

Abstract
Kufor–Rakeb syndrome (KRS) is caused by loss-of-function mutations in ATP13A2 (PARK9) and characterized by juvenile-onset parkinsonism, pyramidal signs, and cognitive decline. Previous studies suggested that PARK9 deficiency causes lysosomal dysfunction and α-synuclein (α-syn) accumulation, whereas PARK9 overexpression suppresses toxicity of α-syn. However, the precise mechanism of PARK9 effect on lysosomes and α-syn has been unknown. Here, we found that overexpressed PARK9 localized to multivesicular bodies (MVBs) in the human H4 cell line. The results from patient fibroblasts showed that loss of PARK9 function leads to decreased number of the intraluminal vesicles in MVBs and diminished release of exosomes into culture media. By contrast, overexpression of PARK9 results in increased release of exosomes in H4 cells and mouse primary cortical neurons. Moreover, loss of PARK9 function resulted in decreased secretion of α-syn into extracellular space, whereas overexpressed PARK9 promotes secretion of α-syn, at least in part via exosomes. Finally, we found that PARK9 regulates exosome biogenesis through functional interaction with the endosomal sorting complex required for transport machinery. Together, these data suggest the involvement of PARK9 in the biogenesis of exosomes and α-syn secretion and raise a possibility that disruption of these pathways in patients with KRS contributes to the disease pathogenesis.
Keywords: α-synuclein, ATP13A2, exosome, multivesicular body, PARK9, Parkinson's disease
Introduction
Loss-of-function mutations in ATP13A2/PARK9 cause Kufor–Rakeb syndrome (KRS), a rare autosomal recessive disorder characterized by early onset parkinsonism and other neurological symptoms, such as dementia and pyramidal signs (Najim al-Din et al., 1994; Ramirez et al., 2006). PARK9 belongs to P-type ATPase super family to which no specific substrates have been assigned (Axelsen and Palmgren, 1998). PARK9 is expressed predominantly in CNS, especially in dopaminergic neurons of substantia nigra (Ramirez et al., 2006). Initial studies revealed that overexpression of PARK9 reduced the toxicity of α-synuclein (α-syn) in yeast cells and rat primary dopaminergic neurons (Gitler et al., 2009). In addition, PARK9 expression levels were increased in postmortem brains from sporadic PD patients (Ramirez et al., 2006; Park et al., 2011; Ramonet et al., 2012). Studies in cultured neurons also demonstrated that transiently expressed wild-type PARK9 localized to acidic vesicles (Ramirez et al., 2006; Tan et al., 2011; Ramonet et al., 2012). We and others have shown that depletion of PARK9 causes lysosomal dysfunction, accumulation of α-syn (Dehay et al., 2012; Usenovic et al., 2012a), and increased sensitivity to zinc (Park et al., 2014; Tsunemi and Krainc, 2014). PARK9-deficiency also leads to mitochondrial dysfunction with decreased ATP synthesis and increased oxidative stress, suggesting that impaired lysosome-autophagy pathway may affect clearance of mitochondria (Grünewald et al., 2012; Gusdon et al., 2012). Furthermore, studies of PARK9-interacting partners implicated its role in vesicular trafficking (Usenovic et al., 2012b).
Together, these studies uncovered the molecular mechanisms of PARK9 function in the context of disease pathogenesis, whereas its physiological function remains largely unknown.
Here, we found that PARK9 localizes to multivesicular bodies (MVBs) and plays a role in secretion of exosomes into extracellular space. Expression levels of PARK9 correlate with the number of exosomes that release α-syn into the medium, suggesting that PARK9 can also alter levels of α-syn expression via exosome-mediated secretion.
Materials and Methods
Plasmids.
Lentiviral plasmids expressing short hairpin plasmid RNA (shRNA) targeting human ATP13A2/PARK9, mouse Atp13A2/PARK9, mouse Tsg101, scrambled control shRNA, and a lentiviral plasmid expressing human ATP13A2 (PARK9) were used for viral packaging (Tsunemi and Krainc, 2014).
Cell culture.
Human neuroglioma cell line (H4)-expressing wild-type α-syn under the control of a tetracycline inducible promoter was used as described previously (Mazzulli et al., 2011).
Primary dermal fibroblasts taken from male KRS patients carrying a homozygous mutation (1550C>T; MUT1) and compound heterozygous mutations (3176 T>G, 3253 delC; MUT2) in PARK9 were used (Tsunemi and Krainc, 2014). Mouse embryonic cortical neurons or neuro2A cells were cultured and gene silencing was conducted as described previously (Tsunemi et al., 2012; Tsunemi and Krainc, 2014). Sphingomyelinase (S8633; Sigma-Aldrich) was used for enhancing sphingomyelinase activity.
Confocal imaging.
The cells were fixed and stained as described previously (Tsunemi et al., 2012) with the antibodies; anti-LAMP1 (Santa Cruz Biotechnology), anti-CD63 (Developmental Studies Hybridoma Bank), or anti-lysobisphosphatidic acid (LBPA; Echelon). H4 cells were incubated with Cascade Blue-conjugated dextran (Invitrogen) for 48 h before fixation. Confocal imaging was conducted on the Leica TCS SPE confocal system with Leica DMI 4000B CSQ inverted microscope equipped with an ACS APO 63× (1.3 numerical aperture) oil-immersion objective. For quantification analysis, 10,000 cells/well were plated in triplication and fluorescence intensity was measured using SpectraMax i3 multimode microplate reader (Molecular Devices). DAPI was used to normalize intensities for CD63 or LBPA.
Electron microscopy.
The cells were fixed with 2.5% glutaraldehyde in PBS, pH7.4, for 1 h. After postfixed with 1% osmium tetroxide in PBS, the cells were dehydrated through a series of graded ethanols from 50 to 70%, and stained with 1% uranyl acetate in 70% ethanol. The cells were further dehydrated to 100% ethanol, and embedded in LX112 resin (Ladd), which was polymerized. The resin blocks were thin sectioned with Ultracut UCT microtome (Leica) at 70 nm. Transmission electron microscopic analysis was conducted with an FEI Tecnai Spirit G2 transmission electron microscope.
Cryo-immunogold electron microscopy.
After fixed with 4.0% paraformaldehyde plus 0.2% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.4 (Electron Microscopy Sciences) for 1 h, the cells were pelleted. The pellet was mixed with warm 2.0% agarose and cut into small pieces, which were then cryoprotected in 2.3 m sucrose in PBS. Ultrathin sections were cut on a Leica EM FCS at −80°C. Double-immunogold staining was performed by incubating with mouse-anti-CD63 for 1 h at RT, followed by 1 h on drops of goat-anti-mouse 10 nm IgG gold (Ted Pella). The grids were incubated on drops of the second primary, rabbit-anti-V5, rinsed, and then incubated on drops of goat-anti-rabbit 15 nm IgG gold.
Exosome isolation.
Exosomes were purified from 20 ml of cell-conditioned media. A basic differential centrifugation method (200 × g for 5 min, 1200 × g for 10 min, and 16,500 × g for 30 min), followed by ultracentrifugation at 110,000 × g for 60 min (Optima-MAX ultracentrifuge, with TLA110 rotor and Optiseal tubes, Beckman Coulter) was conducted. After washed in PBS, the exosomes were collected by a centrifugation at 110,000 × g for 60 min.
Nanoparticle tracking analysis.
Analyses of vesicles present in the medium were conducted by nanoparticle tracking system, using the NanoSight LM10 system (NanoSight), configured with a 405 nm laser and a high-sensitivity digital camera system (OrcaFlash2.8, Hamamatsu C11440, NanoSight). Videos were recorded and analyzed using the NTA-software (v2.3). Samples were administered and recorded under controlled flow, using the NanoSight syringe pump and script control system.
Western blotting.
Western blotting was conducted as previously described (Tsunemi et al., 2012; Schultheis et al., 2013). We used anti-α-syn antibody (C-20; Santa Cruz Biotechnology), anti-neural specific enolase (NSE) antibody (no. 16625; Polysciences), anti-Alix antibody (1A12; Santa Cruz Biotechnology), anti-Tsg101 antibody (4A10; GeneTex), and anti-Flotillin1 antibody (610821; BD Transduction Laboratories).
αSynuclein ELISA.
The 96-well half-area high binding ELISA plates (Costar) were coated with anti-syn42 mouse monoclonal antibody (BD Transduction Laboratories) at 4°C overnight shaking, and then blocked with 2% BSA/PBS (blocking buffer). The media were concentrated with centrifugal filters (Amicon Ultra 3K device), and the exosomes permeabilized by adding SDS to reach the final concentration of 0.02%. The samples that were ran in triplicates were incubated overnight at 4°C with primary anti α-syn rabbit polyclonal antibody (S9500–01E; USBiological), followed by incubation with the secondary anti-rabbit antibody (Jackson ImmunoResearch). The reaction was initiated by adding TMB One Component HRP Microwell Substrate (SurModics) and stopped by 450 nm Liquid Stop Solution (SurModics). Plates were read at 450 nm with SpectraMax i3 multimode microplate reader (Molecular Devices). All the data were in linear range from 10 to 500 pg/ml.
Statistical analysis.
All data were prepared for analysis with standard spreadsheet software (Microsoft Excel). Statistical analysis was performed by one-way ANOVA post hoc Tukey test or Student t test. All error bars represent SEM in figures.
Results
Loss-of-function of PARK9 leads to decreased density of intraluminal vesicles in MVBs
Although PARK9 has been shown to localize to acidic vesicles, the nature of these vesicles has not been elucidated in detail (Ramirez et al., 2006). To further examine PARK9 localization, we expressed PARK9-V5 expression plasmid in human neuroglioma H4 cells (Mazzulli et al., 2011) and conducted double-immunostaining using antibodies against V5 and vesicular markers. Cascade Blue-conjugated dextran was used for late endosome and lysosome marker (Short et al., 2010). As previously reported, PARK9 colocalized with LAMP1-positive vesicles, suggesting localization to late endosomes and lysosomes. (Mane et al., 1989; Fig. 1A, left). Using antibody against CD63, a more specific marker for multivesicular regions of the late endosomes (Kobayashi et al., 2000), we found that PARK9 also localized to multivesicular endosomes (MVEs; Fig. 1A, right). To confirm these findings, immuno-EM with antibodies against V5-PARK9 and CD63 was performed. A few PARK9-V5-labeled gold particles were associated with CD63-positive vesicles (Fig. 1B), suggesting that PARK9 localizes, at least in part, to MVBs, possibly to the outer membrane of the vesicles. Using the CD63 antibody, we next examined the morphology of MVEs in mutant PARK9 cells by examining patient (MUT1 and MUT2) and control fibroblasts (WT1 and WT2; Fig. 1C). These experiments revealed that PARK9 mutant cells exhibit significantly increased number and/or size of MVEs (Fig. 1C,D). This was confirmed with another MVE marker, LBPA (Kobayashi et al., 1998; Fig. 1E). To further analyze these MVEs, we performed EM analysis that revealed densely packed intraluminal vesicles (ILVs) in WT cells (Fig. 1F, left), whereas ILVs were very sparse in PARK9 mutant cells (Fig. 1F, right), suggesting that PARK9 may control the number of ILVs in MVBs.
Figure 1.
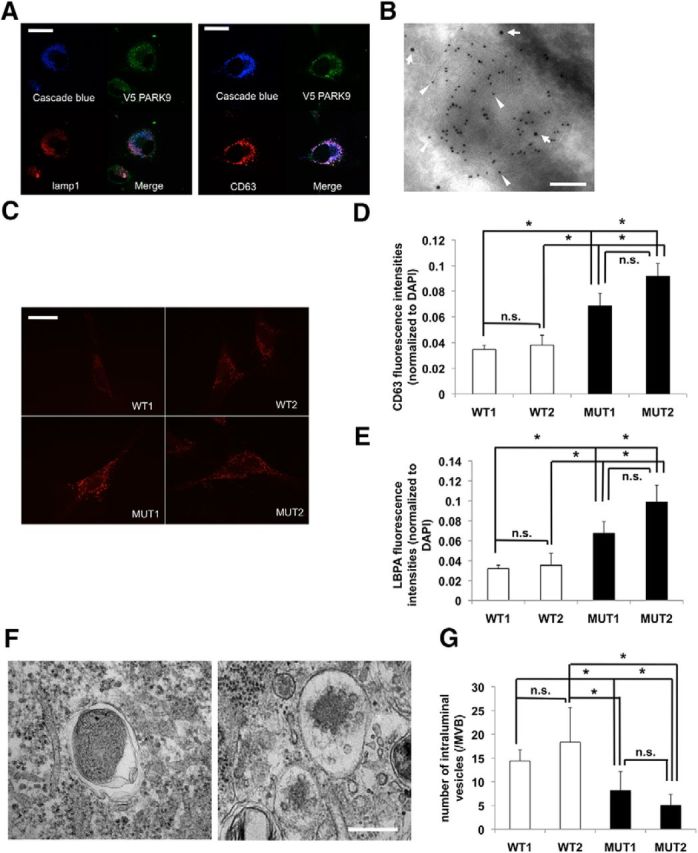
PARK9 localizes to MVBs. A, Left, Representative images of LAMP1 immunostaining of PARK9–V5-expressing neuroglioma H4 cells. A, Right, Representative images of CD63 immunostaining of PARK9–V5-expressing H4 cells. Cascade Blue-conjugated dextran was used for the late endosome-lysosome marker. Scale bar, 10 μm. B, Representative immuno-EM image of multivesicular body from PARK9–V5-expressing H4 cells. Double-staining was conducted using anti-CD63 antibody (10 nm gold particles, indicated by arrowheads) and anti-V5 antibody (15 nm gold particles, indicated by arrows). Scale bar, 50 nm. At least 30 cells from six sections were examined in three independent experiments. C, Representative images of CD63 immunostaining of two wild-type (WT) and two PARK9-mutant (MUT) fibroblast lines. Scale bar, 20 μm. D, Quantification of CD63 immunostaining intensity is shown after normalization to DAPI immunointensity (n = 3, *p < 0.03). E, Quantification of LBPA immunostaining intensity is shown after normalization to DAPI (n = 3, *p < 0.03). The values represent mean ± SEM from three independent experiments. F, Representative EM images of MVBs from WT (left) and MUT (right) fibroblasts from the examination of 20 cells/section in four sections. Scale bar, 100 nm. G, Quantification analysis of the number of ILVs in MBV from each fibroblasts (n = 30–50/cells, *p < 0.05). Data analysis was performed by one-way ANOVA and post hoc Tukey test.
PARK9 regulates exosome biogenesis
Previous studies showed that MVBs fuse with plasma membrane and release ILVs that are referred to as exosomes in extracellular medium (Simons and Raposo, 2009). Because PARK9 mutant cells contain fewer ILVs, we asked whether the number of exosomes is affected by PARK9 mutation. After isolating exosomes by conventional centrifugation, we performed Western blotting using established markers, Alix, flotillin1, Tsg101, or CD63 to confirm the identity of collected exosomes. These results suggested that exosomes from PARK9 mutant fibroblasts were fewer compared with wild-type fibroblasts (Fig. 2A). Then, we further analyzed exosomes by nanoparticle tracking analysis (NTA; Grünewald et al., 2012) to simultaneously quantify the size and the number of exosomes. As shown in Figure 2B, PARK9 mutant fibroblasts released small, but significantly lower number of exosomes compared with wild-type fibroblasts. These results were consistent with the finding of fewer ILVs in MVBs.
Figure 2.

Exosome production is regulated by PARK9. A, Immunoblot analysis of exosome marker proteins (Alix, flotillin1, Tsg101, and CD63) in exosomes from wild-type and PARK9 mutant fibroblasts. Similar results were obtained in two other experiments. B, Quantification analysis of exosome release in the media from wild-type and PARK9 mutant fibroblasts (*p < 0.03). Data were analyzed by one-way ANOVA and post hoc Tukey test. C, Quantification analysis of exosome release in the media from mock transected and PARK9 overexpressed H4 cells (n = 3, *p < 0.01). D, Immunoblot analysis of exosome marker proteins (Alix, flotillin1, Tsg101, and CD63) in exosomes from Mock-transfected and PARK9-overexpressed H4 cells. Results were confirmed by two additional experiments. E, Quantification analysis of exosome release in the media from scrambled- (Scrb) and PARK9 shRNA-treated primary cortical neurons (n = 3, *p < 0.01). F, Quantification analysis of exosome release in the media from mock-transected and PARK9-overexpressed primary cortical neurons (n = 3, *p < 0.01). The values represent mean ± SEM from three independent experiments. Data were analyzed by Student t test (D, E).
Next, we hypothesized that PARK9 may be involved in the secretion of exosomes. In support of this hypothesis, we found that overexpression of PARK9 led to increased number of exosomes compared with vector transfected control (Fig. 2C). This result was confirmed by the Western blot analysis using exosome markers (Fig. 2D). To confirm these findings in primary neurons, we silenced PARK9 expression with shRNA and examined exosome release compared with neurons treated with scrambled shRNA. These experiments revealed a significantly decreased exosome release from PARK9-silenced neurons (Fig. 2E). Conversely, lentiviral overexpression of PARK9 in primary neurons exhibited increased exosome release compared with vector-transfected neurons (Fig. 2F). Together, these findings further support the notion that PARK9 plays a role in the production of exosomes.
PARK9 promotes secretion of intracellular α-syn into the media via exosomes
Recent evidence suggests that α-syn is at least in part secreted from neurons via exosomes (Emmanouilidou et al., 2010; Alvarez-Erviti et al., 2011; Danzer et al., 2012). Because our data suggest that PARK9 regulates exosome release, and the fact that α-syn accumulates in cell and animal models of KRS (Dehay et al., 2012; Usenovic et al., 2012b; Schultheis et al., 2013), we analyzed α-syn secretion with highly sensitive ELISA (Fig. 3A). To examine whether PARK9 alters α-syn release via exosomes, first, we confirmed our previous data (Usenovic et al., 2012b; Tsunemi and Krainc, 2014), that silencing of PARK9 with shRNA significantly increased α-syn protein levels (Fig. 3C), whereas overexpression of PARK9 reduced the level of α-syn (Fig. 3D). These results were also confirmed by immunocytochemical analysis demonstrating decreased α-syn expression (Fig. 3E).
Figure 3.

PARK9 expression affects intracellular and extracellular levels of α-syn. A, The α-syn ELISA standard curve plotted OD450 (on the y-axis) and α-syn protein concentration (on the x-axis). B, The human PARK9-expression levels in PARK9-silenced and PARK9-overexpressed H4 cells (n = 3, *p < 0.01). C, Top, Immunoblot analysis of α-syn expression in Scrb and PARK9 shRNA-treated H4 cells. Bottom, Quantification of α-syn expression level. After normalization to NSE, the expression of α-syn in PARK9-silenced H4 cells was divided by expression in Scrb shRNA-transfected cells (n = 4, *p < 0.03). D, Top, Immunoblot analysis of α-syn expression level in mock and PARK9 overexpressed H4 cells (n = 4, *p < 0.03). Bottom, Quantification analysis of α-syn expression level; (n = 3, *p < 0.03). Data were analyzed by Student t test (C, D). E, Representative images of mock transfected (right) and PARK9 overexpressed H4 cells. PARK9 is shown in green and α-syn is shown in red. Scale bar, 50 nm. F, G, The expression of α-syn in the media (F) and exosomes (G) from H4 cells. The total expression levels of α-syn in the media or exosomes were normalized by total protein amount (n = 3, *p < 0.01). H–K, The expression of α-syn in Triton-soluble fractions (H), SDS-soluble fractions (I), media (J), or exosomes (K) taken from WT and MUT fibroblasts; (n = 3, *p < 0.01). L–N, The expression of α-syn in Triton-soluble fractions (L), SDS-soluble fractions (M), and media (N) taken from PCNs where PARK9 expression was modulated (n = 3, *p < 0.01). Levels were normalized to total protein. The values represent mean ± SEM from three independent experiments. Data analysis was performed by one-way ANOVA and post hoc Tukey test (B, F–N).
Next, we asked whether PARK9 expression levels affect secretion of α-syn. Indeed, we found that PARK9 silencing decreased α-syn levels in cell culture media, whereas overexpression of PARK9 increased α-syn secretion (Fig. 3F). These data suggest that PARK9 has a significant role in α-syn secretion. Then, we asked whether PARK9-mediate α-syn secretion is exosome-dependent or not. When we measured α-syn in exosomes, we observed similar results under these conditions (Fig. 3G), suggesting that PARK9 contributes to α-syn secretion, at least in part, via the exosomes. Based on these findings, we speculated that secretion of α-syn might have influence on the level of intracellular α-syn.
Then, we confirmed our findings using fibroblasts from KRS patients that carry a mutation in PARK9 (Fig. 3H–K). We analyzed α-syn levels in the Triton-soluble fractions, SDS-soluble fractions, media, or exosomes from cultured fibroblasts. Although levels of α-syn in Triton-soluble fractions were not significantly different, we found increased levels of α-syn in SDS-soluble fractions in mutant compared with wild-type fibroblasts (Fig. 3I). Compared with wild-type cells, α-syn was also decreased in either media or exosomes released from mutant cells (Fig. 3J,K). Finally, we analyzed α-syn secretion in primary neurons by modulating expression of PARK9 (Fig. 3L–N). Overexpression of human PARK9 significantly lowered Triton-soluble and SDS-soluble intracellular α-syn (Fig. 3L,M), whereas α-syn in cell media was increased (Fig. 3N). In contrast, silencing of mouse Atp13A2 (PARK9) increased expression of intracellular α-syn (Fig. 3L,M) but decreased α-syn secretion (Fig. 3N). Importantly, this effect was partially rescued by overexpression of human PARK9 in silenced neurons (Fig. 3L,M). Together, these data suggest that decreased secretion of α-syn in PARK9-deficient cells, at least in part, occurs via exosome-mediated α-syn release and highlight the importance of PARK9 in regulation of exosome release. Next, we examined possible mechanisms of PARK9-mediated regulation of exosome biogenesis and release. We have previously reported that PARK9-deficient cells exhibit reduced activity of sphingomyelinase, suggesting impaired ceramide synthesis (Tsunemi and Krainc, 2014). Because ceramide is known to affect exosome biogenesis, we first examined this pathway (Trajkovic et al., 2008). To this end, cells were treated with neutral sphingomyelinase that elevates levels of ceramide and promotes production of exosomes. Using this approach, we observed a significant increase of exosomes in wild-type cells as expected. However, expression of neutral sphingomyelinase also increased exosome production in cells expressing mutant PARK9 (Fig. 4A), suggesting that PARK9 does not interact with the ceramide pathway in regulation of exosomes.
Figure 4.

PARK9-mediated exosome biogenesis requires ESCRT machinery. A, Cells were treated with neutral sphingomyelinase (100 mμ/ml) and exosomes were examined in the media from wild-type and PARK9 mutant fibroblasts (*p < 0.001). Data were analyzed by Student t test. B, The effect of PARK9 overexpression on exosome release in N2A cells with silenced Tsg101 (*p < 0.01, **p < 0.001). The values represent mean ± SEM from three independent experiments. Data analysis was performed by one-way ANOVA and post hoc Tukey test. C, Immunoblot analysis of exosome marker proteins (Alix, flotillin1, Tsg101, and CD63) in exosomes in response to silencing Tsg101 and/or overexpression of PARK9.
Next, we tested whether PARK9 cooperates with endosomal sorting complex required for transport (ESCRT) that plays a major role in exosome generation (Jouvenet, 2012). As expected, silencing of tumor susceptibility gene 101 (Tsg101), one of ESCRT-I proteins, resulted in significant decrease in exosomes release (Fig. 4B). Importantly, whereas PARK9 overexpression increased production of exosomes in wild-type cells, it had no effect in Tsg101 silenced cells, suggesting that intact ESCRT machinery was required for PARK9 effects on exosomes. These results were further confirmed by Western blot analysis showing the changes of exosome markers Alix, flotillin1, Tsg101, and CD63 in response to silencing Tsg101 and/or overexpression of PARK9 (Fig. 4C). These data collectively suggest that PARK9 regulates exosome biogenesis at least in part via functional interactions with the ESCRT pathway.
Discussion
In this study, we found that loss-of-function of PARK9 leads to decreased density of intraluminal vesicles and diminished release of exosomes into culture media, whereas its overexpression promotes the release of exosomes. Moreover, PARK9 also regulates secretion of α-syn into extracellular space, at least in part, via exosomes, suggesting that disruption of these pathways in patients with KRS may contribute to the disease pathogenesis.
Several different types of vesicles are released from cells into extracellular space including exosomes, shedding vesicles, and nanoparticles (Ludwig and Giebel, 2012). Exosomes that have a diameter of 40–100 nm, are derived from MVBs and contain membrane components, proteins, lipids, and microRNA (Simpson et al., 2008). MVBs fuse with the plasma membrane and release ILVs that are then referred to as exosomes (Simpson et al., 2008). Recent studies revealed that ESCRT plays a role in exosome generation (Jouvenet, 2012). For example, ESCRT-0 contains hepatocyte growth factor regulated tyrosine kinase substrate (Hrs) that when silenced reduces ILV formation, resulting in reduced exosome secretion (Bache et al., 2003; Tamai et al., 2010). Hrs recruits an ESCRT-I protein Tsg101, which also results in fewer ILVs and exosomes when silenced (Falguières et al., 2008; Colombo et al., 2013). Importantly, either genetic silencing of Hrs or Tsg101 causes neurodegeneration in mice (Kim et al., 2007; Tamai et al., 2008), underscoring the importance of ESCRT-related exosome generation in neuronal function. Our data suggest that PARK9 also plays a role in the generation and release of exosomes. The results from NTA revealed that PARK9 mutant fibroblasts generate less exosomes than wild-type cells (Fig. 2A,B), whereas in conditions of overexpressed PARK9, exosomes were significantly increased (Fig. 2C,D). These data are consistent with the recent report by Kong et al. (2014), and here we further examined the mechanism of this effect. We found that PARK9 at least in part regulates production of exosomes by interacting with the ESCRT pathway (Fig. 4B,C) and further studies will be required to examine the molecular details of this interaction.
Released exosomes are taken up by neighboring cells via endocytosis (Ludwig and Giebel, 2012), indicating that exosomes mediate cell to cell communications. For example, MHC class II-containing exosomes released from B lymphocytes induce antigen-specific MHC class II-restricted T-cell response (Raposo et al., 1996). Tumor cells also release exosomes that can facilitate tumor growth, invasion, and metastasis (Thuma and Zöller, 2014). Interestingly, recent studies suggested that exosomes play a role in propagation of aggregation-prone proteins (Lai and Breakefield, 2012). For example, exosomes can release amyloid-β peptides and Tau accumulated in MVBs into extracellular space (Rajendran et al., 2006; Saman et al., 2012). Initial studies showed that α-syn could be released via nonconventional exocytosis (Lee et al., 2005) and recent data defined this release is mediated via exosomes (Emmanouilidou et al., 2010; Alvarez-Erviti et al., 2011). We found that PARK9 promotes secretion of α-syn, at least in part, via exosomes. Although α-syn is a cytosolic protein with predominant localization in presynaptic terminals (Jakes et al., 1994), it is also found outside of cells. α-Syn is also detected in CSF and serum from healthy individuals and from PD patients (El-Agnaf et al., 2003; Tokuda et al., 2006), suggesting that α-syn possesses features of secretory proteins. The results from cell culture models showed that secreted α-syn can be taken up by neighboring cells via endocytosis (Desplats et al., 2009) and this uptake is mediated via exosomes (Danzer et al., 2012), whereas mouse studies revealed that injected α-syn fibrils spread between anatomically connected regions (Luk et al., 2012). These results suggest that α-syn can be transmitted from cell to cell and that prion-like seeding mechanism may contribute to PD pathology (Angot et al., 2010; Olanow and Brundin, 2013). Using sensitive ELISA, we observed an increase in α-ayn release in cells overexpressing PARK9 (Fig. 3F,G,N) and a decrease in PARK9 mutant fibroblasts (Fig. 3J,K). Interestingly, the amount α-syn in exosomes correlated with PARK9 expression, suggesting that PARK9 physiologically controls α-syn secretion through exosomes. These findings are in agreement with a recent report by Kong et al. (2014), where PARK9 overexpression decreased intracellular α-syn but increased α-syn secretion in the media. We also found that loss-of-function of PARK9 reduced exosomal α-syn suggesting the relevance of this pathway for disease. It will be of interest to examine the role of PARK9 in propagation of α-syn in vivo.
Previous studies showed that chemical inhibition of lysosomes or trafficking from endosome to lysosome increases α-syn release (Emmanouilidou et al., 2010; Alvarez-Erviti et al., 2011). PARK9-deficiency is known to cause lysosomal dysfunction and α-syn accumulation (Dehay et al., 2012; Usenovic et al., 2012a; Fig. 3D). Therefore, it would be predicted that lysosomal dysfunction caused by PARK9-deficiency results in increased release of exosomes and α-syn. However, α-syn release is decreased in these experiments (Fig. 3F,G), suggesting that the compensatory release of exosomes is inhibited in the presence of PARK9-deficiency. This conclusion is supported by our data demonstrating decreased number of ILVs in MVBs in PARK9-deficient cells. Based on these results, we hypothesize that decreased secretion of α-syn in PARK9-depleted cells further augments intracellular accumulation and toxicity of α-syn. However, further studies will be required to elucidate the precise molecular nature of PARK9 involvement in the function of MVBs in relation to lysosomal function.
In summary, our study implicates PARK9 function in the biogenesis of exosomes and secretion of α-syn. PARK9-mediated α-syn release via exosomes may, at least in part, explain PARK9 protective effect against α-syn toxicity. Together, these data raise a possibility that disruption of these pathways in patients with KRS contributes to the disease pathogenesis.
Footnotes
This work was supported by National Institutes of Health Grant R01NS076054 (D.K.). We thank Katarina Trajkovic for helpful suggestions, Clarissa Valdez and Aarthi Subramanian for technical assistance, Ellen Sapp in Marian DiFiglia lab for assistance with EM, Nadia Atai in Xandra Breakefield lab for assistance with Nanosight, Hyunkyung Jeong for mouse Tsg101 silencing, and John Ryder for assistance with α-syn ELISA.
The authors declare no competing financial interests.
References
- Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011;42:360–367. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Are synucleinopathies prion-like disorders? Lancet Neurol. 2010;9:1128–1138. doi: 10.1016/S1474-4422(10)70213-1. [DOI] [PubMed] [Google Scholar]
- Axelsen KB, Palmgren MG. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol. 1998;46:84–101. doi: 10.1007/PL00006286. [DOI] [PubMed] [Google Scholar]
- Bache KG, Brech A, Mehlum A, Stenmark H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol. 2003;162:435–442. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Moita C, van Niel G, Kowal J, Vigneron J, Benaroch P, Manel N, Moita LF, Théry C, Raposo G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J Cell Sci. 2013;126:5553–5565. doi: 10.1242/jcs.128868. [DOI] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109:9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, Cookson MR, Hardy J, Allsop D. α-Synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003;17:1945–1947. doi: 10.1096/fj.03-0098fje. [DOI] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falguières T, Luyet PP, Bissig C, Scott CC, Velluz MC, Gruenberg J. In vitro budding of intralumenal vesicles into late endosomes is regulated by Alix and Tsg101. Mol Biol Cell. 2008;19:4942–4955. doi: 10.1091/mbc.E08-03-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, Lindquist S. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald A, Arns B, Seibler P, Rakovic A, Münchau A, Ramirez A, Sue CM, Klein C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor–Rakeb syndrome. Neurobiol Aging. 2012;33:1843e1–1843e7. doi: 10.1016/j.neurobiolaging.2011.12.035. [DOI] [PubMed] [Google Scholar]
- Gusdon AM, Zhu J, Van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. 2012;45:962–972. doi: 10.1016/j.nbd.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- Jouvenet N. Dynamics of ESCRT proteins. Cell Mol Life Sci. 2012;69:4121–4133. doi: 10.1007/s00018-012-1035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BY, Olzmann JA, Barsh GS, Chin LS, Li L. Spongiform neurodegeneration-associated E3 ligase Mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol Biol Cell. 2007;18:1129–1142. doi: 10.1091/mbc.E06-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 1998;392:193–197. doi: 10.1038/32440. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Vischer UM, Rosnoblet C, Lebrand C, Lindsay M, Parton RG, Kruithof EK, Gruenberg J. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol Biol Cell. 2000;11:1829–1843. doi: 10.1091/mbc.11.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA. Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum Mol Genet. 2014;23:2816–2833. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- Lai CP, Breakefield XO. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front Physiol. 2012;3:228. doi: 10.3389/fphys.2012.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig AK, Giebel B. Exosomes: small vesicles participating in intercellular communication. Int J Biochem Cell Biol. 2012;44:11–15. doi: 10.1016/j.biocel.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mane SM, Marzella L, Bainton DF, Holt VK, Cha Y, Hildreth JE, August JT. Purification and characterization of human lysosomal membrane glycoproteins. Arch Biochem Biophys. 1989;268:360–378. doi: 10.1016/0003-9861(89)90597-3. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najim al-Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor–Rakeb syndrome. Acta Neurol Scand. 1994;89:347–352. doi: 10.1111/j.1600-0404.1994.tb02645.x. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Brundin P. Parkinson's disease and alpha synuclein: is Parkinson's disease a prion-like disorder? Mov Disord. 2013;28:31–40. doi: 10.1002/mds.25373. [DOI] [PubMed] [Google Scholar]
- Park JS, Mehta P, Cooper AA, Veivers D, Heimbach A, Stiller B, Kubisch C, Fung VS, Krainc D, Mackay-Sim A, Sue CM. Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor–Rakeb syndrome, a form of early-onset parkinsonism. Hum Mutat. 2011;32:956–964. doi: 10.1002/humu.21527. [DOI] [PubMed] [Google Scholar]
- Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM. Parkinson's disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet. 2014;23:2802–2815. doi: 10.1093/hmg/ddt623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- Ramonet D, Podhajska A, Stafa K, Sonnay S, Trancikova A, Tsika E, Pletnikova O, Troncoso JC, Glauser L, Moore DJ. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum Mol Genet. 2012;21:1725–1743. doi: 10.1093/hmg/ddr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, Geuze HJ. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287:3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short JR, Knox C, Dorrington RA. Subcellular localization and live-cell imaging of the Helicoverpa armigera stunt virus replicase in mammalian and Spodoptera frugiperda cells. J Gen Virol. 2010;91:1514–1523. doi: 10.1099/vir.0.020156-0. [DOI] [PubMed] [Google Scholar]
- Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, Dickson DW, Mazzulli JR, Bardgett ME, Haik KL, Ekhator O, Chava AK, Howard J, Gannon M, Hoffman E, Chen Y, Prasad V, Linn SC, Tamargo RJ, Westbroek W, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013;22:2067–2082. doi: 10.1093/hmg/ddt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Raposo G. Exosomes: vesicular carriers for intercellular communication. Curr Opin Cell Biol. 2009;21:575–581. doi: 10.1016/j.ceb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Simpson RJ, Jensen SS, Lim JW. Proteomic profiling of exosomes: current perspectives. Proteomics. 2008;8:4083–4099. doi: 10.1002/pmic.200800109. [DOI] [PubMed] [Google Scholar]
- Tamai K, Toyoshima M, Tanaka N, Yamamoto N, Owada Y, Kiyonari H, Murata K, Ueno Y, Ono M, Shimosegawa T, Yaegashi N, Watanabe M, Sugamura K. Loss of hrs in the central nervous system causes accumulation of ubiquitinated proteins and neurodegeneration. Am J Pathol. 2008;173:1806–1817. doi: 10.2353/ajpath.2008.080684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai K, Tanaka N, Nakano T, Kakazu E, Kondo Y, Inoue J, Shiina M, Fukushima K, Hoshino T, Sano K, Ueno Y, Shimosegawa T, Sugamura K. Exosome secretion of dendritic cells is regulated by hrs, an ESCRT-0 protein. Biochem Biophys Res Commun. 2010;399:384–390. doi: 10.1016/j.bbrc.2010.07.083. [DOI] [PubMed] [Google Scholar]
- Tan J, Zhang T, Jiang L, Chi J, Hu D, Pan Q, Wang D, Zhang Z. Regulation of intracellular manganese homeostasis by Kufor–Rakeb syndrome-associated ATP13A2 protein. J Biol Chem. 2011;286:29654–29662. doi: 10.1074/jbc.M111.233874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuma F, Zöller M. Outsmart tumor exosomes to steal the cancer initiating cell its niche. Semin Cancer Biol. 2014;28C:39–50. doi: 10.1016/j.semcancer.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Tokuda T, Salem SA, Allsop D, Mizuno T, Nakagawa M, Qureshi MM, Locascio JJ, Schlossmacher MG, El-Agnaf OM. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson's disease. Biochem Biophys Res Commun. 2006;349:162–166. doi: 10.1016/j.bbrc.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brügger B, Simons M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- Tsunemi T, Krainc D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. 2014;23:2791–2801. doi: 10.1093/hmg/ddt572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012a;32:4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, Knight AL, Ray A, Wong V, Brown KR, Caldwell GA, Caldwell KA, Stagljar I, Krainc D. Identification of novel ATP13A2 interactors and their role in alpha-synuclein misfolding and toxicity. Hum Mol Genet. 2012b;21:3785–3794. doi: 10.1093/hmg/dds206. [DOI] [PMC free article] [PubMed] [Google Scholar]