:63. doi: 10.1097/00006454-200101000-00013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Validation of the specificity of PADRE-tetramer staining. Mice were immunized with PADREQ11 (0.05 mM co-assembled with 1.95 mM Q11), and the draining lymph nodes were collected after 7 days. Cells were stained for CD4, lineage markers (i.e. a “dump” channel for CD8 T cells, B cells, dendritic cells, and macrophages) and either the PADRE-MHC tetramer (a) or the control CLIP tetramer (b). Only the PADRE-MHC tetramer stained the cells.
Figure S2. A T cell epitope is required for the boost. Mice were initially immunized with co-assembled E214-Q11 (E-Q11)/PADRE-Q11 (1 mM E214-Q11 + 0.05 mM PADRE-Q11 + 0.95 mM Q11), and then boosted with either E-Q11/PADRE-Q11 co-assemblies (same concentrations as in primary immunization), EQ11 assembled alone (1 mM E214-Q11 + 1 mM Q11), or phosphate-buffered saline. Only the group that received PADRE-Q11 in the booster solution showed a booster response. N = 5 mice per group, analyzed by 2-way ANOVA. Error bars show standard deviation.
Table S1. Peptides used in this paper. All peptides were purified by HPLC, and their masses were confirmed by MALDI (within 2 m/z of the predicted molecular weight + 1 H+).