

Abstract
Bacterial surface colonizers are subject to a variety of physical stresses. During the colonization of human epithelia such as on the skin or the intestinal mucosa, bacteria mainly have to withstand the mechanical stress of being removed by fluid flow, scraping, or epithelial turnover. To that end, they express a series of molecules to establish firm attachment to the epithelial surface, such as fibrillar protrusions (pili) and surface-anchored proteins that bind to human matrix proteins. In addition, some bacteria – in particular gut and urinary tract pathogens – use internalization by epithelial cells and other methods such as directed inhibition of epithelial turnover to ascertain continued association with the epithelial layer. Furthermore, many bacteria produce multi-layered agglomerations called biofilms with a sticky extracellular matrix, providing additional protection from removal. This review will give an overview over the mechanisms human bacterial colonizers have to withstand physical stresses with a focus on bacterial adhesion.
Keywords: Adhesion, Staphylococcus, Streptococcus, Pseudomonas aeruginosa, Escherichia coli, Helicobacter pylori
Introduction
In their natural environment, bacteria have to cope with a variety of physical stresses. Most bacteria live in a surface-attached form. During colonization they thus first need to make sure that attachment is established. Later on, they must defend themselves against forces that may detach them from the surface, such as mechanical stresses exerted by fluid flow or any kind of scraping. To that end, many bacteria “encapsulate” themselves in a sticky matrix called biofilm (Costerton et al., 1995). Bacteria that live in the environment or on abiotic man-made surfaces often establish attachment only by hydrophobic or electrostatic interaction. Bacteria that colonize animal or human tissue additionally use specific mechanisms of adhesion to host proteins or other macromolecules for attachment. The molecular factors underlying the subsequent cellular agglomeration phase are in principle similar between colonizers of animals and those of abiotic surfaces and involve molecules that cause intercellular aggregation. In multi-species communities, they may include interaction with the surfaces of other colonizing organisms.
Other physical and physico-chemical stresses that may be harmful to bacteria during the colonization of surfaces are osmotic stress, ultraviolet radiation, and shifts in pH value, to name but a few. Bacteria living in environments with sun exposure need mechanisms to withstand UV rays, while skin bacteria often experience shifts in salt concentration. Extreme pH values may be encountered for example by bacteria living in the stomach.
In the present review, molecular mechanisms that bacteria have developed to withstand physical stresses during colonization will be reviewed. As this is a vast field, these molecular principles will be explained using examples from bacteria colonizing humans. Furthermore, because colonization is a frequent source of infections, especially those bacteria will be addressed that are pathogens or opportunistic pathogens. As for osmotic, UV, and acid stresses, the reader is referred to dedicated reviews. This review will primarily address mechanisms of attachment, aggregation, and evasion of epithelial turnover of human bacterial pathogens, as these are the most important mechanisms that ensure bacterial colonization under the constant pressure of mechanical removal. Furthermore, it should be noted that colonizers of humans are also exposed to a variety of mechanisms of innate and adapted host defense and have developed specific resistance mechanisms to cope with those (Fedtke et al., 2004).
Colonization of the skin
The skin is the largest human organ and is colonized by a wide variety of microorganisms (Roth & James, 1988). Many occupy specific niches that differ in many aspects, such as regarding humidity and the chemical composition of host-produced secretions. The skin environment is in general dry and acidic, but more moist conditions are found in glands and hair follicles. The surface of the skin is composed of keratinocytes, which are terminally differentiated at the most exposed layers. This surface is disrupted by invaginations – sebaceous, apocrine and eccrine (sweat) glands and hair follicles (Wysocki, 1995). Eccrine glands secrete water and electrolytes, and contribute to the acidification of the skin. Apocrine glands, found mostly in the armpits and anogenital regions, produce viscous secretions. Sebaceous glands are connected to hair follicles and produce sebum. Sebum is an oily/waxy matter containing for example triglycerides that are degraded by propionibacteria to produce free fatty acids, which also contribute to the acidic pH of the skin surface (Ingham et al., 1981, Christensen & Bruggemann, 2013). Notably, the density of glands differs significantly between different areas of the skin; and those with low density such as arms and legs are in general dryer and contain a lower density of bacteria. In addition to moisture, differences in temperature may lead to differences in the degree of bacterial skin colonization.
The skin is host to a wide variety of colonizing bacteria. According to both classical, culture-based studies as well as recent metagenomic investigations, Propionibacterium ssp. dominates in sebaceous sites and staphylococci and corynebacteria in moist areas (Grice & Segre, 2011). 16S rRNA metagenomic sequencing analyses revealed a great variety of bacteria colonizing the dry areas of the skin, which includes many Gram-negative species. However, bacterial colonization in these areas is overall lower than at the moist sites.
Among the propionibacteria, Propionibacterium acnes is the most important skin colonizer, together with Propionibacterium avidum and Propionibacterium granulosum (Mak et al., 2013). It is well known as a contributor to the development of the skin disease, acne, although the mechanistic details of how P. acnes promotes acne are not well understood and P. acnes may not be involved in all cases of acne (Williams et al., 2012, Shaheen & Gonzalez, 2013). In addition to acne, P. acnes may occasionally be involved in opportunistic infections such as endocarditis or osteomyelitis (Jakab et al., 1996, Soderquist et al., 2010). These may involve biofilms, judging from the observation that P. acnes is able to form biofilms in-vitro (Ramage et al., 2003, Coenye et al., 2008). The molecular components of P. acnes biofilms, however, are largely unknown. Notably, P. acnes is also attributed a beneficial role in skin colonization, as its acidic fermentation products lower the skin pH, thereby preventing colonization of harmful pathogens such as Staphylococcus aureus (Cogen et al., 2008).
Staphylococci are widely regarded as the most important colonizers of the human skin, both in terms of frequency and sources for infection (Kloos & Schleifer, 1986). There is a certain preference of specific coagulase-negative staphylococci (CoNS) species for the site of colonization (Kloos & Musselwhite, 1975). The main species colonizing various areas of the skin are Staphylococcus epidermidis and Staphylococcus hominis. CoNS are opportunistic pathogens and S. epidermidis, for example, is the most frequent cause of catheter-related infections (Rogers et al., 2009). However, the involvement of CoNS in such infections may be regarded “accidental” rather than representing a clear pathogenesis program (Otto, 2009), and is likely due chiefly to the shear abundance of CoNS on the skin and their extraordinary capacity to colonize tissue surfaces, as will be discussed in detail further below. In contrast, the coagulase-positive species S. aureus is commonly regarded as a major and dangerous human pathogen (Lowy, 1998), although about one third of the population is colonized non-symptomatically by S. aureus in the nares and rectal areas (Wertheim et al., 2005). Of note, S. aureus non-symptomatic colonization is correlated with a higher chance of subsequent infection (von Eiff et al., 2001).
Except for the species Corynebacterium diphteriae, corynebacteria are commonly innocuous bacteria found widespread in nature; some species may colonize humans (Evaldson et al., 1982). Occasionally, some corynebacterial species may cause opportunistic infections in immune-suppressed patients (Lipsky et al., 1982). Several studies have addressed a potential bacterial competition between S. aureus and corynebacteria in the human nose. In general, there appears to be a negative correlation between the abundance of corynebacteria – as well as P. acnes and S. epidermidis – and S. aureus colonization, indicating bacterial interference (Frank et al., 2010).
In the following, the mechanistic underpinnings of bacterial skin colonization and bacterial resistance to physical stresses in that environment will be discussed. Most of what we know about these mechanisms is derived from investigations on S. epidermidis and S. aureus, which is why there will be a focus on those bacteria.
Attachment
One of the main physical stresses that skin bacteria have to cope with is of mechanical nature: they need to establish firm adhesion to the skin or mucous surfaces in order not to be scraped off or washed away. While initial, long-range adhesion may be mediated by rather non-specific hydrophobic or electrostatic interactions between bacterial and host surface structures, for short-range, firm adhesion bacteria express specific surface-attached binding proteins. In case of the skin but also many other epithelia, these primarily bind to human matrix proteins, such as fibronectin, collagen, elastin, keratin, vitronectin, etc. The present review will focus on the bacterial components of these interacting partners; for a review on the binding structures of human matrix proteins see Chagnot et al. (Chagnot et al., 2012). In compromised skin, there may be additional host proteins involved in wound healing, such as fibrinogen, to which the bacteria can bind. Bacterial surface proteins that bind human matrix proteins are collectively called MSCRAMMS (microbial surface components recognizing adhesive matrix molecules) (Patti et al., 1994).
Animal studies on staphylococci have used almost exclusively infection rather than colonization models. This is because, unfortunately, there are no easy skin colonization models with which the roles of bacterial adhesion molecules can be tested. The only skin colonization model deemed to appropriately reflect human skin colonization is the pig skin model, which is only rarely used, owing to the difficult requirements for having pigs as test animals. It has been applied more recently predominantly to investigate colonization of pigs as hosts for livestock-associated MRSA (Moodley et al., 2011, Giotis et al., 2012). Therefore, most of what we know about the bacterial components that ascertain binding to host matrix proteins stems from in-vitro tests that use matrix protein-coated abiotic surfaces, such as present in microtiter plates. Furthermore, models of biofilm-associated, often medical device-related, infections with opportunistic pathogens such as S. epidermidis allow conclusions on the importance of bacterial surface proteins in tissue binding. This is based on the notion that indwelling medical devices are soon coated with matrix proteins and matrix proteins in tissue are at least similar in composition to those expressed on the skin surface. Commercially available ex-vivo skin models have not yet been used to investigate the role of bacterial surface proteins in colonization to a considerable extent. Also, there is but one published study on the use of human volunteers to study the role of, for example, S. epidermidis factors in skin colonization, which addressed the exopolysaccharide polysaccharide intercellular adhesin (PIA) (Rogers et al., 2008). Such human volunteer studies have not been used yet to investigate S. epidermidis adhesion proteins.
The immense importance that adhesion to skin tissue, or host tissue in general, has for skin bacteria such as S. epidermidis, is reflected by the great number of surface binding proteins that these bacteria express. S. epidermidis has at least 18 genes for such proteins and the corresponding protein products show considerable functional redundancy as for their human binding partners (Bowden et al., 2005, Gill et al., 2005). S. aureus has at least 29 surface proteins (Gill et al., 2005), but some of those have other, at least primary, functions in S. aureus physiology, such as the immune evasion factor protein A (Forsgren & Nordstrom, 1974). Most adhesins in staphylococci and other bacteria have similar architectures (Fig. 1). In addition to the common N-terminal secretion sequence and the C-terminal sequences important for cell wall anchoring, which will be discussed in the following, they contain characteristic repeat sequences, whose role often is to form an extended domain stretching through the cell wall, and dedicated domains to interact with their respective binding partners. The latter sit at the tip of the repeat domains, exposed at the bacterial surface.
Fig. 1. Common structure of MSCRAMMs.
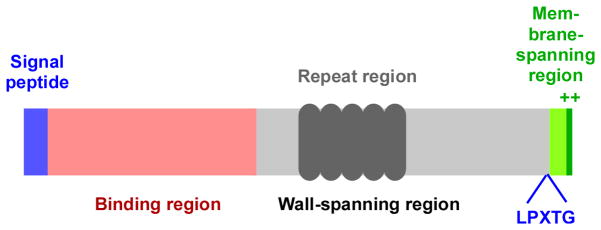
MSCRAMMs are composed of an N-terminal signal peptide region, which in most cases is recognized by the canonical Sec secretion system, triggering export and removal of the signal peptide. The C-terminal region contains the sortase recognition sequence LPXTG followed by a membrane-spanning and positively charged short cytoplasmic sequence. The latter two are removed by sortase, while the main protein part is being anchored to the cell wall. The grey region spans the cell wall, often containing repeat regions, while the N-terminal (red) region is exposed at the surface of the cell and functions to interact with human matrix proteins. Specific MSCRAMMS can differ significantly in details, for example by containing several different repeat and binding regions.
Most staphylococcal surface proteins, as well as those of most other Gram-positive bacteria, are covalently bound to peptidoglycan by the activities of the sortase enzyme family (Mazmanian et al., 1999, Marraffini et al., 2006). The archetypical member of this family is sortase A (SrtA), which catalyzes anchoring of almost all covalently anchored surface proteins in staphylococci and many in other bacteria (Mazmanian et al., 1999) (Fig. 2). Sortases recognize a C-terminal consensus sequence of the amino acid sequence LPXTG, which is present in the to-be-anchored protein just before a membrane-spanning and an ultimate short cationic sequence domain at the very C-terminus. This reaction occurs on the extracellular surface of the bacterial cytoplasmic membrane after the protein in question has been secreted by the Sec secretion system, cleaving off its N-terminal secretion signal sequence. Sortase then cleaves the LPXTG sequence between the conserved threonine and glycine amino acids and links the carboxy group of the threonine residue to the amino group present in peptidoglycan cross bridges (in most staphylococci, these are composed of five glycine residues).
Fig. 2. Mechanism of secretion and anchoring of MSCRAMMs to the Gram-positive cell wall.
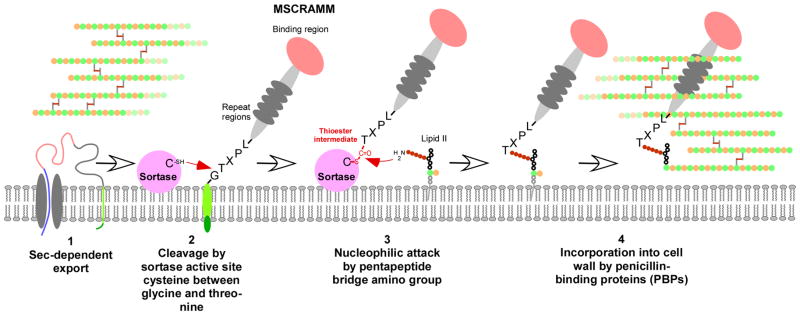
(1) The MSCRAMM protein is secreted by the Sec system, as most other secreted proteins. (However, some MSCRAMMs have dedicated secretion systems, such as S. pyogenes Srr proteins.) This also removes the signal peptide. (2) The active site cysteine thiol of the surface-attached sortase attacks and cleaves between the glycine and threonine residues of the MSCRAMM’s LPXTG motif. (3) The sortase-MSCRAMM thioester-linked intermediate undergoes nucleophilic attack at the thioester carbon by the amino group of an exposed amino acid moiety of the lipid II cell wall biosynythesis precursor (the exposed amino acid of what otherwise would become the peptidoglycan pentapeptide cross bridge in peptidoglycan). (4) The MSCRAMM-lipid II complex is then incorporated by penicillin-binding (i.e., cell wall biosynthesis) proteins into the peptidoglycan network.
S. epidermidis encodes 12 LPTXG-anchored surface proteins (Gill et al., 2005). Only some of those have been characterized in more detail. One, SdrG, belongs to the family of proteins showing a domain composed of serine/aspartate (SD) repeats (McCrea et al., 2000); hence the name Sdr. Similar to other repeat regions, the SD region is assumed to function as an extended protein domain, which stretches through the bacterial peptidoglycan, exposing distal binding domains for the interaction with human matrix proteins. The fibrinogen-binding SD repeat protein SdrG (Davis et al., 2001) is probably the most intensively studied S. epidermidis surface binding protein. Binding of a ligand to SdrG follows a “dock, lock, and latch” mechanism (Ponnuraj et al., 2003). SdrG is necessary and sufficient for the binding of S. epidermidis to immobilized fibrinogen in vitro (Hartford et al., 2001). It has been analyzed as a potential vaccine for S. epidermidis infections (Rennermalm et al., 2004). Of note, its expression is dependent on an in-vivo environment (Sellman et al., 2008).
Another LPXTG-anchored surface protein of S. epidermidis that has been analyzed in great detail is the accumulation-associated protein Aap (Hussain et al., 1997). Aap forms fibrils on the cell surface whose polymerization is dependent on zinc and proteolytic removal of specific domains (Rohde et al., 2005, Banner et al., 2007, Conrady et al., 2008). Aap is likely involved in surface adhesion as well as in making intercellular contacts that promote cellular agglomeration.
MSCRAMMs of S. aureus comprise clumping factors A and B, which mediate adhesion to fibrinogen (Boden & Flock, 1989, McDevitt et al., 1994, Ni Eidhin et al., 1998), three SD repeat proteins, an Aap homologue, and a series of other proteins whose biological functions are not yet understood (Gill et al., 2005). ClfB also binds to a keratin, cytokeratin 10, and may thus play a critical role in the colonization of mucosal surfaces (Walsh et al., 2004). Some MSCRAMMS may be encoded on mobile genetic elements. The SasX protein of S. aureus was shown to facilitate adhesion to epithelial cells, biofilm formation, and nasal colonization (Li et al., 2012). Notably, its presence is linked to the epidemiological success of certain lineages of methicillin-resistant S. aureus (MRSA) in hospital infections.
Other staphylococcal surface binding proteins fulfill functions very similar to those facilitated by LPXTG-anchored proteins, but they are only attached to the bacterial surface in a non-covalent fashion. For example, bi-functional autolysins/adhesins mediate binding to a series of matrix proteins including vitronectin, in addition to having a crucial, primary role in shaping the staphylococcal cell wall (Heilmann, 2011). The major autolysins (Atl) of S. epidermidis, S. aureus and other staphylococci belong to this family, as do the S. epidermidis protein Aae and S. aureus Aaa (Heilmann et al., 1997, Heilmann et al., 2003, Heilmann et al., 2005). S. epidermidis produces two SD repeat proteins other than SdrG, namely SdrF and SdrH, which are both non-covalently anchored (McCrea et al., 2000). SdrF is present not in all, but many S. epidermidis strains, and was demonstrated to bind collagen and have a premier role in facilitating attachment to ventricular assist device drivelines (Arrecubieta et al., 2009). The S. aureus fibronectin binding proteins FnBpA, FnBpB, and Ebh, the fibrinogen–binding proteins Efb and Fib, the collagen-binding protein Cna, and the matrix protein binding proteins Embbp and Eap (Map) also belong to the non-covalently anchored surface protein family (Gill et al., 2005).
Thus, it is evident that S. aureus and S. epidermidis produce a vast variety of host matrix binding proteins with pronounced functional redundancy and a series of various binding functions to ascertain binding to host skin and mucosal surfaces. Interestingly, the quorum-sensing (QS) regulator accessory gene regulator (Agr) regulates expression of many staphylococcal surface proteins in a negative fashion (Vandenesch et al., 1991, Novick et al., 1995). This has so far been primarily interpreted as being related to the function of Agr during the establishment of an infection. Low activity of Agr would lead to enhanced production of these binding proteins when they are needed to establish binding to host tissues, whereas with an increase in the bacterial population, Agr would become more strongly expressed, leading to a shift from the expression of binding proteins to that of toxins and degradative exoenzymes (Novick & Geisinger, 2008). How the activity of Agr during the colonization of epithelia and mucosal surfaces relates to that observed in stationary in-vitro cultures, where it is high, is not very clear. We may assume that it is rather low, as also indicated by its overall relatively low activity in in-vitro biofilms (Vuong et al., 2004), which to a certain extent may resemble the in-vivo colonization status. Finally, recent research indicates that the impact of Agr on the expression of important MSCRAMMS in clinical strains may not always follow the scheme of negative regulation derived from studies with laboratory strains (Cheung et al., 2011).
Many staphylococcal strains are known to cause infections on indwelling medical devices (Otto, 2008). Although the establishment of infectious biofilms on such devices is assumed to occur on a host-matrix protein-derived “conditioning film” on the device surface, direct adhesion of the bacteria to the abiotic plastic surface may also occur occasionally. In that case, the hydrophobicity of the bacterial cell surface mainly determines adhesion characteristics. For example, teichoic acids and DNA originating from lysed cells (“extracellular DNA”, eDNA) were described to facilitate adhesion to plastic surfaces in staphylococci (Gross et al., 2001, Mann et al., 2009). Whether such non-specific interaction plays a role in the attachment to human tissue is poorly understood.
There are also proteins that have been attributed specific roles in the adhesion to abiotic surface in staphylococci. For example, the bifunctional autolysin/adhesin AtlE of S. epidermidis was shown to impact adhesion to plastic in vitro (Heilmann et al., 1997). This is likely due to the significant change in hydrophobicity that accompanies expression of this highly abundant surface protein. In general, the absence or presence of highly abundant molecules, such as eDNA or teichoic acids, changes the physico-chemical structure of the cell surface to a great extent; and thus changes in binding may be due also to secondary effects. For example, changes in teichoic acid structure mediated by D-alanylation impact binding of autolysin to their surface, exemplifying how secondary effects may contribute to the impact of polymeric macromolecules to surface attachment (Peschel et al., 2000).
For many Gram-positive skin colonizers, the basic mechanisms of attachment likely resemble those identified for staphylococci, at least in principle (Fig. 3). Not much is known about the specific mechanisms of surface attachment in the other frequent skin colonizers, corynebacteria and propionibacteria. Corynebacteria as well as several other Gram-positive and Gram-negative bacteria form pili (fimbriae) (Mandlik et al., 2008), and in that capacity are distinguished form staphylococci. Pili are a premier bacterial means to colonize host tissue and form biofilms. Much of what we know on pilus assembly in Gram-positive bacteria stems from work on C. diphteriae, which is likely applicable also for other corynebacteria and pili-forming Gram-positives (such as streptococci). In C. diphteriae, it was shown that both the “housekeeping” sortase SrtA described above and a pilin-specific sortase are responsible for pilus assembly (Ton-That & Schneewind, 2003, Swaminathan et al., 2007). Notably, pili appear to be responsible for a certain degree of tissue tropism of attachment (Dramsi et al., 2006, Mandlik et al., 2007).
Fig. 3. Molecules participating in adhesion and extracellular matrix formation in Gram-positive bacteria.

Proteins can be attached to the bacterial surface in three ways: covalent linking (MSCRAMM), non-covalent attachment (such as by interaction with surface non-protein polymers such as teichoic acids), and as lipoproteins (with an N-terminal fatty acid-containing membrane anchor). A lipoprotein, due to its mode of attachment, sits close to the cytoplasmic bacterial membrane and may not extend through the thick Gram-positive cell wall to mediate adhesion. Consequently, lipoproteins do not appear to play an important role in surface attachment in staphylococci or streptococci. Pili (fimbriae) can protrude for up to ~ 3 μM from the surface and are attached like MSCRAMMs. Exopolysaccharides, important constituents of the extracellular matrix and crucial for biofilm formation, can be neutral, acidic, or basic. Interactions between proteins, the negatively charged teichoic acids and extracellular DNA (eDNA), and exopolysacharides of positive charge likely are of key importance for matrix formation. WTA, wall teichoic acids; LTA, lipoteichoic acids.
Only very few studies have looked at adhesins in P. acnes. One such study discovered a fibronectin binding protein of P. acnes using in-vitro binding experiments (Yu et al., 1997). However, some recent studies used genomic analyses and surface proteome-based detection to gain more insight into putative attachment molecules of propionibacteria. P. granulosum produces pili-like appendices, likely produced from genes homologous to the pilin biosynthetic genes of corynebacteria. P. acnes and P. avidum lack such structures and the corresponding genes. Similarly, only P. granulosum has a gene cluster coding for multiple sortases. Several surface proteins were detected in surface proteome analyses of these three species; however, there are no functional studies yet as to their relevance in host tissue attachment (Brzuszkiewicz et al., 2011, Mak et al., 2013).
Intercellular aggregation
Adhesion to host tissue only provides resistance to mechanical stress for the first layer of adhering microorganisms. To form entire colonies on the skin, agglomerating bacteria must have means to stick to each other. This agglomeration is commonly described as the second step of bacterial biofilm formation; and the mechanisms underlying agglomeration have been thoroughly investigated by biofilm researchers (O’Toole, 2003) (Fig. 4). This most notably includes research on staphylococci, for which the details of biofilm formation and agglomeration are probably best understood among Gram-positive pathogenic bacteria (Otto, 2008).
Fig. 4. Biofilm development.

Biofilm development has three principal phases: attachment, maturation, and detachment. Bacteria can reach a surface either by active motion (such as P. aeruginosa) or passively (such as staphylococci). Attachment occurs via a multitude of specific adhesins and non-specific adhesive forces. Afterwards, cells proliferate and produce extracellular matrix. Disruptive forces are important for the three-dimensional structure of the biofilm, which contains channels important for nutrient delivery to deeper biofilm layers, and ultimately for the detachment of cells or cell clusters from the biofilm.
In general, many different macromolecules make up the extracellular matrix that holds bacteria together in biofilms. These include proteins, exopolysaccharides, eDNA, and in Gram-positive bacteria, teichoic acids. Many of these molecules have other primary functions and their contributions to biofilm formation may be regarded as secondary and accessory, which is evident for example in the case of DNA. Others appear to have evolved specifically for their function in cell agglomeration, such as presumably many matrix exopolysaccharides.
Many staphylococci, but also several other biofilm-forming bacteria such as E. coli, produce a biofilm exopolysaccharide, polysaccharide intercellular adhesin (PIA, also called poly-N-acetylglucosamine, PNAG, or PGA in E. coli) (Mack et al., 1996, Cramton et al., 1999, Allignet et al., 2001, Wang et al., 2004). PIA/PNAG is a homopolymer of N-acetylglucosamine residues, of which about a quarter are de-acetylated (Mack et al., 1996) (Fig. 5). PIA/PNAG is the product of a biosynthetic locus called ica, which contains two genes, icaA and icaD, whose membrane-spanning protein products form an N-acetylglucosamine polymerase (Heilmann et al., 1996, Gerke et al., 1998). The growing N-acetylglucosamine polymer is then exported, presumably by the membrane channel protein IcaD, and de-acetylated by the cell surface located protein IcaB (Vuong et al., 2004). De-acetylation gives PIA/PNAG a positive net charge, which is crucial for its function in cell-to-cell adhesion and biofilm formation in vitro and in vivo (Vuong et al., 2004). Likely, it interacts with other cell surface polymers, such as teichoic acids or eDNA, which often are polyanionic. How exactly teichoic acids and eDNA contribute to agglomeration is not exactly understood. In addition to interacting with the cationic PIA/PNAG they may serve as a scaffold for cationic adhesion proteins.
Fig. 5. Examples of bacterial surface polymers.

Wall teichoic acids (WTA) and lipoteichoic acids (LTA) occur in Gram-positive bacteria. The structures found in S. aureus are shown. WTA is linked via a phosphodiester bond to an N-acetyl muramic acid residue in peptidoglycan. The 2 OH-group of N-acetylmannosamine is phosphodiester linked to three units of (D-alanylated) glycerol-phosphates, to which ~ 40 units of ribitol phosphates (substituted with D-alanine and/or GlcNAc) are linked. In LTA, a diacylglycerol moiety, which is linked to a β-1,6-connected diglucosyl part, forms the membrane anchor. Approximately 40 units of glycerol phosphate are linked to the 6-hydroxyl group of the second glucosyl part. Position 2 of the glycerol phosphate residues is in part substituted either with D-alanine or D-alanine ester and glycosyl (mostly GlcNAc) residues. GlcNAc, N-acetyl-glucosamine. Alginate (of P. aeruginosa) is composed of guluronic acid (GulUA) and mannuronic acid (ManUA) monomers. PIA/PNAG (S. aureus, S. epidermidis, other staphylococci and some other bacteria) is a homopolymer of partially de-acetylated N-acetyl-glucosamine units in β-1-6 linkage.
While PIA/PNAG certainly is an important factor in staphylococcal agglomeration, there are strains that form biofilms in vitro and can be isolated from biofilm infections, but which do not have the ica genes (Kogan et al., 2006, Rohde et al., 2007). In those strains, proteins appear to be primarily responsible for aggregation. Many different surface proteins have been attributed functions in staphylococcal biofilm formation and agglomeration, such as protein A (Merino et al., 2009), the S. aureus surface proteins SasC and SasG (Schroeder et al., 2009, Geoghegan et al., 2010), extracellular matrix binding protein (Embp) (Lasa & Penades, 2006, Christner et al., 2010), biofilm-associated protein (Bap), and the fibronectin-binding proteins FnbpA and FnbpB (O’Neill et al., 2008). The accumulation-associated protein Aap of S. epidermidis, of which SasG is the S. aureus homologue, has been studied in detail and was already mentioned above. Interestingly, there appear to be strain-specific differences in the preferred mode of aggregation, inasmuch as S. aureus strains more often rely on proteins and S. epidermidis strains usually form PIA/PNAG-dependent biofilms (Rohde et al., 2007).
As mentioned above, the roles of these factors were for the most part not directly analyzed using in-vivo colonization models. One study using human volunteers somewhat surprisingly showed that PIA/PNAG production is negatively correlated with skin colonization (Rogers et al., 2008). However, this finding is in agreement with studies showing that presence of the ica genes is higher in infectious than commensal (colonizing) strains of S. epidermidis (Ziebuhr et al., 1997, Gu et al., 2005, Yao et al., 2005) – then again, studies whose setup and conclusions have been criticized (Rohde et al., 2004). Clearly, whether findings from biofilm research suggesting a role of a factor in aggregation during biofilm-associated infection imply that such a factor is beneficial also for colonization has to be analyzed more critically and directly in the future.
Colonization of the digestive tract
Intestine
The intestine has a single-layer epithelium, which is renewed every four to five days by shedding. In the small intestine, tight junctions link epithelial cells, making the epithelial layer virtually impermeable. The intestinal mucosa is highly folded. In particular, the luminal surface of the mucosa of the small intestine is covered by a number of finger-like projections called villi, which are 0.5–1.5 mm in length. Invaginations next to the villi are called crypts (Fig. 6).
Fig. 6. Mechanisms of gut pathogens to colonize the intestinal mucosa and evade epithelial turnover.

Gut pathogens use a variety of mechanisms to attach to the intestinal epithelium and evade epithelial shedding and turnover. Many pathogens use pili to attach to the epithelium. Many also breach tight junctions to gain access to deeper epithelial layers. Several, such as Shigella, cause cell cycle arrest, thereby delaying epithelial turnover. H. pylori and others obtain similar results by delaying apoptosis. L. monocytogenes and others trigger internalization into epithelial cells and some such as Shigella afterwards force matrix adhesion.
About 100 trillion bacteria live in the gut, which is ten times more the number of cells in the human body. According to recent findings from microbiome research, the human intestine contains 500 to 1000 species, most of which belong to the Firmicutes (mostly Clostridium) or Bacteroidetes (mostly Bacteroides) (Guarner & Malagelada, 2003). In addition to Clostridium ssp. and Bacteroides ssp., many gut bacteria belong to the species Fusobacterium, Eubacterium, Ruminococcus, Peptococcus, Peptostreptococcus, and Bifidobacterium. To a lesser extent, Escherichia and Lactobacillus are found. The benign gut microflora has several important functions, including fermentation, production of vitamins, and prevention of the outgrowth of potentially harmful bacteria (Sommer & Backhed, 2013). The colonization mechanisms used by these normal gut colonizers is poorly understood, but recent research indicates that they involve biofilm formation (Donelli et al., 2012). In contrast, mechanisms to withstand the physical stress of being removed from epithelia by mechanic forces or epithelial turnover are well understood in a selected series of gut pathogens, such as Shigella or enterohaemorrhagic E. coli (EHEC), which will be focused on here. As will be described in the following, these mechanisms extend considerably from the mere establishment of adhesion and include, for example, sophisticated subversion of the epithelial renewal process (Fig. 6). Nevertheless, the first steps of intestinal colonization certainly also involve attachment; for example, shiga-toxin producing E. coli adhesion to intestinal epithelial cells is dependent on the expression of fimbrial adhesins (pili) (Lloyd et al., 2012). Pili-dependent adhesion will be discussed in the part on urinary pathogens.
Shigella uses a type III secretion system (IpaB) to cause cell cycle arrest in intestinal crypt progenitor cells, thereby reducing the epithelial turnover rate (Iwai et al., 2007) (Fig. 6). Many other pathogens achieve the same outcome by mechanisms aimed to delay apoptosis in epithelial cells (Kim et al., 2010). Listeria monocytogenes and other pathogens make use of the formation of gaps in the gut mucosa that occur during the normal process of epithelial cell shedding to induce internalization (Barbuddhe & Chakraborty, 2009). Once internalized, intracellular bacterial gut pathogens may subvert the normal epithelial shedding process. For example, Shigella secretes a protein, OspE, via the type III secretion system that interacts with host integrin-linked kinase (ILK), resulting in increased levels of surface β1-integrin and stabilized focal adhesion complexes (Kim et al., 2009). This mechanism ultimately leads to a reinforcement of the infected epithelial cells to the extracellular matrix of the basal lamina. It is likely shared by a series of other intestinal pathogens, which also express OspE, such as EHEC and Salmonella. Furthermore, many gut pathogens use a series of mechanisms to breach tight junctions between epithelial cells, likely primarily in order to gain nutrients, but also to facilitate tighter interactions with the epithelium (Kim et al., 2010).
Stomach
The mucosa of the stomach is coated with a thick, continually secreted layer of mucus to protect surrounding tissue from the acidic conditions inside the stomach. This creates an environment that is very harsh for bacteria to colonize. Therefore, the stomach has for a long time been believed not to contain bacterial colonizers, until the microaerophilic Gram-negative bacterium H. pylori was identified to live in that environment (Marshall & Warren, 1984). Later on, more bacterial colonizers of the stomach were identified, such as Deinococcus radiodurans (Bik et al., 2006), but here there will be a focus on H. pylori, as this bacterium is far better studied. In some populations, the frequency of H. pylori colonization can reach 80%; and ~ 50% of all people are thought to be colonized with H. pylori. H. pylori colonization can be considered an infection, although most people are asymptomatic (Costa et al., 2009).
The majority of H. pylori cells are found in the gastric mucus, but – similar to pathogens of the lower intestinal tract – adhesion to epithelia is considered a prerequisite for infection. Several H. pylori adhesion proteins have been described. The best-characterized is BabA, which attaches to the fucosylated Leb blood antigen (Boren et al., 1993, Aspholm-Hurtig et al., 2004). SabA, another H. pylori adhesin, mediates binding to sialylated glycoconjugates expressed during inflammation in gastric tissue (Mahdavi et al., 2002), but also to the matrix protein laminin and to neutrophils and erythrocytes (Unemo et al., 2005, Walz et al., 2005, Aspholm et al., 2006). H. pylori contains further adhesins, whose role in attachment are not as well understood and whose interaction partners have not yet been identified. The most intensively studied H. pylori virulence factor, the Cag type IV secretion system with its effector molecule CagA subverts intracellular host signaling, leading to changes in cell growth, motility, and alteration of tight junctions, thus in principle similar to mechanism described above used by pathogens of the lower intestinal tract (Fischer et al., 2009) (Fig. 7).
Fig. 7. H. pylori mechanisms important during colonization of the stomach.

H. pylori has a series of extraordinarily potent mechanisms to withstand the acid conditions in the human stomach. Attachment is facilitated by several adhesins, including most notably BabA and SabA. The H. pylori Cag type IV secretion system with its effector CagA leads to a subversion of epithelial cell signaling, causing a variety of processes ensuring pathogen survival in the epithelium.
The pH of the stomach is about 2; therefore, H. pylori needs efficient mechanisms to cope with acid stress. The most important such mechanism is the production of an extraordinarily potent urease, whose catalytic capacity exceeds those of other bacteria, which is likely why H. pylori has the almost unique capacity to colonize the human stomach (Mobley et al., 1995). As further acid resistance systems, H. pylori also has two amidases (AmiE, AmiF) that produce ammonia (Skouloubris et al., 1997, Skouloubris et al., 2001) and an arginase producing urea and ornithine from arginine hydrolyzation (RocF) (Zabaleta et al., 2004) (Fig. 7). All those H. pylori ammonia-producing acid resistance systems are up-regulated by low pH via the ArsSR two-component system (Loh & Cover, 2006, Loh et al., 2010).
Other than H. pylori, several enteropathogenic bacteria show considerable acid resistance to be able to pass through the upper intestinal tract. In E. coli and some other gammaproteobacteria such as Vibrio or Salmonella, acid resistance systems are mainly amino acid decarboxylases (Zhao & Houry, 2010). These systems consist of the decarboxylase, which is induced by low pH and removes carbon dioxide from amino acids such as arginine, lysine or glutamate, and a specific antiporter. The decarboxylation reaction consumes a cytoplasmic proton. The subsequent export of the decarboxylation product with the concomitant import of the amino acid educt effectively removes that proton from the cytoplasm and transports it to the periplasmic space.
Colonization of the urinary tract
The urinary tract consists of the kidneys, ureters, bladder, and urethra. The urinary tract is normally sterile and only colonized in the case of an infection. An infection of the lower urinary tract is called a simple cystitis (a bladder infection) and a pyeolonephritis if the kidneys are infected. Urinary tract infections (UTIs) are very frequent and occur much more often in women than men. In the U. S. alone, ~ 8 million UTIs occur each year in otherwise healthy young women. UTIs have a high rate of recurrence (~ 30%), which is to a great part due to efficient mechanisms that UTI pathogens have to colonize the urinary tract. By far the most frequent pathogen involved with UTIs is E. coli (80–90% of uncomplicated UTIs), followed by Staphylococcus saprophyticus (Svanborg & Godaly, 1997). Here, there will be a focus on E. coli, as adhesion mechanisms of pathogenic staphylococci were already described in the part on skin and mucosal colonization.
Uropathogenic E. coli (UPEC) express many toxins and other virulence factors, some of which attack the urinary epithelium. For example, the secreted autotransporter (SAT) compromises gap junctions of bladder and kidney epithelia, thereby escaping epithelial turnover in a way discussed above for gut pathogens (Guyer et al., 2002). However, the best-studied factors ascertaining colonization of UPEC in the urinary tract are adhesins (Wright & Hultgren, 2006). UPEC express a series of adhesins, which belong to one of three families of pili found in Gram-negative bacteria (type I pili, type 4 pili, curli) or are outer membrane proteins. Different types of pili are the most important surface structures facilitating attachment of UPEC.
Type I pili are assembled by the chaperone-usher pathway, the most widespread pilus biogenesis pathway in Gram-negative bacteria (Waksman & Hultgren, 2009) (Fig. 8). Important in UPEC are the type 1, P, F1C, S, and Afa/Dr adhesins (Wright & Hultgren, 2006). The genetic loci responsible for the production of these adhesins share a common composition: they often contain regulatory genes, followed by a major subunit gene, a cytoplasmic chaperone, an outer membrane usher, and the minor and adhesin subunit genes. Type 1 and P pili will be discussed in more detail here.
Fig. 8. Structure of pili of the chaperon-usher-secreted type (P and type 1 pili).

Type I pili of Gram-negative bacteria are secreted and linked to the outer membrane by the chaperone-usher pathway. Type 1 and P pili are the most important examples, shown here. PapC and FimC represent the ushers, while PapD/PapH and FimC, respectively, are the chaperones. The main part of the pilus is made of and linked to the outer membrane by the PapA and FimA protein units, of which ~ 1000 assemble. PapG and FimH are the adhesin molecules situated on the tip of the pilus. Between the PapA or FimA multimers and the PapG/FimH tips, type 1 and P pili differ somewhat in structure, with the type 1 pilus having additional subunits (see graph).
P pili show the strongest association with severe UTI in epidemiological studies (Bergsten et al., 2005). They are encoded by the pap (pyelonephritis-associated pili) gene cluster (Hull et al., 1981, Vaisanen et al., 1981). P pili are ~ 7 nm wide and consist of PapA subunits. The ~ 2-nm wide tip is made of PapE subunits, to which the PapG adhesin is linked via the PapF adaptor. The fibrillum is joined to the rod via PapK (Lindberg et al., 1987, Hultgren et al., 1993, Dodson et al., 2001) (Fig. 8). Like all UPEC pili the PapG adhesin recognizes a specific sugar epitope on host epithelial receptors (Lindberg et al., 1987). In addition to providing a means to establish attachment, P pili are required for triggering the innate immune response in the urinary tract (Frendeus et al., 2000, Frendeus et al., 2001).
Type 1 pili are about 1 μM long, 7 nm wide and occur typically in a number of around 1000 copies/bacterium (Hahn et al., 2002). They are encoded by the fim gene cluster, which is present in UPEC and apathogenic E. coli. Most of the type 1 pilus (fimbrium) is composed of the major subunit FimA. The adhesin FimH sits at the tip and recognizes a variety of receptors by binding to D-mannose moieties and structurally similar peptide epitopes (Waksman & Hultgren, 2009) (Fig. 8). Binding to uroplakins 1a and 1b likely mediates attachment of type 1 pili to the bladder epithelium (Wu et al., 1996).
Type IV pili are more widespread than type I pili and are expressed by a series of Gram-negative (and Gram-positive) bacteria. They are similar in shape to type I pili, but are secreted by a type II secretion system. Similar to type I pili, they are involved in tissue colonization as well as co-aggregation (Craig & Li, 2008, Pelicic, 2008).
As a second step of colonization during UTI, UPEC form intracellular bacterial communities (IBCs), also described as “intracellular biofilms” (Hunstad & Justice, 2010). Similar to many intracellular gut pathogens, UPEC invade epithelial cells to form intracellular agglomerations, thereby evading epithelial turnover and ascertaining long-term persistence. In fact, IBC formation lets UPEC persist for months in the bladder without bacteria being detectable in the urine. Type 1 pili (but not P pili) are needed for internalization (Martinez et al., 2000). However, it is poorly understood whether IBCs resemble extracellular E. coli biofilms on the structural level; the similarity is only based on microscopic evaluation. Type 1 pili might also play a role in the formation of IBCs, in addition to their roles in adhesion and internalization, given that they are crucial contributors to in-vitro biofilm formation.
Colonization of the mouth and respiratory tract
The upper respiratory tract (consisting of the nasal cavity, pharynx, and larynx) is colonized by a multitude of bacteria. Asymptomatic colonization of the nose by S. aureus, S. epidermidis, and other bacteria was already discussed in the chapter on skin colonization. Colonization of the mouth and throat is often accompanied by disease, such as the formation of dental plaque leading to caries, periodontal disease, etc., or in the case of streptococcal pharyngitis (“Strep throat”).
Dental plaque is a multi-species agglomeration in biofilm mode (ten Cate, 2006). Notably, unlike other surfaces in the human body, the teeth do not have a shedding epithelium. Therefore, bacteria can persist for a long time on teeth without the need for mechanisms to overcome epithelial turnover processes. According to recent metagenomic studies, ~ 25,000 bacterial species can be found in the mouth, of which ~ 1,000 make up dental biofilm, although this has been stressed to possibly represent an over-estimation (Keijser et al., 2008, Zaura et al., 2009, Nyvad et al., 2013). Among the dental plaque-forming bacteria, Streptococcus mutans and relatives, as well as a series of anaerobic bacteria such as Fusobacterium and several actinobacteria dominate. Recent studies comparing the oral microbiome from individuals with and without caries revealed that some bacterial genera, such as Streptococcus, are associated with disease (Jiang et al., 2013). Streptococcal species that colonize the oral cavity mostly belong to the mitis (comprising among others Streptococcus oralis, Streptococcus gordonii, S. mitis, and Streptococcus pneumoniae) and mutans (comprising among others S. mutans, Streptococcus sobrinus, Streptococcus downei) groups. The latter are all associated with dental caries (Seminario et al., 2005). Group A streptococci (including most notably Streptococcus pyogenes) are the normal cause of streptococcal pharyngitis (Wessels, 2011), but they can also cause much more severe infections, such as streptococcal flesh-eating syndrome (necrotizing fasciitis) (Olsen & Musser, 2010).
The lower respiratory tract (consisting of the trachea, bronchi, and lungs) is commonly only colonized by bacteria in the case of disease, and colonization is frequently followed by infection (Bogaert et al., 2004). Here again, streptococci play an eminent role. S. pneumoniae is a premier cause of pneumonia (Dockrell et al., 2012). Due to the predominant importance of streptococci in asymptomatic and particularly, infectious colonization of the respiratory tract, this chapter will focus on how streptococci adhere to respiratory epithelia and form biofilms as present in dental plaque. A second part of this chapter will present principles and adhesion mechanisms of P. aeruginosa, a Gram-negative bacterium with an important role in the pathogenesis of cystic fibrosis (CF).
Streptococcal adhesion and colonization
The basic principles of streptococcal adhesion resemble those described above for staphylococci, inasmuch as adhesion is to a great extent due to surface-attached proteins. The most important streptococcal adhesins will be described here. Additionally, streptococcal adhesion mechanisms that are not present in staphylococci will be presented, such as pili. Finally, streptococcal molecules, such as specific exopolysaccharides, that contribute to intercellular aggregation will be discussed. Colonization by group B streptococci such as during dental plaque formation is very difficult to mimic in animal infection models. Therefore, most information on streptococcal adhesion and colonization mechanisms is derived from in-vitro and ex-vivo work. For the more pathogenic S. pyogenes and S. pneumoniae, much information on acute infection has been derived from animal infection models, which used a series of different animals ranging from mice to non-human primates for modeling pharyngitis (Virtaneva et al., 2005, Chiavolini et al., 2008). However, the use of animal models to specifically study colonization is rare also for these bacteria.
Attachment of streptococci, similar to other surface colonizers, follows a two-step model, in which initial, long-range attachment is mediated by hydrophobic or electrostatic interactions via cell surface polymers, such as teichoic acids, and by molecules such as pili that extend from the cell surface. In a second step, shorter-range interactions are accomplished by surface binding proteins (such as MSCRAMMs) (Nobbs et al., 2009) (Fig. 9).
Fig. 9. Two-step model of streptococcal tissue attachment.
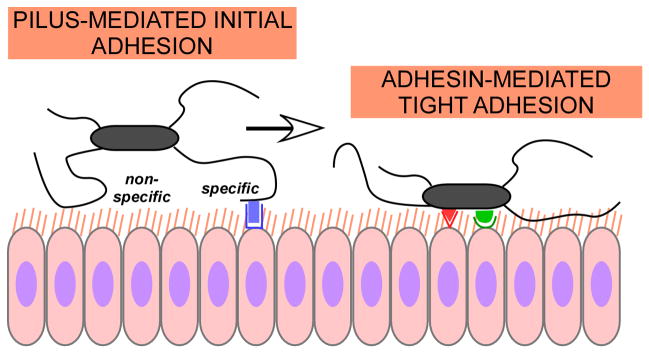
Streptococcal adhesion follows a two-step model, which in principle is valid also for other Gram-positive pathogens that have pili and attach to epithelial tissues. Long-range attachment is accomplished predominantly by pili, via non-specific (electrostatic/hydrophobic) or specific, pilus adhesin-mediated interaction. Then, short-range interactions by specific bacterial adhesins facilitate firm attachment to the epithelial cells.
The role of teichoic acids in surface colonization as a first step of how Gram-positive colonizers may adhere to surfaces has already been discussed for staphylococci. Both the direct interaction of these polyanions with oppositely charged tissue components as well as teichoic acids representing a scaffold to which non-covalently anchored surface adhesins bind may be involved. In addition, lipoteichoic acids of S. pneumoniae and other respiratory pathogens contain phosphorylcholine, which mediates bacterial adherence to the receptor for platelet-activating factor (rPAF) (Cundell et al., 1995, Gisch et al., 2013).
What mainly distinguishes streptococci from staphylococci during long-range attachment is the formation of pili. Pili, up to 10 nm in diameter, may extend up to 3 μM from the cell surface. Among the streptococci, pili are found in the pathogenic S. pyogenes, S. agalactiae and S. pneumoniae, while they are rare among the oral streptococci (Nobbs et al., 2009). The genetic loci of pathogenic streptococci encoding pilus biosynthesis proteins are termed PI. Several PI may be found in one species, up to nine in S. pyogenes, for example (Falugi et al., 2008). The formation and composition of Gram-positive pili were already discussed for corynebacteria above. Streptococcal pili were found to mediate adhesion to a wide variety of host tissues (Maisey et al., 2007, Nobbs et al., 2009). In addition, they may be required for internalization, thereby contributing to the escape from epithelial turnover, and for bacterial agglomeration. Formation of capsule, for example in S. pneumoniae, prevents entrapment in the nasal mucus and thus assists initial attachment to the epithelial layer (Nelson et al., 2007); however capsule formation is down-regulated afterwards, as it would prevent tighter interaction with epithelial structures (Weiser et al., 1994).
After initial attachment is accomplished, a tighter association with host tissues is mediated via surface adhesins (Nobbs et al., 2009). Similar to staphylococci, there are a great number of covalently anchored surface adhesins in streptococci, reflecting the immense importance of these proteins for streptococcal colonization. There is a great functional redundancy, ascertaining efficient tissue attachment. Notably, surface adhesin genes are subject to a high degree of genetic variation, leading to both non-synonymous point mutations and variability in the number of repeats in the repeat regions. This is assumed to create variations in their functional capabilities (Nobbs et al., 2009).
The M proteins are likely the best-studied streptococcal adhesins. M proteins are predominantly in an α-helical conformation, forming coiled-coil dimers, and have an extended conformation at the cell surface (Nilson et al., 1995). The antiparallel interactions found in the M-protein N-terminal region may play a role in bacterial aggregation (Frick et al., 2000). The hypervariable regions may bind a series of human plasma proteins and immunoglobulins. The B region binds fibrinogen, albumin, and IgG, while the C region binds factor H and CD46 in addition to albumin. There is a high degree of variation among the different M proteins, especially in the N-terminal hypervariable but also the B and C regions. These variations affect binding capabilities and thus lead to a different binding behavior dependent on the type of M protein (Nobbs et al., 2009). In addition to several M proteins that may bind fibronectin, streptococci express a large series of other fibronectin-binding proteins. S. pyogenes has at least 11 fibronectin-binding proteins. Collectively, they ascertain attachment to host cells via binding to the extracellular matrix (Kreikemeyer et al., 2004).
AgI/II proteins are found frequently in oral streptococci. They are all composed of A, V, and P domains, which bind host macromolecules such as fibronectin, laminin, and collagen. A major receptor for AgI/II proteins is gp340 (formerly called salivary agglutinin), which is found on the teeth, providing an important means for oral streptococci to establish attachment leading to dental plaque (Rundegren, 1986). Interestingly, gp340 also interacts with pili of S. pyogenes (Edwards et al., 2008). AgI/II proteins may also bind other oral microorganisms, giving them an additional role in bacterial interaction in dental plaque microbial communities (Lamont et al., 1991, Jenkinson & Demuth, 1997).
Serine-rich repeat proteins (Srr) are glycoproteins that are present in oral and pathogenic streptococci with the exception of S. pyogenes. They contain serine-rich repeats in ~ 75% of the whole protein. Srr proteins are crucial in establishing binding to epithelial cells and platelets by interacting with sialic acid and oligosaccharide-containing receptors (Takahashi et al., 2002). Interestingly, Srr proteins are secreted via a dedicated secretion system, a second Sec-dependent system called SecA2, emphasizing their importance for bacterial physiology (Takamatsu et al., 2004).
Similar to the bifunctional autolysins/adhesins of staphylococci, streptococci produce anchorless adhesins that commonly have an original enzymatic function not related to adhesion. Many are glycolytic enzymes released to the extracellular milieu probably by cell lysis. Surface-bound glycerylaldehyde-3-phosphate dehydrogenase (GAPDH) appears to be an important adhesin in many streptococci, binding to many extracellular targets including plasmin, plasminogen, fibrinogen, and fibronectin. Another glycolytic enzyme, α-enolase, is a major streptococcal plasmin- and plasminogen binding protein (Nobbs et al., 2009). Finally, some streptococci express surface-associated exoglycosidases, such as the neuraminidase, galactosidase, and N-acetylglucosaminidase of S. pneumoniae. These enzymes help to “unmask” sugar moieties on the epithelial surface for interaction with bacterial binding proteins (King et al., 2006).
The second step of bacterial biofilm formation, intercellular aggregation, is again in principle similar in streptococci to what has been discussed above for staphylococci. It involves surface proteins, exopolysacharides, etc., and is controlled by several regulatory, including QS systems (Nobbs et al., 2009). However, what distinguishes streptococcal from staphylococcal biofilms found in the human body is that many staphylococcal biofilm-associated infections, in particular those developing on indwelling medical devices, involve mono-species biofilms, while streptococci contribute to multi-species biofilms, such as most notably dental plaque. Interactions with other bacteria and their secreted products therefore play an eminent role in streptococcal biofilm formation.
Exopolysacharides are of key importance in streptococcal biofilms. Oral streptococci secrete enzymes that synthesize fructans and glucans, which bind to proteins on the bacterial cell surface. S. mutans secretes several glucosyltransferases that produce glucans of different water solubility. The water-insoluble glucans contribute to adherence and the building of bacterial communities (Bowen & Koo, 2011). Polysaccharide capsules in pathogenic streptococci often have a function in resisting phagocytosis by human white blood cells, but some exopolysaccharides may also contribute to binding other bacteria in biofilm communities. For example, galactose and galactosamine-containing exopolysaccharides of some oral streptococci mediate binding to the fimbrial lectins of Actinomyces species (Mergenhagen et al., 1987).
Colonization of the cystic fibrosis lung
Colonization of the lower respiratory tract only occurs in the case of an infection. For this to happen, the patient must be in an immune-compromised status, have an underlying, for example viral, infection, or be genetically predisposed. Cystic fibrosis (CF, also called mucoviscidosis) is a hereditary disease characterized by abnormal transport of chloride and sodium ions across epithelia. CF is caused by a mutation in the gene coding for the protein cystic fibrosis transmembrane conductance regulator (CFTR), which regulates chloride and sodium transport across membranes. The most severe manifestations of CF are seen in the lungs, which in CF patients show the formation of highly viscous material, prone to colonization by bacterial pathogens. The most common colonizers of the CF lung are P. aeruginosa, S. aureus, and Burkholderia cepacia, of which during the lifetime of a CF patient, P. aeruginosa usually increases in relative abundance (Harrison, 2007).
Mechanisms of attachment and the formation of aggregates, or biofilms, are crucial for the survival of P. aeruginosa in the lungs. P. aeruginosa is a model bacterium for biofilm formation and most principles of bacterial biofilm formation were described first in P. aeruginosa (Hoiby et al., 2010). While the evidence is mostly circumstantial, it is commonly believed that P. aeruginosa forms biofilms in CF lungs (Singh et al., 2000). Therefore, mechanisms of P. aeruginosa biofilm formation that are observed in-vitro are presumed to occur during colonization of the CF lung. However, it is often difficult to judge what relevance they have for the in-vivo situation; especially as P. aeruginosa infection of the CF lung is difficult to mimic in animal models (Joo & Otto, 2012).
Attachment of P. aeruginosa to the lung epithelium is believed to be mainly mediated by two lectins that bind to alpha-D-galactose (P. aeruginosa lectin I, PA-IL) and L-fucose (P. aeruginosa lectin II, PA-IIL) on the glycocalyx of the human cells (Imberty et al., 2004). Furthermore, type 4 pili and flagella were shown to bind to host N-glycans and heparan sulfate chains of heparan sulfate proteoglycans, respectively, at the surface of polarized epithelium (Bucior et al., 2012). Extracellular DNA (eDNA) and the Psl exopolysacharide were also implicated in attachment processes, but these results are mostly derived from in-vitro experiments on abiotic surfaces. Both eDNA and Psl are probably rather involved in aggregation than attachment processes sensu stricto. Finally, pili and flagella are involved in attachment processes in an indirect fashion, as they give motility to the bacterium and thus facilitate their transport to a surface (Joo & Otto, 2012).
Aggregation of P. aeruginosa to multicellular infectious communities, the second step in biofilm formation, predominantly involves several exopolysaccharides. P. aeruginosa produces three exopolysaccharides, the glucose-rich Pel polysaccharide, the mannose-rich Psl polysaccharide, and alginate, which is composed of guluronic acid and mannuronic acid monomers (Friedman & Kolter, 2004, Joo & Otto, 2012). Alginate is overproduced during CF infection, a phenotype easily distinguishable on agar plates in the laboratory, which is referred to as “mucoid” (Martin et al., 1993). However, the involvement of alginate in P. aeruginosa in-vivo biofilms is controversial (Yang et al., 2012).
The regulation of P. aeruginosa biofilm factors has been given considerable attention. For example, at least three QS systems regulate biofilm formation in P. aeruginosa in a complicated fashion (Joo & Otto, 2012). Similar to findings achieved in S. aureus and S. epidermidis, recent research in P. aeruginosa suggests that QS regulates the structuring of a biofilm and detachment of clusters from a biofilm, a process important for the dissemination of a biofilm-associated infection and its systemic spread (Bjarnsholt et al., 2010, Otto, 2013). Interestingly, in all these pathogens naturally occurring mutants in QS systems accumulate over time and can be isolated form infectious biofilms (Vuong et al., 2004, Traber et al., 2008, Bjarnsholt et al., 2010). These mutants appear to have increased capacity to colonize and form extended biofilms, while they likely lost the capacity for dissemination (Otto, 2013).
Conclusions and outlook
To overcome the problem of mechanical removal from human tissues, often exacerbated by epithelial turnover (except for on the teeth), bacteria have invented a series of adhesion mechanisms. These range from initial interactions by non-specific physical forces to long-range adhesion by protrusions such as pili and short-range, firm interactions mediated by bacterial surface proteins. Aggregation and encapsulation in an extracellular matrix (biofilm formation) further helps to prevent removal. In addition, many bacteria use internalization to persist in epithelial cells and thereby withstand epithelial shedding. In specific environments, bacteria also need to cope with additional stresses, such as a highly acidic environment in the stomach.
While we have a fairly good understanding of bacterial adhesins and biofilm formation as it occurs in vitro, the in-vivo relevance of many findings is still unclear, owing to the fact that colonization of many human epithelia is often difficult to mimic in animal infection models. Frequently, we need to extrapolate from the ex-vivo analysis of the binding capacities of adhesins to immobilized matrix proteins or from infection models (such as in the case of staphylococcal infections), which may not correctly represent asymptomatic colonization. Furthermore, it is becoming increasingly clear that in most situations, bacteria not only interact with human epithelia during colonization, but also with sometimes up to thousands of other bacterial species. For example, we are only beginning to understand how interactions between the surface molecules of different bacteria can strengthen a bacterial multi-species biofilm or how “probiotic”, benign bacteria prevent colonization with potential pathogens. Significant advances in the interaction of “benign” colonizers with potential pathogens are currently being made in the cases of skin and intestinal colonization (Stecher & Hardt, 2008, Naik et al., 2012, Stecher et al., 2013). Both the setup of better animal colonization models and experiments aimed at the analysis of inter-bacterial interactions will be needed to complete our view of bacterial colonization in vivo.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), U.S. National Institutes of Health (NIH). The author wishes to thank Dr. Hwang-Soo Joo for help with the chemical structures presented in Figure 5.
Bibliography
- Allignet J, Aubert S, Dyke KG, El Solh N. Staphylococcus caprae strains carry determinants known to be involved in pathogenicity: a gene encoding an autolysin-binding fibronectin and the ica operon involved in biofilm formation. Infect Immun. 2001;69:712–718. doi: 10.1128/IAI.69.2.712-718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrecubieta C, Toba FA, von Bayern M, Akashi H, Deng MC, Naka Y, Lowy FD. SdrF, a Staphylococcus epidermidis surface protein, contributes to the initiation of ventricular assist device driveline-related infections. PLoS Pathog. 2009;5:e1000411. doi: 10.1371/journal.ppat.1000411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspholm M, Olfat FO, Norden J, et al. SabA is the H. pylori hemagglutinin and is polymorphic in binding to sialylated glycans. PLoS Pathog. 2006;2:e110. doi: 10.1371/journal.ppat.0020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspholm-Hurtig M, Dailide G, Lahmann M, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305:519–522. doi: 10.1126/science.1098801. [DOI] [PubMed] [Google Scholar]
- Banner MA, Cunniffe JG, Macintosh RL, Foster TJ, Rohde H, Mack D, Hoyes E, Derrick J, Upton M, Handley PS. Localized tufts of fibrils on Staphylococcus epidermidis NCTC 11047 are comprised of the accumulation-associated protein. J Bacteriol. 2007;189:2793–2804. doi: 10.1128/JB.00952-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbuddhe SB, Chakraborty T. Listeria as an enteroinvasive gastrointestinal pathogen. Curr Top Microbiol Immunol. 2009;337:173–195. doi: 10.1007/978-3-642-01846-6_6. [DOI] [PubMed] [Google Scholar]
- Bergsten G, Wullt B, Svanborg C. Escherichia coli, fimbriae, bacterial persistence and host response induction in the human urinary tract. Int J Med Microbiol. 2005;295:487–502. doi: 10.1016/j.ijmm.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnsholt T, Jensen PO, Jakobsen TH, et al. Quorum sensing and virulence of Pseudomonas aeruginosa during lung infection of cystic fibrosis patients. PLoS One. 2010;5:e10115. doi: 10.1371/journal.pone.0010115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden MK, Flock JI. Fibrinogen-binding protein/clumping factor from Staphylococcus aureus. Infect Immun. 1989;57:2358–2363. doi: 10.1128/iai.57.8.2358-2363.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert D, van Belkum A, Sluijter M, Luijendijk A, de Groot R, Rumke HC, Verbrugh HA, Hermans PW. Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet. 2004;363:1871–1872. doi: 10.1016/S0140-6736(04)16357-5. [DOI] [PubMed] [Google Scholar]
- Boren T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- Bowden MG, Chen W, Singvall J, Xu Y, Peacock SJ, Valtulina V, Speziale P, Hook M. Identification and preliminary characterization of cell-wall-anchored proteins of Staphylococcus epidermidis. Microbiology. 2005;151:1453–1464. doi: 10.1099/mic.0.27534-0. [DOI] [PubMed] [Google Scholar]
- Bowen WH, Koo H. Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Res. 2011;45:69–86. doi: 10.1159/000324598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzuszkiewicz E, Weiner J, Wollherr A, et al. Comparative genomics and transcriptomics of Propionibacterium acnes. PLoS One. 2011;6:e21581. doi: 10.1371/journal.pone.0021581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucior I, Pielage JF, Engel JN. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PLoS Pathog. 2012;8:e1002616. doi: 10.1371/journal.ppat.1002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagnot C, Listrat A, Astruc T, Desvaux M. Bacterial adhesion to animal tissues: protein determinants for recognition of extracellular matrix components. Cell Microbiol. 2012;14:1687–1696. doi: 10.1111/cmi.12002. [DOI] [PubMed] [Google Scholar]
- Cheung GY, Wang R, Khan BA, Sturdevant DE, Otto M. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect Immun. 2011;79:1927–1935. doi: 10.1128/IAI.00046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavolini D, Pozzi G, Ricci S. Animal models of Streptococcus pneumoniae disease. Clin Microbiol Rev. 2008;21:666–685. doi: 10.1128/CMR.00012-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen GJ, Bruggemann H. Bacterial skin commensals and their role as host guardians. Benef Microbes. 2013:1–15. doi: 10.3920/BM2012.0062. [DOI] [PubMed] [Google Scholar]
- Christner M, Franke GC, Schommer NN, et al. The giant extracellular matrix-binding protein of Staphylococcus epidermidis mediates biofilm accumulation and attachment to fibronectin. Mol Microbiol. 2010;75:187–207. doi: 10.1111/j.1365-2958.2009.06981.x. [DOI] [PubMed] [Google Scholar]
- Coenye T, Honraet K, Rossel B, Nelis HJ. Biofilms in skin infections: Propionibacterium acnes and acne vulgaris. Infect Disord Drug Targets. 2008;8:156–159. doi: 10.2174/1871526510808030156. [DOI] [PubMed] [Google Scholar]
- Cogen AL, Nizet V, Gallo RL. Skin microbiota: a source of disease or defence? Br J Dermatol. 2008;158:442–455. doi: 10.1111/j.1365-2133.2008.08437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrady DG, Brescia CC, Horii K, Weiss AA, Hassett DJ, Herr AB. A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci U S A. 2008;105:19456–19461. doi: 10.1073/pnas.0807717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AC, Figueiredo C, Touati E. Pathogenesis of Helicobacter pylori infection. Helicobacter. 2009;14(Suppl 1):15–20. doi: 10.1111/j.1523-5378.2009.00702.x. [DOI] [PubMed] [Google Scholar]
- Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. Microbial biofilms. Annu Rev Microbiol. 1995;49:711–745. doi: 10.1146/annurev.mi.49.100195.003431. [DOI] [PubMed] [Google Scholar]
- Craig L, Li J. Type IV pili: paradoxes in form and function. Curr Opin Struct Biol. 2008;18:267–277. doi: 10.1016/j.sbi.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramton SE, Gerke C, Schnell NF, Nichols WW, Gotz F. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun. 1999;67:5427–5433. doi: 10.1128/iai.67.10.5427-5433.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. 1995;377:435–438. doi: 10.1038/377435a0. [DOI] [PubMed] [Google Scholar]
- Davis SL, Gurusiddappa S, McCrea KW, Perkins S, Hook M. SdrG, a fibrinogen-binding bacterial adhesin of the microbial surface components recognizing adhesive matrix molecules subfamily from Staphylococcus epidermidis, targets the thrombin cleavage site in the Bbeta chain. J Biol Chem. 2001;276:27799–27805. doi: 10.1074/jbc.M103873200. [DOI] [PubMed] [Google Scholar]
- Dockrell DH, Whyte MK, Mitchell TJ. Pneumococcal pneumonia: mechanisms of infection and resolution. Chest. 2012;142:482–491. doi: 10.1378/chest.12-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson KW, Pinkner JS, Rose T, Magnusson G, Hultgren SJ, Waksman G. Structural basis of the interaction of the pyelonephritic E. coli adhesin to its human kidney receptor. Cell. 2001;105:733–743. doi: 10.1016/s0092-8674(01)00388-9. [DOI] [PubMed] [Google Scholar]
- Donelli G, Vuotto C, Cardines R, Mastrantonio P. Biofilm-growing intestinal anaerobic bacteria. FEMS Immunol Med Microbiol. 2012;65:318–325. doi: 10.1111/j.1574-695X.2012.00962.x. [DOI] [PubMed] [Google Scholar]
- Dramsi S, Caliot E, Bonne I, Guadagnini S, Prevost MC, Kojadinovic M, Lalioui L, Poyart C, Trieu-Cuot P. Assembly and role of pili in group B streptococci. Mol Microbiol. 2006;60:1401–1413. doi: 10.1111/j.1365-2958.2006.05190.x. [DOI] [PubMed] [Google Scholar]
- Edwards AM, Manetti AG, Falugi F, Zingaretti C, Capo S, Buccato S, Bensi G, Telford JL, Margarit I, Grandi G. Scavenger receptor gp340 aggregates group A streptococci by binding pili. Mol Microbiol. 2008;68:1378–1394. doi: 10.1111/j.1365-2958.2008.06220.x. [DOI] [PubMed] [Google Scholar]
- Evaldson G, Heimdahl A, Kager L, Nord CE. The normal human anaerobic microflora. Scand J Infect Dis Suppl. 1982;35:9–15. [PubMed] [Google Scholar]
- Falugi F, Zingaretti C, Pinto V, et al. Sequence variation in group A Streptococcus pili and association of pilus backbone types with lancefield T serotypes. J Infect Dis. 2008;198:1834–1841. doi: 10.1086/593176. [DOI] [PubMed] [Google Scholar]
- Fedtke I, Gotz F, Peschel A. Bacterial evasion of innate host defenses--the Staphylococcus aureus lesson. Int J Med Microbiol. 2004;294:189–194. doi: 10.1016/j.ijmm.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Fischer W, Prassl S, Haas R. Virulence mechanisms and persistence strategies of the human gastric pathogen Helicobacter pylori. Curr Top Microbiol Immunol. 2009;337:129–171. doi: 10.1007/978-3-642-01846-6_5. [DOI] [PubMed] [Google Scholar]
- Forsgren A, Nordstrom K. Protein A from Staphylococcus aureus: the biological significance of its reaction with IgG. Ann N Y Acad Sci. 1974;236:252–266. doi: 10.1111/j.1749-6632.1974.tb41496.x. [DOI] [PubMed] [Google Scholar]
- Frank DN, Feazel LM, Bessesen MT, Price CS, Janoff EN, Pace NR. The human nasal microbiota and Staphylococcus aureus carriage. PLoS One. 2010;5:e10598. doi: 10.1371/journal.pone.0010598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frendeus B, Godaly G, Hang L, Karpman D, Lundstedt AC, Svanborg C. Interleukin 8 receptor deficiency confers susceptibility to acute experimental pyelonephritis and may have a human counterpart. J Exp Med. 2000;192:881–890. doi: 10.1084/jem.192.6.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frendeus B, Wachtler C, Hedlund M, Fischer H, Samuelsson P, Svensson M, Svanborg C. Escherichia coli P fimbriae utilize the Toll-like receptor 4 pathway for cell activation. Mol Microbiol. 2001;40:37–51. doi: 10.1046/j.1365-2958.2001.02361.x. [DOI] [PubMed] [Google Scholar]
- Frick IM, Morgelin M, Bjorck L. Virulent aggregates of Streptococcus pyogenes are generated by homophilic protein-protein interactions. Mol Microbiol. 2000;37:1232–1247. doi: 10.1046/j.1365-2958.2000.02084.x. [DOI] [PubMed] [Google Scholar]
- Friedman L, Kolter R. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J Bacteriol. 2004;186:4457–4465. doi: 10.1128/JB.186.14.4457-4465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan JA, Corrigan RM, Gruszka DT, Speziale P, O’Gara JP, Potts JR, Foster TJ. Role of surface protein SasG in biofilm formation by Staphylococcus aureus. J Bacteriol. 2010;192:5663–5673. doi: 10.1128/JB.00628-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerke C, Kraft A, Sussmuth R, Schweitzer O, Gotz F. Characterization of the N-acetylglucosaminyltransferase activity involved in the biosynthesis of the Staphylococcus epidermidis polysaccharide intercellular adhesin. J Biol Chem. 1998;273:18586–18593. doi: 10.1074/jbc.273.29.18586. [DOI] [PubMed] [Google Scholar]
- Gill SR, Fouts DE, Archer GL, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giotis ES, Loeffler A, Knight-Jones T, Lloyd DH. Development of a skin colonization model in gnotobiotic piglets for the study of the microbial ecology of meticillin-resistant Staphylococcus aureus ST398. J Appl Microbiol. 2012;113:992–1000. doi: 10.1111/j.1365-2672.2012.05397.x. [DOI] [PubMed] [Google Scholar]
- Gisch N, Kohler T, Ulmer AJ, Muthing J, Pribyl T, Fischer K, Lindner B, Hammerschmidt S, Zahringer U. Structural reevaluation of Streptococcus pneumoniae Lipoteichoic acid and new insights into its immunostimulatory potency. J Biol Chem. 2013;288:15654–15667. doi: 10.1074/jbc.M112.446963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9:244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross M, Cramton SE, Gotz F, Peschel A. Key role of teichoic acid net charge in Staphylococcus aureus colonization of artificial surfaces. Infect Immun. 2001;69:3423–3426. doi: 10.1128/IAI.69.5.3423-3426.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Li H, Li M, Vuong C, Otto M, Wen Y, Gao Q. Bacterial insertion sequence IS256 as a potential molecular marker to discriminate invasive strains from commensal strains of Staphylococcus epidermidis. J Hosp Infect. 2005;61:342–348. doi: 10.1016/j.jhin.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361:512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- Guyer DM, Radulovic S, Jones FE, Mobley HL. Sat, the secreted autotransporter toxin of uropathogenic Escherichia coli, is a vacuolating cytotoxin for bladder and kidney epithelial cells. Infect Immun. 2002;70:4539–4546. doi: 10.1128/IAI.70.8.4539-4546.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn E, Wild P, Hermanns U, Sebbel P, Glockshuber R, Haner M, Taschner N, Burkhard P, Aebi U, Muller SA. Exploring the 3D molecular architecture of Escherichia coli type 1 pili. J Mol Biol. 2002;323:845–857. doi: 10.1016/s0022-2836(02)01005-7. [DOI] [PubMed] [Google Scholar]
- Harrison F. Microbial ecology of the cystic fibrosis lung. Microbiology. 2007;153:917–923. doi: 10.1099/mic.0.2006/004077-0. [DOI] [PubMed] [Google Scholar]
- Hartford O, O’Brien L, Schofield K, Wells J, Foster TJ. The Fbe (SdrG) protein of Staphylococcus epidermidis HB promotes bacterial adherence to fibrinogen. Microbiology. 2001;147:2545–2552. doi: 10.1099/00221287-147-9-2545. [DOI] [PubMed] [Google Scholar]
- Heilmann C. Adhesion mechanisms of staphylococci. Adv Exp Med Biol. 2011;715:105–123. doi: 10.1007/978-94-007-0940-9_7. [DOI] [PubMed] [Google Scholar]
- Heilmann C, Hussain M, Peters G, Gotz F. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol Microbiol. 1997;24:1013–1024. doi: 10.1046/j.1365-2958.1997.4101774.x. [DOI] [PubMed] [Google Scholar]
- Heilmann C, Hartleib J, Hussain MS, Peters G. The multifunctional Staphylococcus aureus autolysin aaa mediates adherence to immobilized fibrinogen and fibronectin. Infect Immun. 2005;73:4793–4802. doi: 10.1128/IAI.73.8.4793-4802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilmann C, Schweitzer O, Gerke C, Vanittanakom N, Mack D, Gotz F. Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol Microbiol. 1996;20:1083–1091. doi: 10.1111/j.1365-2958.1996.tb02548.x. [DOI] [PubMed] [Google Scholar]
- Heilmann C, Thumm G, Chhatwal GS, Hartleib J, Uekotter A, Peters G. Identification and characterization of a novel autolysin (Aae) with adhesive properties from Staphylococcus epidermidis. Microbiology. 2003;149:2769–2778. doi: 10.1099/mic.0.26527-0. [DOI] [PubMed] [Google Scholar]
- Hoiby N, Ciofu O, Bjarnsholt T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 2010;5:1663–1674. doi: 10.2217/fmb.10.125. [DOI] [PubMed] [Google Scholar]
- Hull RA, Gill RE, Hsu P, Minshew BH, Falkow S. Construction and expression of recombinant plasmids encoding type 1 or D-mannose-resistant pili from a urinary tract infection Escherichia coli isolate. Infect Immun. 1981;33:933–938. doi: 10.1128/iai.33.3.933-938.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultgren SJ, Abraham S, Caparon M, Falk P, St Geme JW, 3rd, Normark S. Pilus and nonpilus bacterial adhesins: assembly and function in cell recognition. Cell. 1993;73:887–901. doi: 10.1016/0092-8674(93)90269-v. [DOI] [PubMed] [Google Scholar]
- Hunstad DA, Justice SS. Intracellular lifestyles and immune evasion strategies of uropathogenic Escherichia coli. Annu Rev Microbiol. 2010;64:203–221. doi: 10.1146/annurev.micro.112408.134258. [DOI] [PubMed] [Google Scholar]
- Hussain M, Herrmann M, von Eiff C, Perdreau-Remington F, Peters G. A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infect Immun. 1997;65:519–524. doi: 10.1128/iai.65.2.519-524.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imberty A, wimmerova M, Mitchell EP, Gilboa-Garber N. Structures of the lectins from Pseudomonas aeruginosa: insight into the molecular basis for host glycan recognition. Microbes Infect. 2004;6:221–228. doi: 10.1016/j.micinf.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Ingham E, Holland KT, Gowland G, Cunliffe WJ. Partial purification and characterization of lipase (EC 3.1.1.3) from Propionibacterium acnes. J Gen Microbiol. 1981;124:393–401. doi: 10.1099/00221287-124-2-393. [DOI] [PubMed] [Google Scholar]
- Iwai H, Kim M, Yoshikawa Y, et al. A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell. 2007;130:611–623. doi: 10.1016/j.cell.2007.06.043. [DOI] [PubMed] [Google Scholar]
- Jakab E, Zbinden R, Gubler J, Ruef C, von Graevenitz A, Krause M. Severe infections caused by Propionibacterium acnes: an underestimated pathogen in late postoperative infections. Yale J Biol Med. 1996;69:477–482. [PMC free article] [PubMed] [Google Scholar]
- Jenkinson HF, Demuth DR. Structure, function and immunogenicity of streptococcal antigen I/II polypeptides. Mol Microbiol. 1997;23:183–190. doi: 10.1046/j.1365-2958.1997.2021577.x. [DOI] [PubMed] [Google Scholar]
- Jiang W, Zhang J, Chen H. Pyrosequencing analysis of oral microbiota in children with severe early childhood dental caries. Curr Microbiol. 2013;67:537–542. doi: 10.1007/s00284-013-0393-7. [DOI] [PubMed] [Google Scholar]
- Joo HS, Otto M. Molecular basis of in vivo biofilm formation by bacterial pathogens. Chem Biol. 2012;19:1503–1513. doi: 10.1016/j.chembiol.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, ten Cate JM, Crielaard W. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 2008;87:1016–1020. doi: 10.1177/154405910808701104. [DOI] [PubMed] [Google Scholar]
- Kim M, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Sasakawa C. Bacterial interactions with the host epithelium. Cell Host Microbe. 2010;8:20–35. doi: 10.1016/j.chom.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Kim M, Ogawa M, Fujita Y, Yoshikawa Y, Nagai T, Koyama T, Nagai S, Lange A, Fassler R, Sasakawa C. Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature. 2009;459:578–582. doi: 10.1038/nature07952. [DOI] [PubMed] [Google Scholar]
- King SJ, Hippe KR, Weiser JN. Deglycosylation of human glycoconjugates by the sequential activities of exoglycosidases expressed by Streptococcus pneumoniae. Mol Microbiol. 2006;59:961–974. doi: 10.1111/j.1365-2958.2005.04984.x. [DOI] [PubMed] [Google Scholar]
- Kloos W, Schleifer KH. In: Staphylococcus. Bergey’s Manual of Systematic Bacteriology. PHASSM, MES, JGH, editors. Williams & Wilkins; Baltimore: 1986. [Google Scholar]
- Kloos WE, Musselwhite MS. Distribution and persistence of Staphylococcus and Micrococcus species and other aerobic bacteria on human skin. Appl Microbiol. 1975;30:381–385. doi: 10.1128/am.30.3.381-395.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan G, Sadovskaya I, Chaignon P, Chokr A, Jabbouri S. Biofilms of clinical strains of Staphylococcus that do not contain polysaccharide intercellular adhesin. FEMS Microbiol Lett. 2006;255:11–16. doi: 10.1111/j.1574-6968.2005.00043.x. [DOI] [PubMed] [Google Scholar]
- Kreikemeyer B, Klenk M, Podbielski A. The intracellular status of Streptococcus pyogenes: role of extracellular matrix-binding proteins and their regulation. Int J Med Microbiol. 2004;294:177–188. doi: 10.1016/j.ijmm.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Lamont RJ, Demuth DR, Davis CA, Malamud D, Rosan B. Salivary-agglutinin-mediated adherence of Streptococcus mutans to early plaque bacteria. Infect Immun. 1991;59:3446–3450. doi: 10.1128/iai.59.10.3446-3450.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasa I, Penades JR. Bap: a family of surface proteins involved in biofilm formation. Res Microbiol. 2006;157:99–107. doi: 10.1016/j.resmic.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Li M, Du X, Villaruz AE, et al. MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat Med. 2012;18:816–819. doi: 10.1038/nm.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg F, Lund B, Johansson L, Normark S. Localization of the receptor-binding protein adhesin at the tip of the bacterial pilus. Nature. 1987;328:84–87. doi: 10.1038/328084a0. [DOI] [PubMed] [Google Scholar]
- Lipsky BA, Goldberger AC, Tompkins LS, Plorde JJ. Infections caused by nondiphtheria corynebacteria. Rev Infect Dis. 1982;4:1220–1235. doi: 10.1093/clinids/4.6.1220. [DOI] [PubMed] [Google Scholar]
- Lloyd SJ, Ritchie JM, Torres AG. Fimbriation and curliation in Escherichia coli O157:H7: a paradigm of intestinal and environmental colonization. Gut Microbes. 2012;3:272–276. doi: 10.4161/gmic.20661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh JT, Cover TL. Requirement of histidine kinases HP0165 and HP1364 for acid resistance in Helicobacter pylori. Infect Immun. 2006;74:3052–3059. doi: 10.1128/IAI.74.5.3052-3059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh JT, Gupta SS, Friedman DB, Krezel AM, Cover TL. Analysis of protein expression regulated by the Helicobacter pylori ArsRS two-component signal transduction system. J Bacteriol. 2010;192:2034–2043. doi: 10.1128/JB.01703-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- Mack D, Fischer W, Krokotsch A, Leopold K, Hartmann R, Egge H, Laufs R. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol. 1996;178:175–183. doi: 10.1128/jb.178.1.175-183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdavi J, Sonden B, Hurtig M, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisey HC, Hensler M, Nizet V, Doran KS. Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J Bacteriol. 2007;189:1464–1467. doi: 10.1128/JB.01153-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak TN, Schmid M, Brzuszkiewicz E, Zeng G, Meyer R, Sfanos KS, Brinkmann V, Meyer TF, Bruggemann H. Comparative genomics reveals distinct host-interacting traits of three major human-associated propionibacteria. BMC Genomics. 2013;14:640. doi: 10.1186/1471-2164-14-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandlik A, Swierczynski A, Das A, Ton-That H. Corynebacterium diphtheriae employs specific minor pilins to target human pharyngeal epithelial cells. Mol Microbiol. 2007;64:111–124. doi: 10.1111/j.1365-2958.2007.05630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandlik A, Swierczynski A, Das A, Ton-That H. Pili in Gram-positive bacteria: assembly, involvement in colonization and biofilm development. Trends Microbiol. 2008;16:33–40. doi: 10.1016/j.tim.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann EE, Rice KC, Boles BR, Endres JL, Ranjit D, Chandramohan L, Tsang LH, Smeltzer MS, Horswill AR, Bayles KW. Modulation of eDNA release and degradation affects Staphylococcus aureus biofilm maturation. PLoS ONE. 2009;4:e5822. doi: 10.1371/journal.pone.0005822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Dedent AC, Schneewind O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol Mol Biol Rev. 2006;70:192–221. doi: 10.1128/MMBR.70.1.192-221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- Martin DW, Schurr MJ, Mudd MH, Govan JR, Holloway BW, Deretic V. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc Natl Acad Sci U S A. 1993;90:8377–8381. doi: 10.1073/pnas.90.18.8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, Hultgren SJ. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000;19:2803–2812. doi: 10.1093/emboj/19.12.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Liu G, Ton-That H, Schneewind O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science. 1999;285:760–763. doi: 10.1126/science.285.5428.760. [DOI] [PubMed] [Google Scholar]
- McCrea KW, Hartford O, Davis S, Eidhin DN, Lina G, Speziale P, Foster TJ, Hook M. The serine-aspartate repeat (Sdr) protein family in Staphylococcus epidermidis. Microbiology. 2000;146 (Pt 7):1535–1546. doi: 10.1099/00221287-146-7-1535. [DOI] [PubMed] [Google Scholar]
- McDevitt D, Francois P, Vaudaux P, Foster TJ. Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol Microbiol. 1994;11:237–248. doi: 10.1111/j.1365-2958.1994.tb00304.x. [DOI] [PubMed] [Google Scholar]
- Mergenhagen SE, Sandberg AL, Chassy BM, Brennan MJ, Yeung MK, Donkersloot JA, Cisar JO. Molecular basis of bacterial adhesion in the oral cavity. Rev Infect Dis. 1987;9(Suppl 5):S467–474. doi: 10.1093/clinids/9.supplement_5.s467. [DOI] [PubMed] [Google Scholar]
- Merino N, Toledo-Arana A, Vergara-Irigaray M, Valle J, Solano C, Calvo E, Lopez JA, Foster TJ, Penades JR, Lasa I. Protein A-mediated multicellular behavior in Staphylococcus aureus. J Bacteriol. 2009;191:832–843. doi: 10.1128/JB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley HL, Island MD, Hausinger RP. Molecular biology of microbial ureases. Microbiol Rev. 1995;59:451–480. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley A, Latronico F, Guardabassi L. Experimental colonization of pigs with methicillin-resistant Staphylococcus aureus (MRSA): insights into the colonization and transmission of livestock-associated MRSA. Epidemiol Infect. 2011;139:1594–1600. doi: 10.1017/S0950268810002888. [DOI] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, et al. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337:1115–1119. doi: 10.1126/science.1225152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AL, Roche AM, Gould JM, Chim K, Ratner AJ, Weiser JN. Capsule enhances pneumococcal colonization by limiting mucus-mediated clearance. Infect Immun. 2007;75:83–90. doi: 10.1128/IAI.01475-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Eidhin D, Perkins S, Francois P, Vaudaux P, Hook M, Foster TJ. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol Microbiol. 1998;30:245–257. doi: 10.1046/j.1365-2958.1998.01050.x. [DOI] [PubMed] [Google Scholar]
- Nilson BH, Frick IM, Akesson P, Forsen S, Bjorck L, Akerstrom B, Wikstrom M. Structure and stability of protein H and the M1 protein from Streptococcus pyogenes. Implications for other surface proteins of gram-positive bacteria. Biochemistry. 1995;34:13688–13698. doi: 10.1021/bi00041a051. [DOI] [PubMed] [Google Scholar]
- Nobbs AH, Lamont RJ, Jenkinson HF. Streptococcus adherence and colonization. Microbiol Mol Biol Rev. 2009;73:407–450. doi: 10.1128/MMBR.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP, Geisinger E. Quorum sensing in staphylococci. Annu Rev Genet. 2008;42:541–564. doi: 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- Novick RP, Projan SJ, Kornblum J, Ross HF, Ji G, Kreiswirth B, Vandenesch F, Moghazeh S. The agr P2 operon: an autocatalytic sensory transduction system in Staphylococcus aureus. Mol Gen Genet. 1995;248:446–458. doi: 10.1007/BF02191645. [DOI] [PubMed] [Google Scholar]
- Nyvad B, Crielaard W, Mira A, Takahashi N, Beighton D. Dental caries from a molecular microbiological perspective. Caries Res. 2013;47:89–102. doi: 10.1159/000345367. [DOI] [PubMed] [Google Scholar]
- O’Neill E, Pozzi C, Houston P, Humphreys H, Robinson DA, Loughman A, Foster TJ, O’Gara JP. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J Bacteriol. 2008;190:3835–3850. doi: 10.1128/JB.00167-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole GA. To build a biofilm. J Bacteriol. 2003;185:2687–2689. doi: 10.1128/JB.185.9.2687-2689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RJ, Musser JM. Molecular pathogenesis of necrotizing fasciitis. Annu Rev Pathol. 2010;5:1–31. doi: 10.1146/annurev-pathol-121808-102135. [DOI] [PubMed] [Google Scholar]
- Otto M. Staphylococcal biofilms. Curr Top Microbiol Immunol. 2008;322:207–228. doi: 10.1007/978-3-540-75418-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcus epidermidis - the ‘accidental’ pathogen. Nat Rev Microbiol. 2009;7:555–567. doi: 10.1038/nrmicro2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu Rev Med. 2013;64:175–188. doi: 10.1146/annurev-med-042711-140023. [DOI] [PubMed] [Google Scholar]
- Patti JM, Allen BL, McGavin MJ, Hook M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol. 1994;48:585–617. doi: 10.1146/annurev.mi.48.100194.003101. [DOI] [PubMed] [Google Scholar]
- Pelicic V. Type IV pili: e pluribus unum? Mol Microbiol. 2008;68:827–837. doi: 10.1111/j.1365-2958.2008.06197.x. [DOI] [PubMed] [Google Scholar]
- Peschel A, Vuong C, Otto M, Gotz F. The D-alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob Agents Chemother. 2000;44:2845–2847. doi: 10.1128/aac.44.10.2845-2847.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnuraj K, Bowden MG, Davis S, Gurusiddappa S, Moore D, Choe D, Xu Y, Hook M, Narayana SV. A “dock, lock, and latch” structural model for a staphylococcal adhesin binding to fibrinogen. Cell. 2003;115:217–228. doi: 10.1016/s0092-8674(03)00809-2. [DOI] [PubMed] [Google Scholar]
- Ramage G, Tunney MM, Patrick S, Gorman SP, Nixon JR. Formation of Propionibacterium acnes biofilms on orthopaedic biomaterials and their susceptibility to antimicrobials. Biomaterials. 2003;24:3221–3227. doi: 10.1016/s0142-9612(03)00173-x. [DOI] [PubMed] [Google Scholar]
- Rennermalm A, Nilsson M, Flock JI. The fibrinogen binding protein of Staphylococcus epidermidis is a target for opsonic antibodies. Infect Immun. 2004;72:3081–3083. doi: 10.1128/IAI.72.5.3081-3083.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers KL, Rupp ME, Fey PD. The presence of icaADBC is detrimental to the colonization of human skin by Staphylococcus epidermidis. Appl Environ Microbiol. 2008;74:6155–6157. doi: 10.1128/AEM.01017-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers KL, Fey PD, Rupp ME. Coagulase-negative staphylococcal infections. Infect Dis Clin North Am. 2009;23:73–98. doi: 10.1016/j.idc.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Rohde H, Kalitzky M, Kroger N, Scherpe S, Horstkotte MA, Knobloch JK, Zander AR, Mack D. Detection of virulence-associated genes not useful for discriminating between invasive and commensal Staphylococcus epidermidis strains from a bone marrow transplant unit. J Clin Microbiol. 2004;42:5614–5619. doi: 10.1128/JCM.42.12.5614-5619.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde H, Burdelski C, Bartscht K, Hussain M, Buck F, Horstkotte MA, Knobloch JK, Heilmann C, Herrmann M, Mack D. Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol Microbiol. 2005;55:1883–1895. doi: 10.1111/j.1365-2958.2005.04515.x. [DOI] [PubMed] [Google Scholar]
- Rohde H, Burandt EC, Siemssen N, et al. Polysaccharide intercellular adhesin or protein factors in biofilm accumulation of Staphylococcus epidermidis and Staphylococcus aureus isolated from prosthetic hip and knee joint infections. Biomaterials. 2007;28:1711–1720. doi: 10.1016/j.biomaterials.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Roth RR, James WD. Microbial ecology of the skin. Annu Rev Microbiol. 1988;42:441–464. doi: 10.1146/annurev.mi.42.100188.002301. [DOI] [PubMed] [Google Scholar]
- Rundegren J. Calcium-dependent salivary agglutinin with reactivity to various oral bacterial species. Infect Immun. 1986;53:173–178. doi: 10.1128/iai.53.1.173-178.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder K, Jularic M, Horsburgh SM, et al. Molecular characterization of a novel Staphylococcus aureus surface protein (SasC) involved in cell aggregation and biofilm accumulation. PLoS One. 2009;4:e7567. doi: 10.1371/journal.pone.0007567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellman BR, Timofeyeva Y, Nanra J, Scott A, Fulginiti JP, Matsuka YV, Baker SM. Expression of Staphylococcus epidermidis SdrG increases following exposure to an in vivo environment. Infect Immun. 2008;76:2950–2957. doi: 10.1128/IAI.00055-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminario A, Broukal Z, Ivancakova R. Mutans streptococci and the development of dental plaque. Prague Med Rep. 2005;106:349–358. [PubMed] [Google Scholar]
- Shaheen B, Gonzalez M. Acne sans P. acnes. J Eur Acad Dermatol Venereol. 2013;27:1–10. doi: 10.1111/j.1468-3083.2012.04516.x. [DOI] [PubMed] [Google Scholar]
- Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature. 2000;407:762–764. doi: 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- Skouloubris S, Labigne A, De Reuse H. Identification and characterization of an aliphatic amidase in Helicobacter pylori. Mol Microbiol. 1997;25:989–998. doi: 10.1111/j.1365-2958.1997.mmi536.x. [DOI] [PubMed] [Google Scholar]
- Skouloubris S, Labigne A, De Reuse H. The AmiE aliphatic amidase and AmiF formamidase of Helicobacter pylori: natural evolution of two enzyme paralogues. Mol Microbiol. 2001;40:596–609. doi: 10.1046/j.1365-2958.2001.02400.x. [DOI] [PubMed] [Google Scholar]
- Soderquist B, Holmberg A, Unemo M. Propionibacterium acnes as an etiological agent of arthroplastic and osteosynthetic infections--two cases with specific clinical presentation including formation of draining fistulae. Anaerobe. 2010;16:304–306. doi: 10.1016/j.anaerobe.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Sommer F, Backhed F. The gut microbiota--masters of host development and physiology. Nat Rev Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- Stecher B, Hardt WD. The role of microbiota in infectious disease. Trends Microbiol. 2008;16:107–114. doi: 10.1016/j.tim.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Stecher B, Berry D, Loy A. Colonization resistance and microbial ecophysiology: using gnotobiotic mouse models and single-cell technology to explore the intestinal jungle. FEMS Microbiol Rev. 2013;37:793–829. doi: 10.1111/1574-6976.12024. [DOI] [PubMed] [Google Scholar]
- Svanborg C, Godaly G. Bacterial virulence in urinary tract infection. Infect Dis Clin North Am. 1997;11:513–529. doi: 10.1016/s0891-5520(05)70371-8. [DOI] [PubMed] [Google Scholar]
- Swaminathan A, Mandlik A, Swierczynski A, Gaspar A, Das A, Ton-That H. Housekeeping sortase facilitates the cell wall anchoring of pilus polymers in Corynebacterium diphtheriae. Mol Microbiol. 2007;66:961–974. doi: 10.1111/j.1365-2958.2007.05968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Konishi K, Cisar JO, Yoshikawa M. Identification and characterization of hsa, the gene encoding the sialic acid-binding adhesin of Streptococcus gordonii DL1. Infect Immun. 2002;70:1209–1218. doi: 10.1128/IAI.70.3.1209-1218.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu D, Bensing BA, Sullam PM. Genes in the accessory sec locus of Streptococcus gordonii have three functionally distinct effects on the expression of the platelet-binding protein GspB. Mol Microbiol. 2004;52:189–203. doi: 10.1111/j.1365-2958.2004.03978.x. [DOI] [PubMed] [Google Scholar]
- ten Cate JM. Biofilms, a new approach to the microbiology of dental plaque. Odontology. 2006;94:1–9. doi: 10.1007/s10266-006-0063-3. [DOI] [PubMed] [Google Scholar]
- Ton-That H, Schneewind O. Assembly of pili on the surface of Corynebacterium diphtheriae. Mol Microbiol. 2003;50:1429–1438. doi: 10.1046/j.1365-2958.2003.03782.x. [DOI] [PubMed] [Google Scholar]
- Traber KE, Lee E, Benson S, Corrigan R, Cantera M, Shopsin B, Novick RP. agr function in clinical Staphylococcus aureus isolates. Microbiology. 2008;154:2265–2274. doi: 10.1099/mic.0.2007/011874-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unemo M, Aspholm-Hurtig M, Ilver D, Bergstrom J, Boren T, Danielsson D, Teneberg S. The sialic acid binding SabA adhesin of Helicobacter pylori is essential for nonopsonic activation of human neutrophils. J Biol Chem. 2005;280:15390–15397. doi: 10.1074/jbc.M412725200. [DOI] [PubMed] [Google Scholar]
- Vaisanen V, Elo J, Tallgren LG, Siitonen A, Makela PH, Svanborg-Eden C, Kallenius G, Svenson SB, Hultberg H, Korhonen T. Mannose-resistant haemagglutination and P antigen recognition are characteristic of Escherichia coli causing primary pyelonephritis. Lancet. 1981;2:1366–1369. doi: 10.1016/s0140-6736(81)92796-3. [DOI] [PubMed] [Google Scholar]
- Vandenesch F, Kornblum J, Novick RP. A temporal signal, independent of agr, is required for hla but not spa transcription in Staphylococcus aureus. J Bacteriol. 1991;173:6313–6320. doi: 10.1128/jb.173.20.6313-6320.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtaneva K, Porcella SF, Graham MR, et al. Longitudinal analysis of the group A Streptococcus transcriptome in experimental pharyngitis in cynomolgus macaques. Proc Natl Acad Sci U S A. 2005;102:9014–9019. doi: 10.1073/pnas.0503671102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. N Engl J Med. 2001;344:11–16. doi: 10.1056/NEJM200101043440102. [DOI] [PubMed] [Google Scholar]
- Vuong C, Kocianova S, Yao Y, Carmody AB, Otto M. Increased colonization of indwelling medical devices by quorum-sensing mutants of Staphylococcus epidermidis in vivo. J Infect Dis. 2004;190:1498–1505. doi: 10.1086/424487. [DOI] [PubMed] [Google Scholar]
- Vuong C, Kocianova S, Voyich JM, Yao Y, Fischer ER, DeLeo FR, Otto M. A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J Biol Chem. 2004;279:54881–54886. doi: 10.1074/jbc.M411374200. [DOI] [PubMed] [Google Scholar]
- Waksman G, Hultgren SJ. Structural biology of the chaperone-usher pathway of pilus biogenesis. Nat Rev Microbiol. 2009;7:765–774. doi: 10.1038/nrmicro2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh EJ, O’Brien LM, Liang X, Hook M, Foster TJ. Clumping factor B, a fibrinogen-binding MSCRAMM (microbial surface components recognizing adhesive matrix molecules) adhesin of Staphylococcus aureus, also binds to the tail region of type I cytokeratin 10. J Biol Chem. 2004;279:50691–50699. doi: 10.1074/jbc.M408713200. [DOI] [PubMed] [Google Scholar]
- Walz A, Odenbreit S, Mahdavi J, Boren T, Ruhl S. Identification and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays. Glycobiology. 2005;15:700–708. doi: 10.1093/glycob/cwi049. [DOI] [PubMed] [Google Scholar]
- Wang X, Preston JF, 3rd, Romeo T. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J Bacteriol. 2004;186:2724–2734. doi: 10.1128/JB.186.9.2724-2734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser JN, Austrian R, Sreenivasan PK, Masure HR. Phase variation in pneumococcal opacity: relationship between colonial morphology and nasopharyngeal colonization. Infect Immun. 1994;62:2582–2589. doi: 10.1128/iai.62.6.2582-2589.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005;5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- Wessels MR. Clinical practice. Streptococcal pharyngitis. N Engl J Med. 2011;364:648–655. doi: 10.1056/NEJMcp1009126. [DOI] [PubMed] [Google Scholar]
- Williams HC, Dellavalle RP, Garner S. Acne vulgaris. Lancet. 2012;379:361–372. doi: 10.1016/S0140-6736(11)60321-8. [DOI] [PubMed] [Google Scholar]
- Wright KJ, Hultgren SJ. Sticky fibers and uropathogenesis: bacterial adhesins in the urinary tract. Future Microbiol. 2006;1:75–87. doi: 10.2217/17460913.1.1.75. [DOI] [PubMed] [Google Scholar]
- Wu XR, Sun TT, Medina JJ. In vitro binding of type 1-fimbriated Escherichia coli to uroplakins Ia and Ib: relation to urinary tract infections. Proc Natl Acad Sci U S A. 1996;93:9630–9635. doi: 10.1073/pnas.93.18.9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki AB. A review of the skin and its appendages. Adv Wound Care. 1995;8:53–54. 56–62. 64 passim. [PubMed] [Google Scholar]
- Yang L, Hengzhuang W, Wu H, Damkiaer S, Jochumsen N, Song Z, Givskov M, Hoiby N, Molin S. Polysaccharides serve as scaffold of biofilms formed by mucoid Pseudomonas aeruginosa. FEMS Immunol Med Microbiol. 2012;65:366–376. doi: 10.1111/j.1574-695X.2012.00936.x. [DOI] [PubMed] [Google Scholar]
- Yao Y, Sturdevant DE, Villaruz A, Xu L, Gao Q, Otto M. Factors characterizing Staphylococcus epidermidis invasiveness determined by comparative genomics. Infect Immun. 2005;73:1856–1860. doi: 10.1128/IAI.73.3.1856-1860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JL, Mansson R, Flock JI, Ljungh A. Fibronectin binding by Propionibacterium acnes. FEMS Immunol Med Microbiol. 1997;19:247–253. doi: 10.1111/j.1574-695X.1997.tb01094.x. [DOI] [PubMed] [Google Scholar]
- Zabaleta J, McGee DJ, Zea AH, Hernandez CP, Rodriguez PC, Sierra RA, Correa P, Ochoa AC. Helicobacter pylori arginase inhibits T cell proliferation and reduces the expression of the TCR zeta-chain (CD3zeta) J Immunol. 2004;173:586–593. doi: 10.4049/jimmunol.173.1.586. [DOI] [PubMed] [Google Scholar]
- Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009;9:259. doi: 10.1186/1471-2180-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Houry WA. Acid stress response in enteropathogenic gammaproteobacteria: an aptitude for survival. Biochem Cell Biol. 2010;88:301–314. doi: 10.1139/o09-182. [DOI] [PubMed] [Google Scholar]
- Ziebuhr W, Heilmann C, Gotz F, Meyer P, Wilms K, Straube E, Hacker J. Detection of the intercellular adhesion gene cluster (ica) and phase variation in Staphylococcus epidermidis blood culture strains and mucosal isolates. Infect Immun. 1997;65:890–896. doi: 10.1128/iai.65.3.890-896.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]