

Abstract
Hyperoxia has been uniformly efficacious in experimental focal cerebral ischemia. However, pilot clinical trials have showed mixed results slowing its translation in patient care. To explain the discordance between experimental and clinical outcomes, we tested the impact of endothelial dysfunction, exceedingly common in stroke patients but under-represented in experimental studies, on the neuroprotective efficacy of normobaric hyperoxia. We used hyperlipidemic apolipoprotein E knock-out and endothelial nitric oxide synthase knock-out mice as models of endothelial dysfunction, and examined the effects of normobaric hyperoxia on tissue perfusion and oxygenation using high-resolution combined laser speckle and multispectral reflectance imaging during distal middle cerebral artery occlusion. In normal wild-type mice, normobaric hyperoxia rapidly and significantly improved tissue perfusion and oxygenation, suppressed peri-infarct depolarizations, reduced infarct volumes, and improved neurological function. In contrast, normobaric hyperoxia worsened perfusion in ischemic brain and failed to reduce infarct volumes or improve neurological function in mice with endothelial dysfunction. These data suggest that the beneficial effects of hyperoxia on ischemic tissue oxygenation, perfusion, and outcome are critically dependent on endothelial nitric oxide synthase function. Therefore, vascular risk factors associated with endothelial dysfunction may predict normobaric hyperoxia nonresponders in ischemic stroke. These data may have implications for myocardial and systemic circulation as well.
Keywords: cerebral blood flow, focal cerebral ischemia, laser speckle imaging, multispectral reflectance imaging, optical imaging, viability threshold
Introduction
Improving brain tissue oxygenation by supplemental O2 inhalation, either through a high-flow mask (i.e., normobaric) or in a pressure chamber (i.e., hyperbaric), is a potential therapeutic strategy in acute stroke. In contrast to hyperbaric hyperoxia (HBO), which has limited feasibility in the hyperacute setting, normobaric hyperoxia (NBO) can be rapidly initiated in the field within minutes after stroke onset. Although animal studies have firmly established the neuroprotective efficacy of NBO in ischemic stroke (Flynn and Auer, 2002; Singhal et al., 2002a; Kim et al., 2005; Shin et al., 2007; W. Liu et al., 2011), pilot human studies have yielded contradictory results (Anderson et al., 1991; Nighoghossian et al., 1995; Rusyniak et al., 2003; Singhal et al., 2005; Chiu et al., 2006). While one pilot NBO trial suggested an improvement in outcome (Singhal et al., 2005), a second and larger study failed to show a benefit (Padma et al., 2010). In a large multicenter retrospective cohort, arterial hyperoxia was independently associated with higher mortality in ventilated stroke patients (Rincon et al., 2014). Trials of HBO in acute ischemic stroke were equally disappointing. In one pilot HBO trial, there was no benefit, and in two others normoxic groups appeared to fare better than the hyperoxic groups (Anderson et al., 1991; Rusyniak et al., 2003). Indeed, a randomized trial of NBO was terminated early due to higher mortality in the hyperoxia group (http://clinicaltrials.gov/show/NCT00414726). Although worse outcomes were judged to be unrelated to hyperoxia treatment per se, data clearly suggest a discord between animal studies and clinical trials.
Protective mechanism(s) of NBO in ischemic stroke are poorly understood. In addition to the well documented improvement in O2 availability and metabolism in ischemic tissue (S. Liu et al., 2004; Shin et al., 2007; Singhal et al., 2007; Baskerville et al., 2011; Sun et al., 2011), hyperoxia has been reported to preserve the blood–brain barrier and diminish ischemic edema (Veltkamp et al., 2005; W. Liu et al., 2009b, 2011), implicating a vascular endothelial site of action. Indeed, we and others have shown that NBO rapidly improves ischemic tissue perfusion as one mechanism of neuroprotection (Nakajima et al., 1983; Jimènez et al., 2001; Shin et al., 2007; Baskerville et al., 2011; Wu et al., 2012). How supplemental O2 augments tissue perfusion to provide an acute cerebral hemodynamic benefit is unknown.
Stroke often occurs in individuals with hypertension and hyperlipidemia, which are associated with endothelial dysfunction defined as deficient vascular reactivity due to insufficient endothelial production of nitric oxide. While endothelial dysfunction is exceedingly common in stroke patients enrolled in clinical trials, it is rarely represented in animal studies of stroke. Therefore, we hypothesized that improved ischemic tissue perfusion afforded by hyperoxia is dependent on normal endothelial function and critical for tissue outcome. We tested this using endothelial nitric oxide synthase knock-out (eNOS KO) and hyperlipidemic apolipoprotein E knock-out (ApoE KO) mice as models of endothelial dysfunction (Bonthu et al., 1997; d'Uscio et al., 2001a; Kitayama et al., 2007). Systemic and cerebrovascular endothelial dysfunction have already been extensively characterized in the ApoE KO, including cranial window preparations directly interrogating the pial arteries and arterioles that are critical for collateral flow in focal ischemia (Bonthu et al., 1997; Laursen et al., 2001; d'Uscio et al., 2001a,b; Ozaki et al., 2002; Kitayama et al., 2007). We recently showed reduced resting cerebral blood flow (CBF), impaired cerebrovascular reflexes, and enlarged perfusion defects in the ApoE KO on high-fat diet (HFD; Ayata et al., 2013). HFD alone has also been shown to impair endothelial function in otherwise normal rats (W. Li et al., 2013). The eNOS KO, by the very nature of the genetic defect, also has impaired endothelial function (P. L. Huang et al., 1995). The data we present herein using these animal models strongly suggest that normal endothelial function is requisite for NBO efficacy in focal ischemia, and may explain the clinical failure of NBO.
Materials and Methods
Experimental animals.
All experimental procedures were performed in accordance with the Guide for Care and Use of Laboratory Animals (NIH Publication No. 85-23, 1996), and were approved by the institutional review board (MGH Subcommittee on Research Animal Care, SRAC). Wild-type (C57BL/6J), and ApoE KO and eNOS KO mice on a C57BL/6J genetic background (The Jackson Laboratory), were housed under diurnal lighting conditions and allowed food and tap water ad libitum. We used only male mice to build upon the previous mutant studies of endothelial dysfunction, and to avoid the potentially confounding effects of female gonadal hormones on stroke outcome (Hurn and Macrae, 2000) at this proof-of-concept stage. A subgroup of wild-type mice, and all ApoE KO mice, were fed a Western-type HFD (42% of total calories from fat; 0.15% cholesterol; Harlan Teklad) for 8 weeks starting at 4 weeks of age. We have recently shown that ApoE KO at this age and HFD duration develops early stage atherosclerotic lesions limited to scattered subendothelial foam cells in carotid arteries, and mild fatty streaks in the proximal aortic arch, without flow-limiting stenoses, which might pose a physical barrier and reduce the perfusion pressure as a confounder (Ayata et al., 2013).
Distal middle cerebral artery occlusion.
Mice were anesthetized with 2% isoflurane (in 70% N2O and 30% O2). Femoral artery was catheterized for the measurement of BP (ETH 400 transducer amplifier). Rectal temperature was kept at 36.8–37.1°C using a thermostatically controlled heating mat (FHC). Mice were paralyzed (pancuronium bromide, 0.4 mg/kg/h, i.p.), mechanically ventilated (CWE, SAR-830), and placed in a stereotaxic frame (David Kopf). In survival experiments to measure infarct volume at 48 h, morbidity and mortality precluded neuromuscular paralysis, intubation, and ventilation. Arterial blood gases and pH were measured in all mice every 30 min (Corning 178 blood gas/pH analyzer; Ciba Corning Diagnostics). These data were continuously recorded using a data acquisition and analysis system (PowerLab; AD Instruments) and stored in a computer. Mice were allowed to stabilize for 30 min after surgical preparation.
Distal middle cerebral artery occlusion (dMCAO) was induced as previously described (Ayata et al., 2013). After general surgical preparation, mice were placed in a stereotaxic frame, and skull surface was prepared for optical imaging. The temporalis muscle was separated from the temporal bone and a burr hole (2 mm diameter) was drilled under saline cooling over the distal middle cerebral artery (MCA) just above the zygomatic arch. The dura was kept intact and MCA was occluded using a microvascular clip.
We studied the spatiotemporal characteristics of CBF changes during dMCAO using laser speckle flowmetry (LSF) noninvasively through intact skull, as described in detail previously (Ayata et al., 2004). The severity of CBF deficit was analyzed two-dimensionally by quantifying the area of the hypoperfused cortex using a thresholding paradigm (pixels with residual CBF ≤20% or ≤30% of the pre-ischemic baseline, expressed in mm2). In addition, we studied changes in tissue oxygenation using multispectral reflectance imaging, as previously described in detail (Shin et al., 2007; Yuzawa et al., 2012). A region of interest was placed in ischemic penumbra to measure changes in oxyhemoglobin and deoxyhemoglobin concentrations over time during dMCAO. Hemoglobin saturation (satHb) was calculated as oxyhemoglobin/(oxyhemoglobin + deoxyhemoglobin) and expressed as percentage of baseline.
The impact of NBO on infarct volume was determined in a separate group of mice without intubation or mechanical ventilation, since these surgical procedures significantly increase morbidity and mortality during the recovery period. To determine infarct volume, microvascular clip was carefully removed after 75 min of dMCAO, and successful reperfusion confirmed using LSF; animals without reperfusion or with hemorrhage during clip removal were excluded from the study. Mice were killed 48 h after ischemia. In pilot studies we determined that 48 h time point captures the maximum infarct volume after dMCAO and reduces the confounding effect of ischemic brain edema that becomes significant at 72 h (data not shown). After dMCAO, whole brains were incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC; Sigma) solution (1 g/50 ml saline) for 60 min. After photographing the hemispheric surface, brains were cut into 1-mm-thick coronal sections, and infarct area at each section was measured and integrated along the anteroposterior axis to calculate infarct volume (mm3). Relatively small infarct volumes in this pure cortical stroke model precluded correction for ischemic swelling (i.e., indirect method). The viability threshold for CBF was calculated by spatially overlapping the laser speckle flow maps and the TTC infarct, and determining the residual CBF at the infarct edge, as described in detail previously (Eikermann-Haerter et al., 2012; Ayata et al., 2013).
Imaging was started 5 min before dMCAO and continued uninterrupted for 75 min. Normoxia group was maintained on 30% O2, whereas in the NBO group, the fraction of oxygen in inspired air was increased to 100% 15 min after dMCAO. In survival experiments to assess infarct volume, mice were allowed to breathe room air at the time of reperfusion at 75 min, and killed 48 h after dMCAO onset.
Filament middle cerebral artery occlusion.
A nylon monofilament was inserted into the internal carotid artery via the external carotid artery followed by reperfusion after 60 min under isoflurane anesthesia (2.5% induction, 1.5% maintenance, in 70% N2O and 30% O2) and laser Doppler monitoring as previously described (Eikermann-Haerter et al., 2012). Mice were then placed in a temperature-controlled incubator with easy access to food and water. Neurological outcomes were graded 24 h after reperfusion as follows: 0, normal; 1, forepaw monoparesis; 2, circling to right; 3, falling to right; 4, no spontaneous walking and depressed consciousness; and 5, death. In addition, a composite neurological score was adopted to assess spontaneous activity (0–3), limb movement (0–3), climbing (0–3), and balance (0–3) for a total score of 12 in normal mice, as described previously (Parra et al., 2002). Brains were cut into 1-mm-thick coronal sections and incubated in 2% TTC solution for 30 min. Infarct volume was calculated by subtracting the volume of the ipsilateral noninfarcted tissue from the contralateral hemisphere at each section level integrated along the anteroposterior axis (i.e., indirect method) to eliminate the contribution of ischemic swelling. The 24 h time point was chosen for assessment after filament MCAO (fMCAO) due to relatively high mortality after 24 h in this model.
Experimental groups and data analysis.
For dMCAO, normoxia or NBO was instituted in WT, WT on HFD (WT + HFD), ApoE KO on HFD (ApoE KO + HFD), and eNOS KO. For fMCAO, normoxia or NBO was instituted in WT and eNOS KO. In the absence of suitable pilot data, initial sample sizes empirically targeted at least 80% power to detect a 35% difference between the groups with an SD of 25% of the mean. Final sample sizes were based on power calculations in these initial cohorts. Mice were randomly assigned (coin flip) to normoxia or hyperoxia groups. HFD caused characteristic fur changes that precluded effective blinding during data acquisition; allocation was concealed during subsequent endpoint assessments. A priori exclusion criteria included technical failure to successfully occlude (i.e., CBF ≤20% of baseline) or reperfuse (i.e., CBF ≥50% of baseline) the middle cerebral artery without hemorrhage. In the eNOS KO only, we experienced difficulty in inserting the filament to achieve successful occlusion. As a result, 13 of 25 mice were excluded due to insufficient CBF reduction or subarachnoid hemorrhage as technical failures.
Data are expressed as box-whisker plots in which individual data points, median, 25–75% and full ranges, and the mean (+) are shown, or as mean ± SEM. Statistical comparisons were done using one-way or two-way ANOVA for repeated measures followed by Fisher's protected LSD; p < 0.05 was considered statistically significant.
Results
Systemic physiology
NBO raised arterial pO2 to a range of 300–400 mmHg, but otherwise did not significantly impact other systemic physiological parameters (Fig. 1). Arterial BP was significantly higher in ApoE KO + HFD and eNOS KO groups compared with WT. Otherwise, arterial pH, pCO2, and pO2 were all within normal limits. Last, HFD significantly increased body weight in both WT and ApoE KO mice, and eNOS KO showed a small but significant reduction in body weight compared with WT.
Figure 1.
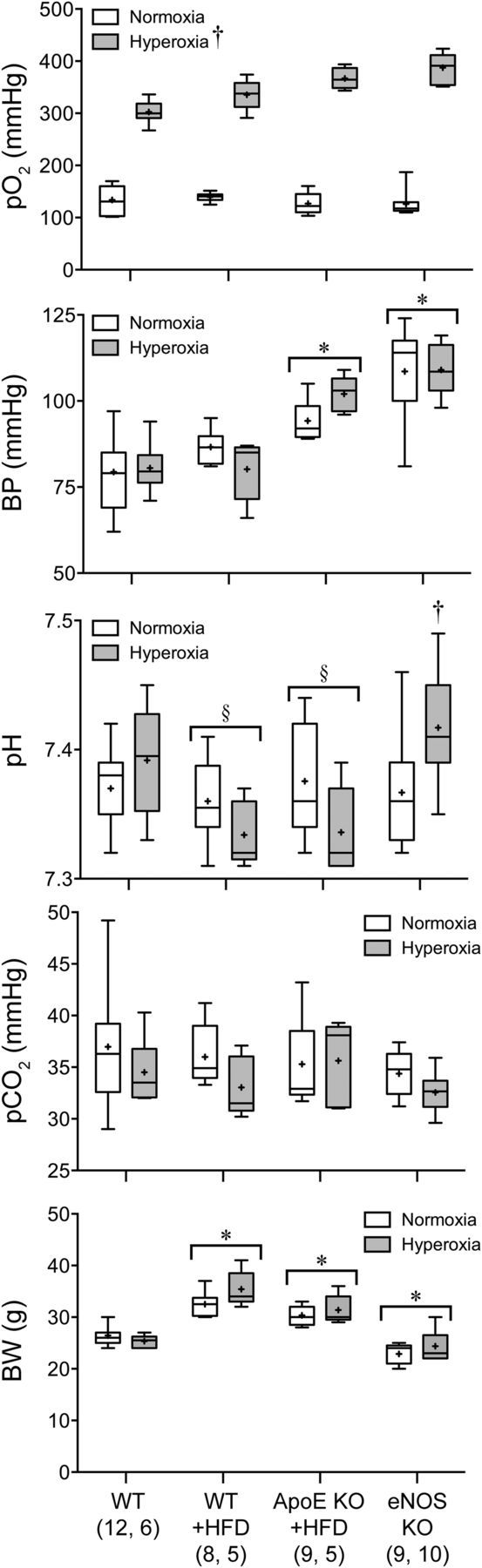
Systemic physiological parameters. Data are expressed as box-whisker plots in which median (horizontal line), 25–75% and full ranges (box-whisker), and mean (+) are shown. BW, body weight. †p < 0.05 versus normoxia; *p < 0.05 versus all other animal groups; §p < 0.05 versus eNOS KO. Two-way ANOVA followed by Fisher's LSD. Sample sizes are shown under each group.
CBF and oxygenation
Upon dMCAO, a focal area of hypoperfusion developed over the dorsolateral cortex (Fig. 2A). Institution of NBO in WT mice improved ischemic tissue perfusion and diminished the area of perfusion defect by almost 50% (Fig. 2B, green arrows), as described previously (Shin et al., 2007). In all other groups, NBO failed to improve the perfusion, and indeed markedly worsened it in eNOS KO mice (∼40% increase in the area of perfusion defect; Fig. 2B, red arrows).
Figure 2.
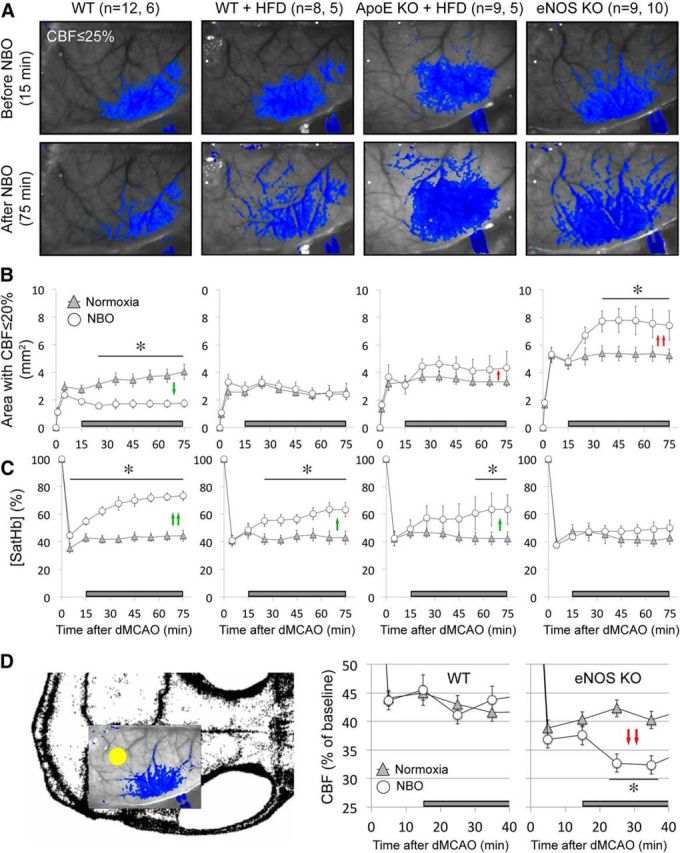
NBO worsens CBF deficit after dMCAO in eNOS−/− and ApoE−/−. A, Representative speckle contrast images taken just before and 60 min after NBO (15 and 75 min after dMCAO, respectively) in WT, WT on HFD (WT + HFD), ApoE KO on HFD (ApoE KO + HFD), and eNOS KO mice. Superimposed in blue are pixels with residual CBF ≤25%. NBO reduced the area of perfusion defect in WT, but increased it in ApoE KO + HFD and in eNOS KO. Imaging field position is shown in D. B, Time course of changes in area of cortex with severe CBF deficit (residual CBF ≤20%) in normoxic and NBO mice. NBO stabilized the area of perfusion defect in WT (green arrows), but abruptly worsened it in eNOS KO and to a lesser extent in ApoE KO + HFD (red arrows). Using a residual CBF threshold of 30% or 40% yielded similar conclusions (data not shown). Time 0 indicates dMCAO and horizontal bar represents 100% O2 starting 15 min after dMCAO. C, Time course of changes in satHb within an ROI placed within the penumbra. NBO rapidly and significantly increased cortical oxygenation in WT, and to a lesser extent in WT + HFD and ApoE KO + HFD groups (green arrows), but failed to do so in eNOS KO, presumably because of significantly worsening perfusion negated increased O2 delivery (see A and B). D, Mouse skull showing the position of imaging field over the right hemisphere, and the ROI used to measure CBF changes in mildly ischemic cortex (yellow dot). NBO did not change perfusion in mildly ischemic cortex away from the ischemic core in WT mice, but abruptly reduced it in eNOS KO (red arrows). *p < 0.05 versus normoxia group, two-way ANOVA for repeated measures followed by Fisher's LSD. Vertical error bars indicate ± SEM. Sample sizes are shown next to each group in A.
In WT mice, NBO significantly improved ischemic tissue oxygenation (Fig. 2C, green arrows). This effect was diminished in WT and ApoE KO mice on HFD, and completely absent in the eNOS KO. The hemodynamic improvement in the WT and worsening in the eNOS KO were not a consequence of reverse steal from the mildly ischemic cortex, where NBO did not alter CBF in the WT, and significantly reduced it in the eNOS KO (Fig. 2D, red arrows).
Infarct volume
Consistent with its effects on tissue perfusion and oxygenation, NBO reduced infarct volume by almost 50% when measured 48 h after dMCAO (Fig. 3A,B). In all other groups, NBO did not significantly reduce infarct volume, and indeed tended to worsen it in the eNOS KO.
Figure 3.
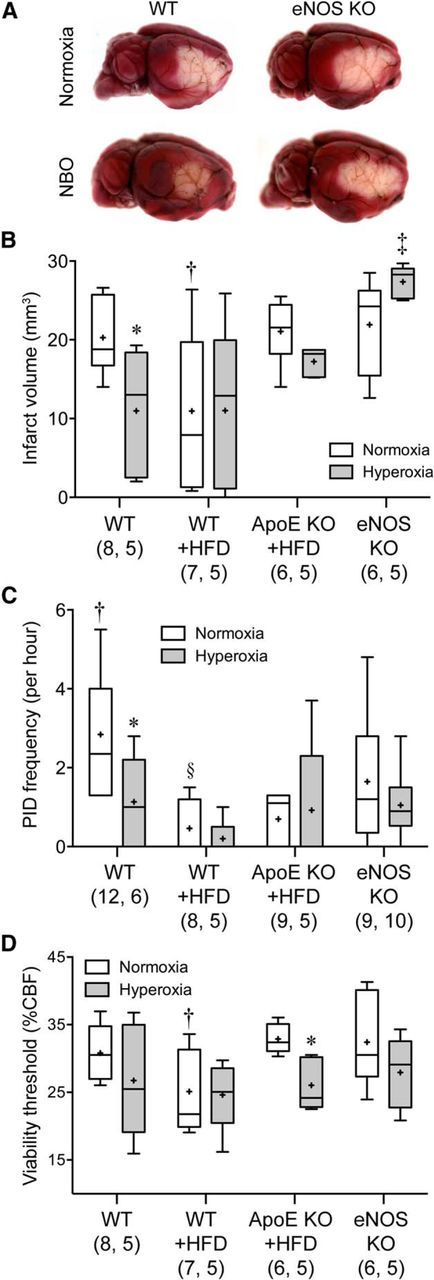
The cerebroprotective effect of NBO is diminished in ApoE KO and abolished in eNOS KO mice. A, Representative topical TTC-stained brains showing the dorsolateral cortical infarct 48 h after 75 min transient dMCAO in WT and eNOS KO. B, NBO significantly reduced infarct volume in the WT, but not in WT + HFD or ApoE KO + HFD groups. Indeed there was a trend for larger infarct volumes after NBO in eNOS KO. Of note, HFD decreased infarct volumes significantly in WT mice. C, The frequency of PIDs was also reduced by NBO in WT mice only, as well as by HFD in WT and ApoE KO mice. D, Hyperoxia decreased the minimum critical CBF level required to survive 60 min dMCAO, suggesting that enhanced cortical oxygenation was still neuroprotective, but tissue outcome was offset by worsening perfusion in ApoE KO + HFD and eNOS KO groups. Data are expressed as box-whisker plots in which median (horizontal line), 25–75% and full ranges (box-whisker), and mean (+) are shown. *p < 0.05 versus normoxia; †p < 0.05 versus all other normoxic groups; ‡p < 0.05 versus all other hyperoxic groups; §p < 0.05 versus normoxic WT and eNOS KO; two-way ANOVA followed by Fisher's LSD. Sample sizes are shown under each group.
Peri-infarct depolarizations
We have previously shown that NBO suppresses peri-infarct depolarizations (PIDs) as an additional mechanism of neuroprotection in focal cerebral ischemia (Shin et al., 2007). Therefore, we tested whether NBO still inhibits PID occurrence in the presence of endothelial dysfunction. We detected PIDs using the characteristic CBF transients on laser speckle imaging as a sensitive and specific surrogate (Shin et al., 2006, 2007; Strong et al., 2006; Eikermann-Haerter et al., 2012). In the WT, NBO once again reduced PID frequencies (Fig. 3C). This effect was diminished in all other groups, in part due to a direct effect of HFD on PID frequency (see below).
Viability threshold
To determine whether improved tissue perfusion by NBO is solely responsible for reduced infarct volumes, we calculated the CBF threshold for viability in each group. NBO decreased the viability threshold as a treatment effect across the entire cohort. However, in post hoc analysis statistical significance was reached only in the ApoE KO + HFD group. These data suggest that in the presence of NBO, cortical tissue requires lower levels of CBF to survive 75 min of dMCAO (Fig. 3D).
Neurological function
To test whether the effect of NBO on infarct volumes is also reflected in functional outcomes, we performed 1 h transient fMCAO and examined the motor function 24 h later. In WT mice, NBO significantly improved both the infarct volumes and neurological function compared with normoxic group (Fig. 4). In contrast, NBO failed to reduce the infarct volumes and tended to worsen the neurological function in eNOS KO mice.
Figure 4.
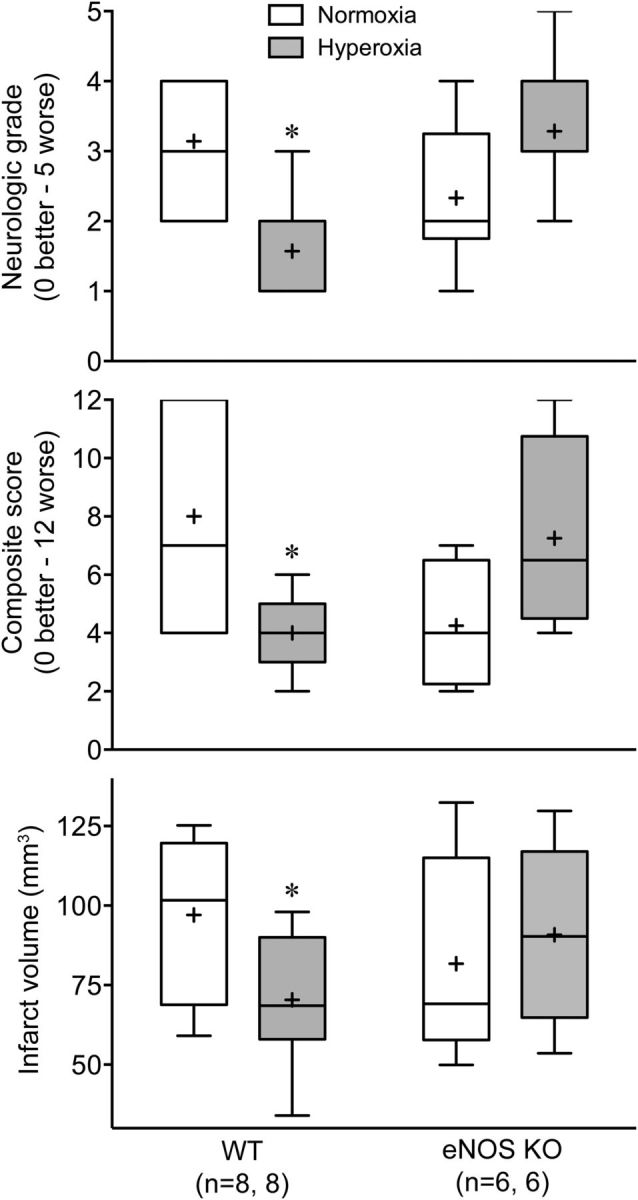
NBO improves neurological and tissue outcome after fMCAO in WT but not in eNOS KO mice. Data are expressed as box-whisker plots in which median (horizontal line), 25–75% and full ranges (box-whisker), and mean (+) are shown. *p ≤ 0.05 versus normoxia; two-way ANOVA followed by Fisher's LSD.
Effects of HFD alone on various outcome measures
We observed that HFD affected several outcome measures in both WT and ApoE KO mice. The characteristic expansion of perfusion defect over time was absent in cohorts on HFD (Fig. 2B), likely related to the suppression of PID occurrence in these groups (Fig. 3C). As a result, HFD was associated with smaller infarct volumes in WT mice (Fig. 3B).
Discussion
A large body of preclinical data suggests that NBO is neuroprotective in stroke via multiple converging mechanisms including improved tissue oxygenation, perfusion and metabolism, inhibition of harmful peri-infarct spreading depolarizations, and preservation of blood–brain barrier and reduced brain edema (Branston et al., 1976; Singhal et al., 2002a, 2007; S. Liu et al., 2006; Henninger et al., 2007; Shin et al., 2007; W. Liu et al., 2008, 2009b, 2011; Yuan et al., 2010; Michalski et al., 2012). However, pilot clinical trials to date have yielded disappointing results, some even suggesting potential harm (Anderson et al., 1991; Nighoghossian et al., 1995; Rusyniak et al., 2003; Singhal et al., 2005; Chiu et al., 2006; Rincon et al., 2014; also see http://clinicaltrials.gov/show/NCT00414726). Our data implicate vascular endothelium as a critical site of action for NBO, and show that eNOS dysfunction is capable of abrogating the neuroprotective effects of NBO in focal cerebral ischemia. These data provide a plausible explanation for the discordance between strong neuroprotective efficacy in animal studies and lack of it in pilot clinical trials conducted thus far. Stroke patients are often elderly with multiple vascular risk factors associated with endothelial dysfunction. Therefore, NBO may be ineffective, and even harmful in those patients with severe endothelial dysfunction. Unfortunately, small sample sizes in previous trials preclude subgroup analyses to test this. Conversely, one can speculate that NBO may achieve its full neuroprotective potential in young patients, particularly in pediatric stroke. The possibility is worth exploring clinically.
Several studies thus far have shown that NBO can indeed augment or preserve tissue perfusion (Singhal et al., 2002a, 2005; S. Liu et al., 2006; Shin et al., 2007; Wu et al., 2012). The mechanism of improved ischemic tissue perfusion by NBO is unknown, but may include enhanced NO production by eNOS through O2 substrate availability, and suppression of PIDs and their vasoconstrictive effects on penumbral circulation (Shin et al., 2007). These mechanisms, however, could not explain the worsening of tissue perfusion in eNOS KO, and to a lesser extent in ApoE KO, during NBO, as neither tissue oxygenation (Fig. 2C) nor the frequency of PIDs (Fig. 3C) was adversely affected by NBO in the mutants. It is possible that eNOS dysfunction (e.g., cofactor deficiency) may lead to enhanced free radical production by eNOS (Dumitrescu et al., 2007) during NBO in the ApoE KO. However, such a mechanism cannot explain the findings in eNOS KO where eNOS is not expressed.
As an alternative mechanism to explain improved perfusion in the WT, NBO can cause mild vasoconstriction in normal brain potentially redirecting blood from nonischemic cortex where it is not needed to the ischemic brain (i.e., reverse steal or Robin Hood effect). However, NBO did not change CBF in nonischemic brain in the WT, and indeed rapidly worsened it in the eNOS KO (Fig. 2D), arguing against reverse steal as a relevant mechanism in this model.
Of note, in contrast to the perceived role of eNOS in the pathophysiology of stroke (Endres et al., 2004), we did not find larger infarcts after dMCAO or worse neurological outcomes after fMCAO in eNOS KO mice compared with WT in this study. Indeed, a review of literature shows that infarct volumes in eNOS KO range from ∼20% smaller to ∼20% larger compared with WT controls after fMCAO (Z. Huang et al., 1996; Kilic et al., 2005; Rikitake et al., 2005; Hiroi et al., 2006). Our data also show that both ApoE KO on HFD and eNOS KO have higher BPs. eNOS KO is known to be hypertensive (P. L. Huang et al., 1995; Wood et al., 2013), and we and others have recently reported higher BPs in the ApoE KO mice on HFD (Ayata et al., 2013; Cleverley et al., 2013; Pelham et al., 2013; Alfaidi et al., 2014; Mai et al., 2014). Hypertension was observed in other hyperlipidemic animal models as well (McCalden et al., 1987), although not all studies agree (Kitayama et al., 2007). Mechanisms are unknown, but may involve eNOS dysfunction (Rossitch et al., 1991; d'Uscio et al., 2001a, b; Kitayama et al., 2007), possibly via reciprocal changes in eNOS phosphorylation at the positive and negative regulatory sites (i.e., reduced S1176 and increased T494 phosphorylation, respectively; Ayata et al., 2013).
Last, our data suggest that HFD exerts a protective effect in acute focal cerebral ischemia and implicates suppression of PIDs as a mechanism. We have previously reported a similar effect where 8 weeks of HFD yielded smaller infarcts and lower viability thresholds compared with 4 weeks of HFD in WT mice (Ayata et al., 2013). This may be related to the ketogenic properties of HFD, exerting direct anti-excitatory and neuroprotective effects (Puchowicz et al., 2008). The direct cerebrovascular effects of HFD in nonhyperlipidemic models have been variable in literature. For example, 8 weeks of HFD in WT rats enlarged infarcts after fMCAO and markedly increased hemorrhagic transformation despite the absence of hyperlipidemia (Li et al., 2013). Enlarged infarcts were also reported in another study after 10 weeks of HFD in rats (Deutsch et al., 2009). However, in aged mice HFD tended to reduce infarcts after dMCAO compared with regular diet (Dhungana et al., 2013), and 1 month HFD did not increase infarct volumes in a rat endothelin injection model of focal ischemia (Langdon et al., 2011). While apparently contradictory, differences in species and experimental model may explain these discrepancies. For example, dMCAO and ET-1 injection models yield relatively small cortical infarcts and rarely any hemorrhagic transformation; the latter may have contributed to the larger infarct sizes after fMCAO in rats on HFD. In addition, rats develop fewer PIDs than mice, and therefore, the protective effect of HFD may be milder in rats. Altogether, data suggest that the direct effects of HFD on stroke outcome may be species and model dependent.
In summary, considering (1) the ease with which NBO can be implemented rapidly in the field, (2) uniform proof of efficacy in animal models including clinically relevant endpoints such as MRI (Singhal et al., 2002b, Singhal et al., 2007; S. Liu et al., 2006; Henninger et al., 2007; Shin et al., 2007; W. Liu et al., 2008), and (3) safety data dispelling concerns related to hemorrhage or free radical production (Singhal et al., 2002b; Kim et al., 2005; S. Liu et al., 2006; W. Liu et al., 2008, 2009a; Sun et al., 2010), a better understanding of the mechanisms of failure in clinical trials is needed. Refocusing on physiological therapies is particularly important given the difficulties associated with translating pharmacological therapies in stroke.
Footnotes
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (2014R1A5A2009936), the National Institutes of Health (NS061505 and NS055104), Fondation Leducq, Neuroendovascular Research Fund from the Andrew David Heitman Foundation, and The Ellison Foundation.
The authors declare no competing financial interests.
References
- Alfaidi M, Chamberlain J, Francis S. 212 docosahexaenoic acid but not alpha-linolenic acid ameliorates high fat diet induced atherosclerosis and hypertension in APOE-/- mice via IL-1 mechanism. Heart. 2014;100(Suppl 3):A116–A117. doi: 10.1136/heartjnl-2014-306118.212. [DOI] [Google Scholar]
- Anderson DC, Bottini AG, Jagiella WM, Westphal B, Ford S, Rockswold GL, Loewenson RB. A pilot study of hyperbaric oxygen in the treatment of human stroke. Stroke. 1991;22:1137–1142. doi: 10.1161/01.STR.22.9.1137. [DOI] [PubMed] [Google Scholar]
- Ayata C, Dunn AK, Gursoy Ozdemir Y, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004;24:744–755. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- Ayata C, Shin HK, Dileköz E, Atochin DN, Kashiwagi S, Eikermann-Haerter K, Huang PL. Hyperlipidemia disrupts cerebrovascular reflexes and worsens ischemic perfusion defect. J Cereb Blood Flow Metab. 2013;33:954–962. doi: 10.1038/jcbfm.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskerville TA, Deuchar GA, McCabe C, Robertson CA, Holmes WM, Santosh C, Macrae IM. Influence of 100% and 40% oxygen on penumbral blood flow, oxygen level, and T2*-weighted MRI in a rat stroke model. J Cereb Blood Flow Metab. 2011;31:1799–1806. doi: 10.1038/jcbfm.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonthu S, Heistad DD, Chappell DA, Lamping KG, Faraci FM. Atherosclerosis, vascular remodeling, and impairment of endothelium-dependent relaxation in genetically altered hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 1997;17:2333–2340. doi: 10.1161/01.ATV.17.11.2333. [DOI] [PubMed] [Google Scholar]
- Branston NM, Symon L, Crockard HA. Recovery of the cortical evoked response following temporary middle cerebral artery occlusion in baboons: relation to local blood flow and PO2. Stroke. 1976;7:151–157. doi: 10.1161/01.STR.7.2.151. [DOI] [PubMed] [Google Scholar]
- Chiu EH, Liu CS, Tan TY, Chang KC. Venturi mask adjuvant oxygen therapy in severe acute ischemic stroke. Arch Neurol. 2006;63:741–744. doi: 10.1001/archneur.63.5.741. [DOI] [PubMed] [Google Scholar]
- Cleverley K, Du X, Premecz S, Le K, Zeglinski M, Nicholson T, Goh CY, Lu Y, Anderson HD, Moghadasian MH, Jassal DS. The effects of fish oil consumption on cardiovascular remodeling in ApoE deficient mice. Can J Physiol Pharmacol. 2013;91:960–965. doi: 10.1139/cjpp-2013-0077. [DOI] [PubMed] [Google Scholar]
- Deutsch C, Portik-Dobos V, Smith AD, Ergul A, Dorrance AM. Diet-induced obesity causes cerebral vessel remodeling and increases the damage caused by ischemic stroke. Microvasc Res. 2009;78:100–106. doi: 10.1016/j.mvr.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhungana H, Rolova T, Savchenko E, Wojciechowski S, Savolainen K, Ruotsalainen AK, Sullivan PM, Koistinaho J, Malm T. Western-type diet modulates inflammatory responses and impairs functional outcome following permanent middle cerebral artery occlusion in aged mice expressing the human apolipoprotein E4 allele. J Neuroinflammation. 2013;10:102. doi: 10.1186/1742-2094-10-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu C, Biondi R, Xia Y, Cardounel AJ, Druhan LJ, Ambrosio G, Zweier JL. Myocardial ischemia results in tetrahydrobiopterin (BH4) oxidation with impaired endothelial function ameliorated by BH4. Proc Natl Acad Sci U S A. 2007;104:15081–15086. doi: 10.1073/pnas.0702986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Uscio LV, Smith LA, Katusic ZS. Hypercholesterolemia impairs endothelium-dependent relaxations in common carotid arteries of apolipoprotein e-deficient mice. Stroke. 2001a;32:2658–2664. doi: 10.1161/hs1101.097393. [DOI] [PubMed] [Google Scholar]
- d'Uscio LV, Baker TA, Mantilla CB, Smith L, Weiler D, Sieck GC, Katusic ZS. Mechanism of endothelial dysfunction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2001b;21:1017–1022. doi: 10.1161/01.ATV.21.6.1017. [DOI] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Lee JH, Yuzawa I, Liu CH, Zhou Z, Shin HK, Zheng Y, Qin T, Kurth T, Waeber C, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C. Migraine mutations increase stroke vulnerability by facilitating ischemic depolarizations. Circulation. 2012;125:335–345. doi: 10.1161/CIRCULATIONAHA.111.045096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci. 2004;27:283–289. doi: 10.1016/j.tins.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Flynn EP, Auer RN. Eubaric hyperoxemia and experimental cerebral infarction. Ann Neurol. 2002;52:566–572. doi: 10.1002/ana.10322. [DOI] [PubMed] [Google Scholar]
- Henninger N, Bouley J, Nelligan JM, Sicard KM, Fisher M. Normobaric hyperoxia delays perfusion/diffusion mismatch evolution, reduces infarct volume, and differentially affects neuronal cell death pathways after suture middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 2007;27:1632–1642. doi: 10.1038/sj.jcbfm.9600463. [DOI] [PubMed] [Google Scholar]
- Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, Noma K, Ueki K, Nguyen NH, Scanlan TS, Moskowitz MA, Cheng SY, Liao JK. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci U S A. 2006;103:14104–14109. doi: 10.1073/pnas.0601600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Macrae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000;20:631–652. doi: 10.1097/00004647-200004000-00001. [DOI] [PubMed] [Google Scholar]
- Jimènez A, Arriero MM, López-Blaya A, González-Fernandez F, García R, Fortes J, Millás I, Velasco S, Sánchez De Miguel L, Rico L, Farré J, Casado S, López-Farré A. Regulation of endothelial nitric oxide synthase expression in the vascular wall and in mononuclear cells from hypercholesterolemic rabbits. Circulation. 2001;104:1822–1830. doi: 10.1161/hc3901.095769. [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Matter CM, Lüscher TF, Bassetti CL, Hermann DM. Aggravation of focal cerebral ischemia by tissue plasminogen activator is reversed by 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor but does not depend on endothelial NO synthase. Stroke. 2005;36:332–336. doi: 10.1161/01.STR.0000152273.24063.f7. [DOI] [PubMed] [Google Scholar]
- Kim HY, Singhal AB, Lo EH. Normobaric hyperoxia extends the reperfusion window in focal cerebral ischemia. Ann Neurol. 2005;57:571–575. doi: 10.1002/ana.20430. [DOI] [PubMed] [Google Scholar]
- Kitayama J, Faraci FM, Lentz SR, Heistad DD. Cerebral vascular dysfunction during hypercholesterolemia. Stroke. 2007;38:2136–2141. doi: 10.1161/STROKEAHA.107.481879. [DOI] [PubMed] [Google Scholar]
- Langdon KD, Clarke J, Corbett D. Long-term exposure to high fat diet is bad for your brain: exacerbation of focal ischemic brain injury. Neuroscience. 2011;182:82–87. doi: 10.1016/j.neuroscience.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.CIR.103.9.1282. [DOI] [PubMed] [Google Scholar]
- Li W, Prakash R, Chawla D, Du W, Didion SP, Filosa JA, Zhang Q, Brann DW, Lima VV, Tostes RC, Ergul A. Early effects of high-fat diet on neurovascular function and focal ischemic brain injury. Am J Physiol Regul Integr Comp Physiol. 2013;304:R1001–R1008. doi: 10.1152/ajpregu.00523.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Shi H, Liu W, Furuichi T, Timmins GS, Liu KJ. Interstitial pO2 in ischemic penumbra and core are differentially affected following transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24:343–349. doi: 10.1097/01.WCB.0000110047.43905.01. [DOI] [PubMed] [Google Scholar]
- Liu S, Liu W, Ding W, Miyake M, Rosenberg GA, Liu KJ. Electron paramagnetic resonance-guided normobaric hyperoxia treatment protects the brain by maintaining penumbral oxygenation in a rat model of transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:1274–1284. doi: 10.1038/sj.jcbfm.9600277. [DOI] [PubMed] [Google Scholar]
- Liu W, Sood R, Chen Q, Sakoglu U, Hendren J, Cetin O, Miyake M, Liu KJ. Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J Neurochem. 2008;107:1196–1205. doi: 10.1111/j.1471-4159.2008.05664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Hendren J, Qin XJ, Liu KJ. Normobaric hyperoxia reduces the neurovascular complications associated with delayed tissue plasminogen activator treatment in a rat model of focal cerebral ischemia. Stroke. 2009a;40:2526–2531. doi: 10.1161/STROKEAHA.108.545483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Hendren J, Qin XJ, Shen J, Liu KJ. Normobaric hyperoxia attenuates early blood-brain barrier disruption by inhibiting MMP-9-mediated occludin degradation in focal cerebral ischemia. J Neurochem. 2009b;108:811–820. doi: 10.1111/j.1471-4159.2008.05821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chen Q, Liu J, Liu KJ. Normobaric hyperoxia protects the blood brain barrier through inhibiting Nox2 containing NADPH oxidase in ischemic stroke. Med Gas Res. 2011;1:22. doi: 10.1186/2045-9912-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai A, Du J, Li JM. 155 improved metabolism and endothelial function in Nox2/ApoE double knockout mice under high fat diet. Heart. 2014;100(Suppl 3):A90–A91. doi: 10.1136/heartjnl-2014-306118.155. [DOI] [Google Scholar]
- McCalden TA, Nath RG, Coleman L, Meyer JS. Hypercholesterolemia and the baboon cerebral circulation. Life Sci. 1987;41:447–451. doi: 10.1016/0024-3205(87)90220-7. [DOI] [PubMed] [Google Scholar]
- Michalski D, Hobohm C, Weise C, Pelz J, Heindl M, Kamprad M, Kacza J, Härtig W. Interrelations between blood-brain barrier permeability and matrix metalloproteinases are differently affected by tissue plasminogen activator and hyperoxia in a rat model of embolic stroke. Med Gas Res. 2012;2:2. doi: 10.1186/2045-9912-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S, Meyer JS, Amano T, Shaw T, Okabe T, Mortel KF. Cerebral vasomotor responsiveness during 100% oxygen inhalation in cerebral ischemia. Arch Neurol. 1983;40:271–276. doi: 10.1001/archneur.1983.04050050039004. [DOI] [PubMed] [Google Scholar]
- Nighoghossian N, Trouillas P, Adeleine P, Salord F. Hyperbaric oxygen in the treatment of acute ischemic stroke. A double-blind pilot study. Stroke. 1995;26:1369–1372. doi: 10.1161/01.STR.26.8.1369. [DOI] [PubMed] [Google Scholar]
- Ozaki M, Kawashima S, Yamashita T, Hirase T, Namiki M, Inoue N, Hirata K, Yasui H, Sakurai H, Yoshida Y, Masada M, Yokoyama M. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J Clin Invest. 2002;110:331–340. doi: 10.1172/JCI15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padma MV, Bhasin A, Bhatia R, Garg A, Singh MB, Tripathi M, Prasad K. Normobaric oxygen therapy in acute ischemic stroke: a pilot study in Indian patients. Ann Indian Acad Neurol. 2010;13:284–288. doi: 10.4103/0972-2327.74203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra A, McGirt MJ, Sheng H, Laskowitz DT, Pearlstein RD, Warner DS. Mouse model of subarachnoid hemorrhage associated cerebral vasospasm: methodological analysis. Neurol Res. 2002;24:510–516. doi: 10.1179/016164102101200276. [DOI] [PubMed] [Google Scholar]
- Pelham CJ, Keen HL, Lentz SR, Sigmund CD. Dominant negative PPARgamma promotes atherosclerosis, vascular dysfunction, and hypertension through distinct effects in endothelium and vascular muscle. Am J Physiol Regul Integr Comp Physiol. 2013;304:R690–R701. doi: 10.1152/ajpregu.00607.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchowicz MA, Zechel JL, Valerio J, Emancipator DS, Xu K, Pundik S, LaManna JC, Lust WD. Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J Cereb Blood Flow Metab. 2008;28:1907–1916. doi: 10.1038/jcbfm.2008.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, Moskowitz MA, Liao JK. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. 2005;36:2251–2257. doi: 10.1161/01.STR.0000181077.84981.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon F, Kang J, Maltenfort M, Vibbert M, Urtecho J, Athar MK, Jallo J, Pineda CC, Tzeng D, McBride W, Bell R. Association between hyperoxia and mortality after stroke: a multicenter cohort study. Crit Care Med. 2014;42:387–396. doi: 10.1097/CCM.0b013e3182a27732. [DOI] [PubMed] [Google Scholar]
- Rossitch E, Jr, Alexander E, 3rd, Black PM, Cooke JP. L-arginine normalizes endothelial function in cerebral vessels from hypercholesterolemic rabbits. J Clin Invest. 1991;87:1295–1299. doi: 10.1172/JCI115132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyniak DE, Kirk MA, May JD, Kao LW, Brizendine EJ, Welch JL, Cordell WH, Alonso RJ. Hyperbaric oxygen therapy in acute ischemic stroke: results of the Hyperbaric Oxygen in Acute Ischemic Stroke Trial Pilot Study. Stroke. 2003;34:571–574. doi: 10.1161/01.STR.0000050644.48393.D0. [DOI] [PubMed] [Google Scholar]
- Shin HK, Dunn AK, Jones PB, Boas DA, Moskowitz MA, Ayata C. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab. 2006;26:1018–1030. doi: 10.1038/sj.jcbfm.9600252. [DOI] [PubMed] [Google Scholar]
- Shin HK, Dunn AK, Jones PB, Boas DA, Lo EH, Moskowitz MA, Ayata C. Normobaric hyperoxia improves cerebral blood flow and oxygenation, and inhibits peri-infarct depolarizations in experimental focal ischaemia. Brain. 2007;130:1631–1642. doi: 10.1093/brain/awm071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal AB, Dijkhuizen RM, Rosen BR, Lo EH. Normobaric hyperoxia reduces MRI diffusion abnormalities and infarct size in experimental stroke. Neurology. 2002a;58:945–952. doi: 10.1212/WNL.58.6.945. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Wang X, Sumii T, Mori T, Lo EH. Effects of normobaric hyperoxia in a rat model of focal cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2002b;22:861–868. doi: 10.1097/00004647-200207000-00011. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Benner T, Roccatagliata L, Koroshetz WJ, Schaefer PW, Lo EH, Buonanno FS, Gonzalez RG, Sorensen AG. A pilot study of normobaric oxygen therapy in acute ischemic stroke. Stroke. 2005;36:797–802. doi: 10.1161/01.STR.0000158914.66827.2e. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Ratai E, Benner T, Vangel M, Lee V, Koroshetz WJ, Schaefer PW, Sorensen AG, Gonzalez RG. Magnetic resonance spectroscopy study of oxygen therapy in ischemic stroke. Stroke. 2007;38:2851–2854. doi: 10.1161/STROKEAHA.107.487280. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Bezzina EL, Anderson PJ, Boutelle MG, Hopwood SE, Dunn AK. Evaluation of laser speckle flowmetry for imaging cortical perfusion in experimental stroke studies: quantitation of perfusion and detection of peri-infarct depolarisations. J Cereb Blood Flow Metab. 2006;26:645–653. doi: 10.1038/sj.jcbfm.9600240. [DOI] [PubMed] [Google Scholar]
- Sun L, Zhou W, Mueller C, Sommer C, Heiland S, Bauer AT, Marti HH, Veltkamp R. Oxygen therapy reduces secondary hemorrhage after thrombolysis in thromboembolic cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:1651–1660. doi: 10.1038/jcbfm.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Strelow H, Mies G, Veltkamp R. Oxygen therapy improves energy metabolism in focal cerebral ischemia. Brain Res. 2011;1415:103–108. doi: 10.1016/j.brainres.2011.07.064. [DOI] [PubMed] [Google Scholar]
- Veltkamp R, Siebing DA, Sun L, Heiland S, Bieber K, Marti HH, Nagel S, Schwab S, Schwaninger M. Hyperbaric oxygen reduces blood-brain barrier damage and edema after transient focal cerebral ischemia. Stroke. 2005;36:1679–1683. doi: 10.1161/01.STR.0000173408.94728.79. [DOI] [PubMed] [Google Scholar]
- Wood KC, Cortese-Krott MM, Kovacic JC, Noguchi A, Liu VB, Wang X, Raghavachari N, Boehm M, Kato GJ, Kelm M, Gladwin MT. Circulating blood endothelial nitric oxide synthase contributes to the regulation of systemic blood pressure and nitrite homeostasis. Arterioscler Thromb Vasc Biol. 2013;33:1861–1871. doi: 10.1161/ATVBAHA.112.301068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu O, Lu J, Mandeville JB, Murata Y, Egi Y, Dai G, Marota JJ, Diwan I, Dijkhuizen RM, Kwong KK, Lo EH, Singhal AB. Dynamic functional cerebral blood volume responses to normobaric hyperoxia in acute ischemic stroke. J Cereb Blood Flow Metab. 2012;32:1800–1809. doi: 10.1038/jcbfm.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Liu W, Liu B, Schnell A, Liu KJ. Normobaric hyperoxia delays and attenuates early nitric oxide production in focal cerebral ischemic rats. Brain Res. 2010;1352:248–254. doi: 10.1016/j.brainres.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzawa I, Sakadz̆ić S, Srinivasan VJ, Shin HK, Eikermann-Haerter K, Boas DA, Ayata C. Cortical spreading depression impairs oxygen delivery and metabolism in mice. J Cereb Blood Flow Metab. 2012;32:376–386. doi: 10.1038/jcbfm.2011.148. [DOI] [PMC free article] [PubMed] [Google Scholar]