

Abstract
A mild and efficient o- and p-nitrobenzyl cleavage protocol was developed. o- and p-Nitrobenzyl groups were easily removed from a variety of substrates using 20% aqueous NaOH in methanol at 75 °C, presumably via oxidation at the benzylic position by oxygen dissolved in the solution. These easily introducible and removable nitrobenzyl groups can serve as valuable protecting groups for the synthesis of multifunctional, complex molecules.
Keywords: Protecting group, o-Nitrobenzyl group, p-Nitrobenzyl group, Amide protection, Deprotection
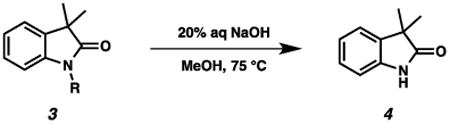
The selection of protection groups is critical to the synthesis of multifunctional complex molecules. Over the past decades, a wide variety of protecting groups have been developed to mask specific functional groups selectively.1,2 However, despite considerable effort, relatively few protecting groups have been developed for the amide N-H (e.g., PMB, TBS, Benzyl, SEM, allyl, etc.).3 Consequently, limitations associated with cleavage of amide protecting groups often arise.4 Herein, we report o- and p-nitrobenzyl groups as easily introducible and removable protecting groups for both R2N-H (including amides) and RO-H groups.
In the course of our efforts toward the synthesis of the polycyclic, complex alkaloid perophoramidine, we serendipitously found that the o-nitrobenzyl group was easily removed by using 20% aqueous NaOH in methanol at 75 °C (Scheme 1).5 o-Nitrobenzyl groups have been used as photocleavable protecting groups for amides and heterocycles such as indoles, benzimidazole, and 6-chlorouracil as well as the more common hydroxyl group.6,7 In addition, a method for removing p-nitrobenzyl groups on hydroxyl groups in two steps by reduction to a p-aminobenzyl group followed by electrochemical oxidation was disclosed by the Kusumoto group.8 To the best of our knowledge, ours was the first example of using simple aqueous NaOH to remove a nitrobenzyl group from a lactam.
Scheme 1.

o-Nitrobenzyl group cleavage on perophoramidine intermediate (1).
To explore the general reactivity of nitrobenzyl group cleavage reactions by using simple aqueous NaOH, we initially chose 3,3-dimethyl oxindole as a test substrate (Table 1). We found that both ortho- and para-nitrobenzyl groups on oxindoles 3a and 3c were smoothly cleaved under our standard conditions (entries 1 and 3). However, a meta-nitrobenzyl group as well as a simple benzyl group on the oxindole nitrogen (3b and 3d) were both unreactive (entries 2 and 4). Notably, the o-nitrobenzyl group was also successfully removed even in the absence of light (entry 5). Interestingly, only trace amounts of product were observed when degassed water and methanol were used (entry 6). Furthermore, we discovered that cleavage of the o-nitrobenzyl group was successful when the degassed water and methanol were purged with oxygen gas (entry 7). We therefore concluded that oxygen was necessary for the reaction, but light was not.
Table 1.
Cleavage of nitrobenzyl groups on 3,3-dimethyloxindole.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Substrate | R | Time (h) | Yleld (%) |
| 1 | 3a | -o-nitrobenzyl | 5 | 69 |
| 2 | 3b | -m-nitrobenzyl | 24 | 0 |
| 3 | 3c | -p-nitrobenzyl | 1.5 | 63 |
| 4 | 3d | -benzyl | 24 | 0 |
| 5a | 3a | -o-nitrobenzyl | 6.5 | 72 |
| 6b | 3a | -o-nitrobenzyl | 12 | trace |
| 7b,c | 3a | -o-nitrobenzyl | 6.5 | 66 |
The reaction was performed in the dark.
Degassed water and methanol were used.
Purged wlth O2.
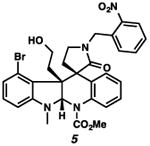
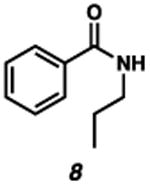
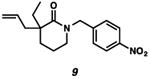
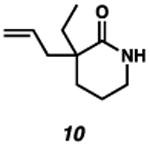
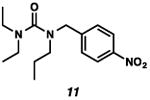
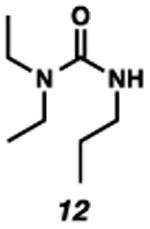
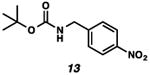
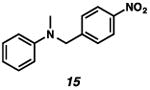
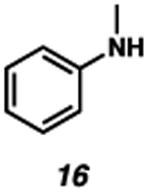
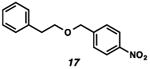
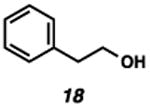
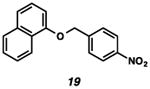
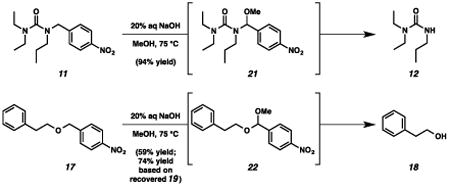
With the standard conditions in hand, we explored the scope of the o- and p-nitrobenzyl group cleavage reactions with various substrates (Table 2). o- and p-Nitrobenzyl protected substrates were easily prepared by a variety of methods, including reductive amination, simple alkylation, EDCI coupling, and acylation. The o-nitrobenzyl group on a highly functionalized communesin F intermediate 5 was smoothly cleaved under our standard conditions in good yield (entry 1).5 p-Nitrobenzyl groups on amide 7 and lactam 9 were also cleaved in moderate yields (entries 2 and 3). The p-nitrobenzyl group on urea 11 was also successfully cleaved and methyl 4-nitrobenzoate was isolated as a by-product (entry 4). An attempt to remove the p-nitrobenzyl group on secondary carbamate 13 proved unsuccessful (entry 5). Additionally, removal of the p-nitrobenzyl group on amine 15 was facile, furnishing aniline 16 in 65% yield (entry 6). Interestingly, 4-nitrobenzaldehyde was isolated as a by-product in this reaction, presumably resulting from the oxidation reaction at the benzylic position by oxygen dissolved in the solution. Furthermore, we successfully removed the p-nitrobenzyl groups from ethers 17 and 19 in moderate yields (entries 7 and 8). This work offers a one-step alternative to previous work by the Kusumoto group.8 In the cases of deprotection reactions from urea 11 and ether 17, the corresponding hemiaminal ether and acetal intermediates were observed (see ref. 9). It is important to note that these deprotection conditions are compatible with free hydroxyl groups, aminals, aryl bromides, methyl carbamates, silyl ethers, and Boc groups present on the substrates.
Table 2.
Scope of the o- and p-nitrobenzyl deprotecion reactions.
| Entry | Substrate | Product | Time (h) | Yield (%) |
|---|---|---|---|---|
| 1 |
|
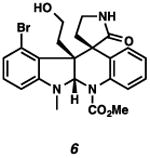
|
4 | 70 |
| 2 |
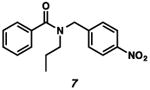
|
|
12 | 42 |
| 3 |
|
|
1 | 55 |
| 4 |
|
|
10.5 | 94a,b |
| 5 |
|
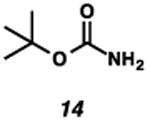
|
6 | 0 |
| 6 |
|
|
2 | 65c |
| 7 |
|
|
32 | 59b (74)d |
| 8 |
|
|
48 | 48 |
Methyl 4-nitrobenzoate was isolated as a by-product.
See Ref. 9 for detail.
4-Nitrobenzaldehyde was isolated as a by-product.
Yield based on recovered starting material.
A representative procedure is as follows: To a 20 mL scintillation vial with a magnetic stir bar were added p-nitrobenzyl protected oxindole 3c (30 mg, 0.10 mmol, 1.0 equiv), MeOH (1.0 mL), and 20% aq NaOH (1.0 mL). The reaction mixture was stirred for 1.5 h at 75 °C. The reaction mixture was then cooled to 23 °C, and extracted with EtOAc (3×2 mL). The organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography to afford 3,3-dimethyloxindole 4 (10.3 mg, 63% yield).
In summary, a mild and efficient deprotection protocol was discovered for the cleavage of o- and p-nitrobenzyl ethers, amides, ureas, and anilines. Since relatively few options for protection of the amide N-H functionality exist, easily introducible and removable o- and p-nitrobenzyl groups could prove useful in the synthesis of alkaloids and biologically active molecules, which possess amide functionalities. In our laboratory, this has already proven to be the case in our formal syntheses of communesin F and perophoramidine. In addition, p-nitrobenzyl protected anilines, ureas, and alcohols were also competent substrates for this transformation, which may further its utility as a mild deprotection method in cases where other conditions fail.
Supplementary Material
Acknowledgments
The authors wish to thank NIH-NIGMS (R01GM080269-01), Amgen, and Caltech for financial support. S.-J.H thanks Fulbright (Foreign Student Program, No. 15111120) and the Ilju Foundation of Education & Culture (Pre-doctoral Research Fellowship) for financial support.
Footnotes
Supplementary Material: Supplementary data associated with this article can be found, in the online version, at XXXXXXXXXXXX.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Wuts PGM, Greene TW. Greene's Protective Groups in Organic Synthesis. 4th. John Wiley & Sons; New York: 2007. [Google Scholar]; (b) Kocienski PJ. Protecting Groups. George Thieme Verlag; Stuttgart and New York: 1994. [Google Scholar]
- 2.Recent examples:Evans V, Mahon MF, Webster RL. Tetrahedron. 2014 doi: 10.1016/j.tet.2014.07.080.Yang Q, Njardarson JT. Tetrahedron Lett. 2013;54:7080–7082. doi: 10.1016/j.tetlet.2013.10.097.Sugiura R, Kozaki R, Kitani S, Gosho Y, Tanimoto H, Nishiyama Y, Morimoto T, Kakiuchi K. Tetrahedron. 2013;69:3984–3990.
- 3.(a) Williams RM, Kwast E. Tetrahedron Lett. 1989;30:451–454. [Google Scholar]; (b) Pagano N, Wong EY, Breiding T, Liu H, Wilbuer A, Bregman H, Shen Q, Diamond SL, Meggers E. J Org Chem. 2009;74:8997–9009. doi: 10.1021/jo901641k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rodríguez-Vázquez N, Salzinger S, Silva LF, Amorín M, Granja JR. Eur J Org Chem. 2013;17:3477–3493. [Google Scholar]; (d) Overman LE, Rosen MD. Angew Chem Int Ed. 2000;39:4596–4599. [PubMed] [Google Scholar]; (e) Würdemann M, Christoffers J. Org Biomol Chem. 2010;8:1894–1898. doi: 10.1039/b922827f. [DOI] [PubMed] [Google Scholar]; (f) Fukuyama T, Frank RK, Jewell CF., Jr J Am Chem Soc. 1980;102:2122–2123. [Google Scholar]
- 4.(a) Hoffmann RW, Breitfelder S, Schlapbach A. Helv Chim Acta. 1996;79:346–352. [Google Scholar]; (b) Hennessy EJ, Buchwald SL. J Org Chem. 2005;70:7371–7375. doi: 10.1021/jo051096o. [DOI] [PubMed] [Google Scholar]; (c) Madin A, O'Donnell CJ, Oh T, Old DW, Overman LE, Sharp MJ. J Am Chem Soc. 2005;127:18054–18065. doi: 10.1021/ja055711h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fetter J, Giang LT, Czuppon T, Lempert K, Kajtár-Peredy M, Czira G. Tetrahedron. 1994;50:4185–4200. [Google Scholar]; (e) Smith AB, III, Haseltine JN, Visnick M. Tetrahedron. 1989;45:2431–2449. [Google Scholar]
- 5.Han SJ, Vogt F, Krishnan S, May JA, Gatti M, Virgil SC, Stoltz BM. Org Lett. 2014;16:3316–3319. doi: 10.1021/ol5013263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Piloto AM, Costa SPG, Goncalves MST. Tetrahedron. 2014;70:650–657. [Google Scholar]; (b) Voelker T, Ewell T, Joo J, Edstrom ED. Tetrahedron Lett. 1998;39:359–362. [Google Scholar]; (c) Snider BB, Busuyek MV. Tetrahedron. 2001;57:3301–3307. [Google Scholar]; (d) Miknis GF, Williams RM. J Am Chem Soc. 1993;115:536–547. [Google Scholar]
- 7.(a) Stutz A, Pitxch S. Synlett. 1999:930–934. [Google Scholar]; (b) Pitsch S. Helv Chim Acta. 1997;80:2286–2314. [Google Scholar]
- 8.Fukase K, Tanaka H, Torii S, Kusumoto S. Tetrahedron Lett. 1990;31:389–392. [Google Scholar]
-
9.From the deprotection reaction of urea 11, hemiaminal ether intermediate 21 was observed in 1 hour under the Standard reaction conditions and intermediate 21 was converted to propyl urea 12 after 10.5 hours. In addition, acetal 22 was observed in 2 hours during the deprotection reaction of ether 17, and the desired phenylethyl alcohol 18 was isolated after 32 hours.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.