

Abstract
Study Design
To test for rare genetic mutations, a cohort of patients with unexplained early onset scoliosis (EOS) was screened using high-density microarray genotyping. A cohort of patients with adolescent idiopathic scoliosis (AIS) was similarly screened, and the results were compared.
Summary of background data
Patients with scoliosis in infancy or early childhood (EOS) are at high risk for progressive deformity and associated problems including respiratory compromise. EOS is frequently associated with genetic disorders, but many patients present with non-specific clinical features and without an associated diagnosis. We hypothesized that EOS in these patients may be caused by rare genetic mutations detectable by next-generation genomic methods.
Methods
We ascertained 24 patients with unexplained EOS from pediatric orthopedic clinics. We genotyped them, along with 39 connecting family members, using the Illumina OmniExpress-12 v1.0 beadchip. Resulting genotypes were analyzed for chromosomal changes, specifically copy number variation (CNV) and absence of heterozygosity (AOH). We screened 482 AIS patients and 744 healthy controls, which were similarly genotyped with the same beadchip, for chromosomal changes identified in the EOS cohort.
Results
Copy number variation (CNV) and absence of heterozygosity (AOH) analyses revealed a genetic diagnosis of chromosome 15q24 microdeletion syndrome in one patient, and maternal uniparental disomy of chromosome 14 in a second patient. Prior genetic testing and clinical evaluations had been negative in both cases. A large novel chromosome 10 deletion was likely causal in a third EOS patient. These mutations identified in the EOS patients were absent in AIS patients and controls, and thus not associated with AIS or found in asymptomatic individuals.
Conclusions
Our data underscore the utility of updated genetic evaluations including high-density microarray-based genotyping and other “next-generation” methods in patients with unexplained EOS, even where prior genetic studies were negative. These data also suggest the intriguing possibility that other mutations detectable by whole genome sequencing, as well as epigenetic effects, await discovery in the EOS population.
Introduction
Early onset scoliosis (EOS), by definition, affects children up to five years of age. In surgical cohorts, reported mortality rates vary but are as high as 18% compared to 0.08% in the general U.S. population [1, 2]. Children with EOS can pose a significant and challenging clinical problem, as they are at risk for pulmonary compromise as well as other growth disturbances [3]. In extreme cases, EOS can lead to thoracic insufficiency syndrome, in which the thorax is unable to support normal lung growth and function [4]. Consequently, intense effort has been given to developing surgical methods and devices that preserve lung function and growth while controlling deformity [5–7]. The pathogenesis of EOS is heterogeneous, as these patients represent numerous underlying diagnoses that generally divide into three classes. One class of EOS is “congenital” scoliosis (CS), where deformity is caused by vertebral anomalies or segmentation defects. Although CS can be clearly heritable, it is often sporadic and may result from gene-environment interactions [8]. A second class of EOS is due to known heritable syndromes, many of which are well-recognized and diagnosed by clinical genetic testing, such as Ehlers-Danlos and Larsen syndrome [9]. However a significant fraction, roughly one-third of surgical cases, is without an identifiable diagnosis and is therefore described as “idiopathic”. Historically, idiopathic scoliosis (IS) has been described by the terms “infantile” (onset ages 0–3 years), “juvenile” (onset ages 4–9 years), or “adolescent” (onset age 10 years or older) [10]. However, EOS nomenclature derives more from the natural history of spinal growth and deformity. Here, we use the term “unexplained EOS” to avoid confusion with previous nomenclature and to include all EOS children who may have associated growth issues but have not been ascribed a clear underlying diagnosis.
Unlike later onset AIS, unexplained EOS rarely presents with positive family history of scoliosis and may affect boys more than girls [11]. The perception of low heritability in EOS has invoked environmental explanations, including fetal crowding in the womb or positioning of the child in the crib [10, 11], but these theories have not been substantiated. For many patients, postnatal disease onset, coupled with particularly malignant deformity progression, argues that EOS is likely to be genetically driven. Although comprehensive population studies are few, the prevalence of unexplained EOS has been cited as less than 1% of the total idiopathic scoliosis population [10]. We hypothesized that EOS could arise from rare de novo mutations, in other words, mutations that are absent in the parents and the general population but present in the affected offspring. We also hypothesized that such mutations are likely to be heterogeneous, that is, to correspond to many different causal genes, reflecting the clinical heterogeneity observed in this population.
Many mutations that would be missed by traditional techniques are discoverable using methods that search the chromosomes more comprehensively. One method, high-density microarray-based genotyping, enables testing of greater than one million single nucleotide polymorphisms (SNPs) spaced across the genome. Measuring SNP content and signal intensity reveals gains and losses of genetic material known as copy number variations, or CNVs, typically at higher resolution than a traditional karyotype [12,13]. Microarray-based genotyping also yields information about SNP heterozygosity. A heterozygous SNP harbors different sequences at the same location, indicative of the inheritance of two chromosomes. Absence of heterozygosity (AOH) may be an indication of genetic aberration. Large, contiguous regions of AOH genome-wide suggests parental consanguinity. AOH in specific chromosomal regions may indicate abnormal chromosomal inheritance, as in Prader-Willi and Angelman syndromes [14], or loss of genetic material due to deletion. These applications of microarray-based genotyping have been shown to increase the likelihood of finding the genetic cause of congenital structural anomalies or neurocognitive disorders, and the method is typically the first-tier genetic testing approach in these populations [15]. Therefore, we assessed a cohort of 24 probands with unexplained EOS using microarray-based genotyping to test the hypothesis of rare causal mutations. Subsequently, we also screened a large cohort of AIS patients and controls to assess whether variation in EOS genes are more generally associated with IS.
Materials and Methods
Study Design
This was a genome-wide microarray genotyping discovery study to assess rare de novo CNVs and regions of AOH in a cohort of 24 EOS cases. A follow-up screening study tested EOS-associated CNVs and regions of AOH in 482 AIS cases and 744 controls.
Cases
The Texas Scottish Rite Hospital for Children (TSRHC) Idiopathic Scoliosis DNA registry was established in 1997. From 1997–2013 there were 87 EOS and 4,292 AIS patients treated in TSRHC pediatric orthopedic clinics. Forty-one EOS and 1,913 AIS probands, plus additional family members, were enrolled into the registry during that time. Informed consent was obtained from all participating research subjects as specified by the University of Texas Southwestern Medical Center Institutional Review Board. Twenty-four unrelated EOS probands and 39 parents were included in the present study. The ethnic composition of the EOS cohort was: Asian/Pacific Islander (1), Hispanic/Latino (4), Black, non-Hispanic (2), White, non-Hispanic (16), and Other (1). Follow-up studies included 482 AIS cases, all non-Hispanic white ethnicity. All affected subjects in these cohorts met the standard criterion for a positive diagnosis of idiopathic scoliosis: lateral deviation from the midline greater than 10 degrees, as measured from standing spinal radiographs, axial rotation toward the side of the deviation, and exclusion of all other etiologies. For the purposes of this study, we elected to require a minimum Cobb angle of 15 degrees, given the known inter/intra-observer variance.
Controls
Unaffected control individuals (N=744) were ascertained from within the local Texas population or non-orthopedic clinics at TSRHC. Informed consent was obtained from each participant as described above. Any individuals with diagnosis of scoliosis, or family history of scoliosis, were excluded by questionnaire in these individuals. DNA was obtained from whole blood.
Microarray-based genotyping and analysis
We used the Illumina OmniExpress-12 v1.0 beadchip (Illumina Inc., San Diego, CA, USA) to genotype single nucleotide polymorphisms (SNPs) in EOS cases and parents, AIS cases, and controls described above. Resulting data were evaluated for AOH and CNVs at a resolution of ~10,000 base pairs (10 kb) using algorithms contained in Illumina Partition (Illumina Inc., San Diego, CA, USA), Partek Segment (Partek Inc., Chesterfield, MO, USA), and Quanti-SNP 2.0 (Wellcome Trust Center for Human Genetics, Oxford, United Kingdom) software packages. Genotypes were aligned with the human reference genome (GRCh37/hg19). We selected all regions of AOH greater than 3 million base pairs (Mb) in EOS probands, as smaller regions of AOH are common in normal populations [16]. We also selected CNVs greater than 10 kb for the same reason, and then excluded those that were found in the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home), as these are likely to be common in normal populations. Clinical testing was performed with Cytogenomic SNP Microarray (CMASNP) (ARUP Laboratories, Salt Lake City, UT).
Results
We performed microarray-based genotyping in 24 EOS probands and 39 connecting family members and searched for chromosomal changes using multiple methods as described. Resulting regions of AOH and CNV were examined in parents to identify those mutations that arose de novo, i.e. that were not present in the parents. Using this approach, we describe three informative patients, two of which were provided genetic diagnoses.
Case 15-1 is a thirteen-year-old Hispanic male who originally presented at age ten with a diagnosis of juvenile IS; records indicated the onset of scoliosis around three years of age (Figure 1a). Birth history included placenta previa and dislocated hips that were treated successfully. The patient had gross motor delays requiring physical therapy since nineteen months of age. He also received speech and occupational therapy, and special education in reading and math. A prior diagnosis of hypertension was reported, and he was treated with hydrochlorothiazide (HCTZ). Surgeries included tonsillectomy, tympanoplasty and myringotomy; the patient was noted to use hearing aids. Clinical genetic and MRI studies were reported to be normal. Ligamentous laxity and low tone were noted. Neurologic evaluation revealed that the patient was <5th percentile (145 cm) in height, >98th percentile (63.7 kg) in weight and 50th percentile (56.5 cm) for fronto-occipital circumference (FOC). Bilateral large ears, flat nasal bridge, triangular shaped face, high forehead, tapered digits and gynecomastia were also noted (Figure 1a). Nasal quality of the voice was noted but was mild, along with somewhat decreased tongue mobility. The patient is left-handed, with normal left-side coordination but had difficulty with right-side movements and tasks. Decreased pain sensation was noted in hands, forearms, feet, and lower legs bilaterally. Follow-up brain MRI revealed mild cerebral atrophy; EMG/nerve conduction studies were normal. Spine MRI revealed scoliosis as expected, fatty filum with normal position of the conus, sacral dimpling, apparent right-sided descending thoracic aorta and possible cardiomegaly. Urodynamic studies were negative and genitalia were noted to be normal. A follow up cardiology consult revealed normal heart structure with mild transverse arch hypoplasia and borderline prolonged QTc. Prior to surgery, the patient’s scoliotic curve measured 70 degrees by the Cobb angle method with a bone age of twelve and one-half years (Figure 1b). Surgical correction included anterior and posterior spinal fusion with instrumentation.
Figure 1. Results for case 15-1.
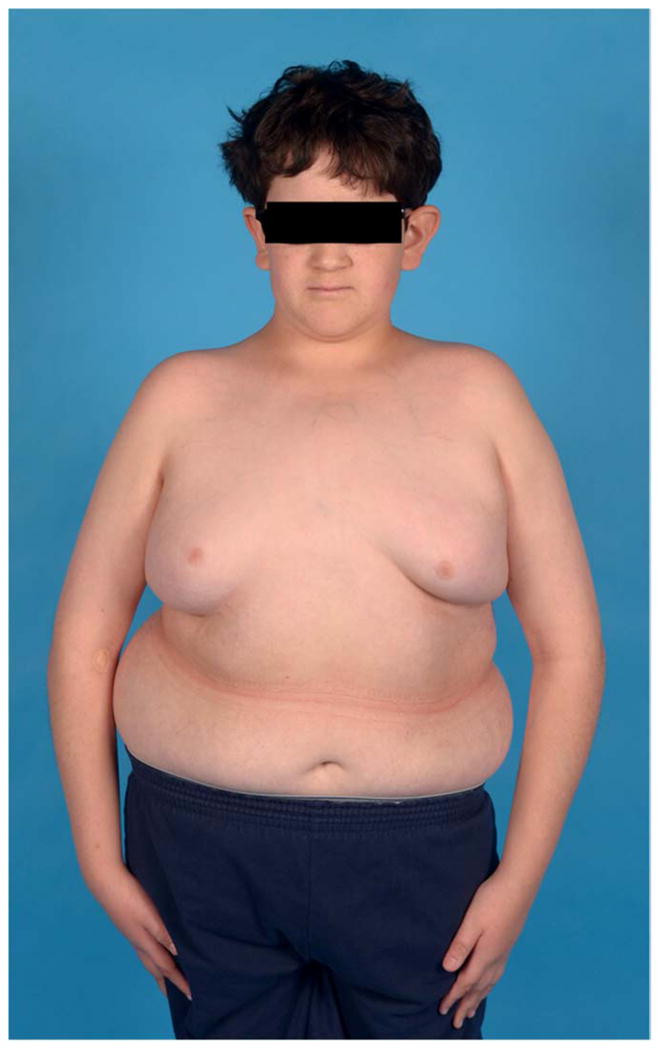

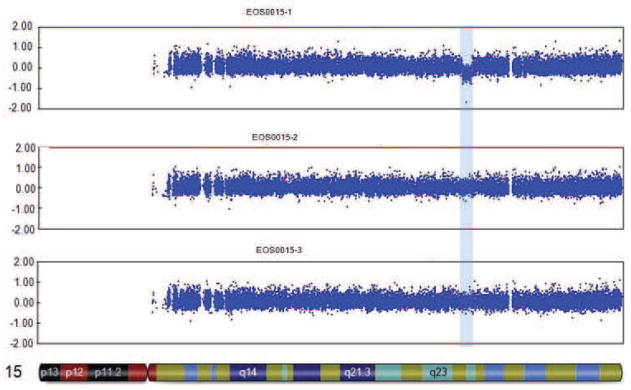

a. Photograph prior to scoliosis surgery, showing low tone, obesity, large ears bilaterally, and flat nasal bridge. b. Posteroanterior and sagittal radiographs showing 70 degree left thoracic scoliosis, with excessive soft tissue shadows (obesity) and mild epiphyseal dysplasia of the femoral heads. c. CNV analysis for chromosome 15, with logR ratio plotted on the Y-axis versus chromosome position on the X-axis. Case 15-1 is noted at top, with mother and father (middle and bottom, respectively). Deleted region is noted by blue shading. d. Case 15-1 deleted interval (red box) compared to deletions reported for other 15q24 microdeletion cases (below). Previously described breakpoint regions mediating non-allelic homologous recombination are noted above. “BP1,2,3,4” notation is as described by Sharp et al.(19); “15q24a,b,c,d,e” notation is as described by El-Hattab et al.(21)
CNV analysis identified a ~1.6Mb deletion on the maternally-inherited chromosome 15q24.1-q24.2; the deletion was not present in the parents (Figure 1c). These results, together with clinical findings, are consistent with a diagnosis of de novo chromosome 15q24 microdeletion syndrome [17]. Figure 1d compares the deletion in our patient to other published 15q24 microdeletion cases in the literature and suggests that our findings may define a new minimal deletion.
Case 29-1 presented at sixteen months of age with a pronounced thoracic scoliosis (Figure 2a). Medical history was unremarkable except for marked hypotonia and failure to thrive that initially required gastrostomy tube feeding. A muscle biopsy performed to evaluate the hypotonia was unremarkable. Genetic evaluations in infancy were negative, with a normal karyotype. Dysmorphic facial features were absent; intellectual abilities were normal. Scoliosis was her most significant and persistent clinical problem (Figure 2b). As a result of her diminutive size, initial management was with intermittent prolonged halo-gravity traction (15 of the 36 months after presentation), followed by bracing. At four years of age, after a final period in halo-traction, she was managed with a “growing rod” construct. Despite her initial failure to thrive, she met her speech and fine-motor milestones on time and has been performing above average at a grade-appropriate level. At age eight she was diagnosed with precocious puberty, which responded to treatment. She had definitive anterior and posterior spinal fusion at age ten years.
Figure 2. Results for case 29-1.
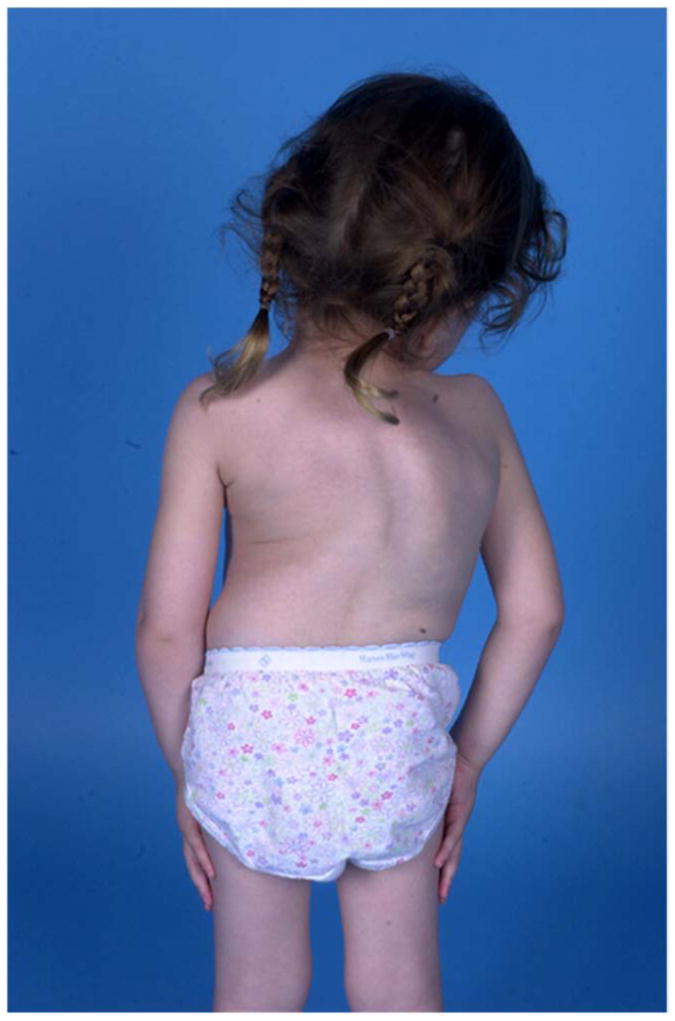

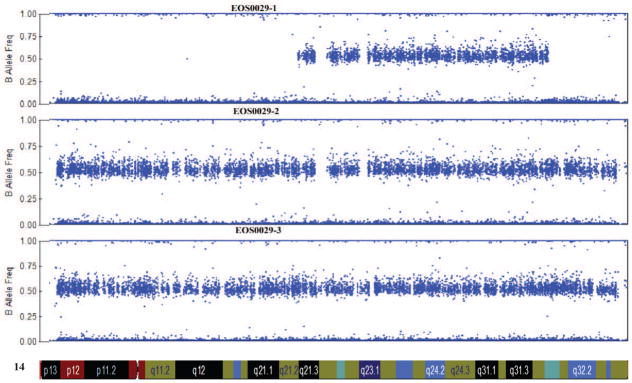
a. Prominent rib hump due to scoliosis is evident. b. Standing posteroanterior and sagittal spinal radiographs for case 29-1 at 16 months of age. c. Microarray genotyping analysis for chromosome 14, where B allele frequency is plotted on the Y-axis versus chromosome position on the X-axis. Large regions displaying loss of heterozygosity are evident at both proximal and distal ends of the chromosome in patient (29-1) as compared to parents (mother, middle, and father, bottom).
Genome-wide SNP microarray analysis of DNA from the patient and her parents revealed extensive AOH with no copy number loss for most of chromosome 14 (Figure 2c) and an intervening region of heterozygosity from 57.2 Mb to 95.3 Mb. Of 76,605 SNPs on chromosome 14 included in the analysis (described in Materials and Methods), 99.85% were inherited from the mother. The overwhelming proportion of maternally-inherited variants suggested the entire chromosome 14 was of maternal origin, a condition known as uniparental disomy (UPD) chromosome 14 (mat), or Temple Syndrome [18, 19]. The patient was subsequently referred to clinical genetics for confirmation of the diagnosis. Clinical methylation analysis confirmed an unmethylated (maternal) pattern and absence of a methylated (paternal) pattern at the MEG3 locus, a site on chromosome 14 known to display different patterns of methylation between maternal and paternal chromosomes [20]. This result is diagnostic for Temple Syndrome [21].
Case 30-1 presented at two and one-half years of age with mild scoliosis of 17 degrees (Figure 3a). Birth and newborn history were normal with the exception of laryngomalacia. At age four, the patient continued to be followed for his scoliosis. Copy number analysis of this patient revealed a ~375 kb deletion at chromosome 10q24.1 but normal chromosomes in both parents (Figure 3b). No other de novo chromosomal variants were otherwise detected by this method. The deleted region encompasses two annotated genes, SLIT1 encoding the slit1 homolog of drosophila, and LCOR encoding ligand-dependent nuclear receptor co-repressor. SLIT1 was of particular interest, as it encodes a ligand for the Robo family of axon guidance molecules that are associated with progressive scoliosis [22]. Interestingly, the patient’s mother did not carry the 10q24.1 deletion but had reported treatment for adolescent-onset IS.
Figure 3. Results for case 30-1.


a. Standing posteroanterior and lateral spinal radiographs for case 30-1 at 2.5 years of age. This patient’s curve has since progressed to >30 degrees. b. Microarray CNV analysis of chromosome 10 for case 30-1. LogR (Y-axis) is plotted versus chromosome position (X-axis). Deletion interval is shown by blue shading, with corresponding physical map in the call-out below.
Of 24 EOS patients evaluated by high-density microarrays, 21 were negative for potentially pathogenic chromosomal changes using our strategy. The three pathogenic changes identified in EOS patients were absent in 482 AIS patients, suggesting that these chromosomal variations are uncommon in general idiopathic scoliosis populations. Likewise, these variants were not identified in 744 healthy controls, supporting the likelihood that these variants are pathogenic in the EOS patients.
Discussion
The etiological underpinnings of EOS, outside of known genetic disorders, are poorly understood. High-density microarray genotyping provided successfully identified de novo mutations, and provided genetic diagnoses in two patients, each with somewhat non-specific clinical features in their early childhood years. The constellation of features in Case 15-1 suggested an underlying syndrome, yet prior genetic studies were normal. Chromosome 15q24 microdeletion syndrome has emerged only recently as a recognizable diagnosis, subsequent to genetic testing with chromosome microarrays [13, 17]. Previous reports describe developmental delay, ear and eye abnormalities, hypotonia, genito-urinary anomalies, craniofacial dysmorphism and minor digital anomalies including proximally implanted thumbs, mild brachydactyly, syndactyly, camptodactyly, and long slender fingers as salient features of the syndrome [23–28]. Other variable clinical features of 15q24 microdeletion syndrome include truncal obesity, nasal speech/hoarse voice, scoliosis, elevated serum triglycerides/lipids, splenomegaly, hepatomegaly, congenital diaphragmatic hernia and multiple cystic lesions of the corpus callosum [23, 24, 26]. “Dislocated hips” was not noted in other reports and may be a new finding, presumably related to general ligamentous laxity. The 15q24 microdeletion critical region is particularly gene-dense, with over 40 annotated transcripts in the interval defined by our patient, and it is therefore challenging to correlate a specific gene with the scoliosis phenotype. Previous genetic studies have implicated pathways of axon growth and guidance [29, 30], and we note at least one such gene, SEMA7, is encoded in the region of interest. Further study is required to assess the contributions of individual genes to the variable features of chromosome 15q24 microdeletion syndrome.
The clinical features of patient 29-1, other than her progressive scoliosis, were unremarkable. Consequently, an underlying chromosomal syndrome was not suspected, a conclusion that seemed to be borne out by extensive prior testing. However, her clinical features are consistent with the salient features of UPD(14)mat (Temple syndrome) that include small size and weight, hypotonia, feeding problems at birth, and precocious puberty. Developmental delay (which she did not exhibit), scoliosis, and truncal obesity are variously reported [18, 31]. The clinical features of UPD(14)mat are proposed to be largely explained by a cluster of imprinted genes on chromosome 14q32.2. Specifically, DLK1 and RTL1 on chromosome 14q32.2 are only expressed from the paternal chromosome; consequently, these genes were “missing” in our patient who carried two copies of the maternal chromosome. Conversely, the GTL2, antisense RTL1 and MEG8 genes on 14q32.2 are only expressed from the maternal chromosome and, consequently, our patient carried twice the dosage of these genes. One patient with bi-parental chromosome 14 inheritance – but apparent loss of imprinting at the DLK1/GTL2 locus – has been described with early onset scoliosis, suggesting that this region is indeed responsible for the spinal deformity in our patient [19], but further study is required to understand its role in scoliosis.
The 375 kb de novo deletion on chromosome 10q24 in patient 30-1 may be pathogenic; however, evidence in additional patients and/or animal models is needed to confirm this. Interestingly, the deletion creates haploinsufficiency of SLIT1, a well-studied axon guidance gene. Specifically, murine Slit proteins prevent axons from growing into ventral brain regions, prevent axons from crossing the brain midline, and participate in channeling axons toward appropriate targets [32]. Slit proteins are ligands for ROBO3, a transmembrane protein that controls midline crossing in vertebrate brain and spinal cord [33]. This pathway has been associated with scoliosis in humans, as apparent loss-of-function mutations in ROBO3 cause recessive horizontal gaze palsy with progressive scoliosis, or HGPPS [21].
The three mutations that we discovered in EOS patients, UPD(14)mat, 15q24 microdeletion, and 10q24 microdeletion, were absent in our larger cohort of AIS cases. This was not surprising, as the ascertainment for AIS would have excluded early onset disease and other clinical associations. However, it is possible that more subtle mutations in the three genomic regions may cause AIS. We also expect that further study of the biochemical pathways involved in EOS will inform our understanding of AIS etiology.
As with the three positive cases, other non-specific clinical features were noted in the 21 cases that were negative by microarray genotyping. These included patent ductus arteriosus/patent foramen ovale, pectus, a large, diffuse port-wine stain, left hip dysplasia, and mild sacral dimpling in five separate cases. Another patient was described as having very mild flat nasal bridge and slightly wide-spaced eyes but no other findings. Other findings in single cases were temporary failure to thrive, temporary hypotonia, ligamentous laxity (two cases), “midline depression”, episodic incontinence, and history of “numbness” that was negative by neurologic examination. Cobb angle measurements of the scoliosis in these children at first presentation ranged from 17 to 67 to degrees; for many, progressive scoliosis was arguably their most distinct feature. Further genetic studies including whole genome sequencing are planned in these patients.
The results of this study suggest that chromosome microarray analysis should be considered for all unexplained EOS cases, as it may be diagnostic in an appreciable fraction of patients. Cases with progressive scoliosis and other non-specific clinical findings may be atypical presentations of known syndromes and, therefore, primary candidates for genetic testing. Our data particularly underscore the need to consider clinical genetic evaluation of EOS patients even when prior genetic studies were negative, as newer testing platforms are more sensitive for discovering alterations such as UPD. Whole genome sequencing (WGS) in particular is emerging as a sensitive genetic testing tool [34]. Indeed WGS of case 29-1 also revealed UPD(14)mat (data not shown). Although a molecular diagnosis may not guide surgical correction, it is a definitive answer and sometimes predictive of future medial complications, such as with precocious puberty in Temple Syndrome. Molecular results are also useful for accurate recurrence risk counseling. Finally, documenting causal molecular changes in EOS will fuel future investigations into the underlying disease etiologies.
Acknowledgments
Funding Resources
We thank each of the patients and families for their participation in the study. This work was supported by the Fondation Cotrel, NIH grant R01HD02973, the Crystal Charity Ball, the Scoliosis Research Society, and Texas Scottish Rite Hospital for Children Research Fund (to C.A.W.).
Footnotes
IRB Approval
University of Texas Southwestern Medical Center Institutional Review Board
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Phillips JH, Knapp DR, Jr, Herrera-Soto J. Mortality and morbidity in early-onset scoliosis surgery. Spine (Phila Pa 1976) 2013;38:324–7. doi: 10.1097/BRS.0b013e31826c6743. [DOI] [PubMed] [Google Scholar]
- 2.Mortality rate, under-5 (per 1,000 live births) World Bank; in press. Available at: http://data.worldbank.org/indicator/SH.DYN.MORT. [Google Scholar]
- 3.Herring JA. Tachdjian’s pediatric orthopaedics. Philadelphia: WB Saunders; 2008. [Google Scholar]
- 4.Karol LA. Early definitive spinal fusion in young children: what we have learned. Clin Orthop Relat Res. 2011;469:1323–9. doi: 10.1007/s11999-010-1622-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell RM, Akbarnia BA, Yazici M, Thompson GH, editors. The Growing Spine: Management of Spinal Disorders in Young Children. Springer-Verlag; New York: 2011. VEPTR Expansion Thoracoplasty; pp. 469–86. in press. [Google Scholar]
- 6.Akbarnia BA, Mundis GM, Jr, Salari P, Yazici M, Thompson GH, et al. The Growing Spine: Management of Spinal Disorders in Young Children. NY: Springer Verlag press; 2010. Dual Growing Rods; pp. 449–68. [Google Scholar]
- 7.Son-Hing GHTJP, Akbarnia BA, Thompson GH, Yazici M, et al. The Growing Spine: Management of Spinal Disorders in Young Children. NY: Springer Verlag press; 2010. Single Growing Rods; pp. 441–8. [Google Scholar]
- 8.Sparrow DB, Chapman G, Smith AJ, et al. A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell. 2012;149:295–306. doi: 10.1016/j.cell.2012.02.054. [DOI] [PubMed] [Google Scholar]
- 9.Herring J, editor. In Tachdjian’s pediatric orthopaedics. Vol. 1 Philadelphia: WB Saunders; 2008. [Google Scholar]
- 10.Akbarnia BA. Management themes in early onset scoliosis. J Bone Joint Surg Am. 2007;89 (Suppl 1):42–54. doi: 10.2106/JBJS.F.01256. [DOI] [PubMed] [Google Scholar]
- 11.Wynne-Davies R. Infantile Idiopathic Scoliosis: Causative Factors, Particularly in the First Six Months of Life. J Bone Joint Surg Br. 1975;57-B:138–41. [PubMed] [Google Scholar]
- 12.Paria N, Copley LA, Herring JA, et al. The impact of large-scale genomic methods in orthopaedic disorders: insights from genome-wide association studies. J Bone J Surg. 2014;96(5):e38, 1–10. doi: 10.2106/JBJS.M.00398. [DOI] [PubMed] [Google Scholar]
- 13.Slavotinek AM. Novel microdeletion syndromes detected by chromosome microarrays. J Hum Genet. 2008;124:1–17. doi: 10.1007/s00439-008-0513-9. [DOI] [PubMed] [Google Scholar]
- 14.Jiang Y, Tsai TF, Bressler J, Beaudet AL. Imprinting in Angelman and Prader-Willi syndromes. Curr Opin Genet Dev. 1998;8(3):334–42. doi: 10.1016/s0959-437x(98)80091-9. [DOI] [PubMed] [Google Scholar]
- 15.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, Smolej-Narancic N, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–72. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cushman LJ, Torres-Martinez W, Cherry AM, et al. A report of three patients with an interstitial deletion of chromosome 15q24. Am J Med Genet A. 2005;137:65–71. doi: 10.1002/ajmg.a.30836. [DOI] [PubMed] [Google Scholar]
- 18.Temple IK, Cockwell A, Hassold T, et al. Maternal uniparental disomy for chromosome 14. J Med Genet. 1991;28:511–14. doi: 10.1136/jmg.28.8.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Temple IK, Shrubb V, Lever M, et al. Isolated imprinting mutation of the DLK1/GTL2 locus associated with a clinical presentation of maternal uniparental disomy of chromosome 14. J Med Genet. 2007;44:637–40. doi: 10.1136/jmg.2007.050807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kagami M, Sekita Y, Nishimura G, et al. Deletions and epimutations affecting the human 14q32. 2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet. 2008;40:237–42. doi: 10.1038/ng.2007.56. [DOI] [PubMed] [Google Scholar]
- 21.Hosoki K, Kagami M, Tanaka T, et al. Maternal uniparental disomy 14 syndrome demonstrates Prader-Willi Syndrome-Like Phenotype. J Pediatr. 2009;155:900–3. doi: 10.1016/j.jpeds.2009.06.045. [DOI] [PubMed] [Google Scholar]
- 22.Jen JC, Chan WM, Bosley TM, et al. Mutations in a human ROBO gene disrupt hindbrain axon pathway crossing and morphogenesis. Science. 2004;304:1509–13. doi: 10.1126/science.1096437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharp AJ, Selzer RR, Veltman JA, et al. (2007) Characterization of a recurrent 15q24 microdeletion syndrome. Hum Mol Genet. 2007;16:567–72. doi: 10.1093/hmg/ddm016. [DOI] [PubMed] [Google Scholar]
- 24.Klopocki E, Graul-Neumann LM, Grieben U, et al. A further case of the recurrent 15q24 microdeletion syndrome, detected by array CGH. Eur J Pediatr. 2008;167:903–8. doi: 10.1007/s00431-007-0616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Hattab AW, Smolarek TA, Walker ME, et al. Redefined genomic architecture in 15q24 directed by patient deletion/duplication breakpoint mapping. Hum Genet. 2009;126:589–602. doi: 10.1007/s00439-009-0706-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masurel-Paulet A, Callier P, Thauvin-Robinet C, et al. Multiple cysts of the corpus callosum and psychomotor delay in a patient with a 3.1 Mb 15q24.1q24. 2 interstitial deletion identified by array-CGH. Am J Med Genet A. 2009;149A:1504–10. doi: 10.1002/ajmg.a.32904. [DOI] [PubMed] [Google Scholar]
- 27.Van Esch H, Backx L, Pijkels E, et al. Congenital diaphragmatic hernia is part of the new 15q24 microdeletion syndrome. Eur J Med Genet. 2009;52:153–6. doi: 10.1016/j.ejmg.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Andrieux J, Dubourg C, Rio M, et al. Genotype-phenotype correlation in four 15q24 deleted patients identified by array-CGH. Am J Med Genet A. 2009;149A:2813–9. doi: 10.1002/ajmg.a.33097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma S, Gao X, Londono D, et al. Genome-wide association studies of adolescent idiopathic scoliosis suggest candidate susceptibility genes. Hum Mol Genet. 2011;20:1456–66. doi: 10.1093/hmg/ddq571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi Y, Kou I, Takahashi A, et al. A genome-wide association study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat Genet. 2011;43:1237–40. doi: 10.1038/ng.974. [DOI] [PubMed] [Google Scholar]
- 31.Ogata T, Kagami M, Ferguson-Smith AC. Molecular mechanisms regulating phenotypic outcome in paternal and maternal uniparental disomy for chromosome 14. Epigenetics. 2008;3:181–7. doi: 10.4161/epi.3.4.6550. [DOI] [PubMed] [Google Scholar]
- 32.Bagri A, Marin O, Plump AS, et al. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 2002;33:233–48. doi: 10.1016/s0896-6273(02)00561-5. [DOI] [PubMed] [Google Scholar]
- 33.Marillat V, Sabatier C, Failli V, et al. The slit receptor Rig-1/Robo3 controls midline crossing by hindbrain precerebellar neurons and axons. Neuron. 2004;43:69–79. doi: 10.1016/j.neuron.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 34.Shashi V, McConkie-Rosell A, Rosell B, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med. 2014;16(2):176–82. doi: 10.1038/gim.2013.99. [DOI] [PubMed] [Google Scholar]